

Abstract
Autoimmune liver diseases (AILD) are rare diseases with a reported prevalence of less than 50 per 100 000 population. As the research landscape and our understanding of AILDs and liver transplantation evolves, there remain areas of unmet needs. One of these areas of unmet needs is prevention of disease recurrence after liver transplantation. Disease recurrence is not an insignificant event because allograft loss with the need for retransplantation can occur. Patients transplanted for AILD are more likely to experience acute rejection compared to those transplanted for non-AILD, and the reason(s) behind this observation is unclear. Tasks for the future include a better understanding of the pathogenesis of AILD, definition of the precise pathogenetic mechanisms of recurrent AILD, and development of strategies that can identify recipients at risk for disease recurrence. Importantly, the role of crosstalk between alloimmune responses and autoimmune responses in AILD is an important area that needs further study.
This article reviews the relevant literature of de novo autoimmune hepatitis, recurrent autoimmune hepatitis, recurrent primary sclerosing cholangitis, and recurrent primary biliary cirrhosis in terms of the clinical entity, the scientific advancements, and future scientific goals to enhance our understanding of these diseases.
A review of the relevant literature of de novo autoimmune hepatitis, recurrent autoimmune hepatitis, recurrent primary sclerosing cholangitis, and recurrent primary biliary cirrhosis in terms of the clinical entity, the scientific advancements and future scientific goals to enhance our understanding of these diseases.
Advances in liver transplantation have resulted in improved survival and better outcomes for most patients with liver disease. However, with these advances and longevity of liver allografts come late graft dysfunction, which is often difficult to diagnose and represents a significant medical management issue. Important causes of late graft dysfunction include de novo autoimmune hepatitis (DAIH) (also described in the literature as plasma cell hepatitis and posttransplant allograft hepatitis) and recurrent autoimmune liver disease (AILD). De novo autoimmune hepatitis was first described in 1998 as a form of late allograft dysfunction that did not result from a recognized cause and is not associated with patients who developed autoimmune hepatitis (AIH) as their primary cause of liver disease.1 It has strong overlapping features with AIH and is seen in 4% to 7% of pediatric and adult liver transplanted patients, mostly pediatric liver transplant recipients.2-6 Autoimmune liver diseases are rare diseases with a reported prevalence of less than 50 per 100000 population; and despite advances in the understanding and treatment of AILD, there still remain areas of unmet needs.
MATERIALS AND METHODS
A comprehensive literature search using PUBMED/MEDLINE was conducted to identify articles in peer-reviewed publications that reported on DAIH, recurrent primary sclerosing cholangitis (rPSC), recurrent AIH (rAIH), and recurrent primary biliary cirrhosis (rPBC). The search was performed on November 1, 2014, for peer-reviewed articles published between 1998 through 2014, and used the following search strategy: AIH, PSC, primary biliary cirrhosis, recurrent PSC, rPBC, recurrent AIH, DAIH, liver transplantation, and acute and chronic rejection. The definition of AIH used was as previously described: elevated aminotransferases in the setting of antinuclear antibody (ANA) or antismooth muscle antibody (ASMA) and anti-liver kidney microsomal antibody (LKMA) of 1:40 or greater, positive soluble liver antigen, serum IgG greater than upper limit of normal, liver histology compatible with AIH, absence of viral hepatitis, and Wilson disease; a score of 6 in the above considered as probable AIH, a score of 7 or greater considered as definite AIH7,8 (of note in children, ANA/SMA ≥ 1:20 and anti-LKM ≥ 1:10 considered as positive).
The definition of DAIH was as previously described: a liver transplant recipient without a history of AILD presenting with unknown etiology of late graft dysfunction. The late graft dysfunction is characterized by elevated aminotransferases typically occurring longer than 2 years after transplant, graft dysfunction not due to any of the following causes: acute and chronic rejection, hepatitis B and C infection, Epstein Barr virus and cytomegalovirus infections, vascular problems, biliary complication, drug toxicity, sepsis, recurrence of primary disease, or posttransplant lymphoproliferative disease; elevated serum immunoglobulin G, positive autoantibody titers: ANA, ASMA, anti-LKM; characteristic biopsy findings of dense lymphocytic portal-tract infiltrate with plasma cells, and interface hepatitis).9,10 Results of the search were then narrowed down to articles and case reports that described new onset AIH, de novo immune hepatitis or plasma cell hepatitis after liver transplantation. A total of 25 peer-reviewed articles reporting DAIH were included in the final review. This overview is therefore limited to DAIH and does not include other causes of allograft dysfunction, such as chronic hepatitis and interface hepatitis that do not fulfill all the criteria for a diagnosis of DAIH.
For recurrent AILD, the definition of rPSC was based on the Mayo Clinic criteria proposed by Graziadei et al,11 which requires: a confirmed diagnosis of PSC before liver transplantation; cholangiograms showing nonanastomotic biliary strictures of the intrahepatic and/or extrahepatic biliary tree with beading and irregularity occurring longer than 90 days after transplantation; or a liver biopsy showing fibrous cholangitis and/or fibro obliterative lesions with or without ductopenia, biliary fibrosis, or biliary cirrhosis, exclusion of hepatic artery thrombosis/stenosis, ductopenic rejections, donor-recipient ABO blood type incompatibility, anastomotic structuring alone, and nonanastomotic strictures before day 90 after liver transplantation.
The definition of rPBC was as previously described12: liver transplantation for confirmed diagnosis of PBC, persistence of serum antimitochondrial antibody, compatible histopathology (portal inflammation, lymphocytic inflammatory infiltrates, lymphocytic cholangitis, epithelioid granulomas), exclusion of differential diagnostic considerations included (hepatitis C infection with lymphocytic cholangitis, drug-induced liver injury, acute cellular rejection, chronic ductopenic rejection, biliary obstruction, graft-versus-host disease). Two of the above 4 are considered as probable diagnosis; 3 of above 4 are considered as definite diagnosis.
Etiology of DAIH
The etiology of DAIH is unclear; however, it is associated with several risk factors, including the number of acute rejection episodes, steroid dependence,9 human lymphocyte antigen DR3 (HLA DR3) phenotype,13 and treatment with pegylated interferon for recurrent hepatitis C in patients who have achieved hepatitis C virus-RNA clearance.14,15 Sex and age of the organ donor have been implicated; organs from older women have been reported as being associated with increased likelihood of developing DAIH.16 Though sex and age as risk factors have not been consistently reported by all centers.9 Interestingly, chronic hepatitis E infection has been described in pediatric liver transplant recipients with persistently elevated serum aminotransferase levels and histological features of portal inflammation and interface hepatitis of unclear etiology; importantly, none of these patients were reported in the publication to fulfill the diagnostic criteria for DAIH17; thus, it is unclear if chronic hepatitis E is a risk factor for the development of DAIH.
