

Significance
Moxifloxacin and other fluoroquinolone antibacterial agents are important antituberculosis therapeutic agents. Fluoroquinolones kill Mycobacterium tuberculosis, the causative agent of tuberculosis, by increasing levels of DNA breaks generated by gyrase, an essential type II topoisomerase that regulates DNA topology. As fluoroquinolone use in antituberculosis regimens is becoming more pronounced, understanding the basis of drug–gyrase interactions and resistance is becoming more important. By using a mechanism-based chemical biology approach, our work identified critical drug features that mediate fluoroquinolone interactions with M. tuberculosis gyrase and determined the biochemical basis for fluoroquinolone resistance caused by the most common clinical mutations in gyrase. These findings allowed us to identify a moxifloxacin derivative that displays enhanced activity against WT gyrase and maintains high activity against clinically relevant resistant enzymes.
Keywords: Mycobacterium tuberculosis, fluoroquinolones, antibiotic resistance, gyrase, complex stability
Abstract
Mycobacterium tuberculosis is a significant source of global morbidity and mortality. Moxifloxacin and other fluoroquinolones are important therapeutic agents for the treatment of tuberculosis, particularly multidrug-resistant infections. To guide the development of new quinolone-based agents, it is critical to understand the basis of drug action against M. tuberculosis gyrase and how mutations in the enzyme cause resistance. Therefore, we characterized interactions of fluoroquinolones and related drugs with WT gyrase and enzymes carrying mutations at GyrAA90 and GyrAD94. M. tuberculosis gyrase lacks a conserved serine that anchors a water–metal ion bridge that is critical for quinolone interactions with other bacterial type II topoisomerases. Despite the fact that the serine is replaced by an alanine (i.e., GyrAA90) in M. tuberculosis gyrase, the bridge still forms and plays a functional role in mediating quinolone–gyrase interactions. Clinically relevant mutations at GyrAA90 and GyrAD94 cause quinolone resistance by disrupting the bridge–enzyme interaction, thereby decreasing drug affinity. Fluoroquinolone activity against WT and resistant enzymes is enhanced by the introduction of specific groups at the C7 and C8 positions. By dissecting fluoroquinolone–enzyme interactions, we determined that an 8-methyl-moxifloxacin derivative induces high levels of stable cleavage complexes with WT gyrase and two common resistant enzymes, GyrAA90V and GyrAD94G. 8-Methyl-moxifloxacin was more potent than moxifloxacin against WT M. tuberculosis gyrase and displayed higher activity against the mutant enzymes than moxifloxacin did against WT gyrase. This chemical biology approach to defining drug–enzyme interactions has the potential to identify novel drugs with improved activity against tuberculosis.
Tuberculosis is a major cause of morbidity and mortality on a global scale and is second only to HIV/AIDS as the most prolific killer among single infectious agents (1). According to the World Health Organization, 9 million people were diagnosed with tuberculosis in 2013, and 1.5 million died from the disease (1). The standard treatment regimen for tuberculosis is a 6-month course that includes a combination of rifampin, isoniazid, pyrazinamide, and ethambutol (2, 3). However, fluoroquinolones are becoming more important in the treatment of tuberculosis and are routinely used in multidrug-resistant cases and in patients who are intolerant of first-line therapy (2, 4, 5). Furthermore, moxifloxacin (a newer-generation fluoroquinolone) has shown promising results as a potential first-line agent as part of the PaMZ regimen (PA-824, moxifloxacin, and pyrazinamide), which currently is in clinical trials (6).
Fluoroquinolones are broad-spectrum antibacterial agents that act by increasing levels of DNA strand breaks generated by type II topoisomerases (7–12). Most bacterial species encode two type II enzymes, gyrase and topoisomerase IV (8, 10, 12–14). In these species, gyrase regulates the superhelical density of the bacterial chromosome and removes torsional stress that is generated ahead of DNA replication forks and transcription complexes, and topoisomerase IV primarily unknots and untangles DNA (11, 15, 16). Mycobacterium tuberculosis, the causative agent of tuberculosis, is unusual in that it encodes only gyrase (17). As a result, this enzyme displays functional properties of both type II topoisomerases (18).
Recent structural (19) and functional (20, 21) studies with topoisomerase IV indicate that quinolones interact with bacterial type II enzymes primarily through a water–metal ion bridge. This bridge is formed by a divalent metal ion that is chelated by the C3/C4 keto acid of the drug and stabilized by four water molecules (19). Two of these water molecules are coordinated by a conserved serine and acidic residue (located four positions downstream) in the A subunit of the enzyme. Substitutions in the residues that anchor the bridge are the most prevalent cause of quinolone resistance (10–13, 22–27). In most species, the serine is mutated far more often than the acidic residue.
In contrast to most bacterial species, M. tuberculosis gyrase contains an alanine (A90) in place of the conserved serine. This situation raises the issue of whether the water–metal ion bridge can be formed or plays a role in mediating quinolone activity in this species. However, the fact that mutations at A90 and the acidic residue (D94) are associated with clinical quinolone resistance in M. tuberculosis (28) suggests that the bridge contributes to quinolone function.
Fluoroquinolones are commonly prescribed for community-acquired pneumonia that is later diagnosed as pulmonary tuberculosis (29). This prior treatment is associated with an increased incidence of fluoroquinolone-resistant disease (30, 31). As the use of fluoroquinolones in treating tuberculosis is becoming more pronounced, understanding the basis of drug–gyrase interactions and resistance is becoming more important. Therefore, we analyzed the interactions of fluoroquinolones and related compounds with WT and resistant mutant M. tuberculosis gyrase. Results indicate that the water–metal ion bridge is partially functional in WT gyrase and that the most common resistance mutations cause a decrease in bridge-mediated drug affinity for the enzyme. In contrast to other species (32, 33), quinolone interactions within the gyrase-cleaved DNA complex depend more heavily on substituents at C7 and C8. Based on an analysis of structure–activity relationships at these two positions, we identified fluoroquinolones that display significantly improved activity against WT and resistant M. tuberculosis gyrase compared with moxifloxacin.
Results and Discussion
Gyrase is a heterotetramer comprised of two subunits, GyrA (which contains the active site tyrosine that cleaves and ligates DNA) and GyrB (which contains the ATPase and metal-binding domains) (8, 10, 12–14). To characterize interactions between quinolones and M. tuberculosis gyrase, we used WT enzyme and GyrAA90S, GyrAA90V, GyrAD94G, and GyrAD94H. Amino acid residues A90 and D94 occur at the positions that, by sequence homology, are predicted to anchor the water–metal ion bridge if it is used to mediate quinolone–enzyme interactions in this species (19–21). The GyrAA90V, GyrAD94G, and GyrAD94H proteins contain three of the most common mutations associated with quinolone resistance in clinical isolates of M. tuberculosis (28). The GyrAA90S protein replaces the alanine found in the WT enzyme with a serine, which is the residue that serves as a bridge anchor in most bacterial type II topoisomerases. The positions of GyrAA90 and GyrAD94 relative to bound quinolones are described in the accompanying paper by Blower et al. (34).
The GyrB subunit of M. tuberculosis gyrase was reannotated based on sequence alignments with 50 other bacterial species (35). It is now believed that the start codon is GTG (rather than ATG), which results in a protein that is 675 aa in length, rather than 714 aa (28). The experiments described in the present paper used GyrB that reflects this updated annotation.
