

Educational Gap
Diagnostic evaluation and management of children with chronic pancytopenia is highly complex. (1)(2)(3) Clinicians should be aware of the subtle signs and symptoms of acquired and congenital childhood pancytopenias to maintain a high index of suspicion, streamline the referral process, and function within their patients’ multidisciplinary medical homes.
Objectives
After completing this article, readers should be able to:
Identify signs and symptoms of pancytopenia.
Identify patients who require emergency referral due to a life-threatening illness.
Discuss the basics of clinical presentation, diagnostics, and management of childhood severe aplastic anemia.
Discuss the basics of clinical presentation, diagnostics, and management of common genetic syndromes associated with pancytopenia.
Understand the basic concepts behind laboratory tests and be aware of which tests are appropriate in specific clinical situations.
Pancytopenia
Pancytopenia is characterized by a decreased number of at least two blood cell lines. Pancytopenia may progress acutely, such as with decreased blood cell counts in fulminant sepsis, disseminated intravascular coagulation, or rapid hemolysis. Alternatively, pancytopenia may evolve insidiously over weeks to months. In this review we focus on the latter group of acquired and congenital pancytopenias in children.
Chronic pancytopenia is a diagnostic challenge. The range of potential causes is bewildering, signs and symptoms overlap substantially, and many diseases presenting with pancytopenia are life-threatening if not recognized and managed properly. Therefore, clinicians must be familiar with clinical scenarios that should prompt evaluation of blood counts and a pediatric hematology referral.
Causes
Pancytopenia may be caused by decreased production or increased pooling or destruction of blood cells. These problems may be congenital or acquired (Tables 1 and 2).
Table 1.
Selected Causes of Acquired Pancytopenia
| Decreased bone marrow function | Leukemia |
| Proliferating leukemic blasts suppress production of healthy blood cells | |
| Aplastic anemia | |
| Nutritional deficiencies (vitamin B12, folic acid) | |
| Vitamin B12 and folic acid are essential for production of nucleotides and, thus, DNA replication. Severe deficiency may cause pancytopenia | |
| Metastatic cancer infiltrating the bone marrow (eg, neuroblastoma, rhabdomyosarcoma) | |
| Fulminant sepsis | |
| Myelodysplastic syndrome | |
| Increased destruction or pooling of blood cells | Splenomegaly |
| Peripheral blood cells are trapped in the enlarged spleen | |
| Paroxysmal nocturnal hemoglobinuria | |
| Complement-mediated destruction of blood cells | |
| Acquired hemophagocytic lymphohistiocytosis | |
| Decreased bone marrow activity due to cytokine storm and hemophagocytosis |
Table 2.
Selected Causes of Congenital Pancytopenia
| Decreased bone marrow function | Fanconi anemia |
| Shwachman-Diamond syndrome | |
| Dyskeratosis congenita | |
| Other inherited bone marrow failure syndromes as a separate line after dyskeratosis congenita | |
| Storage disorders (eg, Gaucher syndrome) | |
| Mitochondrial disorders (eg, Pearson syndrome) | |
| Congenital viral infection (TORCH) | |
| Increased destruction or pooling of blood cells | Autoimmune lymphoproliferative syndrome and other congenital immunodeficiencies |
| Congenital hemophagocytic lymphohistiocytosis | |
| Decreased bone marrow activity due to cytokine storm and hemophagocytosis |
Some children with new-onset pancytopenia are critically ill and require immediate evaluation in an emergency department (Table 3). More frequently, chronic pancytopenia causes subtle but characteristic clinical findings.
Table 3.
Signs and Symptoms of Pancytopenia
| Anemia | Leukopenia, neutropenia | Thrombocytopenia | |
| Common | Pallor, malaise, fatigue, decreased exercise tolerance | None | Increased bruising in unusual locations without obvious trauma, petechiae, recurrent nosebleeds |
| Life-threatening | Hemodynamic instability, hypoxia, loss of consciousness | Febrile neutropenia, sepsis | Life-threatening hemorrhage and intracranial bleeding |
Signs and Symptoms
Case 1: Bert is a previously healthy 4-year-old boy who is seeing his pediatrician for a health supervision visit. His mother mentions that for the last 3 weeks, he has been asking for pain medicines because “his legs are hurting”. Last Saturday Bert missed his baseball practice because he woke up with a nosebleed that stopped after 15 minutes of pressure, but “he was too tired to play anyway.” He spent most of the past week napping. His vital signs are normal except for a heart rate of 128 beats/min. His physical examination reveals a few scattered ecchymoses on shins, forearms, and lower back; dried blood in both nares; and a red, nonblanching pinpoint rash on his cheeks.
Anemia has well-known clinical consequences: malaise, sleepiness, irritability, exercise intolerance, shortness of breath, and pallor. A rapid drop in red blood cell hemoglobin due to acute hemolysis may abruptly trigger clinical symptoms that are reminiscent of a large blood volume loss due to hemorrhage. In contrast, an insidious-onset anemia is associated with relatively mild symptoms as the cardiopulmonary and biochemical compensatory mechanisms activate over time.
Leukopenia impairs the immune system’s ability to fight infections. Neutropenia (low absolute neutrophil count [ANC]) is frequently encountered in clinical practice. Children with severe neutropenia (defined as ANC <500/μL [0.5 × 109/L]) face a high risk of life-threatening bacterial and fungal infections. Fever in a child who has neutropenia is a medical emergency, particularly in the context of acute neutropenia due to cancer chemotherapy or stem cell transplantation. Evidence-based guidelines for the management of febrile neutropenia in children have been developed by an international expert panel. (4) Affected children must be evaluated emergently under the guidance of a pediatric hematologist-oncologist. Blood cultures must be obtained and empiric broad-spectrum antibacterial therapy started immediately.
