

Abstract
Enhanced receptiveness at all synapses on a neuron that receive glutamatergic input is called cell-wide synaptic upscaling. We hypothesize that this type of synaptic plasticity may be critical for long-term memory storage within cortical circuits, a process that may also depend on epigenetic mechanisms, such as covalent chemical modification of DNA. We found that DNA cytosine demethylation mediates multiplicative synaptic upscaling of glutamatergic synaptic strength in cultured cortical neurons. Inhibiting neuronal activity with tetrodotoxin (TTX) decreased the cytosine methylation of and increased the expression of genes encoding glutamate receptors and trafficking proteins, in turn increasing the amplitude but not frequency of miniature excitatory postsynaptic currents (mEPSCs), indicating synaptic upscaling rather than increased spontaneous activity. Inhibiting DNA methyltransferase (DNMT) activity, either by using the small-molecule inhibitor RG108 or by knocking down Dnmt1 and Dnmt3a, induced synaptic upscaling to a similar magnitude as exposure to TTX. Moreover, upscaling induced by DNMT inhibition required transcription; the RNA polymerase inhibitor actinomycin D blocked upscaling induced by DNMT inhibition. Knocking down the cytosine demethylase TET1 also blocked the upscaling effects of RG108. DNMT inhibition induced a multiplicative increase in mEPSC amplitude, indicating that the alterations in glutamate receptor abundance occurred in a coordinated manner throughout a neuron and were not limited to individual active synapses. Our data suggest that DNA methylation status controls transcription-dependent regulation of glutamatergic synaptic homeostasis. Furthermore, covalent DNA modifications may contribute to synaptic plasticity events that underlie the formation and stabilization of memories.
INTRODUCTION
DNA methylation is a powerful and extensively investigated epigenetic mechanism. Until recently, it was thought that once laid down, DNA methylation marks remain unchanged for the lifetime of the organism, although recent studies have challenged this view and indicate that DNA cytosine methylation is actively regulated in nondividing neurons in the adult central nervous system (CNS) (1, 2). In neurons, as in other cells, DNA methylation and attendant changes in chromatin structure are driven by the actions of DNA methyltransferases (DNMTs). “De novo” DNMTs (DNMT3a and 3b) methylate cytosines on a single DNA strand, for example, at novel previously unmethylated sites in the genome. The “maintenance” DNMT (DNMT1) recognizes a hemimethylated C-G dinucleotide (meaning, methylated on only one strand of DNA by a de novo DNMT) and converts the complementary C-G on the opposite strand into a methylated C-G. Through this mechanism, DNA can be perpetually methylated in the face of ongoing turnover of molecular marks. For this reason, many years ago, Francis Crick proposed that a self-perpetuating biochemical autoconversion of methylated DNA might serve as a memory mechanism at the molecular level (3, 4). Because DNA methylation is one of the few biochemical mechanisms known that can perpetuate itself indefinitely in the face of ongoing molecular turnover, it is an intriguing possibility that regulation of DNA methylation plays a role in memory stabilization in the adult nervous system.
Several pieces of evidence are now available that support the idea that active regulation of DNA methylation plays a role in memory function in the adult CNS (5). Work by Levenson et al. (6) demonstrated that general inhibitors of DNMT activity alter DNA methylation in the adult brain and alter the DNA methylation status of the plasticity-promoting genes reelin (Reln) and brain-derived neurotrophic factor (Bdnf). Additional studies demonstrated that de novo DNMT expression is up-regulated in the adult rat hippocampus after contextual fear conditioning and that blocking DNMT activity blocked contextual fear conditioning (7–10). In addition, fear conditioning is associated with rapid methylation and transcriptional silencing of the memory suppressor gene protein phosphatase 1 (PP1) and demethylation and transcriptional activation of the plasticity gene Reln. These findings had the surprising implication that both active DNA methylation and active demethylation might be involved in long-term memory consolidation in the adult CNS. An additional series of studies found that the Bdnf gene locus is also subject to memory-associated changes in DNA methylation and that this effect is regulated by the N-methyl-D-aspartate (NMDA) receptor (11), and that neuronal DNMT-deficient animals have deficits in contextual fear conditioning, Morris maze performance, and hippocampal long-term potentiation (7). Most recently, DNA methylation has been directly implicated as contributing to memory maintenance in the cortex (9), to reward conditioning in the midbrain dopamine circuit (12), and to hippocampal place cell firing pattern stability (2). As a motivator for the current studies, a number of laboratories have of late independently discovered that TET1 oxygenase controls CNS 5-methylcytosine hydroxylation, active DNA demethylation, memory-associated gene transcription, and hippocampus-dependent memory formation (1, 13–21).
These various results support the emerging idea that DNA methylation is dynamically regulated in the adult CNS in response to experience and that active alterations in DNA methylation mediate memory formation and storage in the adult rat hippocampus and cortex. In brief, DNA cytosine methylation is now considered to be a strong candidate as one of the core mechanisms underlying the transcriptional regulation necessary for long-term memory formation and stabilization. However, the cellular and neurophysiologic mechanisms subserving the contribution of epigenetic mechanisms to memory stabilization remain an open question (5, 22).
Because epigenetic mechanisms such as cytosine methylation drive cell-wide transcriptional changes, cell-wide forms of synaptic plasticity are an appealing category of neurophysiologic mechanisms that might be epigenetically targeted and contribute to memory stabilization (22, 23). Synaptic scaling is a form of homeostatic synaptic plasticity (HSP) characterized by cell-wide changes in postsynaptic receptor density. These global modifications in synaptic strength are initiated cell-autonomously over chronic time scales and can occur bidirectionally in response to altered neuronal activity (24–27). Synaptic scaling can occur through multiplicative changes in receptor density that occur simultaneously across an entire complement of synapses in a neuron, over time preserving the distributions of relative synaptic strengths within a single cell (meaning, the weighted contribution of each single synapse to the total cellular complement). During memory formation, multiplicative scaling may provide the cellular stability necessary for the continued storage of experientially obtained site-specific changes in synaptic strength in the face of changes in synaptic strength at other synapses (24, 27). Although it is known that synaptic scaling depends on the transcriptional regulation of multiple genes (26, 28, 29), the mechanisms required to coordinate these large-scale neuron-wide transcriptional changes are unclear.
These considerations imply that a fundamental role for epigenetic mechanisms in memory stabilization and storage might be the control of global synaptic strength via synaptic scaling at the single-neuron level. Specifically, DNA methylation might represent a focal point of control in the large-scale transcriptional regulation necessary for multiplicative synaptic scaling at excitatory CNS synapses. Thus, in the present studies, we investigated whether there is a role for DNMT activity in the activity-dependent upscaling of synaptic strength at glutamatergic synapses in cortical neurons.
RESULTS
Homeostatic upscaling of excitatory strength is associated with altered transcription of the DNA demethylation pathway
We initially sought to investigate the role of DNA methylation in homeostatic synaptic upscaling induced by treatment with tetrodotoxin (TTX) to chronically decrease activity in rat cortical neurons. After a 24-hour TTX treatment of cultured primary cortical neurons, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor–mediated miniature excitatory postsynaptic currents (mEPSCs) were measured from visually identified pyramidal cells using whole-cell electrophysiology (Fig. 1, A to C). Measuring AMPA receptor–mediated mEPSCs enabled the assessment of postsynaptic strength and presynaptic properties of glutamatergic excitatory synaptic transmission. In recorded cells, TTX produced a right shift in the cumulative distribution of mEPSC amplitudes and increased mean mEPSC amplitude in a manner consistent with the increased post-synaptic AMPA receptor density that is associated with homeostatic up-scaling (Fig. 1B) (25, 27). TTX did not affect the mean mEPSC frequency in recorded cells (Fig. 1C), suggesting no change in presynaptic properties.
Fig. 1. Homeostatic upscaling of excitatory strength is associated with alterations in mRNAs for gene products associated with the regulation of DNA methylation.

