

Abstract
Muscular dystrophy (MD) is associated with mutations in genes that stabilize the myofiber plasma membrane, such as through the dystrophin–glycoprotein complex (DGC). Instability of this complex or defects in membrane repair/integrity leads to calcium influx and myofiber necrosis leading to progressive dystrophic disease. MD pathogenesis is also associated with increased skeletal muscle protease levels and activity that could augment weakening of the sarcolemma through greater degradation of cellular attachment complexes. Here, we observed a compensatory increase in the serine protease inhibitor Serpina3n in mouse models of MD and after acute muscle tissue injury. Serpina3n muscle-specific transgenic mice were generated to model this increase in expression, which reduced the activity of select proteases in dystrophic skeletal muscle and protected muscle from both acute injury with cardiotoxin and from chronic muscle disease in the mdx or Sgcd−/− MD genetic backgrounds. The Serpina3n transgene mitigated muscle degeneration and fibrosis, reduced creatine kinase serum levels, restored running capacity on a treadmill and reduced muscle membrane leakiness in vivo that is characteristic of mdx and Sgcd−/− mice. Mechanistically, we show that increased Serpina3n promotes greater sarcolemma membrane integrity and stability in dystrophic mouse models in association with increased membrane residence of the integrins, the DGC/utrophin–glycoprotein complex of proteins and annexin A1. Hence, Serpina3n blocks endogenous increases in the activity of select skeletal muscle resident proteases during injury or dystrophic disease, which stabilizes the sarcolemma leading to less myofiber degeneration and increased regeneration. These results suggest the use of select protease inhibitors as a strategy for treating MD.
Introduction
Muscle degenerative diseases such as muscular dystrophy (MD) are most commonly caused by mutations in genes that are part of the dystrophin–glycoprotein complex (DGC) that otherwise stabilize the myofiber plasma membrane, known as the sarcolemma (1,2). The DGC connects the underlying myofilament proteins to the extracellular matrix (ECM) thereby providing structural support to the sarcolemma to withstand contractile forces (1,2). Downstream of an unstable sarcolemma, MD pathogenesis involves sarcolemmal ruptures with increased intracellular calcium influx that causes myofiber necrosis, leading to activation of the immune response with progressive muscle tissue fibrosis and fatty tissue replacement (1–3). Currently, there is no cure for MD, and while gene therapy to replace the mutant gene would appear to be the most promising approach, major technical hurtles have left the field seeking alternative means to mitigate disease pathogenesis using pharmacological agents that address key mechanistic aspects of myofiber degeneration. For example, muscle tissue from dystrophic humans and mouse models of MD have high levels of extracellular protease activity, presumably associated with infiltrating immune cells (4,5). While many of these proteases are initially induced or activated as part of tissue remodeling that can underlie healing and regeneration, aberrant or excessive activation could actually be detrimental and lead to cellular injury. Indeed, application of broad serine protease inhibitors or anti-fibrotic drugs to modulate matrix metalloproteinase (MMP) activity improved skeletal muscle function in select MD animal models (6,7).
Serine protease inhibitors (serpins) are the largest protease inhibitor super family and Serpina3n (SA3N) is part of the serpina clade, which is the mouse orthologue of human anti-chymotrypsin gene (ACT) (8). While, human ACT is encoded by only one gene, extensive diversification in mice has resulted in 16 replicated serpina genes (9). Elevated expression and activity of Serpina3n has been shown to promote wound healing in several injury models including punctures in the skin, aortic aneurysm and neuron damage (10–13). While, the full range of potential targets of Serpina3n are uncertain, several studies have shown that Serpina3n may accelerate the wound healing process by inhibiting the activity of the serine proteases such as cathepsin G and granzyme B that are associated with inflammation, and MMP9 that is associated with ECM breakdown and remodeling (11,12).
Here, we determined that Serpina3n was uniquely up-regulated in injured skeletal muscle. General serine protease activity was elevated in both MD and acute muscle injury induced by cardiotoxin (CTX) injection, as was expression of Serpina3n. To understand the role of Serpina3n in muscle damage, we created a skeletal muscle-specific transgenic (TG) mouse overexpressing this gene product to mimic its known elevation during disease. Skeletal muscle from Serpina3n TG mice was normal at baseline, and these mice showed a near complete inhibition of the known augmentation of serine protease activity in muscle after CTX injury. The Serpina3n transgene dramatically reduced skeletal muscle pathogenesis in both Sgcd−/− (δ-sarcoglycan deficient) and mdx (dystrophin deficient) mouse models of MD. Mechanistically, we show that Serpina3n overexpression in the extracellular milieu of muscle tissue stabilized the DGC and promoted greater sarcolemmal stability. Hence, Serpina3n itself or a small molecule that produces a similar profile of protease inhibition represents possible new treatments for MD.
Results
Serpina3n is expressed in injured skeletal muscle
In general, MD pathogenesis has been associated with an increase in the activity of several protease systems, including calpain, cysteine proteases, MMPs and serine proteases (4). Here, we examined the serine proteases and observed increased activity from skeletal muscle of both mdx and Sgcd−/− mouse models of MD at 6 weeks, 6 months and 10 months of age compared to wild-type (WT) (Fig. 1A). In our previous gene array studies, we observed a very prominent increase in expression of Serpina3n in mouse models of MD, which is a prominent protease inhibitor (14). We were also aware of studies in neuron and skin injury models showing that elevated Serpina3n expression accelerated healing (10,11,15), hence we hypothesized that Serpina3n induction in MD might be protective and compensatory by reducing destructive protease activity. As confirmation of our initial gene arrays, quantitative polymerase chain reaction (qPCR) and western blotting showed increased expression of Serpina3n in the skeletal muscle of 2–3-month-old mdx mice and Sgcd−/− mice compared with WT (Fig. 1B and C). Deglycosylation was needed to expose the antigens in the protein to the antibody in the western blotting procedure (47 kDa). Similar to tissue injury with ongoing MD, acute muscle injury with CTX injection into the tibialis anterior (TA) muscle of WT mice showed induction of Serpina3n protein at 1, 3 and 5 days after injury, with the greatest expression occurring in the first 3 days as the muscle undergoes degeneration and necrosis (Fig. 1D). Myogenin western blotting showed induction of regenerative fibers at these same time points (Fig. 1D). Hence, in both chronic skeletal muscle disease and in acute injury models, endogenous Serpina3n expression is induced in muscle tissue.
Figure 1.