In the absence of trauma, a low platelet count (thrombocytopenia) does not lead to clinical symptoms until the count decreases below 10 to 20 × 103/μL (10 to 20 × 109/L). Children with severe thrombocytopenia develop spontaneous bruising, bleeding, and petechiae (small subcutaneous capillary hemorrhages). Menstruating females experience heavy and prolonged periods.
Case 1, continued: Bert’s signs and symptoms are concerning for pancytopenia of potentially ominous etiology. Bruised shins are common in active toddlers, but ecchymoses on the forearms and lower back as well as facial petechiae are suggestive of thrombocytopenia. (A possibility of child abuse should always be kept in mind when evaluating a child with an atypical bruising pattern.) Prolonged epistaxis may be due to thrombocytopenia or other coagulation abnormalities. Bert’s decreased activity may be due to anemia or bone pain. His leg pain may be caused by leukemia or infection. A new-onset acute leukemia is high on the differential diagnosis list due to the subacute progression of symptoms and bone pain. This child’s laboratory evaluation begins with a complete blood cell (CBC) count.
An automated CBC count reveals a white blood cell (WBC) count of 600/μL (0.6 × 109/L) with 100% lymphocytes, hemoglobin of 7.3 g/dL (73 g/L), and platelet count of 9 × 103/μL (9 × 109/L). A pediatric hematologist is consulted; she wants to see Bert later this afternoon.
Acquired Pancytopenias
Leukemia
Leukemias are the most common childhood malignancies, representing approximately 30% of cancer in children. Most of the pediatric leukemias are acute lymphoblastic leukemias (ALLs), followed by acute myeloid leukemias (AMLs).
Presentation.
Signs and symptoms of acute childhood leukemia are often relatively mild and nonspecific; thus, it is easy to miss the diagnosis of new leukemia in a general pediatrics setting. Because progressive pancytopenia and bone pain due to proliferation of leukemic blasts in the bone marrow often evolve over weeks to months, it is not unusual to diagnose leukemia in a child who has a 2-month history of leg pain, minor bruising and bleeding, recurrent fevers, and malaise. Therefore, children who present with mild but recurrent “leg and back pain” deserve a careful examination in the office. Most importantly, these children should never be prescribed systemic corticosteroids before excluding the possibility of leukemia. Unsupervised exposure to systemic corticosteroids in patients who have new-onset leukemia may cause rapid death due to tumor lysis if appropriate inpatient supportive care is not provided or it may worsen the patient’s long-term outcome if the leukemic blasts acquire resistance to corticosteroid-based chemotherapy. Therefore, children with “bone pains” and pancytopenia should be evaluated under the guidance of a pediatric hematologist-oncologist or a pediatric rheumatologist, depending on the overall clinical picture.
Diagnosis.
At the initial evaluation, distinguishing acute leukemia from aplastic anemia may be difficult. Although some children with acute leukemia have a high WBC count with circulating blasts, a low or normal WBC value is not uncommon in new-onset leukemias. Furthermore, a substantial number of children with leukemia do not have peripherally circulating blasts. Even if present, peripheral leukemic blasts may not be recognized by automated cell counters. Therefore, a review of a peripheral blood smear by an experienced hematologist is essential.
Some clinical signs and symptoms may be more consistent with leukemia or aplastic anemia, but a bone marrow aspirate/biopsy must be analyzed by a pediatric hematologist-oncologist to establish the correct diagnosis in a child with pancytopenia (Table 4).
Table 4.
Clinical and Laboratory Differences Between Aplastic Anemia and Leukemia
| Complete Blood Cell Counts | Peripheral Blasts | Other Clinical Findings | Bone Marrow | |
| Aplastic anemia | Pancytopenia | Absent | Possible recent history of hepatitis | Uniformly hypocellular; no leukemic blasts are present |
| Leukemia | Pancytopenia or elevated white blood cell count | Often present but may be absent | Bone pain; lymphadenopathy | May be hypo-, hyper-, or normocellular; leukemic blasts present |
A detailed discussion of childhood leukemias is beyond the scope of this review, but excellent reviews have been recently published elsewhere. (5)(6)(7)
Case 1, continued: The hematologist reviews Bert’s blood smear microscopically and is concerned that some lymphocytes have large nuclei with bizarre chromatin condensation patterns, resembling leukemic blasts. Physical examination reveals normal symmetric testicles without evidence of leukemic involvement. A chest radiograph appears normal; no mediastinal mass is visible. A complete metabolic profile documents elevated lactate dehydrogenase (a marker of cell breakdown), but uric acid, potassium, ionized calcium, and creatinine values are all normal, indicating that Bert does not currently have a tumor lysis syndrome. Aggressive intravenous hydration with potassium-free fluids is started.
Bone marrow aspirate reveals sheets of uniformly appearing large cells (Fig 1B). Flow cytometry classifies 90% of these cells as blasts of acute B-cell lymphoblastic leukemia lineage. Bert is admitted to the hospital for further management. His parents are relieved to hear that the cure rate for standard-risk B-cell ALL is higher than 90% with current therapeutic protocols.
Figure 1.

Bone marrow in acute leukemia and severe aplastic anemia. Normal bone marrow (A) contains hematopoietic cells in various stages of maturation. Bone marrow of a patient with acute leukemia (B) is filled with monotonously appearing large leukemic blasts. Bone marrow in severe aplastic anemia (C) is profoundly hypocellular. Aplastic bone marrow is strikingly “empty,” with visible stromal cells but very few hematopoietic cells.