(A) Sample mEPSC records from cortical pyramidal neurons after 24 hours of exposure with control (CTL) or TTX (blue). (B and C) Cumulative probability distributions and mean mEPSC amplitudes (B) and mean mEPSC frequencies (C) from cortical pyramidal neurons treated with TTX. Bar graphs are means ± SEM from cells pooled from at least six experiments for each condition (CTL, n = 21 cells; TTX, n = 15 cells). (B) P < 0.001, Kolmogorov-Smirnov (K-S) test. Inset: *P < 0.001, Mann-Whitney (M-W) test. (D) Rank order plot of 1000 randomly selected mEPSC amplitudes from CTL and TTX (blue values). Fitting the data with y = ax (dashed black line) yielded a slope of 1.29, which was used to scale down amplitudes from TTX-treated cells in (B). The solid black line represents unity. (E) Cumulative probability distributions of scaled-down TTX-induced amplitudes compared to those from CTL cells; P = 0.1024, K-S test. (F) Polymerase chain reaction (PCR) detection of transcript expression relative to CTL after 24-hour TTX treatment. Dashed vertical line represents the average, normalized gene expression values of CTL cells. Data are means ± SEM from three experiments. *P < 0.05, Student’s unpaired t test.
Recently, an improved analytical method for evaluating mEPSC amplitudes was described that is more effective than conventional approaches for discriminating between multiplicative and nonmultiplicative changes (30). Using this approach in combination with other methods (31, 32), we quantitatively assessed the manner by which TTX increased mEPSC amplitudes (Fig. 1, D and E). One thousand randomly selected mEPSC amplitudes from the 10th to 90th percentile of total events from control and TTX groups were rank-ordered and plotted, and a linear relationship between the groups was fitted. We then used the fitted curves to determine the scaling factor (in this case, 1.29) with which to divide the 2119 total amplitudes in the TTX-treated group data, discarding 130 resulting amplitudes that fell below our detection threshold (5.00 pA). Normalized TTX-induced mEPSC amplitudes were not different than those in controls, indicating that a 24-hour TTX treatment multiplicatively increased mEPSC amplitude in recorded pyramidal neurons, suggesting a cell-wide increase in postsynaptic strength (Fig. 1, D and E).
Next, we investigated transcriptional changes associated with homeostatic upscaling. In cultured neurons, a 24-hour TTX treatment increased the transcription of Gria1, encoding an AMPA receptor, and decreased the transcription of the genes Arc (also known as Arg3.1) and Bdnf, encoding the AMPA receptor trafficking protein and a synaptic regulatory protein, respectively (Fig. 1F). These observations confirm that homeostatic up-scaling is associated with targeted transcriptional changes in genes that regulate excitatory synaptic strength (33–35). Additionally, we examined changes in mRNA expression of genes encoding proteins involved in regulating DNA methylation and demethylation (Fig. 1F). Of the nine candidate transcripts we examined, TTX treatment specifically increased the expression of Tet1 and Gadd45b, genes involved in DNA demethylation, neuronal plasticity, and learning and memory (1, 13–21). Together, these results suggest an important link between transcriptional regulation, DNA methylation, and homeostatic synaptic scaling.
Pharmacological DNMT inhibition induces multiplicative upscaling of excitatory strength
We next used a pharmacological approach to directly examine the role of DNA methylation in excitatory synaptic transmission in rat cortical pyramidal neurons. RG108 is a small-molecule, competitive DNMT antagonist that inhibits DNA methylation. RG108 is stable in culture with a half-life of ~20 days at 37°C (36, 37). Previous studies in neurons have shown that RG108 blocks activity-dependent methylation in cultured cells and in vivo (9, 12, 13, 38). We treated cultured cortical pyramidal neurons for 24 hours with 100 μM RG108 and subsequently recorded mEPSCs (Fig. 2, A to C). In recorded cells, RG108 treatment produced a rightward shift in the cumulative distribution of mEPSC amplitudes and increased mean mEPSC amplitude (Fig. 2B). RG108 also increased mean mEPSC frequency in recorded neurons (Fig. 2C). Together, these results demonstrate that chronic, direct pharmacological DNMT inhibition enhances both pre-synaptic and postsynaptic components of excitatory synaptic transmission in pyramidal neurons.
Fig. 2. DNMT inhibition with RG108 multiplicatively upscales excitatory strength.

(A) Sample mEPSC records from cortical pyramidal neurons after 24-hour treatment with control (CTL) or RG108 (red). (B and C) Cumulative probability distributions and mean mEPSC amplitudes (B) and mean mEPSC frequencies (C) from cortical pyramidal neurons treated with RG108. Bar graphs are means ± SEM from cells pooled from at least four experiments for each condition (CTL, n = 12 cells; RG108, n = 12 cells). (B) P < 0.001, K-S test. Inset: *P < 0.001, M-W test. (C) *P < 0.05, Student’s unpaired t test. (D) Rank order plot of 1000 randomly selected mEPSC amplitudes from CTL and RG108-treated cells (red values). Fitting the data with y = ax yielded a scaling factor of 1.23, which was used to scale down amplitudes from RG108-treated cells in (B). (E) Cumulative probability distributions of scaled-down RG108-induced amplitudes compared to those from CTL cells. P = 0.2645, K-S test. Inset: Testing a range of scaling factors (a) between 1.1 and 1.3 revealed that RG108-induced amplitudes from (B) scaled down with a = 1.23 resulted in the highest K-S P value (0.2645) (red dot) compared to CTL.
Using the quantitative approach described above, we found that RG108 increased cumulative mEPSC amplitudes in a multiplicative manner (Fig. 2, D and E). Linear regression of rank-ordered mEPSC amplitudes from control and RG108-treated neurons revealed a scaling factor of 1.23 (Fig. 2D); once adjusted by this scaling factor, amplitudes in RG108-treated neurons were not different than those in control neurons (Fig. 2E). Furthermore, we tested a range of scaling factors between 1.1 and 1.3 (Fig. 2E, inset). RG108-induced amplitudes that were scaled down (normalized) by a factor of 1.23 resulted in the greatest degree of overlap of cumulative amplitude distributions between control and scaled RG108-treated neurons, according to a K-S test, hence validating our analysis. These results demonstrated that chronic, direct pharmacological inhibition of DNA methylation multiplicatively upscales excitatory strength. The multiplicative nature of the upscaling with DNMT inhibition suggests a coordinated up-regulation of the entire neuron-wide complement of synapses, an observation consistent with the idea that the epigenome has the capacity to drive a state change across the entire neuron (10, 11).
Previously, it was shown that inhibiting DNA methylation in cultured hippocampal neurons for 24 hours with the nucleoside analog 5-azacytidine or zebularine decreased mEPSC frequency with no effect on amplitude (39). In our cortical cultures, we were able to replicate this finding after a 24-hour exposure to zebularine (fig. S1). However, nucleoside inhibitors such as zebularine may not be ideally suited to study activity-dependent changes in DNA methylation over chronic time courses in postmitotic neurons because they must be incorporated into DNA and form covalent complexes with DNMTs (40–42), and they are inherently more cytotoxic than RG108 (36, 38, 43). In addition, in a series of control experiments, we observed that antisense oligonucleotide (ASO)–mediated knockdown of the TET1 dioxygenase, a regulator of activity-dependent cytosine demethylation in neurons (1, 14, 15, 19), blocked the capacity of RG108 to induce synaptic scaling (fig. S2). These control data strongly indicate that the effect of RG108 to induce synaptic upscaling is dependent on cytosine demethylation. For these reasons, we used RG108 for the remainder of our pharmacological experiments.