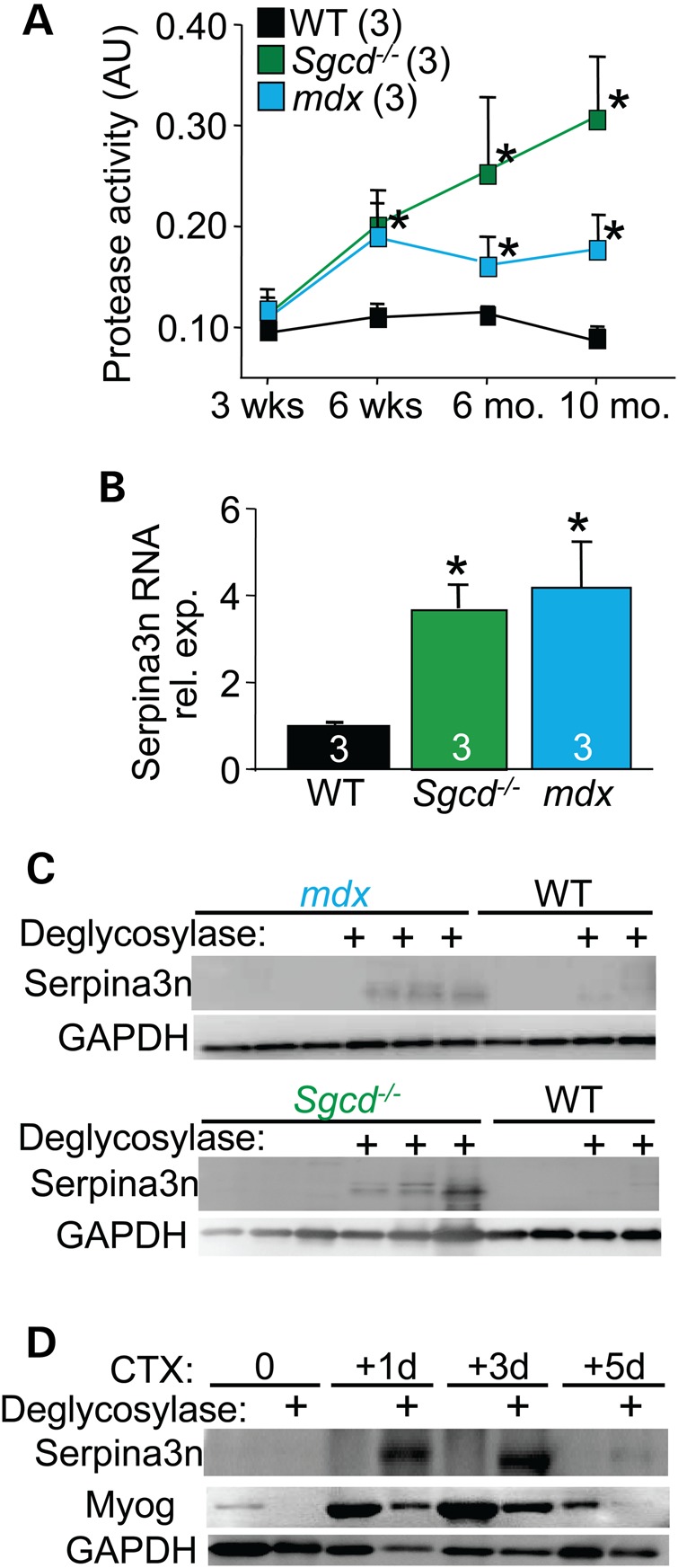
Serpina3n is expressed in injured skeletal muscle. (A) Increased serine protease activity in the quadriceps of mdx and Sgcd−/− mice when compared with WT from 3 weeks to 10 months. of age. The number of mice analyzed is shown. *P < 0.05 versus WT at the same time point. (B) qPCR for Serpina3n mRNA measured from quadriceps of 2–3-month-old mdx and Sgcd−/− mice compared with the WT. *P < 0.05 versus WT. (C) Western blot for Serpina3n expression in the quadriceps of 2–3-month-old mdx and Sgcd−/− mice compared with WT. Deglycosylase (+) treatment was used to unmask the epitope for antibody binding to Serpina3n. GAPDH was used as a processing and loading control. (D) Serpina3n expression in the TA muscle of WT mice at the indicated times in days following acute CTX injury. The ‘+’ signs denote deglycosylase treatment of the protein extracts. Myogenin (Myog) was used as a marker for muscle injury and regeneration and GAPDH was a processing and loading control. All error bars are SEM.
Generation of skeletal muscle-specific Serpina3n TG mice
Increased serine protease activity in the extracellular milieu of skeletal muscle likely arises from secretion of select proteases from inflammatory cells and myofibers (4). While we observed up-regulation of Serpina3n expression in skeletal muscle of dystrophic mice (Fig. 1C), we hypothesized that this effect was only partial and that greater levels of overexpression should more effectively reduce disease-causing protease activity. Hence, we generated skeletal muscle-specific TG mice overexpressing mouse Serpina3n under the control of a modified human skeletal α-actin promoter that was described previously (16) (Fig. 2A). Two Serpina3n TG lines were created: TG-line 1 (high) and TG-line 2 (low) (Fig. 2B). For all subsequent experiments only Serpina3n TG-line 1 were used, which showed robust protein expression in fast skeletal muscles such as the TA, extensor digitorum longus, quadriceps and gastrocnemius (Fig. 2C). Deglycosylation was not needed for western blotting detection with high levels of overexpression. The more slow/oxidative fiber-type muscles, such as the soleus and diaphragm, showed lower levels of Serpina3n overexpression, while the heart showed no transgene expression (Fig. 2C). Expression was not observed in non-TG (NTG) control muscles, even if deglycosylated (Fig. 2C, and data not shown).
Figure 2.

Generation of Serpina3n TG mice. (A) Schematic of the Serpina3n TG construct used to make mice, directed by the skeletal α-actin promoter. pA is the polyadenylation sequence. (B) Western blot analysis for Serpina3n or GAPDH expression from the quadriceps muscle of two separate TG founder lines at 2 months of age. (C) Western blotting for Serpina3n expression in the different skeletal muscles shown, as well as heart from TG-line 1. GAPDH was a loading and processing control. quad, quadriceps; gastroc, gastrocnemius; TA, tibialis anterior; EDL, extensor digitorum longus; diaph, diaphragm; sol, soleus. (D) Protease activity assay from the TA muscle of WT versus Serpina3n TG mice at time 0 or the indicated days after CTX injury. The number of mice analyzed is shown. *P < 0.05 versus NTG. (E) Immunohistochemistry from the quadriceps of NTG or TG mice for Serpina3n expression (green) versus the myofiber outline with laminin staining (red). Nuclei are shown in blue with DAPI staining. (F) Western blot analysis of an enriched extracellular protein fraction from the quadriceps of NTG or TG mice for the indicated proteins. Collagen I was an ECM extract control and α-actinin was a cytosol contamination control, while Ponceau staining assessed loading. (G) Transmission electron microscopy ultrastructural analysis in the quadriceps of TG mice versus NTG (n ≥ 10 sections for each mouse and a total of three separate animals for each genotype). (H) Gravimetric assessment of muscle and heart weights in milligrams, from 2-month-old NTG and TG mice, normalized to tibia length in millimeters. The number of mice used is shown in the graph. Error bars represent SEM.