Enzymatic Activities of WT and Quinolone-Resistant Mutant M. tuberculosis Gyrase.
Before analyzing fluoroquinolone action against the WT and GyrAA90S, GyrAA90V, GyrAD94G, and GyrAD94H mutant M. tuberculosis gyrase proteins, we determined baseline DNA supercoiling and cleavage activities for the enzymes in the absence of drugs (Fig. 1). Each mutant enzyme maintained high DNA supercoiling activity and, in the presence of Ca2+ (used to increase baseline levels of enzyme-mediated DNA scission), cleaved DNA at least as well as WT gyrase. Thus, quinolone resistance does not correlate with a loss of baseline activity for any of the mutant enzymes examined.
Fig. 1.

Catalytic and DNA cleavage activities of WT and mutant M. tuberculosis gyrase in the absence of drugs. (Left) The ability of WT (black), GyrAA90S (A90S, blue), GyrAA90V (A90V, red), GyrAD94H (D94H, green), and GyrAD94G (D94G, yellow) to supercoil relaxed pBR322 plasmid DNA is shown. Gels are representative of three independent experiments. The positions of negatively supercoiled [(-)SC] and relaxed (Rel) DNA controls are indicated. (Right) The ability of the enzymes to cleave negatively supercoiled pBR322 plasmid DNA is shown. Error bars represent the SD of at least three independent experiments.
Effects of Fluoroquinolones and Related Compounds on DNA Cleavage Mediated by WT and Fluoroquinolone-Resistant Mutant M. tuberculosis Gyrase.
The first set of experiments compared the ability of ciprofloxacin and moxifloxacin to induce DNA cleavage by WT gyrase and the mutant enzymes (Fig. 2, Top). The two drugs generated similar levels of cleavage with the WT enzyme. In contrast, moxifloxacin maintained higher activity against the GyrAA90V, GyrAD94G, and GyrAD94H mutant proteins. As shown previously, the D94G and D94H mutations caused a higher level of fluoroquinolone resistance than the A90V mutation (36, 37).
Fig. 2.

Drug-induced DNA cleavage mediated by WT and mutant M. tuberculosis gyrase. The ability of WT (black), GyrAA90S (A90S, blue), GyrAA90V (A90V, red), GyrAD94H (D94H, green), and GyrAD94G (D94G, yellow) to mediate DNA cleavage in the presence of the clinically used fluoroquinolones ciprofloxacin and moxifloxacin (Top Left and Top Right, respectively), the experimental quinazolinediones 8-methyl-3′-(AM)P-dione and 8-H-3′-(AM)P-dione (Middle Left and Bottom Left, respectively), and the experimental fluoroquinolones 8-methyl-3′-(AM)P-FQ and 8-H-3′-(AM)P-FQ (Middle Right and Bottom Right, respectively) is shown. Compound structures are shown in or above their respective panels. Error bars represent the SD of at least three independent experiments.
The aforementioned findings are consistent with the hypothesis that fluoroquinolones interact with WT M. tuberculosis gyrase through a water–metal ion bridge that is anchored primarily by D94. To investigate this possibility, the activity of ciprofloxacin and moxifloxacin against GyrAA90S was examined. The inclusion of the serine residue in place of the alanine provides the potential to reconstitute a fully functional bridge. As seen in Fig. 2 (Top), both fluoroquinolones displayed dramatically higher activity against GyrAA90S than they did against WT gyrase: drug potency (i.e., drug concentration required to induce 50% maximal DNA cleavage) was approximately fivefold higher, and levels of cleavage at 100 µM were approximately twofold higher. Collectively, these findings suggest that there is sufficient conservation of structure such that M. tuberculosis gyrase is capable of using a water–metal ion bridge to mediate fluoroquinolone–enzyme interactions. This conclusion is supported by the structural studies in the accompanying paper by Blower et al. (34).
Quinazolinediones are fluoroquinolone-like compounds that lack the C3/C4 keto acid required to chelate metal ions. In several other species, quinazolinediones containing a 3′-(aminomethyl)pyrrolidinyl [3′-(AM)P] (or a related) group at C7 maintain activity against enzymes containing mutations that disrupt the water–metal ion bridge by forming binding interactions primarily through the C7 substituent (20, 21, 32, 38–42). As seen in Fig. 2 (Middle Left), the activity of 8-methyl-3′-(AM)P-dione is unaffected by mutations that could disrupt or strengthen the water–metal ion bridge in M. tuberculosis gyrase. The activity of 8-methyl-3′-(AM)P-FQ (the fluoroquinolone version of this quinazolinedione) is unaffected by bridge-disrupting mutations but is enhanced upon the introduction of the serine residue at position 90 (Fig. 2, Middle Right). Again, these findings support the hypothesis that clinically relevant fluoroquinolones interact with M. tuberculosis gyrase through the water–metal ion bridge and that D94 likely mediates a partial bridging interaction in the absence of the upstream serine.
Fluoroquinolones that have a C8 substituent generally display better activity against clinical tuberculosis isolates than do derivatives with a C8-H (43, 44). Therefore, 3′-(AM)P-quinazolinedione and -fluoroquinolone derivatives that contained a C8-H were used to examine the influence of the C8 substituent on drug activity (Fig. 2, Bottom). The activity of 8-H-3′-(AM)P-dione was markedly decreased against all of the enzymes, confirming that the C8 methyl group plays an important role in mediating the activity of these compounds. The activity of 8-H-3′-(AM)P-FQ also decreased with each enzyme compared with the C8-methyl version of this fluoroquinolone. The relative activity of 8-H-3′-(AM)P-FQ correlated with the capacity of each enzyme to anchor the water–metal ion bridge.
Taken together, these findings strongly suggest that WT M. tuberculosis gyrase partially supports the water–metal ion bridge and that the bridge and the C7 and C8 substituents play important roles in supporting quinolone activity. Therefore, the contributions of these three drug features to quinolone interactions and activity were analyzed in greater detail.
Role of the Water–Metal Ion Bridge in Facilitating Quinolone Activity Against M. tuberculosis Gyrase.
To determine whether quinolone interactions with M. tuberculosis gyrase are mediated by the water–metal ion bridge, Mg2+ titrations were carried out to assess the effects of A90 mutations on the metal ion concentration required to support DNA cleavage induced by ciprofloxacin (Fig. 3, Left). The amino acid at this position plays a critical role in anchoring the water–metal ion bridge in other bacterial type II enzymes (20, 21, 33). Results with WT gyrase were compared with those of mutant enzymes containing a serine or a valine in place of the alanine at position 90. The GyrAA90S mutant enzyme, which contains the conserved serine residue found in most other bacterial type II enzymes and greatly enhances fluoroquinolone activity (see Fig. 2), required a lower concentration of Mg2+ to achieve half-maximal and maximal levels of quinolone-induced DNA cleavage. In marked contrast, the GyrAA90V mutant enzyme, which causes quinolone resistance (see Fig. 2), required a higher concentration of Mg2+ to achieve half-maximal and maximal levels of quinolone-induced DNA cleavage.
Fig. 3.

Effects of Mg2+ concentration on DNA cleavage mediated by WT and mutant M. tuberculosis gyrase. Results are shown for 10 µM ciprofloxacin (Left) and 10 µM 8-methyl-3′-(AM)P-dione (Right) with WT (black), GyrAA90S (A90S, blue), and GyrAA90V (A90V, red). DNA cleavage for each drug–enzyme pair was normalized to 100% at 6 mM Mg2+ to facilitate direct comparisons. Error bars represent the SD of at least three independent experiments.