DNMT inhibition–dependent upscaling requires neuronal activity
Homeostatic synaptic scaling is driven cell-autonomously in response to chronically altered postsynaptic depolarization (25, 34, 44). We hypothesized that pharmacological DNMT inhibition upscaled excitatory strength in an activity-dependent manner. To examine the underlying neuronal activity required for this effect, we co-incubated cultured cortical neurons with RG108 and APV (D-2-amino-5-phosphonovaleric acid), APV + NBQX (2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide), or TTX for 24 hours. APV, NBQX, and TTX, respectively, antagonize NMDA receptors, AMPA receptors, or suppress Na+-dependent action potential generation. The combination of AMPA receptor and NMDA receptor antagonism inhibits glutamatergic signaling in pyramidal neurons. In recorded pyramidal cells, co-incubation with RG108 and APV significantly increased mEPSC amplitude and frequency, whereas co-incubation with RG108 and APV in the presence of NBQX or TTX did not affect mEPSC amplitude or frequency (Fig. 3, A to C). Given that RG108-induced mEPSC alterations could be independently blocked by antagonism of either AMPA receptors or Na+ channels, these results suggest that upscaling mediated by chronic, direct pharmacological DNMT inhibition requires neuronal activity and suggest a specific role for glutamatergic signaling through AMPA receptors for DNMT inhibition to trigger upscaling.
Fig. 3. Induction of excitatory synaptic scaling by inhibition of DNA methylation is activity-dependent.
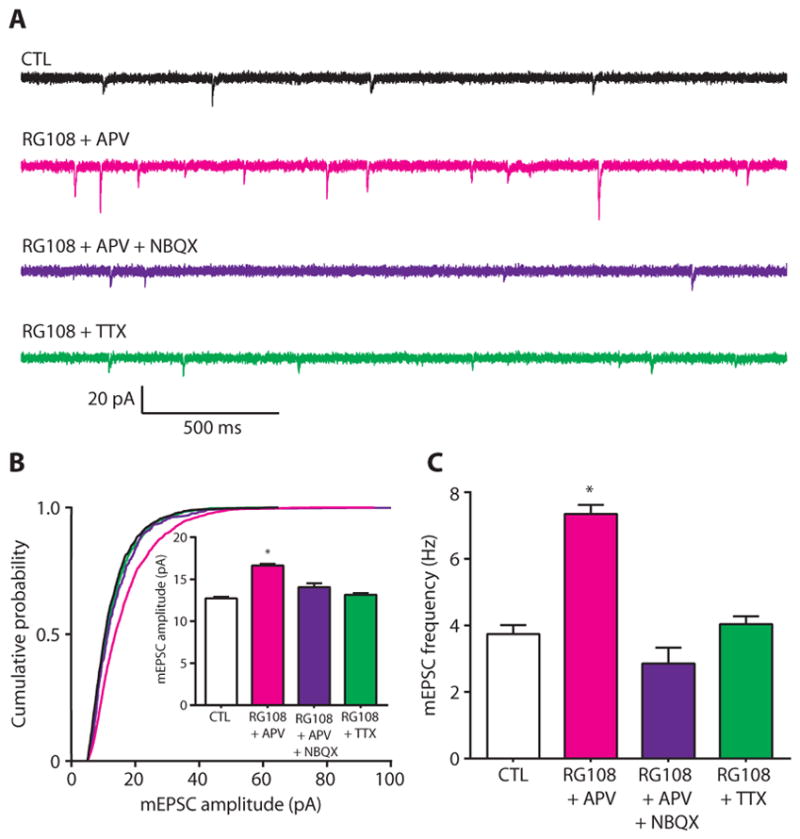
(A) Sample mEPSC traces from cortical pyramidal neurons after 24 hours of exposure to control (CTL), RG108 + APV (magenta), RG018 + APV + NBQX (purple), or RG108 + TTX (green). (B and C) Cumulative probability distributions and mean mEPSC amplitudes (B) and mean mEPSC frequencies (C) from cortical pyramidal neurons treated with RG108 and APV; RG108, APV, and NBQX; or RG108 and TTX. Bar graphs are means ± SEM. Data are cumulative of cells pooled from at least three experiments for each condition (CTL, n = 17 cells; RG108 + APV, n = 9 cells; RG108 + APV + NBQX, n = 6 cells; and RG108 + TTX, n = 12 cells). (B) *P < 0.001 compared to CTL, Kruskal-Wallis (K-W) test followed by Dunn’s test. (C) *P < 0.05 compared to CTL, analysis of variance (ANOVA) followed by Dunnett’s test.
Gene expression is necessary for DNMT inhibition–dependent upscaling
Epigenetic mechanisms, such as DNA methylation, regulate transcription across the genome (13). We hypothesized that chronically inhibiting DNA methylation upscales excitatory strength by altering gene expression. To test this hypothesis, we recorded mEPSCs in cortical pyramidal neurons after a 24-hour co-incubation with the broadly acting transcriptional inhibitor actinomycin D in the presence or absence of RG108. In recorded pyramidal neurons, neither actinomycin D alone nor actinomycin D combined with RG108 had any effect on mEPSC amplitude or frequency (Fig. 4, A to C). These results agree with previous reports showing that actinomycin D treatment alone does not alter basal synaptic transmission (25, 39). The data also demonstrate that the upscaling of excitatory strength mediated by chronic, direct pharmacological DNMT inhibition requires gene transcription. These observations are consistent with our interpretation that DNMT inhibition acts by epigenetically altering gene transcription.
Fig. 4. Transcription inhibition blocks the effect of chronic inhibition of DNA methylation on excitatory transmission.
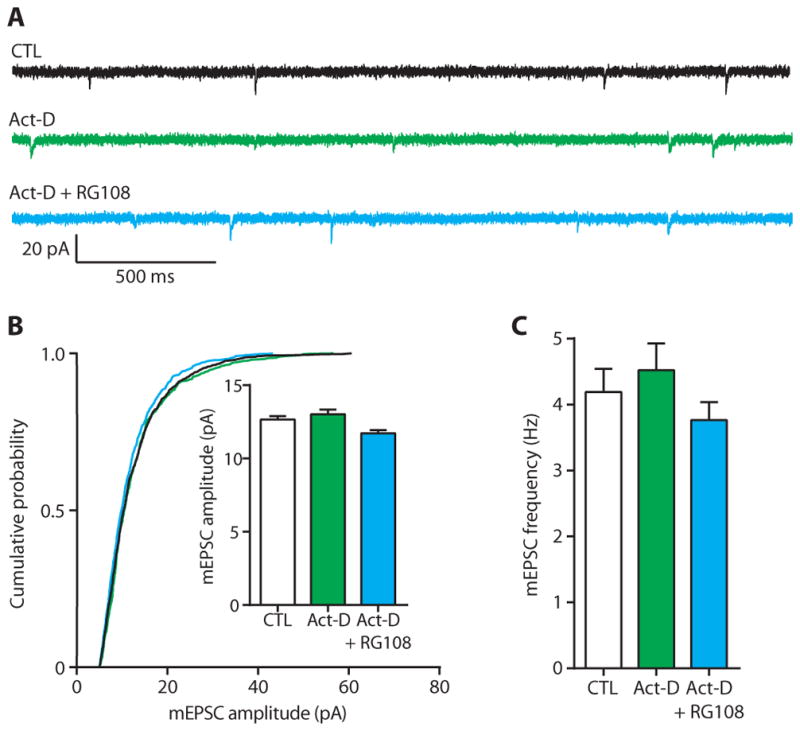
(A) Sample mEPSC traces from cortical pyramidal neurons after 24-hour exposure to control (CTL), actinomycin D (Act-D; green), or Act-D + RG108 (blue). (B and C) Cumulative probability distributions and mean mEPSC amplitudes (B) and mean mEPSC frequencies (C) in cortical pyramidal neurons treated with Act-D alone or co-incubation with RG108. Bar graphs are means ± SEM. Data are cumulative of cells pooled from at least three experiments for each condition (CTL, n = 9 cells; Act-D, n = 4 cells; Act-D + RG108, n = 7 cells). (B) K-W test, P = 0.0059. (C) ANOVA, P = 0.3439.