Pathogenesis of DAIH
One proposed mechanism of the alloimmune response seen in DAIH is related to donor/recipient mismatching across glutathione-S-transferase theta 1 (GSTT1).18-20 Twenty percent of white and 11% to 58% of other ethnic groups possess a genetic deletion at the GSTT1 locus that results in lack of expression of GSTT1, which is a known drug-metabolizing enzyme.21 Lack of expression of GSTT1 by a recipient who receives a graft from a GSTT1 expressing donor is thought to result in immune sensitization of the recipient with resultant development of humoral responses to allograft cells expressing GST. Aguilera et al19 described 6 liver transplant recipients with a recipient/donor combination −/+ for GSTT1−/GSTT1+ with circulating antibodies to GST, all of whom developed DAIH. The GSTT1 mismatching is also an example of how genetic polymorphism may contribute to the pathogenesis of immune-mediated disease and suggests a role for atypical antibody-mediated rejection in the pathogenesis of DAIH.22
Support for the role of donor-specific antibody in the pathogenesis of DAIH comes from reports of positive complement component 4d (C4d) staining in livers of patients with DAIH and donor-specific antibody against GSTT1 (anti-GSTT1).23 The C4d is one of the split products generated during complement activation of the classic and alternative pathways. As it is one of the split products that covalently binds to the surface of cells, specific staining patterns have been useful in identifying patients with antibody-mediated rejection in renal transplantation.24-26 In contrast to the well-established pattern of C4d deposition in renal allografts with antibody-mediated rejection, the pattern of C4d deposition is variable in liver allografts, and there is yet to be a consensus on the relevance of specific staining patterns. Moreover, as the liver is a primary site for the production of local complement factors, staining with C4d may occur in the setting of alternative pathway activation as well. Nonetheless, it does deserve further study especially as it relates to donor-specific antibodies.23
Proposed arguments against DAIH and rejection being the same entity include the absence of bile duct involvement in DAIH, and the degree of plasma cell infiltration and severity of interface hepatitis.10 Additionally, treatment of DAIH differs from classic antirejection regimens. For instance, calcineurin inhibitor dose is typically increased to achieve a higher blood level in acute rejection, whereas a reduction in the calcineurin inhibitor dose has been suggested in DAIH.27,28 Furthermore, established antirejection therapies seem to be ineffective in DAIH patients.27,28 One of the important concepts that has arisen from assessing if DAIH and acute rejection are similar entities is the concept of epitope spreading, which occurs in many immune responses.29,30 Although a narrow range of antigens may initiate early native immune responses, it is recognized that the ensuing tissue damage results in the exposure of additional epitopes (autoantigens) that continue to drive immune response. Thus, an interesting concept is that initial rejection episodes against the graft may elicit tissue damage exposing neoantigens that perpetuate ongoing cellular and humoral responses. It could be of interest to examine whether collateral damage exposes “neo” self-epitopes when donor cells migrate from the graft and are attacked in the periphery potentially illustrating a mechanism via which transplantation could trigger autoimmunity. Finally, perhaps the question to be addressed is if DAIH is indeed a form of rejection whether the target in this form of rejection is the hepatocyte (not the bile duct), similar to what is seen in AIH.
The immune response in DAIH has also been proposed to be a consequence of reactivity to neoantigens with self-sensitization occurring through molecular mimicry; so short peptide sequences from toxic or infectious agents that resemble self-antigens sensitize cells causing aberrant misrecognition of self-antigens as foreign. Molecules that can contribute to such an effect have been labeled the exposome. Some individuals may be genetically predisposed to reactivity of this kind.31
Autoantibodies
Autoantibodies in DAIH are an important diagnostic tool.10 A key question in understanding the pathogenesis of DAIH is the role played by these autoantibodies and their correlation with disease progression. The hypothesis that they function as a marker for disease32 was supported by Avitzur et al33 who showed that positive autoantibodies in children after liver transplantation denoted a higher risk for the development of DAIH over time. Likewise, others have shown that persistence of high titers of ASMA and/or ANA in patients with AIH is associated with disease activity.34 Alternatively, there is a report of patients transplanted for Wilson disease who develop anti–LKM-1 autoantibodies but show no progression to DAIH; however, it is not known how long these patients were followed up, or whether they may have developed DAIH since publication of the initial observation.35
A recent identification of molecular targets of autoantibodies may be helpful in determining whether autoantibodies are in the causal pathway of DAIH. Huguet et al36 used proteomic tools to identify antigens recognized by the atypical LKMA. The proteomic technique consisted of 2-dimensional gel electrophoresis followed by 2-dimensional immunoblotting and subsequent ion trap mass spectrometry. Using sera from 8 patients with DAIH (including 2 with anti-LKMAs), the group identified several 25 kDa peptides, including the carbonic anhydrase isoform III, and β 1 subunit of the proteasome. In addition, they identified molecular targets of the GST families, that is, θ, α, μ, and π. As acknowledged by the authors, these potential targets of anti-LKMAs must be confirmed using monoclonal antibodies or recombinant proteins. They could then potentially be used to aid in evaluating posttransplant allograft dysfunction.
Of interest is the finding that Con A induction of DAIH in an acute rejection rat liver transplant model (Dark Agouti to Lewis) results in the induction of antinuclear antibodies against histone H1 and high mobility group box 1 with prolonged survival.37 Con A administration generates a “bystander hepatitis” not AIH. Additionally, Con A is a lectin that interacts with diverse receptors containing mannose carbohydrates. It is a nonspecific activator of immune cells, preferentially activating innate immune cells in the liver producing a transient acute hepatitis. It is therefore not surprising that ANA appears after such injury. Antinuclear antibodies are also probably the less specific autoantibody in patients with AIH. The relevance of Con A in human DAIH is yet to be established.
Clinical and Laboratory Manifestations
Characteristic of DAIH is a histological picture of interface hepatitis and multilobular collapse associated with increased IgG levels and positive autoantibodies, in the setting of elevated serum aminotransferases.1 As the name implies, cases share immunological, biochemical, and histological features with AIH.38,39 A pattern of centrilobular necroinflammatory activity with plasma cell infiltration has also been described.39 Perhaps contributing to the variable terminology used to describe this condition in the literature is the fact that heterogeneity of IgG and autoantibody levels has been described with some centers reporting DAIH patients with low IgG levels and absent autoantibodies.32 As alluded previously, the role of autoantibodies in DAIH is of interest; particularly, their physiological importance in terms of active involvement in the pathology of DAIH is yet to be determined. Interestingly, IgG 4–positive cases of DAIH have been identified40 which responds to azathioprine and prednisone, with normalization of alanine aminotransferase levels.41 It is worth noting that autoantibodies are frequently present without signs of graft dysfunction, particularly in the pediatric population,32,33,42 and liver biopsy is therefore required to determine the nature of any damage present.6
Features of DAIH are summarized in Table 1.
TABLE 1.
Summary of (i) autoimmune liver diseases known to recur after liver transplantation and (ii) de novo autoimmune hepatitis
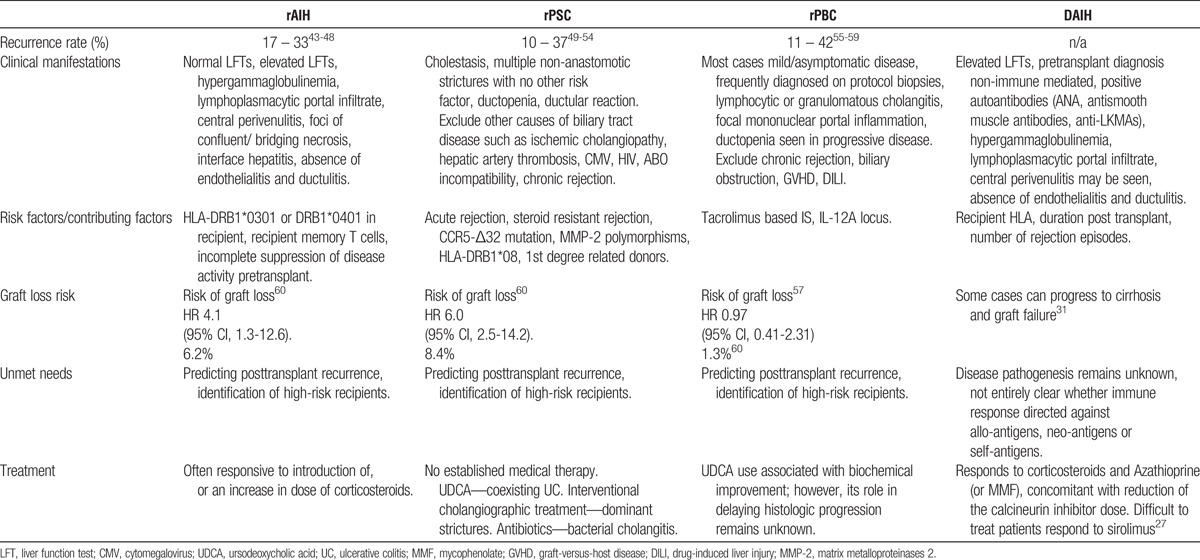
Recurrence of AILD After Liver Transplantation
Autoimmune hepatitis, PSC, and primary biliary cirrhosis recur after liver transplantation43-53,55-59,61-68; disease recurrence is however not an insignificant event because allograft loss with the need for retransplantation can occur. There is therefore opportunity for risk stratification/development of strategies that can identify recipients at risk for disease recurrence. A summary of AILD that recur after liver transplantation is presented in Table 1.