Aplastic Anemia
Presentation
Case 2: Monica is a 16-year-old soccer player who was brought to her local emergency department after she “passed out” during the game. She “woke up on her own” before the paramedics arrived but complains of “feeling dizzy” and indicates that she “has been more tired and pale for a week.” She has always been healthy except for “stomach pain and nausea” 2 months ago. Laboratory studies reveal hemoglobin of 5.1 g/dL (51 g/L), normal mean corpuscular volume, platelet count of 112 × 103/μL (112 × 109/L), WBC count of 13,000/μL (13 × 109/L), ANC of 300/μL (0.30 × 109/L), and no blasts. All other laboratory results are unremarkable. Monica is admitted to the hospital for a red blood cell transfusion and further diagnostic evaluation.
Acquired severe aplastic anemia (SAA) is a rare, life-threatening disorder often caused by autoimmune destruction of hematopoietic stem cells. As the bone marrow stem cells die under the T-cell-directed assault, the production of healthy blood cells falls. The clinical signs and symptoms of anemia, thrombocytopenia, and leukopenia begin to emerge.
Diagnosis.
A careful physical examination and history is imperative because bone marrow suppression may be caused by multiple environmental and genetic factors (Table 5). Bone marrow should be evaluated promptly. In contrast to acute leukemias, bone marrow in aplastic anemia does not contain malignant blasts. Aplastic bone marrow appears “empty” under the microscope; hypocellularity (the paucity of hematopoietic stem cells) can be striking (Fig 1C). SAA is generally defined as hypocellular bone marrow (<25% cellularity) associated with two of three severe cytopenias: ANC lower than 500/μL (0.50 × 109/L), platelet counts lower than 20 × 103/μL (20 × 109/L), low hemoglobin, and absolute reticulocyte count lower than 20 × 103/μL (20 × 109/L). Patients with SAA require transfusions for survival, and their evaluation and clinical management must be expedited to prevent life-threatening complications of pancytopenia.
Table 5.
Causes of Bone Marrow Aplasia
| Drugs | Cytotoxic drugs |
| Chemotherapeutics | |
| Drugs that may cause bone marrow suppression in susceptible individuals | |
| Antiepileptics, antipsychotics, antibiotics | |
| Toxins | Benzene, alcohol |
| Viral infections | Hepatitis, cytomegalovirus, Epstein-Barr virus, parvovirus, human immunodeficiency virus, human herpesvirus-6 |
| Genetic factors | Congenital bone marrow failure syndromes, including Fanconi anemia, Shwachman-Diamond syndrome, dyskeratosis congenita, Pearson syndrome |
The morphology of aplastic bone marrow is nonspecific. The appearance of bone marrow in other bone marrow diseases, including genetic bone marrow failure syndromes, may be identical. A high index of clinical suspicion is necessary to avoid a wrong diagnosis. Immune-mediated SAA is a diagnosis of exclusion. It is established once other causes of bone marrow aplasia, including inherited bone marrow failure syndromes, are ruled out.
Patients with newly diagnosed bone marrow aplasia should be examined by a pediatric hematologist who has expertise in bone marrow failure syndromes. Diagnostic clues are often discovered through a careful physical examination and history taking. Persistent macrocytosis, reflecting stress hematopoiesis, is seen in multiple bone marrow failure syndromes in the absence of hemolysis. Small stature, facial dysmorphism, developmental delay, café-au-lait macules, or foci of cutaneous hypopigmentation are seen in a wide range of congenital bone marrow failure syndromes, including Fanconi anemia (FA). Abnormal teeth and nails as well as oral leukoplakia raise concerns about dyskeratosis congenita. Chronic diarrhea may direct the diagnostic process toward Shwachman-Diamond syndrome. Recurrent fevers or altered mental status should be managed as neutropenic sepsis until proven otherwise, but they are concerning for hemophagocytic lymphohistiocytosis. Thrombosis, dark urine, and recurrent abdominal pain suggest paroxysmal nocturnal hemoglobinuria (PNH) as the cause of pancytopenia.
A detailed family history interview may reveal family members with unexplained “low blood counts,” miscarriages or early childhood deaths, short stature, unusual infections, developmental abnormalities, or early-onset cancers in children or young adults who experienced unusual treatment toxicities. All the aforementioned issues are concerning for an undiagnosed congenital bone marrow failure syndrome associated with cancer predisposition or immunodeficiency. Family history of early-onset breast or pancreatic cancers may be concerning for FA due to disruption of the BRCA branch of the FA signaling network. Family history of pulmonary fibrosis raises suspicion of dyskeratosis congenita.
The clinician must review all laboratory tests performed since the patient’s birth. We commonly find evidence of mild pancytopenia and macrocytosis reported incidentally several months or years before the onset of clinical symptoms, which is concerning for an undiagnosed genetic problem.
The laboratory evaluation of children who have newly diagnosed SAA varies among pediatric hospitals due to lack of evidence-based guidelines. We routinely administer a panel of screening tests to all children with newly diagnosed SAA to exclude common congenital bone marrow failure syndromes. A chromosome breakage test is performed to exclude FA. Flow cytometry should be used to exclude classic PNH. A review of bone marrow studies and chromosome tests can exclude myelodysplasia. Screening tests for Shwachman-Diamond syndrome and dyskeratosis congenita should be considered as well. A comprehensive infectious disease evaluation, including a search for evidence of viral hepatitis, Epstein-Barr virus, cytomegalovirus, parvovirus, and human immunodeficiency virus, should be pursued. Red blood cell folate and serum vitamin B12 as well as copper should be measured to exclude vitamin and micronutrient deficiencies as the cause of failed hematopoiesis. Human leukocyte antigen (HLA) typing of the patient and siblings should be performed because matched-related stem cell transplant plays an essential role in the treatment of aplastic anemia.
Treatment.
Stem cell transplantation and immunosuppression are two excellent therapeutic options for children with immune-mediated SAA.