Combined knockdown of Dnmt1 and Dnmt3a multiplicatively upscales excitatory strength
Currently available DNMT inhibitors, such as RG108, nonselectively inhibit DNMTs (45), raising the question of which DNMT isoform mediates DNMT inhibitor–induced upscaling. We therefore used an ASO-mediated approach to specifically knock down the expression of Dnmt1, Dnmt3a, or both, because these are the two predominant DNMTs in neurons (7). ASOs are synthetic, single-stranded molecules containing chemically modified nucleic acids complementary in sequence to target mRNA (46, 47). Hybridized ASO-mRNA heteroduplexes inhibit expression largely through the recruitment of the endonuclease, RNase H, and subsequent target mRNA degradation (48, 49). Here, we used ASOs containing chemical modifications shown to enhance nuclease resistance and prolong half-life in vitro (50, 51). ASOs designed with similar modifications are absorbed by cultured primary neurons without transfection reagents and inhibit gene expression after a single dose for at least 20 days (52). After an initial screening, an ASO separately targeting each Dnmt transcript was chosen for further experiments (Fig. 5A).
Fig. 5. Combined Dnmt1 and Dnmt3a knockdown multiplicatively upscales excitatory strength.

(A) Depiction of ASO base pairing with target Dnmt mRNA. ASO-binding positions are listed under the target transcript. Underlined ASO sequences represent 2′-OMe–modified nucleotide wings. (B) Bar graphs show relative expression of Dnmt1 and Dnmt3a mRNA after ASO treatment. Data are means ± SEM from three experiments; *P < 0.05 compared to scrambled CTL, ANOVA followed by Dunnett’s test. (C and D) Cumulative probability distributions and mean mEPSC amplitudes (C) and mean mEPSC frequencies (D) in cortical pyramidal neurons treated with Dnmt-targeted ASOs. Bar graphs are means ± SEM. Data are cumulative of cells pooled from at least two experiments for each condition (scrambled CTL, n=16 cells; combined ASOs, n=8 cells; Dnmt1 ASO, n= 5 cells; and Dnmt3a ASO, n= 5 cells). (C) *P< 0.001 compared to scrambled CTL, K-W test followed by Dunn’s test. (D) *P<0.05 compared to scrambled CTL, ANOVA followed by Dunnett’s test. (E) Rank order plot of 1000 randomly selected mEPSC amplitudes from scrambled CTL and combined ASO treatment (orange values). Linear regression yielded a scaling factor of 1.34, which was used to scale down amplitudes from combined ASO-treated cells from (C). (F) Scaled-down amplitudes from combined ASO-treated cells compared to those in scrambled CTL cells; P = 0.1323 compared to scrambled CTL, K-S test.
After confirming ASO selectivity (Fig. 5B), we investigated the effect of Dnmt knockdown on excitatory synaptic transmission in cortical pyramidal neurons. We recorded mEPSCs from pyramidal neurons 5 to 7 days after a single ASO dose in culture. We found that combined knockdown of both Dnmt1 and Dnmt3a produced a right shift in the cumulative distribution of mEPSC amplitudes and increased mEPSC mean amplitude and frequency, whereas knockdown of either Dnmt1 or Dnmt3a alone did not affect mEPSC amplitude or frequency in recorded cells (Fig. 5, C and D). Furthermore, quantitative analysis revealed that combined Dnmt1 and Dnmt3a knockdown resulted in a multiplicative increase in mEPSC amplitude, because the scaled-down, combined knockdown group amplitudes were not different than that of the scrambled control group (Fig. 5, E and F). Together, our results demonstrate that Dnmt1 and Dnmt3a expression can be selectively decreased in cultured primary cortical neurons and that the combined knockdown of these transcripts multiplicatively upscales excitatory strength. Consistent with this idea, while this paper was being copyedited, Yu et al. also published results indicating that active DNA demethylation can control synaptic scaling (53).
DISCUSSION
Chromatin plasticity via posttranslational histone modifications and DNA methylation supports the persistent, global transcriptional regulation necessary for long-term memory. As “cellular correlates of memory,” the regulation of site-specific alterations in synaptic strength via these epigenetic mechanisms has understandably garnered much attention in the field (6, 7, 54–57). It is now evident that long-term, synapse-specific changes are strongly regulated via epigenetic modifications. However, memory is also reliant on transcription-dependent forms of HSP such as synaptic scaling (23, 24, 58). Although mounting evidence has indirectly linked epigenetic mechanisms and HSP (44, 59–61), mechanistic understanding of how these modifications directly influence the global scaling of post-synaptic receptors is lacking. The present study establishes a direct link between DNA methylation and synaptic scaling. Cumulatively, our results strongly suggest a role for activity-dependent DNA methylation and demethylation in the multiplicative upscaling of excitatory strength.
Here, we found a specific association between homeostatic upscaling and the transcription of genes involved in regulating active DNA demethylation in neurons (14, 18). These observations in part prompted our hypothesis of cytosine methylation controlling neuron-wide scaling, along with theoretical considerations that cell-wide changes in the epigenome are particularly well-suited to drive cell-wide functional changes (5, 23). In this regard, we observed that inhibiting DNMT activity or decreasing Dnmt expression was sufficient to induce a multiplicative increase in excitatory synaptic strength, reminiscent of the uniform changes observed with multiplicative homeostatic upscaling. It is noteworthy, however, that homeostatic upscaling represents only one form of HSP. HSP may occur locally, in a connection-specific manner (62, 63), and even at the single-synapse level (64, 65). The inhibition of DNA methylation with RG108 or the combined knockdown of Dnmt1 and Dnmt3a could, in principle, engage any of these less-global cellular mechanisms.
The multiplicative changes in amplitude we found in our analyses seem to exclude local, connection-specific, and single-synapse level changes in AMPA receptor density because these mechanisms would not likely result in such changes (30). However, we observed some nonlinearity in TTX-and RG108-induced scaling at larger mEPSC amplitudes (see Figs. 1D and 2D), which could be consistent with synapse-specific and nonmultiplicative forms of plasticity. Although our results suggest that a largely multiplicative form of synaptic scaling is triggered by TTX and DNMT inhibition in cultured cortical neurons such as we have studied, multiplicative scaling is not a sine qua non for regulation of DNA methylation to act as a controller of homeostatic forms of neural plasticity. Nevertheless, we find the possibility that DNA cytosine methylation drives coordinated, cell-wide, and multiplicative forms of HSP to be quite intriguing.
Further investigation will be necessary to determine the precise molecular mechanisms involved in the regulation of various forms of HSP including TTX-induced synaptic scaling as we studied here. One particularly interesting possibility is that regulation of methylation of the genes for AMPA-subtype glutamate receptors might be involved, as well as methylation of those genes regulating their trafficking. For example, Jayanthi et al. (66) have observed activity-induced alterations in AMPA receptor gene methylation that correlate with glutamate receptor expression in vivo. Previous studies (33–35) as well as our own data have demonstrated that TTX-induced synaptic scaling is associated with altered glutamate receptor gene transcription as well as that of arc, an AMPA receptor trafficking regulator. Moreover, plasticity-regulating genes have recently been discovered to be targets of active DNA demethylation pathways, in the absence of effects on synapse-specific forms of plasticity such as long-term potentiation (1, 19). Thus, activity-dependent AMPA receptor gene promoter methylation might be one specific component of TTX- and DNMT inhibition–induced synaptic scaling, with alterations in arc gene methylation complementing these changes. This specific scenario aside, we anticipate that studies of TTX- and DNMT inhibition–induced changes at the level of the entire neuronal methylome will be necessary to begin to fully model the role of specific cytosine methylation events in homeostatic forms of synaptic plasticity.