Primary sclerosing cholangitis is reported to recur in 10% to 37% of transplanted recipients, a mean of 6 months to 5 years after liver transplantation. It has been hypothesized that enterohepatic lymphocyte recirculation explains the link between PSC and inflammatory bowel disease; so effector T cells generated in lymphoid tissues in the gut during active inflammatory bowel disease persist as long-lived memory cells that recirculate through the liver and can trigger hepatic inflammation under the right conditions, even in the absence of active gut inflammation.69 Interestingly, colectomy before or during liver transplantation has been reported to have a protective effect against rPSC53; however, this protective effect is not consistently reported.52,54,66,70-73
Acute cellular rejection and steroid-resistant acute cellular rejection has been associated with rPSC,52,66,73 and explanations postulated for this association include injury of the biliary epithelium from acute cellular rejection increasing autoimmune epitopes with resultant immune-mediated ductal damage,50 or common factors predisposing to both acute cellular rejection and rPSC, such as a defective mechanism for immune autoregulation.66
Support for the role of the immune system in the development of rPSC is seen in the work reported by op den Dries et al74 where the combination of a loss of function mutation in the CC chemokine receptor 5 (CCR5)-Δ32 with a pretransplant diagnosis of PSC was associated with a significantly higher incidence of nonanastomotic strictures after transplantation. The CCR5 is thought to play a critical role in chemotaxis of regulatory T (Treg) cells to the site of injury.75-77 The CCR5 deficiency is accompanied by an increase of tissue levels of its ligand, CCL5, thus promoting enhanced influx of T cells into tissues by binding to an alternative receptor, CCR1.78-81
Other genetic factors, such as matrix metalloproteinases 2 gene promoter polymorphisms of donor and recipient, together with a pretransplant diagnosis of PSC, are reported to be a risk factor for the development of nonanastomotic strictures after liver transplantation.82 Similarly, some major histocompatibility complex (MHC-II) haplotypes are thought to influence the natural history of PSC after liver transplantation, for example, liver allografts from HLA DR52-positive donors are reported to protect against rPSC,50 and the presence of HLA-DRB1*08 is associated with an increased risk of rPSC.66 Finally, first degree–related donors is associated with more than 3 times the risk of rPSC.83
Autoimmune hepatitis is reported to recur in 17% to 33% of transplanted recipients. Recurrence can be indolent and detected only by surveillance laboratory testing and liver biopsy assessments.47,84,85 Risk factors implicated in recurrence include the susceptibility alleles HLA-DRB1*0301 or DRB1*0401 in the transplant recipient,47 HLA-DR locus mismatching,86 incomplete suppression of disease activity before transplantation, suggesting correlation between proinflammatory mechanisms before transplantation and rAIH.48 It has been suggested that recipient memory T cells play a role in the pathogenesis of rAIH as they recognize conserved autoantigenic peptides expressed by mismatched donor HLA in the allograft, thereby mediating rAIH.12 Regulatory T cell dysfunction is implicated in the pathogenesis of AIH by some groups,87 as such, it has been speculated that immunosuppression used after transplantation may contribute to rAIH by inhibition of autoantigen-specific Treg cells. Inadequate maintenance immunosuppression, especially discontinuation of steroid therapy, has been reported to play a role in disease recurrence,44 indeed optimization of immunosuppression has been reported to successfully treat histological recurrence.88 Conversely, rAIH has also been reported in the background of immunosuppression that is adequate to prevent rejection.89
In primary biliary cirrhosis, up to 30% of patients show features suggestive of recurrence within 5 years of transplantation.90,91 The reported incidence rate is 21% to 37% at 10 years and 43% at 15 years.85 The reported recurrence frequency rate increases with time in part due to different diagnostic criteria and different center policies for protocol biopsies. A big challenge in the diagnosis of rPBC is that recurrence may be present with normal or clinically insignificant elevations of liver tests.56,92-94 Retrospective data suggest an association with tacrolimus-based primary immunosuppression.90 With regard to genetic risk factors, the role of HLA donor-recipient mismatch in rPBC remains controversial.56-58,61 Recently, an association between rPBC and the IL12A locus was reported, suggesting that risk loci for PBC in the native liver might influence the risk of rPBC after liver transplantation, and mechanisms causing PBC in the allograft might be similar to those causing PBC in the native liver.59 The IL-12 signaling results in TH1 polarization of naive T cells; the authors therefore speculate that the mechanism for rPBC may involve either an inappropriate and sustained TH1 response or inefficient TH1 responses to appropriate stimuli as a result of variation in IL-12 signaling.
Proposed Areas for Future Study in DAIH
Fukami et al95 elegantly studied the role an alloimmune response plays in inducing autoimmunity using a murine model of obliterative airway disease. They sought to test whether antibodies developed after transplantation to mismatched donor MHC induces autoimmunity. Anti-MHC class 1 antibodies or control antibodies were administered intrabronchially into native lungs of mice. Animals receiving anti-MHC class 1 but not control antibodies developed a lesion similar to chronic rejection seen after human lung transplantation. Lungs of mice receiving anti-MHC class 1 antibody induced IL-17 as well as de novo antibodies to self-antigens, collagen V, and K α tubulin 1. The IL-17 neutralization resulted in reduction of autoantibody and lesions induced by anti-MHC class 1 antibodies. Their results suggest that antibodies to donor MHC can induce autoimmunity mediated by IL-17.
Interestingly, immunohistochemistry of the de novo liver showed IL-6 positivity within portal tracts as well as IL-17A positivity though to a lesser degree than IL-6 (Figure 1B-C). The IL-6 together with IL-1β and transforming growth factor β is needed to drive naive T cells toward differentiation to the TH17 program.96,97 Taken together, this may suggest a potential role for IL-17 in the perpetuation of chronic inflammation in DAIH.
FIGURE 1.
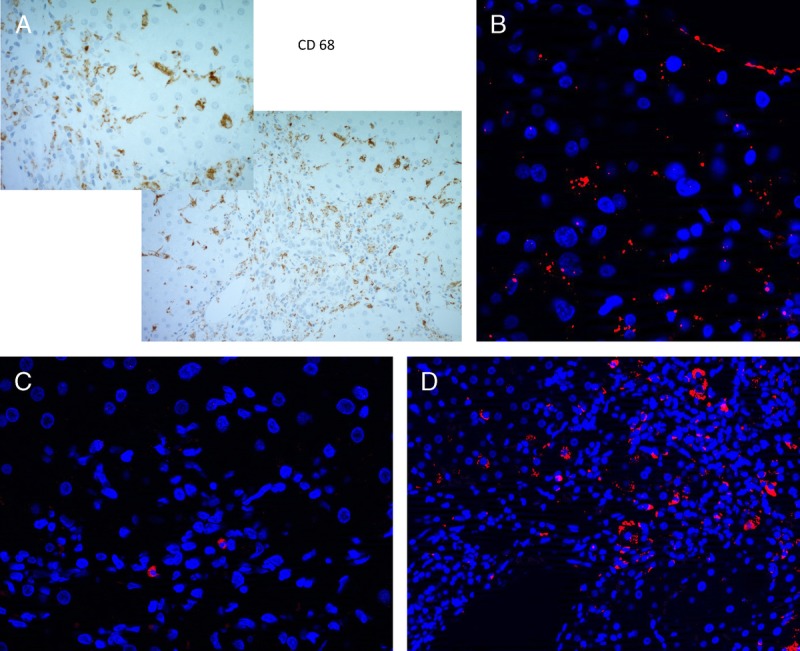
Immunohistochemical staining of paraffin-embedded liver sections from DAIH patients. Immunofluorescence staining of cytokines associated with the Th17 program. Formalin-fixed, paraffin-embedded 4-μ-thick sections from liver biopsies of patients with DAIH were stained for expression of CD 68- and 7-μ-thick sections were stained for expression of IL-6, IL-1β, and IL-17A. A, 200× (insert, 400×) magnification: portal tract with nearby lobule showing numerous CD 68-positive cells. B, 40× magnification shows a cluster of IL-6–positive cells within the portal tract (4’,6-diamidino-2-phenylindole [DAPI], blue; IL-6, orange). C, 40× magnification shows very few IL-1β–positive cells present within the portal tract (DAPI, blue, IL-1β, orange). D, 20× magnification shows several IL-17A–positive cells present within the portal tract (DAPI, blue; IL-17A, orange).