To examine whether transgene-mediated overexpression of Serpina3n produced superior inhibition of endogenous serine protease activity in vivo, we performed acute injury with CTX injection into the TA muscle and then assessed protease activity over 6 days (Fig. 2D). The data show a prominent induction in serine protease activity in NTG TA muscle after injury, which is completely inhibited in the TG mice (Fig. 2D). Consistent with the western blotting results, immunohistochemistry showed minimal expression of endogenous Serpina3n (green) in the healthy NTG muscle, but prominent protein expression in TG muscle that localized to the extracellular region based on its colocalization with laminin (Fig. 2E). However, some intracellular protein expression was also observed in the TG myofibers, likely reflecting a pool of the protein within the secretory pathway that was being processed (Fig. 2E). Western blotting of the extracellular protein fraction isolated from skeletal muscle showed abundant Serpina3n levels in TG compared with NTG mice (Fig. 2F). Muscle histology from mice with skeletal muscle-specific Serpina3n overexpression appeared normal at both 6 weeks (Fig. 3A) and 10 months of age (Supplementary Material, Fig. S1), including no change in muscle ultrastructure at 6 weeks as assessed by transmission electron microscopy (Fig. 2G) and by assessment of skeletal muscle weights (Fig. 2H). Collectively, these data indicate that Serpina3n overexpression potently inhibited increased serine protease activity after acute muscle injury, although this overexpression was of no observable harm to muscle.
Figure 3.
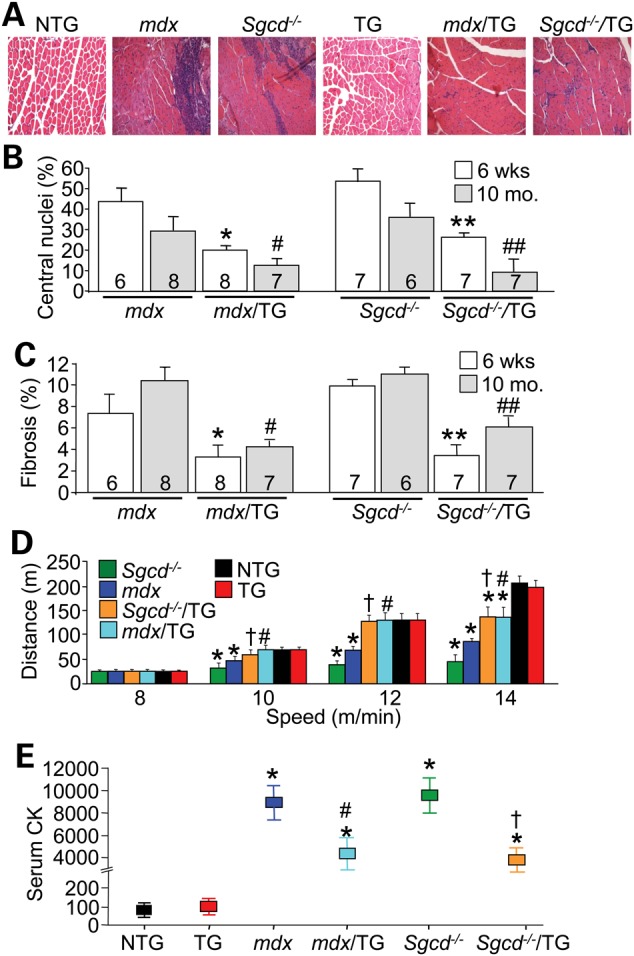
Overexpression of Serpina3n protects against MD. (A) Representative haematoxylin and eosin histological sections from the quadriceps of 6-week-old mice of the indicated genotypes. Magnification is 200×. (B and C) Quantitation of the percentage of myofibers with centrally localized nuclei and fibrotic area from histological sections of the quadriceps in the genotypes shown at 6 weeks and 10 months. of age. The number of mice used is shown in the graph. *P < 0.05 versus mdx at 6 weeks; #P < 0.05 versus mdx at 10 months; **P < 0.05 versus Sgcd−/− at 6 weeks; ##P < 0.05 versus Sgcd−/− at 10 months. (D) Treadmill running performance in 3-month-old mdx/TG or Sgcd−/−/TG compared with mdx or Sgcd−/− mice, with NTG and TG controls. At least six mice were run for each genotypes. *P < 0.05 versus NTG at each speed; #P < 0.05 versus mdx at each speed; †P < 0.05 versus Sgcd−/− at each speed. (E) Serum CK levels in the blood of the indicated genotypes of mice at 3 months of age. *P < 0.05 versus NTG; #P < 0.05 versus mdx; †P < 0.05 versus Sgcd−/−. All error bars represent SEM.
Myofiber-specific overexpression of Serpina3n mitigates MD pathology
To investigate the contribution of Serpina3n as an effector of chronic muscle disease, the Serpina3n TG mouse was crossed with the mdx mouse model of Duchenne MD and Sgcd−/− mice that models limb-girdle MD. Importantly, overexpression of Serpina3n blunted increased activity of the extracellular serine protease cathepsin G and the intracellular cysteine proteases cathepsin B and L, each of which are normally activated in dystrophic muscle from mdx and Sgcd−/− mice, although cysteine protease cathepsin S activity, which is also increased in dystrophic mice, was not reduced by Serpina3n (Supplementary Material, Fig. S2). Associated with this inhibition of select protease activity in skeletal muscle of dystrophic mice, Serpina3n overexpression also significantly reduced histopathology in 6-week-old mdx and Sgcd−/− mice (Fig. 3A–C). Improved muscle histopathology was also observed in older (10 months old) mdx or Sgcd−/− mice containing the Serpina3n transgene (Supplementary Material, Fig. S1), indicating that overexpression of Serpina3n did not merely delay the MD disease progression in these two mouse models (Fig. 3B and C). We also subjected these mice to forced treadmill running to assess whether the observed improvement in histopathology conferred by the muscle-specific Serpina3n transgene correlated with a functional benefit. Both mdx/TG and Sgcd−/−/TG mice showed full restoration of running capacity and endurance on a treadmill at speeds of 10 and 12 m/min, compared with a marked reduced capacity of mdx and Sgcd−/− mice at these speeds (Fig. 3D). Partial protection was also observed in mdx/TG and Sgcd−/−/TG mice at 14 m/min (Fig. 3D). Finally, the Serpina3n transgene also significantly reduced serum creatine kinase (CK) levels in mdx/TG and Sgcd−/−/TG mice compared with mdx and Sgcd−/− mice at 3 months of age, suggesting decreased membrane rupture and ongoing necrosis (Fig. 3E). This profile of healthier skeletal muscle in both mdx/TG and Sgcd−/−/TG mice was also consistent with a reduction in immune cell infiltration as measured by CD45 and Mac3 immunohistochemistry staining, as well as by a general reduction in inflammatory cytokine mRNA levels in muscle from mdx or Sgcd−/− mice that contain the Serpina3n transgene (Supplementary Material, Fig. S3A and B).
Consistent with a decrease in serum CK levels associated with the Serpina3n transgene in the mdx or Sgcd−/− backgrounds, direct assessment of sarcolemmal leakiness in vivo using Evan's blue dye (EBD) technique also showed protection. Mice were injected with EBD for 24 h and then subjected to a period of treadmill running as an added stress to myofiber membranes, after which the mice were sacrificed and muscles were collected. Muscle from both mdx/TG and Sgcd−/−/TG showed significantly reduced EBD myofiber positivity compared with myofibers from mdx or Sgcd−/− mice, while NTG and Serpina3n TG mice showed essentially no positivity, indicating greater membrane stability with the Serpina3n transgene (Fig. 4A and B). To more directly evaluate sarcolemma membrane stability at the single-myofiber level, we also measured the capacity of myofibers isolated from the flexor digitorum brevis (FDB) to be injured and resealed upon laser stimulation in the presence and absence of calcium (Fig. 4C and F). Consistent with increased sarcolemma membrane fragility that is known to occur in dystrophic myofibers (1,2), significantly greater FM1-43 fluorescence dye entry was observed after laser stimulation in the myofibers of mdx and Sgcd−/− mice compared with NTG or TG controls (Fig. 4D and E). However, the presence of the Serpina3n transgene significantly reduced FM1-43 dye fluorescence after laser stimulation in the mdx or Sgcd−/− backgrounds, suggesting less injury and/or greater repair capacity (Fig. 4C–E).