These results demonstrate that the amino acid at position 90 affects the affinity of the divalent metal ion that is chelated by the quinolone and indicate that the water–metal ion bridge is a point of contact between quinolones and M. tuberculosis gyrase. It is likely that the alanine at position 90 in WT gyrase, although it does not serve as a bridge anchor, still allows D94 to anchor the bridge. However, because M. tuberculosis gyrase has only one available amino acid to anchor the bridge, fluoroquinolone interactions are weaker than seen with species that encode two amino acid bridge anchors (20, 21, 33). Substitution of A90 with the larger valine may cause quinolone resistance by interfering with the ability of water molecules to coordinate properly with the Mg2+ or by impeding D94 interactions with the bridge.
As a control for the aforementioned experiment, the effects of A90 mutations on the Mg2+ requirement for DNA cleavage induced by 8-methyl-3′-(AM)P-dione (which does not require a metal ion for enzyme interactions) were determined. In contrast to results with ciprofloxacin, similar levels of Mg2+ were required to achieve half-maximal and maximal cleavage with all three enzymes (Fig. 3, Right).
In other bacterial type II topoisomerases, mutations in bridge-anchoring residues that partially disrupt the water–metal ion bridge often restrict the variety of metal ions that can be used to form the bridge (21, 33). Therefore, we examined the ability of a second divalent metal ion, Mn2+, to support ciprofloxacin activity against WT gyrase and enzymes with mutations at position 90 (Fig. 4). With GyrAA90S, which has a fully intact bridge, the level of ciprofloxacin-induced DNA cleavage was unaffected by the substitution of this metal ion for Mg2+. However, with WT gyrase, which has a partially disrupted bridge, ciprofloxacin activity was decreased approximately twofold. Furthermore, with GyrAA90V, which has a more severely disrupted bridge, the quinolone displayed no ability to enhance enzyme-mediated DNA cleavage in the presence of Mn2+. Therefore, relative levels of resistance seen with enzymes containing mutations in bridge-anchoring residues can be altered by the metal ion used to support this interaction. A stronger water–metal ion bridge allows more latitude with metal ion requirements.
Fig. 4.
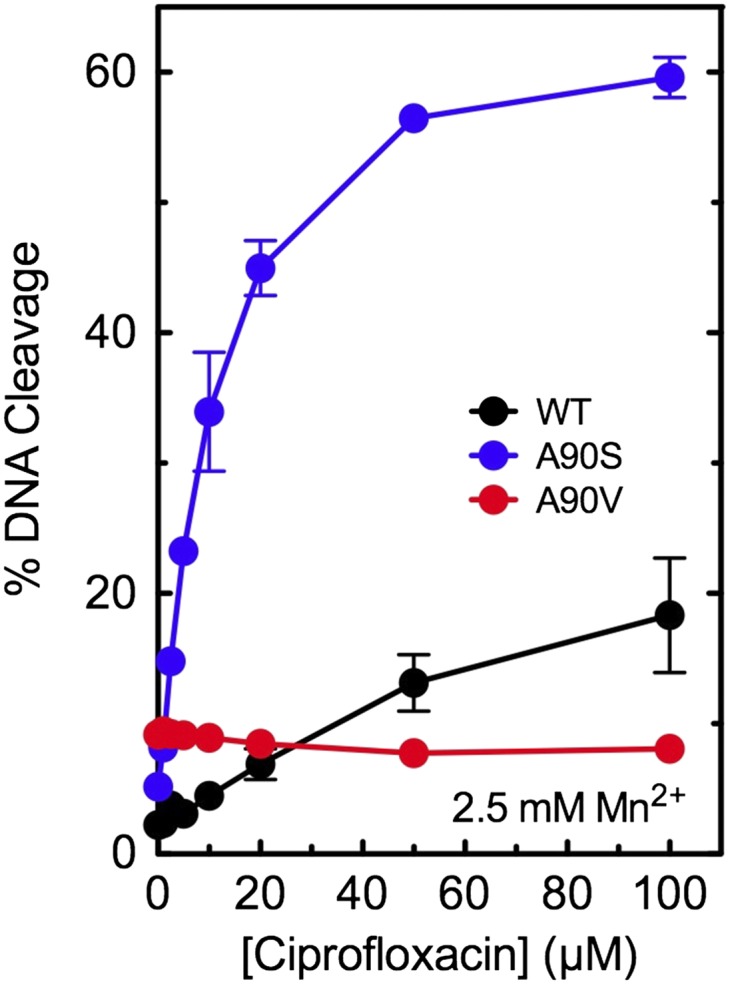
Effects of Mn2+ on drug-induced DNA cleavage mediated by WT and mutant M. tuberculosis gyrase. Results are shown for cleavage mediated by WT (black), GyrAA90S (A90S, blue), and GyrAA90V (A90V, red) in the presence of ciprofloxacin. Assays included 2.5 mM Mn2+, the concentration that yielded maximal enzyme activity, instead of Mg2+. Error bars represent the SD of at least three independent experiments.
The water–metal ion bridge has been shown to play critical roles in mediating fluoroquinolone binding and positioning, but its predominant role appears to differ across species. With Bacillus anthracis topoisomerase IV, it primarily acts as a binding contact (20, 21). However, with Escherichia coli topoisomerase IV, it has little effect on drug affinity and is believed to properly position the drug in a manner that promotes DNA cleavage (33). Therefore, to determine whether the water–metal ion bridge plays a major role in drug–enzyme binding in M. tuberculosis gyrase, we used a competition approach and examined the effects of ciprofloxacin on DNA cleavage induced by 10 µM 8-methyl-3′-(AM)P-dione in two enzymes with impaired bridge function (GyrAA90V and GyrAD94G). In an enzyme with optimal bridge function (GyrAA90S), the fluoroquinolone and quinazolinedione display similar abilities to induce gyrase-mediated DNA cleavage and act with comparable potencies (see Fig. 2, Left, Top, and Middle).
With both resistant mutant enzymes (GyrAA90V and GyrAD94G), ciprofloxacin competed poorly against the bridge-independent quinazolinedione (Fig. 5). Even at a 10-fold molar excess of the quinolone, levels of DNA cleavage decreased by only ∼25%. This finding implies that impaired bridge function decreases the affinity of ciprofloxacin for the enzyme by at least an order of magnitude and strongly suggests that, in M. tuberculosis gyrase, the water–metal ion bridge plays a critical role in mediating quinolone binding.
Fig. 5.