Based on current research evidence from observational studies, a matched-related hematopoietic stem cell transplant is considered the best therapeutic option for children with SAA who have an HLA-matched family donor. With current conditioning regimens and meticulous supportive care, the expected 5-year survival is greater than 90%, but chronic graft-versus-host disease and second malignancies contribute to significant morbidity and mortality in survivors. Therefore, long-term follow-up evaluations with a bone marrow failure clinic are essential.
Patients without a matched-related family donor undergo immunosuppression to stop the T-cell attack on the hematopoietic stem cells. In the United States, the first-line immunosuppressive strategy employs a combination of horse antithymocyte globulin (ATG) and cyclosporine (CSA). A brief course of corticosteroids is used to prevent ATG-induced serum sickness (corticosteroids alone have no role in the treatment of SAA). Approximately two out of three children who have SAA begin to recover their blood counts within 3 months of ATG therapy. These patients remain dependent on red blood cell and platelet transfusions while awaiting blood count recovery. Their risk of life-threatening opportunistic bacterial, viral, and fungal infections may increase even further during that time period due to therapy-induced immunosuppression on top of chronic neutropenia. Therefore, infection prophylaxis plays a vital role, and immediate attention to febrile neutropenia is necessary. Granulocyte colony-stimulating factor may be used to boost neutrophil recovery.
Patients who recover their bone marrow function after immunosuppressive therapy require close long-term follow-up evaluations at a bone marrow failure clinic. Hematopoietic progenitors in a subset of these patients undergo a clonal evolution, which may lead to myelodysplasia and leukemia. These patients are also at risk for PNH, solid tumors, and autoimmune disorders. CSA is tapered very slowly because rapid CSA weaning substantially increases the risk of relapse.
Multi-institutional studies are needed to develop evidence-based guidelines for treatment of children who relapse or never recover bone marrow function after immunosuppression. Accumulating evidence suggests that children with SAA who do not recover counts within 3 months after immunosuppression should be offered an unrelated-matched donor hematopoietic stem cell transplantation if a matched donor is available. Alternative therapeutic strategies are available if transplant is not an option, but they have not been systematically evaluated in clinical trials in the pediatric population. Eltrombopag, a small-molecule thrombopoietin receptor activator, improved hematopoiesis in a significant number of adults with refractory SAA in recent clinical trials. (8) The safety and efficacy of eltrombopag in refractory pediatric aplastic anemia remains to be studied in clinical trials.
Case 2, continued: Bone marrow aspirate and biopsy reveal hypocellular bone marrow with no blasts. Monica does not have an HLA-matched family donor, so she undergoes immunotherapy with ATG and is started on CSA. She is discharged home and returns to the clinic for evaluation and transfusions twice weekly. One month after her immunotherapy, she comes to the emergency department with a temperature to 39.3°C (102.7°F), hypotension, and altered mental status. She is started on broad-spectrum antibiotics and admitted to the pediatric intensive care unit for mechanical ventilation and pressor support. Her blood cultures are positive for Klebsiella pneumoniae. She improves clinically and is successfully extubated after 5 days. Nine weeks after immunosuppression, her transfusion needs begin to decrease in parallel with rising neutrophil and reticulocyte counts. She remains under close clinical surveillance.
Paroxysmal Nocturnal Hemoglobinuria
Presentation.
Case 3: Anna is a 13-year-old girl who was seen by her primary doctor twice over the last 3 weeks because of recurrent “stomach pain” and “headaches.” Two weeks ago she was treated empirically for a presumed “urinary tract infection” because she noticed that “her urine was dark” and her urine dipstick test was positive for hemoglobin. This morning she visits an emergency department because of severe headache and “black urine.” Laboratory results include a WBC count of 1,200/μL (1.2 × 109/L), normal differential count, hemoglobin of 9.9 g/dL (99 g/L), reticulocyte count of 12% (0.12), and platelet count of 28 × 103/μL (28 × 109/L). She has undetectable haptoglobin and elevated lactate dehydrogenase consistent with hemolysis. Her electrolytes, creatinine, uric acid, and coagulation profile are all normal.
PNH is an acquired bone marrow disorder that leads to pancytopenia and thrombosis due to complement-mediated breakdown of blood cells. (9) PNH develops when a hematopoietic stem cell undergoes a somatic mutation in PIG-A, an enzyme essential for the attachment of multiple proteins to the cell membrane through a glycosyl phosphatidylinositide anchor. As a consequence, all blood cells produced by the PIG-A mutant cell do not display the surface signature of GPI-anchored proteins. These proteins include CD55 and CD59, two antigens that protect blood cells from complement-mediated destruction. In PNH, the absence of these proteins on blood cell membranes renders cells susceptible to complement lysis.
Destruction of blood cells causes pancytopenia. Children who have PNH experience recurrent abdominal pain, headaches, and malaise out of proportion to their anemia. Hemoglobinuria, the eponymous sign of PNH, is caused by intravascular breakdown of red blood cells and release of hemoglobin into the bloodstream. Hemoglobinuria in PNH is notable on the first morning void, but it may persist throughout the day; in some patients, hemoglobinuria leads to acute or chronic kidney injury.
PNH-associated thrombosis often leads to life-threatening complications such as arterial or venous thrombosis of the central nervous system. The pathophysiology of thrombosis in PNH is not clear, but the intravascular hemolysis appears to play a crucial role because therapeutic inhibition of complement reduces the risk of thrombosis to normal levels.
Diagnosis.
PNH is diagnosed by flow cytometry tests that detect the CD55/CD59 antigens on the surfaces of blood cells. The size of the “PNH clone” is quantified by measuring the fraction of PNH granulocytes (the PNH red blood cells undergo rapid complement lysis in the bloodstream). The blood of a PNH patient contains a mixture of normal blood cells produced by healthy bone marrow cells and the CD55/CD59-negatve “PNH clone” representing the offspring of the PIG-A-deficient hematopoietic stem cell. The size of the PNH clone has diagnostic and therapeutic implications. In classic PNH, more than 50% of blood cells represent the PNH clone. Bone marrow evaluation may not be needed if the diagnosis of classic PNH is established via flow cytometry. Most patients with PNH have normocellular bone marrow, but PNH may evolve into aplastic anemia.