Another interesting observation is the role Treg cells may play in DAIH. The Treg cells are a subset of T helper cells expressing the canonical marker, forkhead box P3 transcription factor (FoxP3); that negatively regulates the immune response and plays a critical role in maintaining peripheral self-tolerance.98 Their dysfunction has been postulated to play a role in the pathogenesis of several autoimmune disorders.99-103 Similar to some reports in systemic lupus erythematosus,104-107 rheumatoid arthritis,108 and AIH,109 decreased Treg numbers and frequency in peripheral blood have been previously reported in pediatric liver transplant recipients with DAIH110; Of note, decreased Treg numbers and frequency in AIH has not been observed by all groups.111-113 This may be due to lack of a well-defined specific Treg marker in humans and heterogeneity in phenotypes.
A link between epigenetics and various autoimmune diseases has been reported114 DNA methylation is an epigenetic phenomenon: methylation of CpG islands in promoter regions regulates gene transcription and methylation of CpGs leads to gene repression by inhibiting binding to transcription factors. The methylation status of the Treg-specific demethylation region of the FoxP3 noncoding sequence has been shown to regulate expression of the transcription factor, FoxP3; and the demethylation status of CpG islands of FoxP3 is thought to correlate with full suppressive activity of Treg cells.115-117 A proposed area for future study would therefore be investigation of the role epigenetics might play in the pathogenesis of DAIH by examining, for instance, DNA methylation of Treg-specific demethylation region of the FoxP3 noncoding sequence of de novo Treg cells. This would also clarify whether Treg cell dysfunction contributes to the pathogenesis of DAIH.
The above concepts warrant further study; as such, we propose a model (Figure 2) to guide future research into the pathogenesis of DAIH. The model we propose supposes that graft injury may be initiated by acute rejection. The resulting graft injury reveals previously unseen epitopes. The IL-17 would contribute to the inflammatory milieu and lead to the production of antibodies by B cells. The absence of negative regulatory mechanisms possibly contributes to the development of autoimmunity.
FIGURE 2.
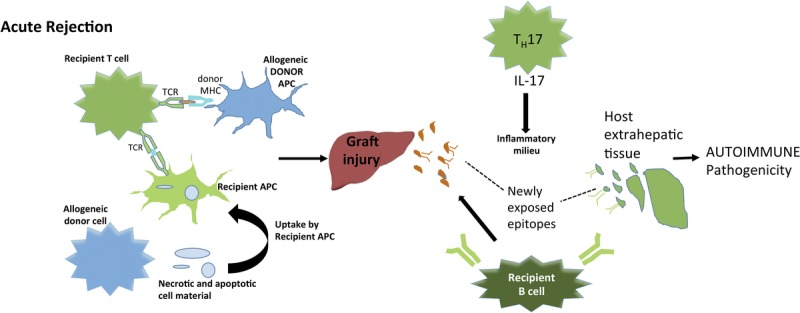
Proposed model for future research into pathogenesis of DAIH. Acute rejection episodes may prime the immune system to mount a self-directed response. Acute rejection is initiated by the large number of recipient T cells that recognize donor alloantigens encoded by MHC.125,126 Alloantigen presentation may be via direct pathway or indirect pathway.127 Graft injury reveals previously unseen epitopes. IL-17 would contribute to an inflammatory milieu and lead to the production of antibodies by B cells. In the absence of other negative regulatory mechanisms, this possibly contributes to the development of autoimmunity.
Proposed Areas for Future Study in Recurrent AILDs
A common theme with AILD is evidence that suggests that patients transplanted for AILD are more likely to experience acute rejection compared to those transplanted for non-AILD. The reason(s) behind this observation are unclear; and how this likelihood to develop rejection may be related to disease recurrence is unknown. Data from the lung transplant field has provided new insights demonstrating that alloimmunity can induce immune responses to self-antigens (autoimmunity) and also a crosstalk between alloimmune and autoimmune responses contribute to the immunopathogenesis of chronic rejection.95 In this context, collagen V is expressed ubiquitously in the lungs and incorporated within collagen I fibrils, making it immunologically protected. Exposure of CoIV to the immune system (after epithelial and endothelial injury from any cause) results in T cell–specific responses to CoIV in a rat model of lung transplantation. Transfer of these CoIV-specific T cells to other transplanted rats (including isografts) results in the development of chronic rejection.118,119 CoIV-specific immune responses have similarly been demonstrated in human lung transplant recipients with chronic rejection120-122; additionally, there is an emerging role of IL-17–mediated immune responses to self-antigens in the pathogenesis of chronic rejection.
In total, these observations suggest that inflammation due to alloimmune responses after transplantation can lead to the development of de novo immune responses against self-antigens. On the flip side, preexisting immune responses to self-antigens can augment the development of alloimmune responses to mismatched donor antigens and both can lead to chronic rejection.123 This lends support for “crosstalk” between alloimmune and autoimmune responses that perpetuate one another, leading to chronic rejection.
Important mediators in the pathogenesis of chronic rejection after transplantation are cellular immune responses to self-antigens. Over the past few years, an emerging role for IL-17 has been observed.95 Although CD4 Th17 cells are a common source of IL-17, NK cells, neutrophils, and γδ-T cells are other sources of IL-17 and may play a potential role in chronic rejection. Indeed, NK cells have emerged as a focus of interest in the transplant field because it is thought they play a role in the immune response in acute and chronic rejection.124
An unmet need in AILD is prevention of disease recurrence after liver transplantation. At present, there is no systematic approach to reduce the risk of disease recurrence, and experience from long-term follow-up studies suggests that graft loss from disease recurrence is not an insignificant problem.60 How the increased likelihood to develop rejection may be related to disease recurrence is unknown. Current hypotheses suggest a crosstalk between inflammatory responses by autoimmune and alloimmune mechanisms may lead to chronic rejection. A proposed link between autoimmune and alloimmune mechanisms after liver transplantation for AILD is suggested. Future studies should investigate if in patients with preexisting immune responses to self-antigens, who undergo liver transplantation, de novo donor-specific antibodies, and antibodies to self-antigens precede the development of rejection and disease recurrence; this may support their use as biomarkers after liver transplantation. Strategies targeted toward prevention, such as the use of antibody depleting regimens, can then be studied. Also, neutralization of IL-17 may represent an important aspect of future therapeutics in preventing recurrent AILD and should be studied. Other opportunities for prevention of disease recurrence after transplantation include identification of high-risk recipients using well-powered genetic and translational studies.
CONCLUSIONS
As the emphasis has shifted from ensuring immediate survival and managing early postoperative complications, the need to address long-term outcomes and the factors that influence these outcomes is urgent. Research into understanding the factors driving late allograft dysfunction could potentially open up new therapeutic options, leading to continued improvement in long-term patient and graft survival and quality of life.
ACKNOWLEDGMENT
The authors thank Adam Arterbery PhD for the immunofluorescence figures.
Footnotes
U.E. was supported by a CTSA grant (UL1 TR000142) from the National Center for Advancing Translational Science (NCATS), a component of the National Institutes of Health and National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under Award Number P30KD034989. C.E. was supported by the National Institute Of Diabetes And Digestive and Kidney Diseases under Award Number T32DK007356.
The authors declare no conflicts of interest.
C.E. participated in the writing of the article. U.E. participated in writing, editing and formatting the article.