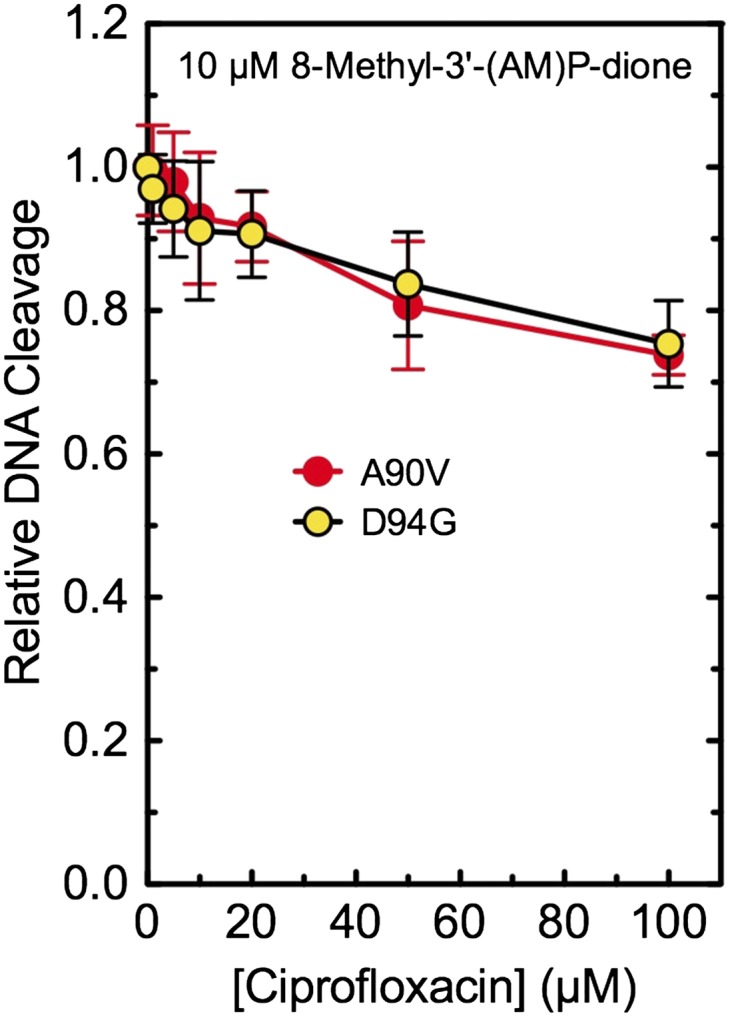
Ability of ciprofloxacin to compete out DNA cleavage induced by 10 µM 8-methyl-3′-(AM)P-dione with mutant M. tuberculosis gyrase. Results are shown for GyrAA90V (A90V, red) and GyrAD94G (D94G, yellow). Both drugs were added to reaction mixtures simultaneously. The low level of cleavage seen in the presence of ciprofloxacin alone was used as a baseline and was subtracted from DNA scission observed in the presence of both drugs. The level of DNA cleavage observed in the presence of the quinazolinedione alone was set to 1.0 to facilitate direct comparisons. Error bars represent the SD of at least three independent experiments.
Enhancing Fluoroquinolone Activity Against WT and Resistant M. tuberculosis Gyrase by Modifying Substituents at C7 and C8.
Previous studies (43, 44) have shown that fluoroquinolones containing a substituent at C8 (in particular, a methoxy group) display higher activity against M. tuberculosis and related organisms. However, the basis for the effects of C8 substitutions on fluoroquinolone activity against M. tuberculosis gyrase is not well understood. Therefore, we compared the activities of fluoroquinolones containing a C8-H, -methyl, or -methoxy group against the WT and resistant enzymes. Two parallel series were examined: one that contained the C7 piperazinyl ring of ciprofloxacin, and one that contained the C7 diazabicyclononyl ring of moxifloxacin.
The first set of experiments examined drug effects against WT gyrase. In general, the inclusion of a C8 substituent enhanced quinolone activity (Fig. 6, Top Left). For the ciprofloxacin-based series, the compound with a C8 methoxy group generated the highest level of DNA cleavage activity. Compared with ciprofloxacin, 8-methoxy-ciprofloxacin (8-methoxy-cipro) induced ∼50% more cleavage at 100 µM drug and displayed a potency that was ∼twofold higher (based on the drug concentration required to induce 50% of the cleavage generated at 100 µM drug). The presence of a C8 methyl group enhanced DNA cleavage, but to a lesser extent than did the methoxy group.
Fig. 6.

Effects of ciprofloxacin- and moxifloxacin-based fluoroquinolones on DNA cleavage mediated by WT and mutant M. tuberculosis gyrase and human topoisomerase IIα. The ability of WT (Top Left), GyrAA90V (A90V, Top Right), and GyrAD94G (D94G, Bottom Left) gyrase and human topoisomerase IIα (hTIIα, Bottom Right) to cleave DNA in the presence of fluoroquinolones containing a C8-H (black), -methyl (blue), or -methoxy (red) group and a C7 piperazinyl (closed symbols) or diazabicyclononyl (open symbols) group is shown. For human topoisomerase IIα, DNA cleavage levels induced by the enzyme in the presence of CP-115,955 or etoposide (dashed lines) are shown for comparison. These latter data are from Aldred et al. (32). The ciprofloxacin and moxifloxacin fluoroquinolone cores are shown at the top. Error bars represent the SD of at least three independent experiments.
A more dramatic effect was seen when the C8 methoxy group of moxifloxacin was replaced with a methyl group. Compared with moxifloxacin (the fluoroquinolone most commonly used to treat tuberculosis) (2, 4–6), 8-methyl-moxifloxacin (8-methyl-moxi) induced approximately twofold more cleavage at 100 µM drug and was approximately twofold more potent. Furthermore, only 10 µM 8-methyl-moxi was required to induce the same level of scission (∼30%) as was seen with 100 µM moxifloxacin. In contrast, 8-H-moxifloxacin (8-H-moxi) displayed an activity similar to that of moxifloxacin.
The second set of experiments examined the effects of the C8 substituent on fluoroquinolone activity against the resistant GyrAA90V and GyrAD94G mutant enzymes (Fig. 6, Top Right and Bottom Left, respectively). When the function of the water–metal ion bridge was disrupted, the C8 substituent had a much greater influence on drug activity. Drugs lacking a C8 substituent (ciprofloxacin and 8-H-moxi) showed little activity against either fluoroquinolone-resistant enzyme. However, in the presence of a C8 methyl or methoxy substituent, high levels of activity were maintained. To this point, 8-methyl-moxi displayed nearly WT activity against GyrAA90V. Moreover, even though its activity against GyrAD94G was decreased, 8-methyl-moxi still induced higher levels of cleavage than did moxifloxacin with WT gyrase. Representative gels showing the effects of moxifloxacin and 8-methyl-moxi on DNA cleavage mediated by the mutant GyrAD94G enzyme are shown in Fig. S1.
Fig. S1.

Effects of moxifloxacin (Top) and 8-methyl-moxi (Bottom) on DNA cleavage mediated by WT (Left) and GyrAD94G mutant gyrase (Right). The positions of nicked (single-stranded breaks), linear (double-stranded breaks), and the negatively supercoiled (SC) substrate are indicated. Ethidium bromide-stained agarose gels are shown and are representative of three independent experiments.
Some fluoroquinolones and related compounds that display high activity against resistant bacterial type II topoisomerases cross-react with human topoisomerase IIα, excluding them from clinical development as antibacterial drugs. For example, CP-115,955 induces higher levels of cleavage with the human enzyme than does etoposide, a commonly prescribed anticancer drug (45, 46) (Fig. 6, Bottom Right). Notably, none of the ciprofloxacin- or moxifloxacin-based compounds used in this study displayed significant activity against human topoisomerase IIα.