Interestingly, many patients with aplastic anemia have a small but detectable PNH clone. According to observational studies, these patients should not be diagnosed with PNH and do not require PNH therapy as long as the size of their PNH clone is less than 50% (usually is much lower) and there are no clinical or laboratory signs or symptoms of PNH. The intriguing relationship between PNH and aplastic anemia is incompletely understood and will be addressed in future studies.
Treatment.
Medical management of PNH has been revolutionized by eculizumab, the monoclonal antibody against the C5 component of the complement cascade. Clinical trials in adult populations showed that eculizumab rapidly inhibits hemolysis in PNH, improves blood counts, and alleviates clinical symptoms. (10) Importantly, eculizumab appears to reverse the risk of thrombosis back to baseline level. However, most patients treated with eculizumab continue to experience mild ongoing hemolysis. This is likely due to alternative complement activation pathways as well as genetic polymorphisms in the complement pathway. Efforts to develop drugs against the alternative pathway are ongoing, and clinical trials are expected soon.
Hematopoietic stem cell transplant is the only cure for PNH. Limited data indicate that a hematopoietic stem cell transplant is a feasible option for selected children and young adults without history of significant comorbidities such as major thrombosis.
Case 3, continued: Flow cytometry demonstrates 86% PNH granulocytes, consistent with the diagnosis of PNH. Scheduled eculizumab infusions are started. After 2 months, Anna is feeling much less fatigued and her abdominal pain has resolved. Her pancytopenia improves and she no longer needs transfusions. She continues to experience occasional morning hemoglobinuria. A donor search is initiated because Anna and her family are interested in a stem cell transplant to cure her PNH.
Congenital Pancytopenias
Fanconi Anemia
Presentation.
Case 4: Donna is a 5-year-old girl who woke up with fever, malaise, abdominal discomfort, and pain on urination. This is Donna’s third urinary tract infection in the last 2 years. She appears ill and pale, and a laboratory evaluation is initiated. CBC count reveals WBC count of 1,100/μL (1.1 × 109/L), ANC of 420/μL (0.42 × 109/L), hemoglobin of 7.6 g/dL (76 g/L), reticulocyte count of 0.1% (0.001), mean corpuscular volume of 108 fL (108 μm3), and platelet count of 90 × 103/μL (90 × 109/L). Donna has been previously “healthy,” but her height and weight have been under the 3rd percentile since birth. Kids at preschool tease Donna because she is smaller than her peers. She has multiple café-au-lait macules on her back. Donna is admitted to the hospital for intravenous antibiotics to manage febrile neutropenia and for further diagnostics.
FA is a rare, genetically complex multiorgan disorder caused by mutations in one of at least 18 genes of the FA signaling network.
The prevalence of FA is estimated at 1 in 100,000 individuals. Progressive bone marrow failure is the most consistent clinical hallmark. The clinical presentation is exceptionally heterogenous; disruption of the FA pathway may cause abnormal development of all organs. (1) Congenital anomalies encountered in FA include but are not limited to short stature, skeletal aberrations such as deformities of thumbs and radii (Fig 2), café-au-lait macules, VACTERL association (vertebral defects, anal atresia, cardiac defects, tracheoesophageal fistula, renal anomalies, and limb abnormalities), and structural malformations of the urogenital tract. Some patients who have multiple congenital anomalies are recognized soon after birth. However, at least one in three patients does not have any visible developmental anomalies. They may not come to medical attention until their school years, when they develop clinically significant pancytopenia due to bone marrow failure. Several patients with unusual or treatment-resistant cancers have been diagnosed with FA as adults, indicating that the prevalence of FA is likely underestimated.
Figure 2.
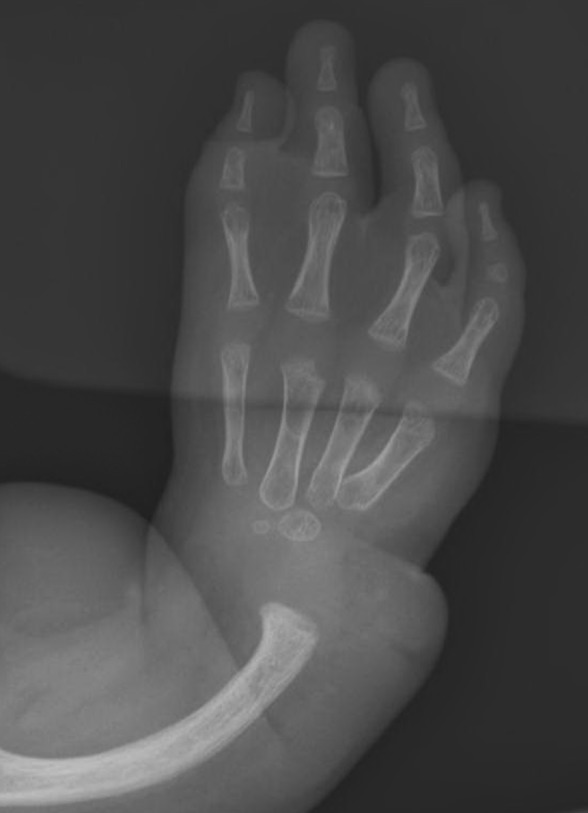
Radial and thumb abnormalities in Fanconi anemia. Absence of the thumb and radius as well as malformed ulna are visible on radiography.