REFERENCES
- 1. Kerkar N, Hadzić N, Davies ET, et al. De-novo autoimmune hepatitis after liver transplantation. Lancet. 1998; 351: 409– 413. [DOI] [PubMed] [Google Scholar]
- 2. Gupta P, Hart J, Millis JM, et al. De novo hepatitis with autoimmune antibodies and atypical histology: a rare cause of late graft dysfunction after pediatric liver transplantation. Transplantation. 2001; 71: 664– 668. [DOI] [PubMed] [Google Scholar]
- 3. Miyagawa-Hayashino A, Haga H, Egawa H, et al. Outcome and risk factors of de novo autoimmune hepatitis in living-donor liver transplantation. Transplantation. 2004; 78: 128– 135. [DOI] [PubMed] [Google Scholar]
- 4. Cho JM, Kim KM, Oh SH, et al. De novo autoimmune hepatitis in Korean children after liver transplantation: a single institution's experience. Transplant Proc. 2011; 43: 2394– 2396. [DOI] [PubMed] [Google Scholar]
- 5. Hernandez HM, Kovarik P, Whitington PF, et al. Autoimmune hepatitis as a late complication of liver transplantation. J Pediatr Gastroenterol Nutr. 2001; 32: 131– 136. [DOI] [PubMed] [Google Scholar]
- 6. Hübscher SG. What is the long-term outcome of the liver allograft? J Hepatol. 2011; 55: 702– 717. [DOI] [PubMed] [Google Scholar]
- 7. Hennes EM, Zeniya M, Czaja AJ, et al. ; International Autoimmune Hepatitis Group. Simplified criteria for the diagnosis of autoimmune hepatitis. Hepatology. 2008; 48: 169– 176. [DOI] [PubMed] [Google Scholar]
- 8. Mieli-Vergani G, Heller S, Jara P, et al. Autoimmune hepatitis. J Pediatr Gastroenterol Nutr. 2009; 49: 158– 164. [DOI] [PubMed] [Google Scholar]
- 9. Venick RS, McDiarmid SV, Farmer DG, et al. Rejection and steroid dependence: unique risk factors in the development of pediatric posttransplant de novo autoimmune hepatitis. Am J Transplant. 2007; 7: 955– 963. [DOI] [PubMed] [Google Scholar]
- 10.Banff Working Group, Demetris AJ, Adeyi O, Bellamy CO, et al. Liver biopsy interpretation for causes of late liver allograft dysfunction. Hepatology. 2006; 44: 489– 501. [DOI] [PubMed] [Google Scholar]
- 11. Graziadei IW, Wiesner RH, Marotta PJ, et al. Long-term results of patients undergoing liver transplantation for primary sclerosing cholangitis. Hepatology. 1999; 30: 1121– 1127. [DOI] [PubMed] [Google Scholar]
- 12. Ilyas JA, O'Mahony CA, Vierling JM. Liver transplantation in autoimmune liver diseases. Best Pract Res Clin Gastroenterol. 2011; 25: 765– 782. [DOI] [PubMed] [Google Scholar]
- 13. Salcedo M, Vaquero J, Bañares R, et al. Response to steroids in de novo autoimmune hepatitis after liver transplantation. Hepatology. 2002; 35: 349– 356. [DOI] [PubMed] [Google Scholar]
- 14. Berardi S, Lodato F, Gramenzi A, et al. High incidence of allograft dysfunction in liver transplanted patients treated with pegylated-interferon alpha-2b and ribavirin for hepatitis C recurrence: possible de novo autoimmune hepatitis? Gut. 2007; 56: 237– 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Aguilera I, Sousa JM, Gómez-Bravo MA, et al. De novo autoimmune hepatitis after interferon treatment in a liver transplant recipient with common variable immunodeficiency. Dig Liver Dis. 2014; 46: 663– 664. [DOI] [PubMed] [Google Scholar]
- 16. Montano-Loza AJ, Vargas-Vorackova F, Ma M, et al. Incidence and risk factors associated with de novo autoimmune hepatitis after liver transplantation. Liver Int. 2012; 32: 1426– 1433. [DOI] [PubMed] [Google Scholar]
- 17. Halac U, Béland K, Lapierre P, et al. Chronic hepatitis E infection in children with liver transplantation. Gut. 2012; 61: 597– 603. [DOI] [PubMed] [Google Scholar]
- 18. Aguilera I, Wichmann I, Sousa JM, et al. Antibodies against glutathione S-transferase T1 (GSTT1) in patients with de novo immune hepatitis following liver transplantation. Clin Exp Immunol. 2001; 126: 535– 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Aguilera I, Sousa JM, Gavilán F, et al. Glutathione S-transferase T1 mismatch constitutes a risk factor for de novo immune hepatitis after liver transplantation. Liver Transpl. 2004; 10: 1166– 1172. [DOI] [PubMed] [Google Scholar]
- 20. Rodriguez-Mahou M, Salcedo M, Fernandez-Cruz E, et al. Antibodies against glutathione S-transferase T1 (GSTT1) in patients with GSTT1 null genotype as prognostic marker: long-term follow-up after liver transplantation. Transplantation. 2007; 83: 1126– 1129. [DOI] [PubMed] [Google Scholar]
- 21. Salinas AE, Wong MG. Glutathione S-transferases—a review. Curr Med Chem. 1999; 6: 279– 309. [PubMed] [Google Scholar]
- 22. Aguilera I, Sousa JM, Gavilan F, et al. Glutathione S-transferase T1 genetic mismatch is a risk factor for de novo immune hepatitis in liver transplantation. Transplant Proc. 2005; 37: 3968– 3969. [DOI] [PubMed] [Google Scholar]
- 23. Aguilera I, Sousa JM, Gavilan F, et al. Complement component 4d immunostaining in liver allografts of patients with de novo immune hepatitis. Liver Transpl. 2011; 17: 779– 788. [DOI] [PubMed] [Google Scholar]
- 24. Collins AB, Schneeberger EE, Pascual MA, et al. Complement activation in acute humoral renal allograft rejection: diagnostic significance of C4d deposits in peritubular capillaries. J Am Soc Nephrol. 1999; 10: 2208– 2214. [DOI] [PubMed] [Google Scholar]
- 25. Regele H, Böhmig GA, Habicht A, et al. Capillary deposition of complement split product C4d in renal allografts is associated with basement membrane injury in peritubular and glomerular capillaries: a contribution of humoral immunity to chronic allograft rejection. J Am Soc Nephrol. 2002; 13: 2371– 2380. [DOI] [PubMed] [Google Scholar]
- 26. Poduval RD, Kadambi PV, Josephson MA, et al. Implications of immunohistochemical detection of C4d along peritubular capillaries in late acute renal allograft rejection. Transplantation. 2005; 79: 228– 235. [DOI] [PubMed] [Google Scholar]
- 27. Floreani A, Liberal R, Vergani D, et al. Autoimmune hepatitis: Contrasts and comparisons in children and adults—a comprehensive review. J Autoimmun. 2013; 46: 7– 16. [DOI] [PubMed] [Google Scholar]
- 28. Mieli-Vergani G, Vergani D. Autoimmune liver diseases in children—what is different from adulthood? Best Pract Res Clin Gastroenterol. 2011; 25: 783– 795. [DOI] [PubMed] [Google Scholar]
- 29. Demetris AJMN, Delaney CP. Overlap between alloimmunity and autoimmunity in the rat and human: evidence for important contributions for dendritic and regulatory. Cells Graft. 2003; 6: 21– 32. [Google Scholar]
- 30. Ciubotariu R, Liu Z, Colovai AI, et al. Persistent allopeptide reactivity and epitope spreading in chronic rejection of organ allografts. J Clin Invest. 1998; 101: 398– 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Czaja AJ. Diagnosis, pathogenesis, and treatment of autoimmune hepatitis after liver transplantation. Dig Dis Sci. 2012; 57: 2248– 2266. [DOI] [PubMed] [Google Scholar]
- 32. Richter A, Grabhorn E, Helmke K, et al. Clinical relevance of autoantibodies after pediatric liver transplantation. Clin Transplant. 2007; 21: 427– 432. [DOI] [PubMed] [Google Scholar]