To further understand the contributions of the C7 and C8 substituents to quinolone activity, we determined the abilities of the two least effective compounds (ciprofloxacin and 8-H-moxi) to compete with the two most effective drugs (8-methoxy-cipro and 8-methyl-moxi; Fig. 7, Left and Right, respectively). The GyrAA90V and GyrAD94G mutant enzymes were used so that the contributions of the C7 and C8 groups would not be obscured by the water–metal ion bridge. A representative gel showing the effects of ciprofloxacin and 8-H-moxi on DNA cleavage mediated by the GyrAD94G mutant gyrase in the presence of 50 µM 8-methoxy-cipro is shown in Fig. S2. Three observations were noted. First, the concentrations of ciprofloxacin and 8-H-moxi required to compete out 50% of the DNA cleavage induced by 50 µM 8-methyl-cipro or 8-methyl-moxi were 100 µM or higher. This finding (which is consistent with the potency data discussed for Fig. 6) indicates that the C8-H derivatives display a decreased affinity for the enzymes compared with 8-methoxy-cipro and 8-methyl-moxi. Thus, a C8 group appears to contribute to drug binding within the enzyme–DNA cleavage complex. Second, compounds bearing a C7 diazabicyclononyl group (as found in moxifloxacin) competed better than those with a C7 piperazinyl substituent (as found in ciprofloxacin), suggesting that moxifloxacin-based, as opposed to ciprofloxacin-based, analogs interact more tightly with M. tuberculosis gyrase. Third, each compound competed less effectively against 8-methyl-moxi than against 8-methoxy-cipro, indicating that 8-methyl-moxi interacts more tightly with the enzyme. This finding is consistent with the observation that 8-methyl-moxi induced higher levels of DNA cleavage with all enzymes tested than did the other fluoroquinolones in these two series (see Fig. 6).
Fig. 7.

Ability of fluoroquinolones with a C8-H to compete out DNA cleavage induced by 50 µM 8-methoxy-cipro or 8-methyl-moxi with mutant M. tuberculosis gyrase. Results are shown for GyrAA90V (A90V, black) and GyrAD94G (D94G, blue). Ciprofloxacin (Cipro, closed symbols) or 8-H-moxi (open symbols) and 8-methoxy-cipro (Left) or 8-methyl-moxi (Right) were added to reaction mixtures simultaneously. The low level of cleavage seen in the presence of the C8-H fluoroquinolones alone was used as a baseline and was subtracted from the DNA scission observed in the presence of both drugs. The level of DNA cleavage observed in the presence of 8-methoxy-cipro or 8-methyl-moxi alone was set to 1.0 to facilitate direct comparisons. Error bars represent the SD of at least three independent experiments.
Fig. S2.

Ability of 50 or 100 µM ciprofloxacin (Cipro) or 50 or 100 µM 8-H-moxi to compete out DNA cleavage induced by 50 µM 8-methoxy-cipro with mutant GyrAD94G Mycobacterium tuberculosis gyrase. Cipro or 8-H-moxi and 8-methoxy-cipro were added to reaction mixtures simultaneously. The low level of cleavage seen in the presence of ciprofloxacin or 8-H-moxi alone is shown at right. Ethidium bromide-stained agarose gels are shown and are representative of three independent experiments.
Finally, to further examine the contributions of the C7 and C8 substituents to quinolone action, we determined the persistence of cleavage complexes formed in the presence of ciprofloxacin, moxifloxacin, 8-methoxy-cipro, and 8-methyl-moxi (Fig. 8). Moxifloxacin induced more stable cleavage complexes than ciprofloxacin with all three enzymes. This finding is consistent with binding studies (47) suggesting that moxifloxacin binds more tightly than ciprofloxacin to M. tuberculosis gyrase. Furthermore, consistent with the competition experiments, compounds containing the C7 diazabicyclononyl group of moxifloxacin induced cleavage complexes that were more stable than those formed with the C7 piperazinyl group of ciprofloxacin with the WT and resistant enzymes. Finally, in all cases, 8-methyl-moxi induced the most stable cleavage complexes. Again, this finding correlates with the high activity of 8-methyl-moxi.
Fig. 8.

Effects of fluoroquinolones on the persistence of cleavage complexes formed by WT and mutant M. tuberculosis gyrase. The stability of ternary enzyme–drug–DNA cleavage complexes formed with WT (Left), GyrAA90V (A90V, Middle), and GyrAD94G (D94G, Right) was determined in the presence of 100 µM ciprofloxacin (Cipro, black), 50 µM moxifloxacin (Moxi, blue), 50 µM 8-methoxy-cipro (red), or 10 µM 8-methyl-moxi (yellow). The data table (Inset, Middle) lists the t1/2 of DNA cleavage complexes formed with each drug–enzyme combination. Initial DNA cleavage-religation reactions were allowed to come to equilibrium and were then diluted 20-fold with reaction buffer. Levels of DNA cleavage at time 0 were set to 100%. Error bars represent the SD of at least three independent experiments.
Summary
As fluoroquinolones become more important for the treatment of tuberculosis and resistance becomes more prevalent, there is a critical need to understand the binding interactions and mechanistic details of quinolone-based agents with M. tuberculosis gyrase to develop new agents with high activity against WT and resistant disease. Thus, we analyzed interactions between quinolone-based agents and WT M. tuberculosis gyrase and resistant mutants. The interaction of current fluoroquinolones with gyrase is governed predominately by a critical water–metal ion bridge. However, in contrast to most bacterial type II enzymes, M. tuberculosis gyrase uses only one amino acid to anchor the bridge. The most common resistance-conferring mutations in gyrase disrupt the bridge interaction between fluoroquinolones and the enzyme. When bridge function is compromised, resistance can be overcome, at least in part, by the introduction of specific groups at the C7 and C8 positions. By dissecting fluoroquinolone–enzyme interactions, we determined that an 8-methyl derivative of moxifloxacin induces high levels of stable cleavage complexes with WT and common fluoroquinolone-resistant mutant enzymes. This chemical biology approach to understanding the basis for fluoroquinolone–enzyme interactions has the potential to guide the future development of novel agents with improved activity against tuberculosis.
Methods
Materials and Enzymes.
Ciprofloxacin and moxifloxacin were obtained from LKT Laboratories. 8-Methyl-cipro, 8-methoxy-cipro, 8-H-moxi, 8-methyl-moxi, 8-H-3′-(AM)P-FQ, 8-H-3′-(AM)P-dione, 8-methyl-3′-(AM)P-FQ, and 8-methyl-3′-(AM)P-dione were synthesized as reported previously (32). Ciprofloxacin-based compounds were stored at −20 °C as 40 mM stock solutions in 0.1 N NaOH and diluted fivefold with 10 mM Tris⋅HCl (pH 7.9) immediately before use. All other compounds were stored at 4 °C as 20 mM stock solutions in 100% DMSO. Table S1 contains the full chemical, library, and abbreviated names for the compounds used in this study. Other chemicals were of analytical reagent grade.
Table S1.