Figure 4.

Serpina3n overexpression protects against sarcolemma membrane damage. (A and B) Representative histological images and quantitation of EBD (red) myofiber staining in the TA muscle following running in the indicated genotypes of mice. WGA-FITC (green) is used as a membrane marker. The number of mice used is shown in the graph. *P < 0.05 versus NTG; #P < 0.05 versus mdx; †P < 0.05 versus Sgcd−/−. (C) Representative images for FM1-43 dye entry (green) from individually isolated FDB myofibers from the indicated genotypes of mice. The top row of fibers were uninjured while the bottom row were all laser injured in the presence of 1.25 mM Ca2+ to cause FM1-43 dye entry as an indication of membrane stability and resealing. (D and E) Quantitation of FM1-43 dye entry after laser injury as shown in (C) from n ≥ 12 fibers for each mouse and a total of four separate animals for each genotype. (F) Representative images for FM1-43 dye entry (green) from isolated myofibers following laser injury in the absence of extracellular Ca2+ (0 Ca2+ buffer supplemented with 0.5 mM EGTA). (G and H) Quantitation of FM1-43 dye entry after laser injury as shown in (F) from n ≥ 9 fibers for each mouse and a total of three separate animals for each genotype. *P < 0.05 versus NTG; #P < 0.05 versus mdx; †P < 0.05 versus Sgcd−/−. All error bars represent SEM.
Removal of calcium from the extracellular solution in this assay further enhanced FM1-43 dye entry in all experimental cohorts after laser stimulation, as calcium is needed for active membrane repair (Fig. 4F–H). Interestingly, overexpression of Serpina3n led to substantially less accumulation of FM1-43 dye in both mdx/TG and Sgcd−/−/TG myofibers when compared with mdx or Sgcd−/− myofibers alone (Fig. 4F–H). This result indicates that overexpression of Serpina3n did not enhance the repair capacity of dystrophic myofibers, which is calcium dependent, but rather provided greater stability to the sarcolemma in the dystrophic myofibers from the onset, suggesting a mechanism whereby serpina3n overexpression leads to greater structural support of the membrane.
Serpina3n stabilizes sarcolemma attachment proteins in dystrophic myofibers
Proteases have been reported to degrade critical attachment proteins that comprise the DGC in dystrophic skeletal muscle or otherwise damage the basal lamina that is required for structural attachments (17,18). Other studies have suggested more direct effects of proteases on the integrity of the DGC and integrin complexes (19–21). Hence, we examined select sarcolemma adhesion complex proteins by both immunohistochemistry and western blotting from membrane-enriched protein fractions (Fig. 5A–C). As expected, expression and membrane localization of key anchoring proteins of the DGC were reduced or absent in skeletal muscle from mdx or Sgcd−/− backgrounds. Levels of dystrophin, β1D- and α7-integrin, α-sarcoglycan, β-sarcoglycan, δ-sarcoglycan, α-dystroglycan and β-dystroglycan were substantially reduced or absent at the sarcolemmal in mdx or Sgcd−/− muscle compared with NTG or TG control (Fig. 5A–C). However, overexpression of Serpina3n resulted in noticeably increased levels or sarcolemmal localization of most of these proteins, as well as an induction of utrophin that could also stabilize the membrane in the absence of dystrophin (22) (Fig. 5A–C). Importantly, mRNA levels for these same proteins were not different (Supplementary Material, Fig. S4). These results suggested that Serpina3n overexpression increased the stability of the sarcolemma and reduced MD pathology by augmenting protein levels of select proteins within the DGC and integrin complexes.
Figure 5.

Overexpression of Serpina3n restores the abundance of sarcolemma adhesion components in two dystrophic mouse models. (A) Immunohistochemistry of transverse quadriceps histological sections or (B) western blotting from quadriceps muscle of membrane protein extracts from mice at 2–3 months of age. The genotypes of mice used are shown with the indicated antibodies. Nuclei are stained in blue with DAPI, and Ponceau was used as a western loading control. Dystr. dystrophin; Utr, utrophin; β1D-Itg, β1D-integrin; α7-Itg, α7-integrin; δ-SG, δ-sarcoglycan; α-DG, α-dystroglycan; β-DG, β-dystroglycan; β-SG, β-sarcoglycan; α-SG; α-sarcoglycan; CaV1.1, L-type calcium channel. (C) Quantitative protein analysis for the indicated proteins in dystrophic muscle following overexpression of Serpina3n. A total of three separate animals for each genotype were used for densitometric analysis. *P < 0.05 versus NTG; #P < 0.05 versus mdx; †P < 0.05 versus Sgcd−/−. All error bars represent SEM.
In addition to modifying the stability of the DGC/utrophin complex and select integrin components within the sarcolemma, Serpina3n overexpression also prevented cleavage of annexin A1 in dystrophic muscle (Fig. 6). Annexin A1 is a membrane repair protein that is known to be highly susceptible to proteolytic cleavage, in particular by serine proteases such as cathepsin G (23,24). Annexin A1 was up-regulated in dystrophic muscle of mdx or Sgcd−/− mice compared with NTG or TG controls, as shown by western blotting of a cytosolic protein fraction from skeletal muscle, or by analysis of mRNA levels (Fig. 6A and B). However, western blotting of a plasma membrane enriched fraction showed cleaved or degraded annexin A1 in muscle from mdx or Sgcd−/− mice (white arrows in Fig. 6A). Remarkably, this degradation of annexin A1 was not observed in the presence of the Serpina3n transgene (Fig. 6A). These results suggest that annexin A1 is induced in dystrophic skeletal muscle, which is likely degraded by proteases that Serpina3n inhibits. Since annexin A1 is a repair and sarcolemma membrane stabilizer protein, these results suggest another mechanism whereby Serpina3a induction could be protective in MD.
Figure 6.

Serpina3n prevents degradation of annexin A1. (A) Western blotting from cytosolic and plasma membrane protein fractions for annexin A1, Serpina3n, troponin I and caveolin 3 from quadriceps of the indicated genotypes of mice at 2–3 months of age. The white arrows show degradation of annexin A1 protein in the mdx and Sgcd−/− membrane protein fractions. Troponin I was a control to show no cytoplasmic protein contamination of the membrane protein fraction, and caveolin 3 was used to show enrichment of membrane proteins versus lower levels in the cytosolic protein fraction. (B) qPCR showed up-regulation of annexin A1 RNA expression in quadriceps of dystrophic mice versus NTG and TG only. The number of mice used for analysis is shown in the graph. *P < 0.05 versus NTG. Error bars represent SEM.