Our results with ASO-mediated knockdown of Dnmt1 and Dnmt3a agree with the finding that DNMT1 and DNMT3a play overlapping roles in adult learning and memory and synaptic plasticity (7). Although ascribed distinct roles, especially in dividing cells (67, 68), the extent to which these enzymes operate nonredundantly is unclear at present. For example, DNMT1 can participate in de novo methylation, and DNMT3a can methylate hemimethylated DNA (69). However, because our ASOs did not achieve complete knockdown, we cannot exclude the possibility that one DNMT may play a more prominent role in multiplicative homeostatic upscaling under physiological conditions. Future studies are needed to dissect the overlapping signaling cascades that regulate synaptic scaling and direct DNMT activity across the genome in an isoform-specific manner.
We interpret our finding that DNMT inhibition causes upscaling as indicating that demethylation-associated secondary transcriptional changes are sufficient to mediate homeostatic upscaling. The capacity of actinomycin D to block RG108-induced upscaling is consistent with this interpretation, as are previous results that RG108 and other DNMT inhibitors can cause demethylation in neurons (9–12). In addition, our observation that knockdown of TET1 dioxygenase can block the synaptic upscaling induced by RG108 is also consistent with our interpretation that DNMT inhibition causes upscaling by causing secondary cytosine demethylation, because TET1 is a known regulator of both active cytosine demethylation and associated transcriptional regulation in neurons (1, 14, 15, 19).
A surprising finding was that DNMT inhibition blocked the ability of TTX to cause upscaling and correspondingly that TTX blocked the ability of DNMT inhibition to itself to cause upscaling: in other words, that when TTX and RG108 were coapplied, no synaptic scaling was observed (Fig. 3). We had anticipated that DNMT inhibition and TTX treatment would occlude each other’s effects, that is, that upscaling would occur with combined treatment that equaled the upscaling caused by either agent alone. One parsimonious explanation for the mutual blockade is as follows. First, DNMT inhibition blocks TTX-induced upscaling because an initial triggering round of increased DNA cytosine methylation is necessary for synaptic inactivity to produce upscaling. Second, the activity dependence (both action potential generation and AMPA receptor activation) of DNMT inhibition by itself to cause upscaling suggests that activity-dependent secondary gene demethylation as a consequence of DNMT inhibition is one component of the mechanism(s) through which 24-hour DNMT blockade causes upscaling.
However, on the basis of our results, we cannot exclude the possibility that DNMT inhibition–induced scaling might be different mechanistically than inactivity-induced homeostatic plasticity. For example, DNMT inhibition causes changes in mEPSC frequency, whereas TTX treatment does not (Figs. 1C and 2C). Thus, one caveat to our interpretation is that there might be an effect of DNMT inhibition–induced demethylation that is mechanistically distinct from “real” inactivity-induced scaling that is produced in response to TTX, in essence that DNMT inhibition might be triggering a mechanism(s) that is acting via a parallel and different methylation-regulated pathway. In this vein, it is important to consider that there may be many routes to homeostatic synaptic scaling in cortical neurons that are physiologically and behaviorally relevant and that are not used in response to TTX. DNMT inhibition might be mimicking one or more of those pathways. However, our results with DNMT inhibition indicates that DNA (de)methylation is capable of triggering synaptic scaling and that this effect is activity- and transcription-dependent, supporting a physiologic role for DNA methylation in controlling cell-wide changes in synaptic function.
Many additional experiments will undoubtedly be required to fully understand the roles of active methylation/demethylation in regulating homeostatic cell-wide plasticity, but it would not be surprising if both methylation and demethylation of multiple gene loci are necessary to coordinately regulate multiplicative cell-wide glutamate receptor homeostasis. Regardless of the particulars of the relevant epigenomic changes, our data clearly implicate DNA methylation/demethylation in the regulation of homeostatic neuroplasticity. Thus, our study suggests that DNA methylation is a mechanism that drives neuronal homeostatic stability. DNMT inhibition in vivo affects the acquisition, consolidation, and long-term maintenance of memory in a region-specific manner (9–12). The present findings imply that these previous results are due not only to the dysregulation of gene expression required for site-specific plasticity but also to the improper induction of homeostatic scaling mechanisms (2). This is especially salient because homeostatic mechanisms influence synaptic plasticity (70–72). Thus, it will be necessary to further determine how these different cellular plasticities are epigenetically regulated on an individual level and how their regulation converges and interacts at the level of the epigenome.
In summary, our studies yield a unifying hypothesis for the means by which epigenetic marks, particularly DNA cytosine methylation, contribute to the stabilization and storage of memory: through controlling neuronal homeostatic plasticity. Sophisticated computational models along with studies in cultured neurons and in vivo have provided a compelling case that HSP is a required mechanism for stable long-term memory storage in the mammalian CNS (24–27). Yet, how the neuron achieves the global cell-wide and genome-wide transcriptional regulation necessary for homeostatic mechanisms to play their stabilizing role in memory has been but sparsely investigated. Our studies suggest that DNA methylation provides the genome-wide breadth, the capacity for molecular self-perpetuation, and the transcription-regulating potency that drives neuronal homeostatic stability in long-term memory consolidation and storage.
MATERIALS AND METHODS
Cell culture
Cultures were prepared from embryonic day 18 rat cortical tissue as described previously (12). Briefly, dissected cortical tissue was digested in papain (Worthington) and dissociated with fire-polished glass pipettes in Neurobasal (Life Technologies) medium supplemented with L-glutamine and B-27 (Life Technologies). The cell suspension was filtered through a 70-μm cell strainer and centrifuged. Cells were resuspended in the supplemented Neurobasal medium at a concentration of 250,000/ml and plated. For biochemical experiments, cells were plated on poly-L-lysine–coated (Sigma-Aldrich) 12-well plates (Corning). For electrophysiology experiments, cells were plated on poly-L-lysine–coated glass coverslips (Menzel-Glaser). Cells were grown in a humidified CO2 (5%) incubator at 37°C. One-half of the medium was changed at 1 to 2 days in culture [days in vitro (DIV)], and cultures were then maintained by changing one-half of the medium every 4 DIV. mEPSCs were recorded after cells matured for 13 to 14 DIV and after 24-hour drug treatments or 5 to 7 days after a single ASO dose. RNA was collected at 13 to 14 DIV after a 24-hour treatment with TTX or 11 to 13 DIV after single-dose ASO treatments.
Drug treatments
Twenty-four–hour treatments of cortical cultures were done at the following concentrations: 0.1% dimethyl sulfoxide (control), 100 μM RG108 (Calbiochem), 1 μM TTX, 10 μM NBQX, 50 μM APV, 1.25 μM actinomycin D (Sigma-Aldrich), and 50 μM zebularine (Sigma-Aldrich).
ASO design, screening, and neuronal treatment
Sequences for rat Dnmt1 (RefSeq ID: NM_053354.3) and Dnmt3a (RefSeq ID: NM_0010039587.1) were retrieved from the National Center for Biotechnology Information database, and candidate ASO sequences were determined using freely available computational online tools (73). Used ASOs were 20-mers constructed on a phosphorothioate-modified backbone, containing a central region of 10 2′-deoxynucliotides flanked 5′ and 3′ by 5 2′-O-methyl (OMe)–modified nucleotide “wings” (“5-10-5 2′-OMe gapmers”). Five to seven candidate ASOs were screened at a single dose after 4 to 7 days’ treatments. We then selected a single ASO targeting each Dnmt mRNA for further testing. Selected ASO sequences were as follows: scrambled control: 5′-mA-mC-mU-mC-mG-C-A-A-C-T-G-C-C-A-C-mU-mC-mU-mC-mC-3′; Dnmt1: 5′-mC-mC-mC-mU-mU-C-C-C-T-T-T-C-C-C-T-mU-mU-mC-mC-mC-3′; and Dnmt3a: 5′-mC-mC-mU-mC-mC-G-G-C-A-T-T-T-C-C-T-mC-mC-mU-mC-mU-3′. A similar approach was used for identifying an ASO targeting Tet1 (RefSeq ID: XM_006256398.2; see fig. S2); the selected ASO sequence was as follows: mU-mG-mC-mC-mG-T-T-C-A-T-C-T-T-C-C-mA-mC-mC-mU-mG. For neuronal treatments, ASOs were resuspended in phosphate-buffered saline to 150 μM, and the stock was added to medium to a final concentration of 1.5 μM.