- 33. Avitzur Y, Ngan BY, Lao M, et al. Prospective evaluation of the prevalence and clinical significance of positive autoantibodies after pediatric liver transplantation. J Pediatr Gastroenterol Nutr. 2007; 45: 222– 227. [DOI] [PubMed] [Google Scholar]
- 34. Couto CA, Bittencourt PL, Porta G, et al. Antismooth muscle and antiactin antibodies are indirect markers of histological and biochemical activity of autoimmune hepatitis. Hepatology. 2014; 59: 592– 600. [DOI] [PubMed] [Google Scholar]
- 35. Lohse AW, Weiler-Norman C, Burdelski M. De novo autoimmune hepatitis after liver transplantation. Hepatol Res. 2007; 37(Suppl 3): S462. [DOI] [PubMed] [Google Scholar]
- 36. Huguet S, Vinh J, Johanet C, et al. Identification by proteomic tool of atypical anti-liver/kidney microsome autoantibodies targets in de novo autoimmune hepatitis after liver transplantation. Ann N Y Acad Sci. 2007; 1109: 345– 357. [DOI] [PubMed] [Google Scholar]
- 37. Nakano T, Goto S, Lai CY, et al. Induction of antinuclear antibodies by de novo autoimmune hepatitis regulates alloimmune responses in rat liver transplantation. Clin Dev Immunol. 2013; 2013: 413928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vergani D, Mieli-Vergani G. Autoimmune hepatitis. Minerva Gastroenterol Dietol. 2004; 50: 113– 123. [PubMed] [Google Scholar]
- 39. Sebagh M, Castillo-Rama M, Azoulay D, et al. Histologic findings predictive of a diagnosis of de novo autoimmune hepatitis after liver transplantation in adults. Transplantation. 2013; 96: 670– 678. [DOI] [PubMed] [Google Scholar]
- 40. Castillo-Rama M, Sebagh M, Sasatomi E, et al. “Plasma cell hepatitis” in liver allografts: identification and characterization of an IgG4-rich cohort. Am J Transplant. 2013; 13: 2966– 2977. [DOI] [PubMed] [Google Scholar]
- 41. Zhao XY, Rakhda MI, Wang TI, et al. Immunoglobulin G4-associated de novo autoimmune hepatitis after liver transplantation for chronic hepatitis B- and C-related cirrhosis and hepatocellular carcinoma: a case report with literature review. Transplant Proc. 2013; 45: 824– 827. [DOI] [PubMed] [Google Scholar]
- 42. Riva S, Sonzogni A, Bravi M, et al. Late graft dysfunction and autoantibodies after liver transplantation in children: preliminary results of an Italian experience. Liver Transpl. 2006; 12: 573– 577. [DOI] [PubMed] [Google Scholar]
- 43. Prados E, Cuervas-Mons V, de la Mata M, et al. Outcome of autoimmune hepatitis after liver transplantation. Transplantation. 1998; 66: 1645– 1650. [DOI] [PubMed] [Google Scholar]
- 44. Milkiewicz P, Hubscher SG, Skiba G, et al. Recurrence of autoimmune hepatitis after liver transplantation. Transplantation. 1999; 68: 253– 256. [DOI] [PubMed] [Google Scholar]
- 45. Ratziu V, Samuel D, Sebagh M, et al. Long-term follow-up after liver transplantation for autoimmune hepatitis: evidence of recurrence of primary disease. J Hepatol. 1999; 30: 131– 141. [DOI] [PubMed] [Google Scholar]
- 46. Reich DJ, Fiel I, Guarrera JV, et al. Liver transplantation for autoimmune hepatitis. Hepatology. 2000; 32(4 Pt 1): 693– 700. [DOI] [PubMed] [Google Scholar]
- 47. González-Koch A, Czaja AJ, Carpenter HA, et al. Recurrent autoimmune hepatitis after orthotopic liver transplantation. Liver Transpl. 2001; 7: 302– 310. [DOI] [PubMed] [Google Scholar]
- 48. Montano-Loza AJ, Mason AL, Ma M, et al. Risk factors for recurrence of autoimmune hepatitis after liver transplantation. Liver Transpl. 2009; 15: 1254– 1261. [DOI] [PubMed] [Google Scholar]
- 49. Goss JA, Shackleton CR, Farmer DG, et al. Orthotopic liver transplantation for primary sclerosing cholangitis. A 12-year single center experience. Ann Surg. 1997; 225: 472– 481 discussion 481-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jeyarajah DR, Netto GJ, Lee SP, et al. Recurrent primary sclerosing cholangitis after orthotopic liver transplantation: is chronic rejection part of the disease process? Transplantation. 1998; 66: 1300– 1306. [DOI] [PubMed] [Google Scholar]
- 51. Graziadei IW, Wiesner RH, Batts KP, et al. Recurrence of primary sclerosing cholangitis following liver transplantation. Hepatology. 1999; 29: 1050– 1056. [DOI] [PubMed] [Google Scholar]
- 52. Campsen J, Zimmerman MA, Trotter JF, et al. Clinically recurrent primary sclerosing cholangitis following liver transplantation: a time course. Liver Transpl. 2008; 14: 181– 185. [DOI] [PubMed] [Google Scholar]
- 53. Alabraba E, Nightingale P, Gunson B, et al. A re-evaluation of the risk factors for the recurrence of primary sclerosing cholangitis in liver allografts. Liver Transpl. 2009; 15: 330– 340. [DOI] [PubMed] [Google Scholar]
- 54. Vera A, Moledina S, Gunson B, et al. Risk factors for recurrence of primary sclerosing cholangitis of liver allograft. Lancet. 2002; 360: 1943– 1944. [DOI] [PubMed] [Google Scholar]
- 55. Neuberger J, Gunson B, Hubscher S, et al. Immunosuppression affects the rate of recurrent primary biliary cirrhosis after liver transplantation. Liver Transpl. 2004; 10: 488– 491. [DOI] [PubMed] [Google Scholar]
- 56. Sanchez EQ, Levy MF, Goldstein RM, et al. The changing clinical presentation of recurrent primary biliary cirrhosis after liver transplantation. Transplantation. 2003; 76: 1583– 1588. [DOI] [PubMed] [Google Scholar]
- 57. Charatcharoenwitthaya P, Pimentel S, Talwalkar JA, et al. Long-term survival and impact of ursodeoxycholic acid treatment for recurrent primary biliary cirrhosis after liver transplantation. Liver Transpl. 2007; 13: 1236– 1245. [DOI] [PubMed] [Google Scholar]
- 58. Manousou P, Arvaniti V, Tsochatzis E, et al. Primary biliary cirrhosis after liver transplantation: influence of immunosuppression and human leukocyte antigen locus disparity. Liver Transpl. 2010; 16: 64– 73. [DOI] [PubMed] [Google Scholar]
- 59. Carbone M, Mells GF, Alexander GJ, et al. Calcineurin inhibitors and the IL12A locus influence risk of recurrent primary biliary cirrhosis after liver transplantation. Am J Transplant. 2013; 13: 1110– 1111. [DOI] [PubMed] [Google Scholar]
- 60. Rowe IA, Webb K, Gunson BK, et al. The impact of disease recurrence on graft survival following liver transplantation: a single centre experience. Transpl Int. 2008; 21: 459– 465. [DOI] [PubMed] [Google Scholar]
- 61. Morioka D, Egawa H, Kasahara M, et al. Impact of human leukocyte antigen mismatching on outcomes of living donor liver transplantation for primary biliary cirrhosis. Liver Transpl. 2007; 13: 80– 90. [DOI] [PubMed] [Google Scholar]
- 62. Gelley F, Zádori G, Görög D, et al. Recurrence of primary sclerosing cholangitis after liver transplantation—the Hungarian experience. Interv Med Appl Sci. 2014; 6: 16– 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Venkat VL, Ranganathan S, Mazariegos GV, et al. Recurrence of primary sclerosing cholangitis in pediatric liver transplant recipients. Liver Transpl. 2014; 20: 679– 686. [DOI] [PubMed] [Google Scholar]