Full chemical, library, and abbreviated names of compounds used in this study
| Name used in this study | Library name | Chemical name |
| Ciprofloxacin | — | 1-Cyclopropyl-6-fluoro-1,4-dihydro-7-(1-piperazinyl)-4-oxo-3-quinolinecarboxylic acid |
| 8-Methyl-cipro | UILI-2–89 | 1-Cyclopropyl-6-fluoro-1,4-dihydro-8-methyl-7-(1-piperazinyl)-4-oxo-3-quinolinecarboxylic acid |
| 8-Methoxy-cipro | UIHS-IIa-101 | 1-Cyclopropyl-6-fluoro-1,4-dihydro-8-methyl-7-(1-piperazinyl)-4-oxo-3-quinolinecarboxylic acid |
| Moxifloxacin | — | 1-Cyclopropyl-6-fluoro-1,4-dihydro-8-methoxy-7-[(4aS,7aS)-octahydro-6H-pyrrolo[3,4-b]pyridin-6-yl]-4-oxo-3-quinolinecarboxylic acid |
| 8-Methyl-moxi | UIHS-IIa-45 | 1-Cyclopropyl-6-fluoro-1,4-dihydro-8-methyl-7-[(4aS,7aS)-octahydro-6H-pyrrolo[3,4-b]pyridin-6-yl]-4-oxo-3-quinolinecarboxylic acid |
| 8-H-moxi | UIHS-IIa-239 | 1-Cyclopropyl-6-fluoro-1,4-dihydro-7-[(4aS,7aS)-octahydro-6H-pyrrolo[3,4-b]pyridin-6-yl]-4-oxo-3-quinolinecarboxylic acid |
| CP-115,955 | CP-115,955 | 1-Cyclopropyl-6-fluoro-1,4-dihydro-7-(4-hydroxyphenyl)-4-oxo-3-quinolinecarboxylic acid |
| 8-Methyl-3′-(AM)P-FQ | UIHS-I-303 | 1-Cyclopropyl-6-fluoro-1,4-dihydro-8-methyl-7-[(3S)-3-(aminomethyl)-1-pyrrolidinyl]-4-oxo-3-quinolinecarboxylic acid |
| 8-Methyl-3′-(AM)P-dione | UIJR1-048, Lot UILI-2–95 | 3-Amino-7-[(3S)-3-(aminomethyl)-1-pyrrolidinyl]-1-cyclopropyl-6-fluoro-8-methyl-2,4(1H,3H)-quinazolinedione |
| 8-H-3′-(AM)P-FQ | UIHS-IIIa-35 | 1-Cyclopropyl-6-fluoro-1,4-dihydro-7-[(3S)-3-(aminomethyl)-1-pyrrolidinyl]-4-oxo-3-quinolinecarboxylic acid |
| 8-H-3′-(AM)P-dione | UIHS-IIa-245 | 3-Amino-7-[(3S)-3-(aminomethyl)-1-pyrrolidinyl]-1-cyclopropyl-6-fluoro-2,4(1H,3H)-quinazolinedione |
The coding regions for residues 2–675 of GyrB and 2–838 of GyrA from M. tuberculosis were amplified from genomic DNA (American Type Culture Collection strain H37Rv) and individually cloned into a pET-28b derivative containing an N-terminal, tobacco etch virus (TEV) protease-cleavable hexahistidine tag. Mutant GyrA vectors were generated by QuikChange (Agilent). Proteins were overexpressed in E. coli strain Rosetta 2 pLysS (EMD Millipore) by growing cells at 30 °C until log phase, then inducing with 1 mM isopropyl-β-d-thiogalactopyranoside for 3 h. Cells were harvested by centrifugation and resuspended in buffer A [20 mM Tris⋅HCl (pH 7.9), 500 mM NaCl, 5 mM imidazole, and 10% (vol/vol) glycerol with protease inhibitors] and then frozen dropwise in liquid nitrogen.
Cells were sonicated and centrifuged, and the clarified lysate was passed over a HisTrap HP column (GE Healthcare). His-tagged protein was eluted with buffer B [20 mM Tris⋅HCl (pH 7.9), 500 mM NaCl, 250 mM imidazole, and 10% glycerol with protease inhibitors] and then concentrated and exchanged into buffer A overnight at 4 °C in the presence of His-tagged TEV protease. This mixture was passed over a HisTrap HP column, and the flow-through was collected, concentrated, and run over an S-300 gel filtration column (GE Healthcare) in buffer C [50 mM Tris⋅HCl (pH 7.9), 500 mM KCl, 2 mM β-mercaptoethanol, and 10% (vol/vol) glycerol]. Peak fractions were pooled, concentrated, and diluted into a high-glycerol variant of buffer C so that the final glycerol concentration reached 30% (vol/vol) before storage at −80 °C.
Human topoisomerase IIα was expressed in yeast and purified as described by Kingma et al. (48).
Negatively supercoiled pBR322 plasmid DNA was prepared from E. coli by using a Plasmid Mega Kit (Qiagen) as described by the manufacturer. Relaxed pBR322 plasmid DNA was generated by treatment with topoisomerase I for 30 min as previously described (49), followed by phenol-chloroform-isoamyl alcohol extraction, ethanol precipitation, and resuspension in 5 mM Tris⋅HCl (pH 8.5) and 500 µM EDTA.
DNA Supercoiling.
DNA supercoiling reactions (20 µL) contained 25 nM WT or mutant gyrase and 5 nM relaxed pBR322 in reaction buffer [10 mM Tris⋅HCl (pH 7.5), 40 mM KCl, 0.1 mg/mL BSA, 6 mM MgCl2, 10% (vol/vol) glycerol, and 0.5 mM DTT] supplemented with 1 mM ATP and were incubated at 37 °C. DNA supercoiling was stopped at times from 0 to 45 min by the addition of 3 µL of 0.77% SDS and 77.5 mM EDTA. Samples were mixed with 2 µL of agarose gel loading buffer [60% (wt/vol) sucrose, 10 mM Tris⋅HCl (pH 7.9), 0.5% bromophenol blue, and 0.5% xylene cyanol FF], heated at 45 °C for 5 min, and subjected to electrophoresis in 1% agarose gels in 100 mM Tris⋅borate (pH 8.3) and 2 mM EDTA. Gels were stained with 0.75 µg/mL ethidium bromide for 30 min. DNA bands were visualized with medium-range UV light and quantified by using an Alpha Innotech digital imaging system.
DNA Cleavage.
DNA cleavage reactions were carried out using the procedure of Fortune and Osheroff as adapted by Aldred et al. (20, 50). Reactions with the bacterial enzyme contained 100 nM WT or mutant gyrase and 10 nM negatively supercoiled pBR322 in a total of 20 µL of reaction buffer. Reactions with the human enzyme contained 110 nM human topoisomerase IIα and 10 nM negatively supercoiled pBR322 in a total of 20 µL of 10 mM Tris⋅HCl (pH 7.9), 5 mM MgCl2, 100 mM KCl, 100 µM EDTA, 25 µM DTT, and 2.5% (vol/vol) glycerol. All reaction mixtures were incubated at 37 °C for 10 min, and enzyme–DNA cleavage complexes were trapped by the addition of 2 µL of 5% (wt/vol) SDS followed by 2 µL of 250 mM EDTA (pH 8.0). Proteinase K (2 µL of a 0.8 mg/mL solution) was added, and samples were incubated at 45 °C for 45 min to digest the enzyme. Samples were mixed with 2 µL of agarose gel loading buffer, heated at 45 °C for 5 min, and subjected to electrophoresis in 1% agarose gels in 40 mM Tris⋅acetate (pH 8.3) and 2 mM EDTA containing 0.5 µg/mL ethidium bromide. DNA bands were visualized as described earlier. DNA cleavage was monitored by the conversion of supercoiled plasmid to linear molecules.
Assays that monitored gyrase-mediated DNA cleavage in the absence of drugs substituted 6 mM CaCl2 for 6 mM MgCl2 in the reaction buffer. Assays that assessed DNA cleavage in the presence of drugs contained 0–100 µM compound. In some reactions, the concentration dependence of MgCl2 was examined or the divalent metal ion was replaced with 2.5 mM MnCl2.