Whole-cell recordings
Whole-cell patch-clamp recordings were made from cultured cortical neurons (13 to 14 DIV). Pyramidal neurons were visually identified using a Zeiss Axiovert 25 inverted microscope. All recordings were performed at room temperature (21° to 23°C) in an extracellular solution containing the following: 120 mM NaCl, 3 mM KCl, 15 mM Hepes, 20 mM glucose, 2 mM CaCl2, and 1mM MgCl2 (pH 7.3), with 1 μM TTX and 20 μM bicuculline to isolate mEPSCs. The patch pipette (3 to 5 megohms) solution contained the following: 125 mM K-gluconate, 10 mM KCl, 10 mM Hepes, 10 mM creatine phosphate, 2 mM Mg–adenosine triphosphate, 0.2 mM Na–guanosine triphosphate, and 0.5 mM EGTA (pH 7.3). Neurons were held at −70 mV, and mEPSCs were collected using an AxoPatch 200B amplifier (Molecular Devices) filtered at 2 kHz. Signals were sampled at 10 kHz and digitized with a Digidata 1200 Series (Molecular Devices). mEPSCs were analyzed using Mini Analysis Program (Synaptosoft) software. All events were verified manually. Series resistance, capacitance, and input resistance were monitored and did not differ among drug treatments.
Multiplicative analysis of mEPSC amplitudes
Multiplicative analyses of mEPSC amplitudes were performed as described (30), with minor modifications. Rank order plots were constructed from 1000 randomly selected mEPSC amplitudes from control and experimental groups. Only amplitudes within the 10th to 90th percentile of events were used to avoid potential deviations of fit due to outliers (31). Treatment group amplitudes were modeled as a linear function, y = ax, of the control group, with the slope of the linear fit, a, representing the scaling factor. The scaling factor was then applied to all treatment group amplitudes. To minimize analytical errors inherent to threshold-limited data (30), scaled-down amplitudes falling below our amplitude detection threshold, typically 5 pA, were discarded, and suprathreshold values were compared to control.
RNA extraction and quantitative real-time PCR
Using the miRNeasy Mini Kit (Qiagen), total RNA was extracted following the manufacturer’s guidelines with the additional RNase-Free DNase (Qiagen) treatment step and eluded in 50 μl of RNase-free water. RNA concentrations were determined spectrophotometrically using the NanoDrop 200c (Thermo Scientific). RNA (300 ng) was then reverse-transcribed into complementary DNA (cDNA) using oligo(dT) and random hexamer primers in the iScript cDNA synthesis kit (Bio-Rad). Quantitative real-time PCR was carried out using a CFX96 Touch Real-Time PCR Detection System (Bio-Rad), SsoAdvanced Universal SYBR Green Supermix (Bio-Rad), and 500 nM intron-spanning primers (table S1). PCR amplifications were performed in triplicate with the following cycling conditions: 95°C for 30 s, followed by 40 cycles of 95°C for 10 s and 60°C for 30 s, followed by real-time melt analysis to verify product specificity. Differential gene expression between samples was determined by the comparative Ct (ΔΔCt) method using glyceraldehyde-3-phosphate dehydrogenase (Gapdh) as an internal control (74, 75).
Statistical analysis
Statistical analyses of mEPSC amplitudes were performed using non-parametric tests with a stringent confidence level of P < 0.001 for significance (30). All other data were analyzed using parametric tests as indicated with P < 0.05 indicating significance. Unless stated otherwise, data are presented as means ± SEM.
Supplementary Material
Acknowledgments
We thank C. Gavin for help with preparing the manuscript, and special thanks to A. Gross, G. Kaas, H. Pruitt, and T. J. Younts for feedback on manuscript drafts. J.D.S. thanks H. Song (Johns Hopkins University) for many helpful discussions. We also thank the anonymous reviewers of previous versions of the manuscript for their helpful suggestions.
Funding: Our work is supported by NIH grants P30-NS047466 (J.J.H.), MH091122, MH57014 (J.D.S.), and DA034681 (J.J.D.); Defense Advanced Research Projects Agency grant HR0011-12-1-0015 (J.D.S.); NIH T32GM008361 and T32NS061788 (support for J.P.M. and M.C.G.-K.); and funds from the Evelyn F. McKnight Brain Research Foundation.
Footnotes
www.sciencesignaling.org/cgi/content/full/8/382/ra61/DC1
Fig. S1. Indirect DNMT inhibition with the nucleoside analog zebularine decreases mEPSC frequency with no effect on amplitude.
Fig. S2. Tet1 knockdown blocks RG108-induced upscaling of excitatory synaptic strength.
Table S1. Primers used in this study.
Author contributions: J.P.M., M.C.G.-K., S.P., J.J.D., J.D.S., and J.J.H. contributed to designing the experiments and interpreting the results. J.P.M., M.C.G.-K., C.H., J.L.P., and S.P. executed the experiments and analyzed data. J.P.M., J.J.H., and J.D.S. wrote the manuscript.
Competing interests: The authors declare that they have no competing financial interests.
REFERENCES AND NOTES
- 1.Kaas GA, Zhong C, Eason DE, Ross DL, Vachhani RV, Ming GL, King JR, Song H, Sweatt JD. TET1 controls CNS 5-methylcytosine hydroxylation, active DNA demethylation, gene transcription, and memory formation. Neuron. 2013;79:1086–1093. doi: 10.1016/j.neuron.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roth ED, Roth TL, Money KM, SenGupta S, Eason DE, Sweatt JD. DNA methylation regulates neurophysiological spatial representation in memory formation. Neuroepigenetics. 2015;2:1–8. doi: 10.1016/j.nepig.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crick F. Memory and molecular turnover. Nature. 1984;312:101. doi: 10.1038/312101a0. [DOI] [PubMed] [Google Scholar]
- 4.Holliday R. Is there an epigenetic component in long-term memory? J Theor Biol. 1999;200:339–341. doi: 10.1006/jtbi.1999.0995. [DOI] [PubMed] [Google Scholar]
- 5.Day JJ, Sweatt JD. DNA methylation and memory formation. Nat Neurosci. 2010;13:1319–1323. doi: 10.1038/nn.2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levenson JM, Roth TL, Lubin FD, Miller CA, Huang IC, Desai P, Malone LM, Sweatt JD. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J Biol Chem. 2006;281:15763–15773. doi: 10.1074/jbc.M511767200. [DOI] [PubMed] [Google Scholar]
- 7.Feng J, Zhou Y, Campbell SL, Le T, Li E, Sweatt JD, Silva AJ, Fan G. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat Neurosci. 2010;13:423–430. doi: 10.1038/nn.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller CA, Campbell SL, Sweatt JD. DNA methylation and histone acetylation work in concert to regulate memory formation and synaptic plasticity. Neurobiol Learn Mem. 2008;89:599–603. doi: 10.1016/j.nlm.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller CA, Gavin CF, White JA, Parrish RR, Honasoge A, Yancey CR, Rivera IM, Rubio MD, Rumbaugh G, Sweatt JD. Cortical DNA methylation maintains remote memory. Nat Neurosci. 2010;13:664–666. doi: 10.1038/nn.2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miller CA, Sweatt JD. Covalent modification of DNA regulates memory formation. Neuron. 2007;53:857–869. doi: 10.1016/j.neuron.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 11.Lubin FD, Roth TL, Sweatt JD. Epigenetic regulation of bdnf gene transcription in the consolidation of fear memory. J Neurosci. 2008;28:10576–10586. doi: 10.1523/JNEUROSCI.1786-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Day JJ, Childs D, Guzman-Karlsson MC, Kibe M, Moulden J, Song E, Tahir A, Sweatt JD. DNA methylation regulates associative reward learning. Nat Neurosci. 