- 64. Miloh T, Anand R, Yin W, et al. Studies of Pediatric Liver Transplantation Research Group Pediatric liver transplantation for primary sclerosing cholangitis. Liver Transpl. 2011; 17: 925– 933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Moncrief KJ, Savu A, Ma MM, et al. The natural history of inflammatory bowel disease and primary sclerosing cholangitis after liver transplantation—a single-centre experience. Can J Gastroenterol. 2010; 24: 40– 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Alexander J, Lord JD, Yeh MM, et al. Risk factors for recurrence of primary sclerosing cholangitis after liver transplantation. Liver Transpl. 2008; 14: 245– 251. [DOI] [PubMed] [Google Scholar]
- 67. Haga H, Miyagawa-Hayashino A, Taira K, et al. Histological recurrence of autoimmune liver diseases after living-donor liver transplantation. Hepatol Res. 2007; 37(Suppl 3): S463– S469. [DOI] [PubMed] [Google Scholar]
- 68. Tamura S, Sugawara Y, Kaneko J, et al. Recurrence of primary sclerosing cholangitis after living donor liver transplantation. Liver Int. 2007; 27: 86– 94. [DOI] [PubMed] [Google Scholar]
- 69. Grant AJ, Lalor PF, Salmi M, et al. Homing of mucosal lymphocytes to the liver in the pathogenesis of hepatic complications of inflammatory bowel disease. Lancet. 2002; 359: 150– 157. [DOI] [PubMed] [Google Scholar]
- 70. Khettry U, Keaveny A, Goldar-Najafi A, et al. Liver transplantation for primary sclerosing cholangitis: a long-term clinicopathologic study. Hum Pathol. 2003; 34: 1127– 1136. [DOI] [PubMed] [Google Scholar]
- 71. Kugelmas M, Spiegelman P, Osgood MJ, et al. Different immunosuppressive regimens and recurrence of primary sclerosing cholangitis after liver transplantation. Liver Transpl. 2003; 9: 727– 732. [DOI] [PubMed] [Google Scholar]
- 72. Brandsaeter B, Schrumpf E, Bentdal O, et al. Recurrent primary sclerosing cholangitis after liver transplantation: a magnetic resonance cholangiography study with analyses of predictive factors. Liver Transpl. 2005; 11: 1361– 1369. [DOI] [PubMed] [Google Scholar]
- 73. Cholongitas E, Shusang V, Papatheodoridis GV, et al. Risk factors for recurrence of primary sclerosing cholangitis after liver transplantation. Liver Transpl. 2008; 14: 138– 143. [DOI] [PubMed] [Google Scholar]
- 74. op den Dries S, Buis CI, Adelmeijer J, et al. The combination of primary sclerosing cholangitis and CCR5-∆Δ32 in recipients is strongly associated with the development of nonanastomotic biliary strictures after liver transplantation. Liver Int. 2011; 31: 1102– 1109. [DOI] [PubMed] [Google Scholar]
- 75. Dobaczewski M, Xia Y, Bujak M, et al. CCR5 signaling suppresses inflammation and reduces adverse remodeling of the infarcted heart, mediating recruitment of regulatory T cells. Am J Pathol. 2010; 176: 2177– 2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Nozaki T, Rosenblum JM, Schenk AD, et al. CCR5 is required for regulation of alloreactive T-cell responses to single class II MHC-mismatched murine cardiac grafts. Am J Transplant. 2009; 9: 2251– 2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wysocki CA, Jiang Q, Panoskaltsis-Mortari A, et al. Critical role for CCR5 in the function of donor CD4+CD25+ regulatory T cells during acute graft-versus-host disease. Blood. 2005; 106: 3300– 3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Locati M, Murphy PM. Chemokines and chemokine receptors: biology and clinical relevance in inflammation and AIDS. Annu Rev Med. 1999; 50: 425– 440. [DOI] [PubMed] [Google Scholar]
- 79. Ajuebor MN, Wondimu Z, Hogaboam CM, et al. CCR5 deficiency drives enhanced natural killer cell trafficking to and activation within the liver in murine T cell–mediated hepatitis. Am J Pathol. 2007; 170: 1975– 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Dawson TC, Beck MA, Kuziel WA, et al. Contrasting effects of CCR5 and CCR2 deficiency in the pulmonary inflammatory response to influenza A virus. Am J Pathol. 2000; 156: 1951– 1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Carr DJ, Ash J, Lane TE, et al. Abnormal immune response of CCR5-deficient mice to ocular infection with herpes simplex virus type 1. J Gen Virol. 2006; 87(Pt 3): 489– 499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ten Hove WR, Korkmaz KS, op den Dries S, et al. Matrix metalloproteinase 2 genotype is associated with nonanastomotic biliary strictures after orthotopic liver transplantation. Liver Int. 2011; 31: 1110– 1117. [DOI] [PubMed] [Google Scholar]
- 83. Egawa H, Ueda Y, Ichida T, et al. Risk factors for recurrence of primary sclerosing cholangitis after living donor liver transplantation in Japanese registry. Am J Transplant. 2011; 11: 518– 527. [DOI] [PubMed] [Google Scholar]
- 84. Duclos-Vallée JC, Sebagh M, Rifai K, et al. A 10 year follow up study of patients transplanted for autoimmune hepatitis: histological recurrence precedes clinical and biochemical recurrence. Gut. 2003; 52: 893– 897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Duclos-Vallee JC, Sebagh M. Recurrence of autoimmune disease, primary sclerosing cholangitis, primary biliary cirrhosis, and autoimmune hepatitis after liver transplantation. Liver Transpl. 2009; 15(Suppl 2): S25– S34. [DOI] [PubMed] [Google Scholar]
- 86. Balan V, Ruppert K, Demetris AJ, et al. Long-term outcome of human leukocyte antigen mismatching in liver transplantation: results of the National Institute of Diabetes and Digestive and Kidney Diseases Liver Transplantation Database. Hepatology. 2008; 48: 878– 888. [DOI] [PubMed] [Google Scholar]
- 87. Longhi MS, Ma Y, Mieli-Vergani G, et al. Aetiopathogenesis of autoimmune hepatitis. J Autoimmun. 2010; 34: 7– 14. [DOI] [PubMed] [Google Scholar]
- 88. Khalaf H, Mourad W, El-Sheikh Y, et al. Liver transplantation for autoimmune hepatitis: a single-center experience. Transplant Proc. 2007; 39: 1166– 1170. [DOI] [PubMed] [Google Scholar]
- 89. O'Grady JG. Phenotypic expression of recurrent disease after liver transplantation. Am J Transplant. 2010; 10: 1149– 1154. [DOI] [PubMed] [Google Scholar]
- 90. Liermann Garcia RF, Evangelista Garcia C, McMaster P, et al. Transplantation for primary biliary cirrhosis: retrospective analysis of 400 patients in a single center. Hepatology. 2001; 33: 22– 27. [DOI] [PubMed] [Google Scholar]
- 91. Carbone M, Neuberger J. Liver transplantation in PBC and PSC: indications and disease recurrence. Clin Res Hepatol Gastroenterol. 2011; 35: 446– 454. [DOI] [PubMed] [Google Scholar]
- 92. Sylvestre PB, Batts KP, Burgart LJ, et al. Recurrence of primary biliary cirrhosis after liver transplantation: histologic estimate of incidence and natural history. Liver Transpl. 2003; 9: 1086– 1093. [DOI] [PubMed] [Google Scholar]
- 93. Abraham SC, Poterucha JJ, Rosen CB, et al. Histologic abnormalities are common in protocol liver allograft biopsies from patients with normal liver function tests. Am J Surg Pathol. 2008; 32: 965– 973. [DOI] [PubMed] [Google Scholar]
- 94. Klein R, Huizenga JR, Gips CH, et al. Antimitochondrial antibody profiles in patients with primary biliary cirrhosis before orthotopic liver transplantation and titres of antimitochondrial antibody-subtypes after transplantation. J Hepatol. 1994; 20: 181– 189. [DOI] [PubMed] [Google Scholar]