FA is a cancer predisposition syndrome. The FA signaling network works in concert to maintain genome fidelity, and disruption of FA genes promotes spontaneous mutagenesis and increases the risk of cancer. (11) Clinical data estimate the lifetime risk of cancer in FA at 20%. AML is the most common FA-associated cancer in early childhood, followed by solid tumors and squamous cell carcinomas in older children.
Diagnosis.
A chromosome breakage test is a sensitive and specific diagnostic tool to screen for FA. It is one of the most useful tests in the diagnostic armamentarium of a pediatric hematologist-oncologist. This test is based on the fact that a low concentration of DNA-crosslinking chemical such as DEB (diepoxybutane) or MMC (mitomycin C) causes chromosome breakage in FA patient cells but not in normal cells.
A positive chromosome breakage test establishes a diagnosis of FA. This is followed by a genetic evaluation to identify the affected gene. A rational approach to genetic diagnostics of FA is essential, given the large number of FA genes. A next-generation parallel sequencing of all FA genes may be pursued. In some patients, multiple variants of unknown significance are identified in several FA genes, and functional complementation studies are required to identify the disease-causing mutation. Alternatively, candidate gene sequencing may be performed first if the patient’s presentation offers sufficient clues to predict what gene may be affected. The genotype-phenotype correlations in FA are only beginning to emerge, but such prediction can be made for selected patients. For example, a child with new-onset FA and a strong family history of early-onset breast cancer may have a biallelic mutation in BRCA2/FANCD1 or one of its binding partners.
Comprehensive guidelines for diagnosis and management of FA are provided by a panel of experts at the Fanconi Anemia Research Fund. Children with newly diagnosed FA should undergo a comprehensive evaluation to screen for clinically silent developmental abnormalities. Creatinine and electrolytes are assessed to screen for kidney dysfunction. Ultrasonographic examination of the abdomen and pelvis is performed to detect renal dysplasia, hydronephrosis, and other functionally important structural aberrations of the urogenital system. The child who has a history of recurrent urinary tract infection should be referred to urology for further studies, such as voiding cystourethrography to determine whether vesicoureteral reflux is present. A formal referral to an endocrinologist is warranted to screen for hypothyroidism, growth hormone deficiency, dyslipidemia, osteopenia, and hyperglycemia. An audiology evaluation is performed to screen for hearing loss. Other tests are guided by the patient’s past medical history.
Treatment.
Supportive care is personalized, based on the results of the baseline evaluation. Most patients experience progressive bone marrow failure over time, but there is substantial variation among patients. Thus, CBC counts are monitored on a scheduled basis to follow the trajectory of pancytopenia. Bone marrow aspirates/biopsies and cytogenetic studies are obtained annually or as clinically needed due to the high risk of myelodysplasia and AML. Multiple observational studies show that hematopoietic stem cell transplantation may be lifesaving for FA patients who develop transfusion-dependent pancytopenia or AML. FA-specific stem cell transplantation protocols must be used because patients are hypersensitive to chemotherapeutics and chemotherapy doses must be minimized to prevent life-threatening complications. Because cancers in FA are extremely difficult to treat, an aggressive surveillance strategy is instituted.
Case 4 continued: Donna’s clinical status quickly improves with intravenous antibiotics, and her ANC increases to 800/μL (0.8 × 109/L). Renal ultrasonography reveals no findings of note. A bone marrow aspirate performed to exclude new-onset leukemia shows hypocellular bone marrow with no blasts. Clinicians presumptively diagnose “viral bone marrow suppression.” One month later, Donna remains healthy, but her follow-up CBC count shows stable pancytopenia, with WBC count of 12,000/μL (12 × 109/L), hemoglobin of 8.1 g/dL (81 g/L), MCV of 106 fL (106 μm3), platelet count of 110 × 103/μL (110 × 109/L), and reticulocyte count of 0.1% (0.001). A chromosome breakage test yields positive results, confirming the clinical suspicion of FA and indicating that Donna’s short stature and frequent infections were due to FA. An FA gene sequencing panel shows two potentially pathogenic mutations in FANCA. Follow-up genetic tests confirm that Donna’s parents are heterozygous carriers of these FANCA mutations. A chromosome breakage test is ordered for Donna’s younger sisters, Kris and Paula. The test result is negative in Paula but positive in Kris, indicating that Kris has undiagnosed FA. A hematologist experienced in treating FA will establish a comprehensive personalized treatment plan for both girls.
Shwachman-Diamond Syndrome
Presentation.
Case 5: Christopher is a 10-month-old boy who has experienced both failure to thrive and feeding dysfunction. During a health supervision visit, his mother mentions that he had diarrhea for the last 3 months. Because he appears pale, a CBC count is ordered. Christopher’s WBC count is 1,200/μL (1.2 × 109/L), ANC is 200/μL (0.2 × 109/L), hemoglobin is 10 g/dL (100 g/L), MCV is 99 fL (99 μm3), and platelet count is 120 × 103/μL (120 × 109/L).
Shwachman-Diamond syndrome (also known as Shwachman-Bodian-Diamond syndrome [SBDS]) is a genetic disorder associated with pancytopenia, exocrine pancreatic insufficiency, skeletal malformations, and developmental delay. The incidence of this syndrome is estimated at 1 in 76,000 individuals. Patients often come to medical attention due to chronic diarrhea and failure to thrive. Most require supplementation with pancreatic enzymes due to exocrine pancreatic insufficiency. The endocrine pancreatic function is normal.
Progressive bone marrow failure often starts in early childhood. Skeletal malformations range from clinically insignificant abnormalities of long bone metaphyses to potentially lethal thoracic abnormalities leading to respiratory failure. Delayed ossification and osteopenia are universal. Although many have SBDS diagnosed in the first postnatal year, the syndrome may be diagnosed in older children because clinical signs and symptoms, as with other inherited bone marrow failure syndromes, are highly heterogenous.
Diagnosis.