Transgenic mice overexpressing Serpina3n showed expression of the protein within the myofibers and the ECM region. To examine the cellular location whereby Serpina3n overexpression might provide protection, presumably by inhibiting one or more proteases, we used human embryonic kidney 293 cells (HEK293) cells and transient transfection assays in culture. A mouse Serpina3n cDNA was subcloned into a 3x-Flag vector (3xFg-S3n) for transfection and Flag detection, which again showed that Serpina3n can also be detected in the intracellular compartment (lysate) and extracellular media of HEK293 cells (Supplementary Material, Fig. S5A and B). Immunocytochemistry of Serpina3n with different organelle/vesicular trafficking markers showed that Serpina3n co-localized with ER (protein disulfide isomerase) and both cis- (GM130) and trans-Golgi (Golgin 97) networks (Supplementary Material, Fig. S5B). Interestingly, Serpina3n selectively colocalized with an endosome marker [mannose 6 phosphate receptor (M6PR)] and a recycling endosome marker (Rab11), but not with lysosomes (Cathepsin D or Lamp 2), nor with a select type of secretory vesicle (Rab3c or Snap25). Although the M6PR traffics cargo to the lysosome, they are not found in the lysosome itself; rather they cycle between the trans-Golgi network and endosomes (25,26). The intracellular localization of Serpina3n within the ER-Golgi-endosome pathway might also provide protection by preventing degradation of sarcolemma adhesion or repair components that themselves are trafficked to the membrane for insertion or secretion through the same processing pathway and vesicles. Collectively, these results suggest at least two locations whereby Serpina3n might provide protection by enhancing sarcolemma membrane stability, either from the extracellular compartment when it is secreted, or from within the cell as these proteins traverse the internal secretory pathway together on their way to sarcolemmal insertion.
Protective effect of Serpina3n following acute CTX injury
Serpina3n was robustly expressed immediately after acute CTX injury (Fig. 1D). To determine whether Serpina3n affects myofiber survival and regeneration, we used CTX to acutely damage and cause focal necrosis of the TA muscles in NTG and TG mice (Fig. 7A). Using embryonic myosin heavy chain (eMHC) as a marker for newly formed and repaired myofibers, we observed its earlier expression (green) in the injured TG muscle (peaked at 5 days post-injury) compared with NTG muscle (peaked at 7 days post-injury), suggesting enhanced regenerative capacity in the TG skeletal muscle (Fig. 7A and B). At 7 days post-injury, eMHC was minimally expressed in the injured TG muscle indicating a more completed regenerative process. Assessment of surface areas of eMHC-positive fibers at 5 days post-injury also showed greater improvement in TG myofibers compared with NTG (Fig. 7C). While enhanced regeneration could be part of the profile associated with the Serpina3n transgene, it is also possible that Serpina3n overexpression renders the muscle less likely to be damaged by CTX in the first place, due to greater stability of the myofibers as characterized throughout our study.
Figure 7.
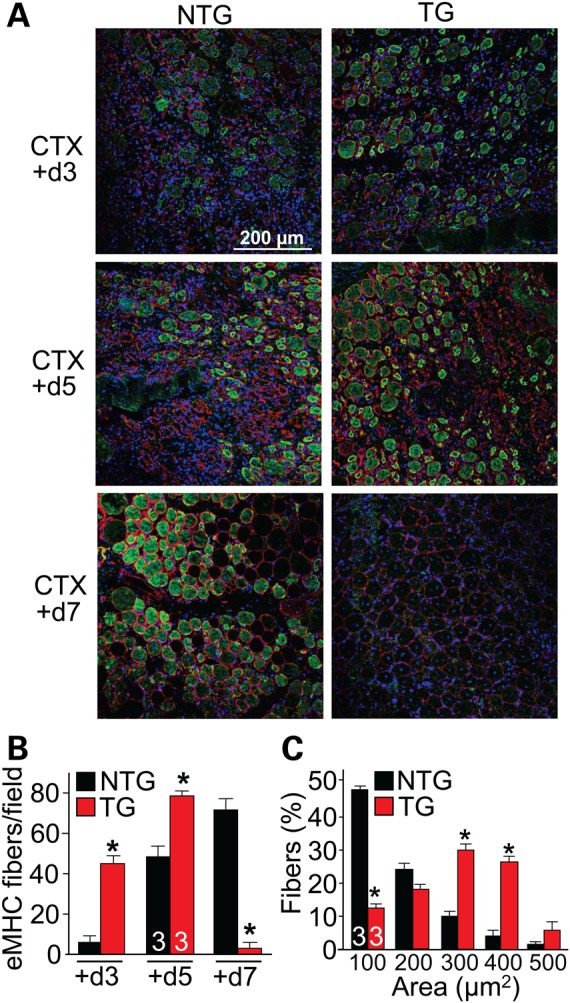
Serpina3n reduces the severity of CTX injury in skeletal muscle. (A) Transverse TA sections were stained with eMHC (green), laminin (red) and DAPI (blue) to detect the regenerative response after CTX injury in NTG and Serpina3n TG mice for the days after injury shown. Newly regenerating myofibers stain for eMHC. (B) Quantification of eMHC-positive fibers in the NTG and TG muscle following the time line of post-CTX injury shown in (A). The number of mice used is shown in the graph. *P < 0.05 versus NTG. (C) Myofiber areas at 5 days post-CTX injury in the TA muscle of NTG and TG mice, separated into ranges. *P < 0.05 versus NTG of the same area range. All error bars represent SEM.
Discussion
The data presented in this study provide genetic evidence supporting the role of protease inhibitors as potential therapeutics in mitigating MD. Indeed, Serpina3n overexpression had a profoundly protective effect in the mdx or Sgcd−/− backgrounds. While it is very difficult to ascertain all the potential proteases that might be regulated by Serpina3n in vivo, our results at least suggest a generalized approach by using broad acting inhibitors of this general class. For example, our results are consistent with an earlier study that used a non-specific serine protease inhibitor, the Bowman–Birk inhibitor, in mdx mice to improve muscle force generation by stabilizing the sarcolemma membrane (7). Our TG approach to overexpress Serpina3n also likely inhibits multiple serine and cysteine proteases that are induced with injury or disease (4,27,28). While some specificity may indeed have been lost with the overexpression approach, it clearly suggested the importance of pathogenic proteases in causing membrane weakening in MD. Other broad acting protease inhibitors were also protective to muscle injury or in the context of MD, such as the use of cromolyn to block mast cell proteases (29,30). Another example was the trypsin-like inhibitor camostat mesilate, which reduced disease manifestations in the mdx mouse model (31).