- 95. Fukami N, Ramachandran S, Saini D, et al. Antibodies to MHC class I induce autoimmunity: role in the pathogenesis of chronic rejection. J Immunol. 2009; 182: 309– 318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Dong C. TH17 cells in development: an updated view of their molecular identity and genetic programming. Nat Rev Immunol. 2008; 8: 337– 348. [DOI] [PubMed] [Google Scholar]
- 97. Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol. 2008; 9: 641– 649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Sakaguchi S, Ono M, Setoguchi R, et al. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev. 2006; 212: 8– 27. [DOI] [PubMed] [Google Scholar]
- 99. Viglietta V, Baecher-Allan C, Weiner HL, et al. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med. 2004; 199: 971– 979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Bonelli M, Savitskaya A, Steiner CW, et al. Phenotypic and functional analysis of CD4+ CD25− Foxp3+ T cells in patients with systemic lupus erythematosus. J Immunol. 2009; 182: 1689– 1695. [DOI] [PubMed] [Google Scholar]
- 101. Franz B, Fritzsching B, Riehl A, et al. Low number of regulatory T cells in skin lesions of patients with cutaneous lupus erythematosus. Arthritis Rheum. 2007; 56: 1910– 1920. [DOI] [PubMed] [Google Scholar]
- 102. Scheinecker C, Bonelli M, Smolen JS. Pathogenetic aspects of systemic lupus erythematosus with an emphasis on regulatory T cells. J Autoimmun. 2010; 35: 269– 275. [DOI] [PubMed] [Google Scholar]
- 103. Sugiyama H, Gyulai R, Toichi E, et al. Dysfunctional blood and target tissue CD4+CD25high regulatory T cells in psoriasis: mechanism underlying unrestrained pathogenic effector T cell proliferation. J Immunol. 2005; 174: 164– 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Lee HY, Hong YK, Yun HJ, et al. Altered frequency and migration capacity of CD4+CD25+ regulatory T cells in systemic lupus erythematosus. Rheumatology (Oxford). 2008; 47: 789– 794. [DOI] [PubMed] [Google Scholar]
- 105. Barath S, Aleksza M, Tarr T, et al. Measurement of natural (CD4+CD25high) and inducible (CD4+IL-10+) regulatory T cells in patients with systemic lupus erythematosus. Lupus. 2007; 16: 489– 496. [DOI] [PubMed] [Google Scholar]
- 106. Baráth S, Soltész P, Kiss E, et al. The severity of systemic lupus erythematosus negatively correlates with the increasing number of CD4+CD25(high)FoxP3+ regulatory T cells during repeated plasmapheresis treatments of patients. Autoimmunity. 2007; 40: 521– 528. [DOI] [PubMed] [Google Scholar]
- 107. Bonelli M, Savitskaya A, von Dalwigk K, et al. Quantitative and qualitative deficiencies of regulatory T cells in patients with systemic lupus erythematosus (SLE). Int Immunol. 2008; 20: 861– 868. [DOI] [PubMed] [Google Scholar]
- 108. Cao D, Malmström V, Baecher-Allan C, et al. Isolation and functional characterization of regulatory CD25brightCD4+ T cells from the target organ of patients with rheumatoid arthritis. Eur J Immunol. 2003; 33: 215– 223. [DOI] [PubMed] [Google Scholar]
- 109. Longhi MS, Ma Y, Bogdanos DP, et al. Impairment of CD4(+)CD25(+) regulatory T-cells in autoimmune liver disease. J Hepatol. 2004; 41: 31– 37. [DOI] [PubMed] [Google Scholar]
- 110. Ekong UD, Mathew J, Melin-Aldana H, et al. Successful resolution of inflammation and increased regulatory T cells in sirolimus-treated post-transplant allograft hepatitis. Pediatr Transplant. 2012; 16: 165– 175. [DOI] [PubMed] [Google Scholar]
- 111. Lin SC, Chen KH, Lin CH, et al. The quantitative analysis of peripheral blood FOXP3-expressing T cells in systemic lupus erythematosus and rheumatoid arthritis patients. Eur J Clin Invest. 2007; 37: 987– 996. [DOI] [PubMed] [Google Scholar]
- 112. Suárez A, López P, Gómez J, et al. Enrichment of CD4+ CD25high T cell population in patients with systemic lupus erythematosus treated with glucocorticoids. Ann Rheum Dis. 2006; 65: 1512– 1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Peiseler M, Sebode M, Franke B, et al. FOXP3+ regulatory T cells in autoimmune hepatitis are fully functional and not reduced in frequency. J Hepatol. 2012; 57: 125– 132. [DOI] [PubMed] [Google Scholar]
- 114. Lu Q. The critical importance of epigenetics in autoimmunity. J Autoimmun. 2013; 41: 1– 5. [DOI] [PubMed] [Google Scholar]
- 115. Floess S, Freyer J, Siewert C, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 2007; 5: e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Baron U, Floess S, Wieczorek G, et al. DNA demethylation in the human FOXP3 locus discriminates regulatory T cells from activated FOXP3(+) conventional T cells. Eur J Immunol. 2007; 37: 2378– 2389. [DOI] [PubMed] [Google Scholar]
- 117. Wang L, Liu Y, Han R, et al. Mbd2 promotes foxp3 demethylation and T-regulatory-cell function. Mol Cell Biol. 2013; 33: 4106– 4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Haque MA, Mizobuchi T, Yasufuku K, et al. Evidence for immune responses to a self-antigen in lung transplantation: role of type V collagen-specific T cells in the pathogenesis of lung allograft rejection. J Immunol. 2002; 169: 1542– 1549. [DOI] [PubMed] [Google Scholar]
- 119. Yasufuku K, Heidler KM, Woods KA, et al. Prevention of bronchiolitis obliterans in rat lung allografts by type V collagen-induced oral tolerance. Transplantation. 2002; 73: 500– 505. [DOI] [PubMed] [Google Scholar]
- 120. Hachem RR, Tiriveedhi V, Patterson GA, et al. Antibodies to K-α 1 tubulin and collagen V are associated with chronic rejection after lung transplantation. Am J Transplant. 2012; 12: 2164– 2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Burlingham WJ, Love RB, Jankowska-Gan E, et al. IL-17-dependent cellular immunity to collagen type V predisposes to obliterative bronchiolitis in human lung transplants. J Clin Invest. 2007; 117: 3498– 3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Tiriveedhi V, Angaswamy N, Brand D, et al. A shift in the collagen V antigenic epitope leads to T helper phenotype switch and immune response to self-antigen leading to chronic lung allograft rejection. Clin Exp Immunol. 2012; 167: 158– 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Bharat A, Saini D, Steward N, et al. Antibodies to self-antigens predispose to primary lung allograft dysfunction and chronic rejection. Ann Thorac Surg. 2010; 90: 1094– 1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Pratschke J, Stauch D, Kotsch K. Role of NK and NKT cells in solid organ transplantation. Transpl Int. 2009; 22: 859– 868. [DOI] [PubMed] [Google Scholar]
- 125. Stefanova I, Dorfman JR, Tsukamoto M, et al. On the role of self-recognition in T cell responses to foreign antigen. Immunol Rev. 2003; 191: 97– 106. [DOI] [PubMed] [Google Scholar]
- 126. Afzali B, Lechler RI, Hernandez-Fuentes MP. Allorecognition and the alloresponse: clinical implications. Tissue Antigens. 2007; 69: 545– 556. [DOI] [PubMed] [Google Scholar]
- 127. Afzali B, Lombardi G, Lechler RI. Pathways of major histocompatibility complex allorecognition. Curr Opin Organ Transplant. 2008; 13: 438– 444. [DOI] [PMC free article] [PubMed] [Google Scholar]