For assays that monitored competition between two drugs, the compounds were added simultaneously to reaction mixtures, and the final concentrations of the compounds are indicated. In these competition assays, the level of cleavage seen with the corresponding concentration of the competing drug (in the absence of the drug held at a constant concentration) was used as a baseline and was subtracted from the cleavage level seen in the presence of both compounds.
Persistence of Gyrase–DNA Cleavage Complexes.
The persistence of gyrase–DNA cleavage complexes established in the presence of drugs was determined by using the procedure of Gentry et al. as adapted by Aldred et al. (20, 51). Initial reaction mixtures contained 500 nM WT or mutant gyrase, 50 nM DNA, and 100 µM ciprofloxacin, 50 µM 8-methoxy-cipro, 50 µM moxifloxacin, or 10 µM 8-methyl-moxi in a total of 20 µL of reaction buffer. Quinolone concentrations that yielded similar levels of DNA cleavage were used. Reaction mixtures were incubated at 37 °C for 10 min and then diluted 20-fold with reaction buffer warmed to 37 °C. Samples (20 µL) were removed at times ranging from 0 to 60 min for the mutant enzymes or 0–180 min for the WT enzyme, and DNA cleavage was stopped with 2 µL of 5% (wt/vol) SDS followed by 2 µL of 250 mM EDTA (pH 8.0). Samples were digested with proteinase K and processed as described earlier for DNA cleavage assays. Levels of DNA cleavage were set to 100% at time 0, and the persistence of cleavage complexes was determined by the loss of the linear reaction product over time.
Acknowledgments
We thank H. Schwanz and G. Li for the preparation of compounds and M. Pendleton and R. Ashley for critical reading of the manuscript. This work was supported by United States Veterans Administration Merit Review Award I01 Bx002198 (to N.O.) and National Institutes of Health Research Grants R01 AI87671 (to R.J.K.), R01 CA077373 (to J.M.B.), and R01 GM033944 (to N.O.). K.J.A. was a trainee under National Institutes of Health Grant T32 CA09582 and T.R.B. was supported by a European Molecular Biology Organization Long-Term Fellowship.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1525055113/-/DCSupplemental.
References
- 1.World Health Organization Tuberculosis. Fact Sheet No 104. 2014 www.who.int/mediacentre/factsheets/fs104/en/Accessed July 21, 2014.
- 2.World Health Organization . Treatment of Tuberculosis: Guidelines. 4th Ed. WHO Press; Geneva: 2010. [PubMed] [Google Scholar]
- 3.TB CARE I . International Standards for Tuberculosis Care. 2014. , Edition 3 (TB CARE I, The Hague) [Google Scholar]
- 4.American Thoracic Society CDC Infectious Diseases Society of America Treatment of tuberculosis. MMWR Recomm Rep. 2003;52(RR-11):1–77, and erratum (2005) 53:1203. [PubMed] [Google Scholar]
- 5.World Health Organization . Guidelines for the Programmatic Management of Drug-Resistant Tuberculosis. WHO Press; Geneva: 2011. [PubMed] [Google Scholar]
- 6.TB Alliance Interactive Portfolio PaMZ. 2015 Available at www.tballiance.org/portfolio/regimen/pamz. Accessed November 24, 2015.
- 7.Hooper DC. Clinical applications of quinolones. Biochim Biophys Acta. 1998;1400(1-3):45–61. doi: 10.1016/s0167-4781(98)00127-4. [DOI] [PubMed] [Google Scholar]
- 8.Hooper DC. Mechanisms of action of antimicrobials: Focus on fluoroquinolones. Clin Infect Dis. 2001;32(Suppl 1):S9–S15. doi: 10.1086/319370. [DOI] [PubMed] [Google Scholar]
- 9.Andriole VT. The quinolones: Past, present, and future. Clin Infect Dis. 2005;41(suppl 2):S113–S119. doi: 10.1086/428051. [DOI] [PubMed] [Google Scholar]
- 10.Drlica K, et al. Quinolones: Action and resistance updated. Curr Top Med Chem. 2009;9(11):981–998. doi: 10.2174/156802609789630947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pommier Y, Leo E, Zhang H, Marchand C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem Biol. 2010;17(5):421–433. doi: 10.1016/j.chembiol.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aldred KJ, Kerns RJ, Osheroff N. Mechanism of quinolone action and resistance. Biochemistry. 2014;53(10):1565–1574. doi: 10.1021/bi5000564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anderson VE, Osheroff N. Type II topoisomerases as targets for quinolone antibacterials: Turning Dr. Jekyll into Mr. Hyde. Curr Pharm Des. 2001;7(5):337–353. doi: 10.2174/1381612013398013. [DOI] [PubMed] [Google Scholar]
- 14.Drlica K, Malik M, Kerns RJ, Zhao X. Quinolone-mediated bacterial death. Antimicrob Agents Chemother. 2008;52(2):385–392. doi: 10.1128/AAC.01617-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levine C, Hiasa H, Marians KJ. DNA gyrase and topoisomerase IV: Biochemical activities, physiological roles during chromosome replication, and drug sensitivities. Biochim Biophys Acta. 1998;1400(1-3):29–43. doi: 10.1016/s0167-4781(98)00126-2. [DOI] [PubMed] [Google Scholar]
- 16.Champoux JJ. DNA topoisomerases: Structure, function, and mechanism. Annu Rev Biochem. 2001;70:369–413. doi: 10.1146/annurev.biochem.70.1.369. [DOI] [PubMed] [Google Scholar]
- 17.Cole ST, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393(6685):537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 18.Aubry A, Fisher LM, Jarlier V, Cambau E. First functional characterization of a singly expressed bacterial type II topoisomerase: The enzyme from Mycobacterium tuberculosis. Biochem Biophys Res Commun. 2006;348(1):158–165. doi: 10.1016/j.bbrc.2006.07.017. [DOI] [PubMed] [Google Scholar]
- 19.Wohlkonig A, et al. Structural basis of quinolone inhibition of type IIA topoisomerases and target-mediated resistance. Nat Struct Mol Biol. 2010;17(9):1152–1153. doi: 10.1038/nsmb.1892. [DOI] [PubMed] [Google Scholar]
- 20.Aldred KJ, et al. Drug interactions with Bacillus anthracis topoisomerase IV: Biochemical basis for quinolone action and resistance. Biochemistry. 2012;51(1):370–381. doi: 10.1021/bi2013905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aldred KJ, McPherson SA, Turnbough CL, Jr, Kerns RJ, Osheroff N. Topoisomerase IV-quinolone interactions are mediated through a water-metal ion bridge: Mechanistic basis of quinolone resistance. Nucleic Acids Res. 2013;41(8):4628–4639. doi: 10.1093/nar/gkt124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Drlica K, Zhao X. DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol Mol Biol Rev. 1997;61(3):377–392. doi: 10.1128/mmbr.61.3.377-392.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Z, et al. Alteration in the GyrA subunit of DNA gyrase and the ParC subunit of DNA topoisomerase IV in quinolone-resistant clinical isolates of Staphylococcus epidermidis. Antimicrob Agents Chemother. 1998;42(12):3293–3295. doi: 10.1128/aac.42.12.3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hooper DC. Mode of action of fluoroquinolones. Drugs. 1999;58(suppl 2):6–10. doi: 10.2165/00003495-199958002-00002. [DOI] [PubMed] [Google Scholar]