While mutations in DGC encoding genes renders the sarcolemmal membrane less stable, the secretion of proteases by infiltrating inflammatory cells or by the myofibers represents another independent mechanism that appears to further aggravate membrane instability through the cleavage of select membrane adhesion complex proteins or components of the underlying basal lamina (19–21). We propose two possible mechanisms of action whereby Serpina3n reduces cleavage of the membrane adhesion complexes to stabilize the myofibers. The first is by inhibiting select extracellular pathogenic proteases secreted by inflammatory cells during injury, which would reduce destabilization of the sarcolemma by blocking extracellular proteases that cleave the extracellular regions of the membrane stabilizing attachment complexes (laminins-DGC and integrins). The second mechanism is that Serpina3n expression inside the cell, within endosomes and vesicles, blocks the activity of select proteases that could be cleaving the membrane stabilizing complexes from inside the cell. For example, overexpression of Serpina3n in dystrophic muscle repressed intracellular proteolytic degradation of annexin A1 in the sarcolemma (Fig. 6) and inhibited several intracellular cathepsins (Supplementary Material, Fig. S2).
Maintenance of annexin A1 at the membrane along with increased membrane levels of the DGC proteins could fully explain the profile of reduced EBD uptake, reduced serum CK levels, and reduced membrane injury with laser stimulation, all of which we observed when Serpina3n was overexpressed in the mdx or Sgcd−/− backgrounds. Integrins can be protective in MD by providing additional membrane stability, such as observed in a severe MD mouse model due to the double deletion of the genes encoding dystrophin and utrophin (32,33). Overexpression of α7β1-integrin was protective in these double null mice (33,34). We observed a very prominent stabilization of both α7 and β1 integrins with Serpina3n overexpression in the mdx or Sgcd−/− backgrounds suggesting another mechanism whereby inhibition of select protease activity with Serpina3n would lead to greater membrane stability.
We also showed that Serpina3n overexpression improved apparent muscle regeneration following acute injury with CTX. Mechanistically, we are not certain if this effect is due to a true augmentation in actual regenerative activity of satellite cells, or if the myofibers themselves are slightly more protected from the full damage due to CTX injection. This later possibility seems plausible given that serine proteases likely play a prominent role in degrading areas of injury, which might extend into only partially injured fibers or even uninjured fibers in the border areas. Regardless, Serpina3n overexpression clearly was protective and allowed the muscle to recover faster after acute CTX injury.
While we were not able to identify the exact proteases that negatively impact membrane stability in MD or with acute injury, Serpina3n is known to block an array of serine proteases such as cathepsin G, Granzyme B, elastase, trypsin, chymotrypsin and MMP9, as well as select cysteine proteases (8,13,15,35). Thus, while transgene-mediated Serpina3n overexpression models the known increase of this protein in MD, this transgene drove considerably higher levels of expression, which we believe is relevant to ‘drug-like’ effect that might inform future therapeutic approaches directed at proteases in general. Indeed, recombinant Serpina3n was used in a mouse model of multiple sclerosis, where it was administered in the tail vein at 25–50 μg each day where it reduced axonal and neuronal injury (13). Hence, use of either Serpina3n itself as a biological treatment, or generation of semi-selective small molecule protease inhibitors would represent a novel therapeutic strategy to attempt in MD, and it may be that less selective inhibition across all classes of proteases proves most efficacious. Finally, we also considered analyzing mice deficient in Serpina3n, but decided against such a strategy because there are 16 clade members within the serpin family in mice, which are variably similar to Serpina3n and would likely compensate for a loss-of-function genetic approach. However, overexpression of Serpina3n showed that antagonizing selective pathogenic proteases in the muscles of dystrophic mice is protective.
Materials and Methods
Ethics statement
All animal procedures and usage were approved by the Institutional Animal Care and Use Committee of the Cincinnati Children's Hospital Medical Center, protocol IACUC2013-0013. No human subjects were used or human tissue or cells.
Animals
The mouse Serpina3n cDNA was cloned in a modified human skeletal α-actin promoter (16) to create skeletal muscle-specific TG mice (FVBN strain). Two TG lines were generated by DNA injection into newly fertilized mouse oocytes. Sgcd–/– and mdx mice were described previously (36,37). To generate the mdx/TG mice, mdx female mice C57Bl/10 were mated with male Serpina3n TG mice (line 1) and first generation males with or without the transgene were analyzed. To generate Sgcd−/−/TG mice, Sgcd−/− females were mated with Serpina3n TG males and siblings from these litters were subsequently mated, and the third generation was analyzed. Comparisons were always made between littermates with a similar mixed background.
Protease activity assay
A serine protease (cathepsin G) activity assay kit was used in either the dystrophic or CTX-injured muscles (Abcam, ab126780). Cysteine protease activity was measured with assay kits for: cathepsin B (Abcam, ab65300); cathepsin L (Abcam, ab65300) and cathepsin S (Abcam, ab65307). Isolated muscles were frozen and ground into a fine powder using a mortar and pestle and sonicated before being adding to ice-cold buffer from the assay kit. The enzymatic assay itself was conducted according to the manufacturer's instructions.
Western blotting and protein preparation from skeletal muscle
Isolated muscles were frozen and ground to a fine powder using a mortar and pestle, sonicated and then added to ice-cold radio-immunoprecipitation assay buffer (140 mM NaCl, 0.1% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 1 mM ethylenediaminetetraacetic acid (EDTA), 0.5 mM ethyleneglycoltetraacetic acid (EGTA) and 50 mM Tris, pH 8.0) with protease inhibitors (Roche). When necessary, 50 μg protein lysate isolated from CTX-injured skeletal muscle, or the quadriceps of Sgcd−/−, mdx and WT mice were treated with protein deglycosylase mix [New England Biolabs (NEB): P6039S] to yield deglycosylated muscle lysates. Equal quantities of untreated and deglycosylated muscle lysate were resolved by SDS–polyacrylamide gel electrophoresis (PAGE) (10%) and transferred to polyvinylidene fluoride membrane and detected by ECF (GE Healthcare; RPN3685). Serpina3n was detected using goat polyclonal antibody from R&D systems (AF-4709; 1:1000); myogenin from Developmental Studies Hybridoma Bank (DSHB) (F5D 1:500), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) from Fitzgerald (10R-G109A 1:10 000).
Extracellular protein fractionation from quadriceps muscle was isolated as described previously (38). In brief, quadriceps muscle was minced and washed with phosphate buffered saline (PBS) followed by a 1 h washing step in 0.5 M NaCl, 10 mM Tris–HCl, pH 7.5 and 25 mM EDTA. Decellularization was performed overnight in 0.1% SDS, 25 mM EDTA, followed by ECM extraction with 4 M guanidine hydrochloride, 50 mM sodium acetate, and 25 mM EDTA at pH 5.8. Proteins were precipitated in 80% ethanol overnight, air dried and subjected to treatment with protein deglycosylation mix (NEB).
Skeletal muscle membrane enrichment protein fractions were prepared following a previous protocol (39). Primary antibodies used included: dystrophin [MANDYS1(3B7) 1:500]; utrophin (DSHB Mancho3-8A4 1:500); α7-integrin (Santa Cruz SC-27706 1:500); β1D-integrin (Chemicon; MAB1900, 1:200); δ-sarcoglycan (Abcam ab137101 1:1000); α-dystroglycan (Millipore 05-593 1:500); β-dystroglycan (MANDAG2 clone 7D11 1:500); β-sarcoglycan (Vector labs VP-B206 1: 500); α-sarcoglycan (DSHB IVD3(1)A9 1:500) and CaV1.1 (Pierce MA3-920 1:1000). Ponceau staining was used as a loading control. Densitometry analysis was quantified with ImageJ software (National Institutes of Health).