2013;16:1445–1452. doi: 10.1038/nn.3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo JU, Ma DK, Mo H, Ball MP, Jang MH, Bonaguidi MA, Balazer JA, Eaves HL, Xie B, Ford E, Zhang K, Ming GL, Gao Y, Song H. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat Neurosci. 2011;14:1345–1351. doi: 10.1038/nn.2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423–434. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo JU, Su Y, Zhong C, Ming GL, Song H. Emerging roles of TET proteins and 5-hydroxymethylcytosines in active DNA demethylation and beyond. Cell Cycle. 2011;10:2662–2668. doi: 10.4161/cc.10.16.17093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129–1133. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma DK, Guo JU, Ming GL, Song H. DNA excision repair proteins and Gadd45 as molecular players for active DNA demethylation. Cell Cycle. 2009;8:1526–1531. doi: 10.4161/cc.8.10.8500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma DK, Jang MH, Guo JU, Kitabatake Y, Chang ML, Pow-Anpongkul N, Flavell RA, Lu B, Ming GL, Song H. Neuronal activity–induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science. 2009;323:1074–1077. doi: 10.1126/science.1166859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rudenko A, Dawlaty MM, Seo J, Cheng AW, Meng J, Le T, Faull KF, Jaenisch R, Tsai LH. Tet1 is critical for neuronal activity-regulated gene expression and memory extinction. Neuron. 2013;79:1109–1122. doi: 10.1016/j.neuron.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sultan FA, Wang J, Tront J, Liebermann DA, Sweatt JD. Genetic deletion of gadd45b, a regulator of active DNA demethylation, enhances long-term memory and synaptic plasticity. J Neurosci. 2012;32:17059–17066. doi: 10.1523/JNEUROSCI.1747-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang RR, Cui QY, Murai K, Lim YC, Smith ZD, Jin S, Ye P, Rosa L, Lee YK, Wu HP, Liu W, Xu ZM, Yang L, Ding YQ, Tang F, Meissner A, Ding C, Shi Y, Xu GL. Tet1 regulates adult hippocampal neurogenesis and cognition. Cell Stem Cell. 2013;13:237–245. doi: 10.1016/j.stem.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sweatt JD. The emerging field of neuroepigenetics. Neuron. 2013;80:624–632. doi: 10.1016/j.neuron.2013.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guzman-Karlsson MC, Meadows JP, Gavin CF, Hablitz JJ, Sweatt JD. Transcriptional and epigenetic regulation of Hebbian and non-Hebbian plasticity. Neuropharmacology. 2014;80:3–17. doi: 10.1016/j.neuropharm.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abbott LF, Nelson SB. Synaptic plasticity: Taming the beast. Nat Neurosci. 2000;3:1178–1183. doi: 10.1038/81453. [DOI] [PubMed] [Google Scholar]
- 25.Ibata K, Sun Q, Turrigiano GG. Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron. 2008;57:819–826. doi: 10.1016/j.neuron.2008.02.031. [DOI] [PubMed] [Google Scholar]
- 26.Turrigiano GG. The self-tuning neuron: Synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391:892–896. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- 28.Siddoway B, Hou H, Xia H. Molecular mechanisms of homeostatic synaptic down-scaling. Neuropharmacology. 2014;78:38–44. doi: 10.1016/j.neuropharm.2013.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang G, Gilbert J, Man HY. AMPA receptor trafficking in homeostatic synaptic plasticity: Functional molecules and signaling cascades. Neural Plast. 2012;2012:825364. doi: 10.1155/2012/825364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim J, Tsien RW, Alger BE. An improved test for detecting multiplicative homeostatic synaptic scaling. PLOS One. 2012;7:e37364. doi: 10.1371/journal.pone.0037364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim J, Alger BE. Reduction in endocannabinoid tone is a homeostatic mechanism for specific inhibitory synapses. Nat Neurosci. 2010;13:592–600. doi: 10.1038/nn.2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Soares C, Lee KF, Nassrallah W, Béïque JC. Differential subcellular targeting of glutamate receptor subtypes during homeostatic synaptic plasticity. J Neurosci. 2013;33:13547–13559. doi: 10.1523/JNEUROSCI.1873-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shepherd JD, Rumbaugh G, Wu J, Chowdhury S, Plath N, Kuhl D, Huganir RL, Worley PF. Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron. 2006;52:475–484. doi: 10.1016/j.neuron.2006.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thiagarajan TC, Lindskog M, Tsien RW. Adaptation to synaptic inactivity in hippocampal neurons. Neuron. 2005;47:725–737. doi: 10.1016/j.neuron.2005.06.037. [DOI] [PubMed] [Google Scholar]
- 35.Rutherford LC, Nelson SB, Turrigiano GG. BDNF has opposite effects on the quantal amplitude of pyramidal neuron and interneuron excitatory synapses. Neuron. 1998;21:521–530. doi: 10.1016/s0896-6273(00)80563-2. [DOI] [PubMed] [Google Scholar]
- 36.Brueckner B, Garcia Boy R, Siedlecki P, Musch T, Kliem HC, Zielenkiewicz P, Suhai S, Wiessler M, Lyko F. Epigenetic reactivation of tumor suppressor genes by a novel small-molecule inhibitor of human DNA methyltransferases. Cancer Res. 2005;65:6305–6311. doi: 10.1158/0008-5472.CAN-04-2957. [DOI] [PubMed] [Google Scholar]
- 37.Stresemann C, Brueckner B, Musch T, Stopper H, Lyko F. Functional diversity of DNA methyltransferase inhibitors in human cancer cell lines. Cancer Res. 2006;66:2794–2800. doi: 10.1158/0008-5472.CAN-05-2821. [DOI] [PubMed] [Google Scholar]
- 38.LaPlant Q, Vialou V, Covington HE, III, Dumitriu D, Feng J, Warren BL, Maze I, Dietz DM, Watts EL, Iñiguez SD, Koo JW, Mouzon E, Renthal W, Hollis F, Wang H, Noonan MA, Ren Y, Eisch AJ, Bolaños CA, Kabbaj M, Xiao G, Neve RL, Hurd YL, Oosting RS, Fan G, Morrison JH, Nestler EJ. Dnmt3a regulates emotional behavior and spine plasticity in the nucleus accumbens. Nat Neurosci. 2010;13:1137–1143. doi: 10.1038/nn.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nelson ED, Kavalali ET, Monteggia LM. Activity-dependent suppression of miniature neurotransmission through the regulation of DNA methylation. J Neurosci. 2008;28:395–406. doi: 10.1523/JNEUROSCI.3796-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Creusot F, Acs G, Christman JK. Inhibition of DNA methyltransferase and induction of Friend erythroleukemia cell differentiation by 5-azacytidine and 5-aza-2′-deoxycytidine. J Biol Chem. 1982;257:2041–2048. [PubMed] [Google Scholar]
- 41.Santi DV, Norment A, Garrett CE. Covalent bond formation between a DNA-cytosine methyltransferase and DNA containing 5-azacytosine. Proc Natl Acad Sci USA. 1984;81:6993–6997. doi: 10.1073/pnas.81.22.6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou L, Cheng X, Connolly BA, Dickman MJ, Hurd PJ, Hornby DP. Zebularine: A novel DNA methylation inhibitor that forms a covalent complex with DNA methyltransferases. J Mol Biol. 2002;321:591–599. doi: 10.1016/S0022-2836(02)00676-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jüttermann R, Li E, Jaenisch R. Toxicity of 5-aza-2′-deoxycytidine to mammalian cells is mediated primarily by covalent trapping of DNA methyltransferase rather than DNA demethylation. Proc Natl Acad Sci USA. 1994;91:11797–11801. doi: 10.1073/pnas.91.25.11797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blackman MP, Djukic B, Nelson SB, Turrigiano GG. A critical and cell-autonomous role for MeCP2 in synaptic scaling up. J Neurosci. 2012;32:13529–13536. doi: 10.1523/JNEUROSCI.3077-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Erdmann A, Halby L, Fahy J, Arimondo PB. Targeting DNA methylation with small molecules: What’s next? J Med Chem. 2015;58:2569–2583. doi: 10.1021/jm500843d. [DOI] [PubMed] [Google Scholar]