Approximately 90% patients clinically diagnosed with SBDS have autosomal recessive mutations in the SBDS gene. The SBDS gene localizes to the nucleolus and the mitotic spindle in a cell cycle-dependent manner to regulate ribosome assembly in interphase and chromosome segregation during mitosis.
The evaluation of pancytopenia begins with a bone marrow examination to exclude leukemia. Chronic pancytopenia in combination with hypocellular bone marrow, history of chronic diarrhea, and skeletal abnormalities should direct the diagnostic process toward SBDS. However, the absence of these physical findings does not exclude the syndrome; some affected patients have subclinical pancreatic insufficiency and no skeletal abnormalities. Quantification of age-appropriate pancreatic enzyme concentrations in serum assists with the diagnosis. Low serum trypsinogen values are seen in affected children younger than age 3 years, but low serum isoamylase values are more sensitive in older children. SBDS gene sequencing confirms the diagnosis and allows genetic counseling for most patients. A skeletal survey helps detect bone abnormalities. Pancreatic imaging reveals replacement of exocrine pancreas with fatty tissue.
Treatment.
Expert guidelines for diagnosis and management of SBDS have been published. (12) The complex medical needs of affected children should be coordinated by an expert hematologist-oncologist. Blood counts should be followed regularly because these children may progress into severe transfusion-dependent bone marrow failure. A significant number of patients require a stem cell transplant to restore normal hematopoiesis. Because the risk of myelodysplasia and AML is significantly higher compared to the general population, a bone marrow evaluation, including karyotype and MDS (myelodysplastic syndrome) genetic fluorescence in situ hybridization screening panel, is performed yearly. Pancreatic enzyme supplementation is managed by a pediatric gastroenterology specialist with expertise in management of SBDA.
Case 5 continued: A bone marrow evaluation reveals a hypocellular bone marrow with no blasts. Results of a chromosome breakage test are negative, excluding FA. Laboratory tests show extremely low serum trypsinogen values consistent with exocrine pancreatic insufficiency, which explains Christopher’s diarrhea. He is referred to gastroenterology to initiate pancreatic enzyme supplementation. Skeletal survey does not reveal any bone abnormalities other than delayed ossification of carpal bones, widened distal ulnae and radii, and generalized osteopenia. A Shwachman-Diamond gene sequencing reveals biallelic mutations in the SBDS gene that had been described as disease-associated. Genetic counseling is provided to the family. A multidisciplinary management plan is developed with collaboration among pediatric hematology, clinical genetics, pediatric gastroenterology, and an endocrinologist.
Other Inherited Bone Marrow Failure Syndromes
Other inherited bone marrow failure syndromes are briefly summarized in Table 6. Because not all bone marrow failure-associated genes have been identified, the disease-causing genes are never identified for a substantial number of patients who have chronic pancytopenia and developmental abnormalities. Advances in clinical exome/genome sequencing facilitate our diagnostic efforts and guide our personalized therapeutic strategies for these complex and fascinating patients.
Table 6.
Selected Inherited Bone Marrow Failure Syndromes*
| Fanconi anemia (FA) | Genetically and clinically heterogenous syndrome of progressive bone marrow failure accompanied by a wide variety of heterogenous developmental abnormalities, such as growth failure, missing or dysmorphic thumbs and radii, craniofacial abnormalities, VACTERL association, renal or urogenital dysplasia, and multiple café-au-lait macules. FA is caused by germline mutation of one of at least 18 FA genes that have been identified to date. FA is associated with a high risk of acute leukemias and solid tumors due to genomic instability. May have autosomal recessive or X-linked inheritance, depending on the affected gene. |
| Laboratory screening tool: chromosome breakage test (positive result is diagnostic of FA). | |
| Shwachman-Diamond syndrome | Progressive pancytopenia associated with failure to thrive, severe chronic diarrhea due to exocrine pancreatic insufficiency, skeletal abnormalities, and increased risk of myelodysplasia and leukemia. Autosomal recessive inheritance. |
| Laboratory screening tool: age-adjusted pancreatic enzyme concentrations. | |
| Dyskeratosis congenita (DC) | Genetically heterogenous disease that causes bone marrow failure and predisposition to cancer in addition to dystrophy of teeth and nails, oral leukoplakia, and reticulated hyperpigmentation of the skin. DC may cause pulmonary fibrosis, immunodeficiency, and liver disease. DC is caused by mutation of one of at least nine genes essential for the maintenance of telomeres (the DNA/protein complexes localized at the tips of chromosomes). Without telomerase, telomeres shorten with each cell division, eventually leading to decreased stem cell proliferation and genomic instability. Affected patients have a high risk of cancer and may require a stem cell transplant due to progressive bone marrow failure. Depending on the affected gene, DC may have autosomal recessive, autosomal dominant, or X-linked inheritance. |
| Laboratory screening tool: telomere length measurement in lymphocytes. | |
| Pearson syndrome | Genetic disorder caused by large deletions of mitochondrial DNA. Often diagnosed in the neonatal period due to growth failure, developmental delay, macrocytic anemia (frequently evolving into pancytopenia), metabolic acidosis, and exocrine pancreatic insufficiency. Bone marrow evaluation reveals multiple vacuoles in the hematopoietic cells. Most patients die in early childhood; survivors develop severe neuromuscular signs and symptoms due to mitochondrial dysfunction. |
| Cartilage-hair hypoplasia | Genetic syndrome with diverse features of short-limbed dwarfism, sparse and fine hair, immunodeficiency, autoimmunity, gastrointestinal dysfunction, pancytopenia, and predisposition to hematopoietic malignancies. This rare autosomal recessive disorder is caused by mutations in the RMRP (mitochondrial RNA-processing endonuclease) gene. Hematopoietic stem cell transplant may be used to cure immunodeficiency. |
| Congenital amegakaryocytic thrombocytopenia | Rare autosomal recessive disorder caused by mutations in the thrombopoietin receptor (MPL). Patients often present with severe thrombocytopenia at birth (which may lead to life-threatening hemorrhage) and reduced number of megakaryocytes in the bone marrow. Progressive bone marrow failure occurs in early childhood, indicating the importance of thrombopoietin for normal hematopoiesis. A hematopoietic stem cell transplant may be needed. |
Specific genetic tests are available for all the genetic syndromes listed in the table. Useful laboratory tools are highlighted where available for specific bone marrow failure syndromes.