Cytosolic and plasma membrane fractionation for annexin A1 was prepared using a membrane protein extraction kit (BioVision K268-50). Primary antibodies used included: annexin A1 (Santa Cruz SC-11387); caveolin 3 (BD Biosciences 610420 1:1000) and troponin I (Cell Signaling 4002S 1:2000).
RNA expression analysis
RNA was extracted from muscles using the RNeasy Kit according to manufacturer's instructions (Qiagen) and reverse transcribed using SuperScript® III Reverse Transcriptase (Invitrogen 18080-044). Expression differences were analyzed by real-time qPCR using SYBR green (Applied Biosystems). Primer sequences for quantified RNA expression were: Serpina3n (F: 5′GGGATGATCAAGGAACTGGTCT and R: 5′CCGCGTAGAACTCAGACTTGAA); utrophin (F: 5′ATTTTGCCAAACATCCTCGGC and R: 5′GACTGGGAGGGGTCATAGTG); α7-integrin (F: 5′CTGCTGTGGAAGCTGGGATTC and R: 5′CTCCTCCTTGAACTGCTGTCG; β1-integrin (F: 5′ATGCCAAATCTTGCGGAGGAAT and R: 5′TTTGCTGCGATTGGTGACATT); α-sarcoglycan (F: 5′GCAGCAGTAACTTGGATACCTC and R: 5′AAAGGATGCACAAACACACGA); β-sarcoglycan (F: 5′AGCACAACAGCAATTTCAAAGC and R: 5′AGGAGGACGATCACGCAGAT); dystroglycan primer sequences were ordered from the Qiagen quantitect primer assay (Cat no.QT00100562); annexin A1 (F: 5′GCCTTGCACAAAGCTATCATGG and R: 5′GCATTGGTCCTCTTGGTAAGAA); α1-interferon (F: 5′ACCCAGCAGATCCTGAACAT and R: 5′AATGAGTCTAGGAGGGTTGTATTCC); β-interferon (F: 5′CACAGCCCTCTCCATCAACTA and R: 5′CATTCCGAATGTTCGTCCT); γ-interferon (F: 5′ATCTGGAGGAACTGGCAAAA and R: 5′TTCAAGACTTCAAAGAGTCTGAGGTA); interleukin-2 (F: 5′GCTGTTGATGGACCTACAGGA and R: 5′TTCAATTCTGTGGCCTGCTT); and tumor necrosis factor-α (F: 5′CGCTCTTCTGTCTACTGAACTT and R: 5′GATGAGAGGGAGGCCATT).
Histological analysis and immunohistochemistry
Muscles were paraffin-embedded and 6 μm histological sections were cut at the center of the muscle and stained with haematoxylin and eosin or Masson's trichrome. Pictures were taken at a magnification of 200×, and fibers in the quadriceps muscle were counted for central nucleation with the ImageJ software. Interstitial fibrotic regions were quantified using MetaMorph 7.1 (Molecular Devices) as percentage of blue area stained by Masson's trichrome. Immunohistochemistry for eMHC was performed on cryosections within optimal cutting temperature compound (OCT)-embedded muscles using the antibody BF-G6 from DSHB. Antibody for laminin was purchased from Sigma-Aldrich (L9393). Unless otherwise indicated, the same antibodies were used for western blotting and immunohistochemistry. Primary antibodies used include: dystrophin (Abcam ab15277 1:200); utrophin (DSHB Mancho3-8A4 1:50); δ-sarcoglycan (NovaCastra D-SARC-CE 1:100); α-dystroglycan (EMD Millipore 05-593 1:50); β-sarcoglycan (B-SARC-CE 1:100; α-sarcoglycan (NovaCastra A-SARC-CE 1:100); CD45 (BD Biosciences 550539 1:200); Mac3 (BD Pharmingen 550292 1:200). Wheat germ agglutinin-fluorescein isothiocyanate (WGA-FITC) (Sigma-Aldrich; L4895) was used at 50 μg/ml to show fiber outlines in the EBD immunohistochemistry experiments and Wheat germ agglutinin-tetramethylrhodamine (Sigma-Aldrich; L5266) was used at 100 μg/ml in the images with CD45 and Mac3 staining. Nuclei were stained blue with 1 mg/ml 4′,6-diamidino-2-phenylindole (DAPI) diluted to 1:5000.
EBD uptake and involuntary running
Mice were injected with EBD (i.p., 10 mg/ml, 0.1 ml/10 g body weight) and subjected to forced treadmill running for 30 min, and there after sacrificed 24 h later and quadriceps removed and embedded in OCT and snap-frozen in liquid nitrogen as previously described (40). Approximately 2–3-month-old mice were acclimatized to the treadmill (Omni-Pacer LC4/M; Columbus Instruments International) for 10 min at a speed of 6 m/min prior to shock grid activation. The speed was progressively increased by 2 m/min every 3 min until a maximum speed of 14 m/min was attained. Running capacity was assessed by the maximum running endurance time before exhaustion. Exhaustion was determined by the animal remaining on the shock grid for more than 10 consecutive seconds.
CTX injury
CTX derived from Naja mossambica was dissolved in sterile saline (Sigma-Aldrich; C9759). The TA muscle was injected with 100 μl of a 10 μM CTX solution through a 26-gauge needle.
Enzymatic dissociation of myofibers and laser sarcolemma membrane stimulation
FDB muscles were surgically removed and incubated in 0 Ca2+ Tyrode buffer (140 mM NaCl, 5 mM KCl, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 2 mM MgCl2 (pH 7.2) containing 2 mg/ml type I collagenase (Sigma-Aldrich, C-0130) for 50 min at 37°C. After two washes in isotonic Tyrode buffer containing 2.5 mM Ca2+, muscle fibers were gently dissociated by several passages through a series of micropipette tips of gradually decreasing diameter. Enzymatically dissociated FDB fibers were plated onto Mattek glass-bottomed dishes containing 1.25 mM Ca2+ isotonic Tyrode. To induce damage to the muscle fiber, a 5 × 5 pixel area of the sarcolemma membrane was irradiated with 80 mW at 351/364 nm wavelength for 10 s using a Nikon A1 confocal microscope equipped with a 60× water immersion lens, with 2.5 μM FM1-43 dye (molecular probes) present in the extracellular Tyrodes solution. Images were captured at 5 s intervals following damage. The mean fluorescence intensity in the damage area was quantified by the ImageJ software (National Institutes of Health). Ca2+ free analysis was performed by replacing the 1.25 mM Ca2+ isotonic Tyrode with 0 Ca2+ Tyrode buffer supplemented with 0.5 mM EGTA.