- 46.Chan JH, Lim S, Wong WS. Antisense oligonucleotides: From design to therapeutic application. Clin Exp Pharmacol Physiol. 2006;33:533–540. doi: 10.1111/j.1440-1681.2006.04403.x. [DOI] [PubMed] [Google Scholar]
- 47.Myers KJ, Dean NM. Sensible use of antisense: How to use oligonucleotides as research tools. Trends Pharmacol Sci. 2000;21:19–23. doi: 10.1016/s0165-6147(99)01420-0. [DOI] [PubMed] [Google Scholar]
- 48.Dash P, Lotan I, Knapp M, Kandel ER, Goelet P. Selective elimination of mRNAs in vivo: Complementary oligodeoxynucleotides promote RNA degradation by an RNase H-like activity. Proc Natl Acad Sci USA. 1987;84:7896–7900. doi: 10.1073/pnas.84.22.7896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Walder RY, Walder JA. Role of RNase H in hybrid-arrested translation by antisense oligonucleotides. Proc Natl Acad Sci USA. 1988;85:5011–5015. doi: 10.1073/pnas.85.14.5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eckstein F. Phosphorothioate oligodeoxynucleotides: What is their origin and what is unique about them? Antisense Nucleic Acid Drug Dev. 2000;10:117–121. doi: 10.1089/oli.1.2000.10.117. [DOI] [PubMed] [Google Scholar]
- 51.McKay RA, Miraglia LJ, Cummins LL, Owens SR, Sasmor H, Dean NM. Characterization of a potent and specific class of antisense oligonucleotide inhibitor of human protein kinase C-α expression. J Biol Chem. 1999;274:1715–1722. doi: 10.1074/jbc.274.3.1715. [DOI] [PubMed] [Google Scholar]
- 52.Carroll JB, Warby SC, Southwell AL, Doty CN, Greenlee S, Skotte N, Hung G, Bennett CF, Freier SM, Hayden MR. Potent and selective antisense oligonucleotides targeting single-nucleotide polymorphisms in the Huntington disease gene / allele-specific silencing of mutant huntingtin. Mol Ther. 2011;19:2178–2185. doi: 10.1038/mt.2011.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu H, Su Y, Shin J, Zhong C, Guo JU, Weng YL, Gao F, Geschwind DH, Coppola G, Ming GL, Song H. Tet3 regulates synaptic transmission and homeostatic plasticity via DNA oxidation and repair. Nat Neurosci. 2015;18:836–843. doi: 10.1038/nn.4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bi G, Poo M. Synaptic modification by correlated activity: Hebb’s postulate revisited. Annu Rev Neurosci. 2001;24:139–166. doi: 10.1146/annurev.neuro.24.1.139. [DOI] [PubMed] [Google Scholar]
- 55.Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJ, Zhou Y, Wang X, Mazitschek R, Bradner JE, DePinho RA, Jaenisch R, Tsai LH. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459:55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Malenka RC, Bear MF. LTP and LTD: An embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 57.Sui L, Wang Y, Ju LH, Chen M. Epigenetic regulation of reelin and brain-derived neurotrophic factor genes in long-term potentiation in rat medial prefrontal cortex. Neurobiol Learn Mem. 2012;97:425–440. doi: 10.1016/j.nlm.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 58.Nelson SB, Turrigiano GG. Strength through diversity. Neuron. 2008;60:477–482. doi: 10.1016/j.neuron.2008.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Corrêa SA, Hunter CJ, Palygin O, Wauters SC, Martin KJ, McKenzie C, McKelvey K, Morris RG, Pankratov Y, Arthur JS, Frenguelli BG. MSK1 regulates homeostatic and experience-dependent synaptic plasticity. J Neurosci. 2012;32:13039–13051. doi: 10.1523/JNEUROSCI.0930-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nelson ED, Kavalali ET, Monteggia LM. MeCP2-dependent transcriptional repression regulates excitatory neurotransmission. Curr Biol. 2006;16:710–716. doi: 10.1016/j.cub.2006.02.062. [DOI] [PubMed] [Google Scholar]
- 61.Qiu Z, Sylwestrak EL, Lieberman DN, Zhang Y, Liu XY, Ghosh A. The Rett syndrome protein MeCP2 regulates synaptic scaling. J Neurosci. 2012;32:989–994. doi: 10.1523/JNEUROSCI.0175-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim J, Tsien RW. Synapse-specific adaptations to inactivity in hippocampal circuits achieve homeostatic gain control while dampening network reverberation. Neuron. 2008;58:925–937. doi: 10.1016/j.neuron.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee KJ, Queenan BN, Rozeboom AM, Bellmore R, Lim ST, Vicini S, Pak DT. Mossy fiber-CA3 synapses mediate homeostatic plasticity in mature hippocampal neurons. Neuron. 2013;77:99–114. doi: 10.1016/j.neuron.2012.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hou Q, Gilbert J, Man HY. Homeostatic regulation of AMPA receptor trafficking and degradation by light-controlled single-synaptic activation. Neuron. 2011;72:806–818. doi: 10.1016/j.neuron.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee MC, Yasuda R, Ehlers MD. Metaplasticity at single glutamatergic synapses. Neuron. 2010;66:859–870. doi: 10.1016/j.neuron.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jayanthi S, McCoy MT, Chen B, Britt JP, Kourrich S, Yau HJ, Ladenheim B, Krasnova IN, Bonci A, Cadet JL. Methamphetamine downregulates striatal glutamate receptors via diverse epigenetic mechanisms. Biol Psychiatry. 2014;76:47–56. doi: 10.1016/j.biopsych.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fuks F, Burgers WA, Godin N, Kasai M, Kouzarides T. Dnmt3a binds deacetylases and is recruited by a sequence-specific repressor to silence transcription. EMBO J. 2001;20:2536–2544. doi: 10.1093/emboj/20.10.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Leonhardt H, Page AW, Weier HU, Bestor TH. A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell. 1992;71:865–873. doi: 10.1016/0092-8674(92)90561-p. [DOI] [PubMed] [Google Scholar]
- 69.Kim GD, Ni J, Kelesoglu N, Roberts RJ, Pradhan S. Co-operation and communication between the human maintenance and de novo DNA (cytosine-5) methyltransferases. EMBO J. 2002;21:4183–4195. doi: 10.1093/emboj/cdf401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Arendt KL, Sarti F, Chen L. Chronic inactivation of a neural circuit enhances LTP by inducing silent synapse formation. J Neurosci. 2013;33:2087–2096. doi: 10.1523/JNEUROSCI.3880-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Roth-Alpermann C, Morris RG, Korte M, Bonhoeffer T. Homeostatic shutdown of long-term potentiation in the adult hippocampus. Proc Natl Acad Sci USA. 2006;103:11039–11044. doi: 10.1073/pnas.0600894103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thiagarajan TC, Lindskog M, Malgaroli A, Tsien RW. LTP and adaptation to inactivity: Overlapping mechanisms and implications for metaplasticity. Neuropharmacology. 2007;52:156–175. doi: 10.1016/j.neuropharm.2006.07.030. [DOI] [PubMed] [Google Scholar]
- 73.Owczarzy R, Tataurov AV, Wu Y, Manthey JA, McQuisten KA, Almabrazi HG, Pedersen KF, Lin Y, Garretson J, McEntaggart NO, Sailor CA, Dawson RB, Peek AS. IDT SciTools: A suite for analysis and design of nucleic acid oligomers. Nucleic Acids Res. 2008;36:W163–W169. doi: 10.1093/nar/gkn198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 75.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.