- 25.Price LB, et al. In vitro selection and characterization of Bacillus anthracis mutants with high-level resistance to ciprofloxacin. Antimicrob Agents Chemother. 2003;47(7):2362–2365. doi: 10.1128/AAC.47.7.2362-2365.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morgan-Linnell SK, Becnel Boyd L, Steffen D, Zechiedrich L. Mechanisms accounting for fluoroquinolone resistance in Escherichia coli clinical isolates. Antimicrob Agents Chemother. 2009;53(1):235–241. doi: 10.1128/AAC.00665-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bansal S, Tandon V. Contribution of mutations in DNA gyrase and topoisomerase IV genes to ciprofloxacin resistance in Escherichia coli clinical isolates. Int J Antimicrob Agents. 2011;37(3):253–255. doi: 10.1016/j.ijantimicag.2010.11.022. [DOI] [PubMed] [Google Scholar]
- 28.Maruri F, et al. A systematic review of gyrase mutations associated with fluoroquinolone-resistant Mycobacterium tuberculosis and a proposed gyrase numbering system. J Antimicrob Chemother. 2012;67(4):819–831. doi: 10.1093/jac/dkr566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grossman RF, Hsueh PR, Gillespie SH, Blasi F. Community-acquired pneumonia and tuberculosis: Differential diagnosis and the use of fluoroquinolones. Int J Infect Dis. 2014;18:14–21. doi: 10.1016/j.ijid.2013.09.013. [DOI] [PubMed] [Google Scholar]
- 30.Long R, et al. Empirical treatment of community-acquired pneumonia and the development of fluoroquinolone-resistant tuberculosis. Clin Infect Dis. 2009;48(10):1354–1360. doi: 10.1086/598196. [DOI] [PubMed] [Google Scholar]
- 31.Devasia RA, et al. Fluoroquinolone resistance in Mycobacterium tuberculosis: The effect of duration and timing of fluoroquinolone exposure. Am J Respir Crit Care Med. 2009;180(4):365–370. doi: 10.1164/rccm.200901-0146OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aldred KJ, et al. Overcoming target-mediated quinolone resistance in topoisomerase IV by introducing metal-ion-independent drug-enzyme interactions. ACS Chem Biol. 2013;8(12):2660–2668. doi: 10.1021/cb400592n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aldred KJ, et al. Role of the water-metal ion bridge in mediating interactions between quinolones and Escherichia coli topoisomerase IV. Biochemistry. 2014;53(34):5558–5567. doi: 10.1021/bi500682e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blower TR, Williamson BH, Kerns RJ, Berger JM. Crystal structure and stability of gyrase–fluoroquinolone cleaved complexes from Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2016;113:1706–1713. doi: 10.1073/pnas.1525047113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Camus JC, Pryor MJ, Médigue C, Cole ST. Re-annotation of the genome sequence of Mycobacterium tuberculosis H37Rv. Microbiology. 2002;148(pt 10):2967–2973. doi: 10.1099/00221287-148-10-2967. [DOI] [PubMed] [Google Scholar]
- 36.Xu C, Kreiswirth BN, Sreevatsan S, Musser JM, Drlica K. Fluoroquinolone resistance associated with specific gyrase mutations in clinical isolates of multidrug-resistant Mycobacterium tuberculosis. J Infect Dis. 1996;174(5):1127–1130. doi: 10.1093/infdis/174.5.1127. [DOI] [PubMed] [Google Scholar]
- 37.Aubry A, et al. Novel gyrase mutations in quinolone-resistant and -hypersusceptible clinical isolates of Mycobacterium tuberculosis: Functional analysis of mutant enzymes. Antimicrob Agents Chemother. 2006;50(1):104–112. doi: 10.1128/AAC.50.1.104-112.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tran TP, et al. Structure-activity relationships of 3-aminoquinazolinediones, a new class of bacterial type-2 topoisomerase (DNA gyrase and topo IV) inhibitors. Bioorg Med Chem Lett. 2007;17(5):1312–1320. doi: 10.1016/j.bmcl.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 39.German N, Malik M, Rosen JD, Drlica K, Kerns RJ. Use of gyrase resistance mutants to guide selection of 8-methoxy-quinazoline-2,4-diones. Antimicrob Agents Chemother. 2008;52(11):3915–3921. doi: 10.1128/AAC.00330-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pan XS, Gould KA, Fisher LM. Probing the differential interactions of quinazolinedione PD 0305970 and quinolones with gyrase and topoisomerase IV. Antimicrob Agents Chemother. 2009;53(9):3822–3831. doi: 10.1128/AAC.00113-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oppegard LM, et al. Comparison of in vitro activities of fluoroquinolone-like 2,4- and 1,3-diones. Antimicrob Agents Chemother. 2010;54(7):3011–3014. doi: 10.1128/AAC.00190-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Malik M, et al. Fluoroquinolone and quinazolinedione activities against wild-type and gyrase mutant strains of Mycobacterium smegmatis. Antimicrob Agents Chemother. 2011;55(5):2335–2343. doi: 10.1128/AAC.00033-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dong Y, Xu C, Zhao X, Domagala J, Drlica K. Fluoroquinolone action against mycobacteria: Effects of C-8 substituents on growth, survival, and resistance. Antimicrob Agents Chemother. 1998;42(11):2978–2984. doi: 10.1128/aac.42.11.2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao BY, Pine R, Domagala J, Drlica K. Fluoroquinolone action against clinical isolates of Mycobacterium tuberculosis: Effects of a C-8 methoxyl group on survival in liquid media and in human macrophages. Antimicrob Agents Chemother. 1999;43(3):661–666. doi: 10.1128/aac.43.3.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hande KR. Etoposide: Four decades of development of a topoisomerase II inhibitor. Eur J Cancer. 1998;34(10):1514–1521. doi: 10.1016/s0959-8049(98)00228-7. [DOI] [PubMed] [Google Scholar]
- 46.Baldwin EL, Osheroff N. Etoposide, topoisomerase II and cancer. Curr Med Chem Anticancer Agents. 2005;5(4):363–372. doi: 10.2174/1568011054222364. [DOI] [PubMed] [Google Scholar]
- 47.Kumar R, Madhumathi BS, Nagaraja V. Molecular basis for the differential quinolone susceptibility of mycobacterial DNA gyrase. Antimicrob Agents Chemother. 2014;58(4):2013–2020. doi: 10.1128/AAC.01958-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kingma PS, Greider CA, Osheroff N. Spontaneous DNA lesions poison human topoisomerase IIα and stimulate cleavage proximal to leukemic 11q23 chromosomal breakpoints. Biochemistry. 1997;36(20):5934–5939. doi: 10.1021/bi970507v. [DOI] [PubMed] [Google Scholar]
- 49.Fortune JM, et al. DNA topoisomerases as targets for the anticancer drug TAS-103: DNA interactions and topoisomerase catalytic inhibition. Biochemistry. 1999;38(47):15580–15586. doi: 10.1021/bi991792g. [DOI] [PubMed] [Google Scholar]
- 50.Fortune JM, Osheroff N. Merbarone inhibits the catalytic activity of human topoisomerase IIα by blocking DNA cleavage. J Biol Chem. 1998;273(28):17643–17650. doi: 10.1074/jbc.273.28.17643. [DOI] [PubMed] [Google Scholar]
- 51.Gentry AC, et al. Interactions between the etoposide derivative F14512 and human type II topoisomerases: Implications for the C4 spermine moiety in promoting enzyme-mediated DNA cleavage. Biochemistry. 2011;50(15):3240–3249. doi: 10.1021/bi200094z. [DOI] [PMC free article] [PubMed] [Google Scholar]