Summary.
On the basis of strong evidence, chronic pancytopenia in children may have a variety of acquired and congenital causes.
On the basis of consensus, primary clinicians play a key role in identifying children who have pancytopenia. A high index of suspicion is indispensable because signs and symptoms of low blood cell counts are often nonspecific. Children with malaise, fatigue, pallor, bone pain, lymphadenopathy, easy bruising and bleeding, recurrent fevers or mouth ulcers, or dizziness should be screened with a complete blood cell count and differential count.
On the basis of observational studies, children with acute leukemias occasionally present with pancytopenia and no leukemic blasts in peripheral blood.
On the basis of strong evidence, acquired aplastic anemia is caused by autoimmune destruction of hematopoietic cells in the bone marrow. It is currently treated by stem cell transplantation or antithymocyte globulin/cyclosporine-based immunosuppression after inherited bone marrow failure syndromes are excluded.
On the basis of strong evidence, paroxysmal nocturnal hemoglobinuria (PNH) is an acquired disorder that renders blood cells susceptible to complement-mediated destruction. PNH presents as pancytopenia, hemoglobinuria manifesting as dark urine, abdominal pain, and high risk of life-threatening thrombosis. Symptoms of PNH are successfully alleviated with the anticomplement antibody eculizumab (although clinical trials in children are needed to establish the efficacy and safety of eculizumab in childhood PNH). Stem cell transplantation provides a curative option.
On the basis of observational studies, children with inherited bone marrow failure syndromes may present with subtle developmental abnormalities before the onset of clinically significant pancytopenia. Thus, patients with failure to thrive, skeletal abnormalities that include radial ray dysplasia/thumb abnormalities, craniofacial dysmorphism, VACTERL syndrome, nail/tooth abnormalities, multiple café-au-lait macules, foci of skin hypo/hyperpigmentation, or oral leukoplakia should be screened via a complete blood cell count with differential count.
On the basis of consensus, children with pancytopenia require prompt referral to a pediatric hematologist-oncologist with expertise in bone marrow failure for further diagnostics and management.
Acknowledgements
We are grateful to Diana Rains, MT, ACSP, SH, for sharing her collection of bone marrow slides (Figs. 1 and 2). We are indebted to patients and families treated in our Pediatric Bone Marrow Failure Clinic for their continued generous support of our research.
Footnotes
AUTHOR DISCLOSURE
Dr. Sharma has disclosed no financial relationships relevant to this article. Dr. Nalepa has disclosed that research in his laboratory is supported through NIH Indiana Pediatric Scientist Award, a research grant from the Heroes Foundation, and Barth Syndrome Fund via Riley Children’s Foundation. This commentary does contain a discussion of an unapproved/investigative use of a commercial product/device.
References
- 1.Auerbach AD. Fanconi anemia and its diagnosis. Mutat Res. 2009;668(1-2):4–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Williams DA, Bennett C, Bertuch A, et al. Diagnosis and treatment of pediatric acquired aplastic anemia (AAA): an initial survey of the North American Pediatric Aplastic Anemia Consortium (NAPAAC). Pediatr Blood Cancer. 2014;61(5):869–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hartung HD, Olson TS, Bessler M. Acquired aplastic anemia in children. Pediatr Clin North Am. 2013;60(6):1311–1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lehrnbecher T, Phillips R, Alexander S, et al. ; International Pediatric Fever and Neutropenia Guideline Panel. Guideline for the management of fever and neutropenia in children with cancer and/or undergoing hematopoietic stem-cell transplantation. J Clin Oncol. 2012;30(35):4427–4438 [DOI] [PubMed] [Google Scholar]
- 5.Gamis AS, Alonzo TA, Perentesis JP, Meshinchi S; COG Acute Myeloid Leukemia Committee. Children’s Oncology Group’s 2013 blueprint for research: acute myeloid leukemia. Pediatr Blood Cancer. 2013;60(6):964–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hunger SP, Loh ML, Whitlock JA, et al. ; COG Acute Lymphoblastic Leukemia Committee. Children’s Oncology Group’s 2013 blueprint for research: acute lymphoblastic leukemia. Pediatr Blood Cancer. 2013;60(6):957–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Inaba H, Greaves M, Mullighan CG. Acute lymphoblastic leukaemia. Lancet. 2013;381(9881):1943–1955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olnes MJ, Scheinberg P, Calvo KR, et al. Eltrombopag and improved hematopoiesis in refractory aplastic anemia. N Engl J Med. 2012;367(1):11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parker CJ. Paroxysmal nocturnal hemoglobinuria. Curr Opin Hematol. 2012;19(3):141–148 [DOI] [PubMed] [Google Scholar]
- 10.Hillmen P, Hall C, Marsh JC, et al. Effect of eculizumab on hemolysis and transfusion requirements in patients with paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2004;350(6):552–559 [DOI] [PubMed] [Google Scholar]
- 11.Kottemann MC, Smogorzewska A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature. 2013;493(7432):356–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dror Y, Donadieu J, Koglmeier J, et al. Draft consensus guidelines for diagnosis and treatment of Shwachman-Diamond syndrome. Ann N Y Acad Sci. 2011;1242:40–55 [DOI] [PubMed] [Google Scholar]