Immunocytochemistry in HEK cells
The mouse Serpina3n cDNA was subcloned into a 3xFg vector from Sigma to generate 3xFg-S3n that was then transfected into HEK293 cells. Approximately 48 h post-transfection, cells lysates and concentrated media were subjected to SDS–PAGE and blotted for Serpina3n. HEK293 cells were plated onto glass bottomed dishes, fixed with 4% paraformaldehyde, blocked with 2% bovine serum albumin dissolved in 0.1% Triton X-100 in PBS buffer. The cells were stained with Serpina3n (green) and different organelle/vesicular trafficking markers (red). Antibodies used for cell staining included protein disulfide isomerase (Abcam ab5484); GM130 (BD Biosciences 610822); Golgin97 (Abcam ab95375); M6PR (Abcam ab32815); Rab11 (Abcam ab95375); Rab3c (Abcam ab3336); cathepsin D (Abcam ab75852); Lamp2 (Abcam ab18528). All antibodies were diluted 1:100. Nuclei were stained blue with a 1:5000 dilution of 1 mg/ml DAPI.
Statistics
All results are presented as mean ± SEM. Statistical analysis was performed with unpaired two-tailed t-test (for two groups) and one-way analysis of variance with a Bonferroni correction (for groups of three or more). P-values of <0.05 were considered significant.
Supplementary Material
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
Funding
This work was supported by grants from the National Institutes of Health (J.D.M.) and the Howard Hughes Medical Institute (J.D.M.). T.S. is funded by the DFG—German Research Foundation (SCHI 1290/1-1) and O.K. is funded by Postdoctoral fellowship from the American Heart Association (15POST25480009).
Supplementary Material
References
- 1.Gumerson J.D., Michele D.E. (2011) The dystrophin-glycoprotein complex in the prevention of muscle damage. J. Biomed. Biotechnol., 2011, 210797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lapidos K.A., Kakkar R., McNally E.M. (2004) The dystrophin glycoprotein complex: signaling strength and integrity for the sarcolemma. Circ. Res., 94, 1023–1031. [DOI] [PubMed] [Google Scholar]
- 3.Burr A.R., Molkentin J.D. (2015) Genetic evidence in the mouse solidifies the calcium hypothesis of myofiber death in muscular dystrophy. Cell Death Differ., 22, 1402–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hollinger K., Selsby J.T. (2013) The physiological response of protease inhibition in dystrophic muscle. Acta. Physiol. (Oxf.), 208, 234–244. [DOI] [PubMed] [Google Scholar]
- 5.Mantle D., Preedy V.R. (2002) Adverse and beneficial functions of proteolytic enzymes in skeletal muscle. An overview. Adverse Drug React. Toxicol. Rev., 21, 31–49. [DOI] [PubMed] [Google Scholar]
- 6.Li H., Mittal A., Makonchuk D.Y., Bhatnagar S., Kumar A. (2009) Matrix metalloproteinase-9 inhibition ameliorates pathogenesis and improves skeletal muscle regeneration in muscular dystrophy. Hum. Mol. Genet., 18, 2584–2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morris C.A., Selsby J.T., Morris L.D., Pendrak K., Sweeney H.L. (2010) Bowman-Birk inhibitor attenuates dystrophic pathology in mdx mice. J. Appl. Physiol. (1985), 109, 1492–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Horvath A.J., Irving J.A., Rossjohn J., Law R.H., Bottomley S.P., Quinsey N.S., Pike R.N., Coughlin P.B., Whisstock J.C. (2005) The murine orthologue of human antichymotrypsin: a structural paradigm for clade A3 serpins. J. Biol. Chem., 280, 43168–43178. [DOI] [PubMed] [Google Scholar]
- 9.Forsyth S., Horvath A., Coughlin P. (2003) A review and comparison of the murine alpha1-antitrypsin and alpha1-antichymotrypsin multigene clusters with the human clade A serpins. Genomics, 81, 336–345. [DOI] [PubMed] [Google Scholar]
- 10.Hoffmann D.C., Textoris C., Oehme F., Klaassen T., Goppelt A., Romer A., Fugmann B., Davidson J.M., Werner S., Krieg T. et al. (2011) Pivotal role for alpha1-antichymotrypsin in skin repair. J. Biol. Chem., 286, 28889–28901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han Y.P., Yan C., Garner W.L. (2008) Proteolytic activation of matrix metalloproteinase-9 in skin wound healing is inhibited by alpha-1-antichymotrypsin. J. Invest. Dermatol., 128, 2334–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ang L.S., Boivin W.A., Williams S.J., Zhao H., Abraham T., Carmine-Simmen K., McManus B.M., Bleackley R.C., Granville D.J. (2011) Serpina3n attenuates granzyme B-mediated decorin cleavage and rupture in a murine model of aortic aneurysm. Cell Death Dis., 2, e209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haile Y., Carmine-Simmen K., Olechowski C., Kerr B., Bleackley R.C., Giuliani F. (2015) Granzyme B-inhibitor serpina3n induces neuroprotection in vitro and in vivo. J. Neuroinflammation, 12, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Millay D.P., Goonasekera S.A., Sargent M.A., Maillet M., Aronow B.J., Molkentin J.D. (2009) Calcium influx is sufficient to induce muscular dystrophy through a TRPC-dependent mechanism. Proc. Natl Acad. Sci. USA, 106, 19023–19028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vicuna L., Strochlic D.E., Latremoliere A., Bali K.K., Simonetti M., Husainie D., Prokosch S., Riva P., Griffin R.S., Njoo C. et al. (2015) The serine protease inhibitor SerpinA3N attenuates neuropathic pain by inhibiting T cell-derived leukocyte elastase. Nat. Med., 21, 518–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brennan K.J., Hardeman E.C. (1993) Quantitative-analysis of the human alpha-skeletal actin gene in transgenic mice. J. Biol. Chem., 268, 719–725. [PubMed] [Google Scholar]
- 17.Yamada H., Saito F., Fukuta-Ohi H., Zhong D., Hase A., Arai K., Okuyama A., Maekawa R., Shimizu T., Matsumura K. (2001) Processing of beta-dystroglycan by matrix metalloproteinase disrupts the link between the extracellular matrix and cell membrane via the dystroglycan complex. Hum. Mol. Genet., 10, 1563–1569. [DOI] [PubMed] [Google Scholar]
- 18.Lattanzi G., Muntoni F., Sabatelli P., Squarzoni S., Maraldi N.M., Cenni V., Villanova M., Columbaro M., Merlini L., Marmiroli S. (2000) Unusual laminin alpha2 processing in myoblasts from a patient with a novel variant of congenital muscular dystrophy. Biochem. Biophys. Res. Commun., 277, 639–642. [DOI] [PubMed] [Google Scholar]
- 19.Badorff C., Lee G.H., Lamphear B.J., Martone M.E., Campbell K.P., Rhoads R.E., Knowlton K.U. (1999) Enteroviral protease 2A cleaves dystrophin: evidence of cytoskeletal disruption in an acquired cardiomyopathy. Nat. Med., 5, 320–326. [DOI] [PubMed] [Google Scholar]
- 20.Liu J., Gurpur P.B., Kaufman S.J. (2008) Genetically determined proteolytic cleavage modulates alpha7beta1 integrin function. J. Biol. Chem., 283, 35668–35678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Agrawal S., Anderson P., Durbeej M., van Rooijen N., Ivars F., Opdenakker G., Sorokin L.M. (2006) Dystroglycan is selectively cleaved at the parenchymal basement membrane at sites of leukocyte extravasation in experimental autoimmune encephalomyelitis. J. Exp. Med., 203, 1007–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blake D.J., Tinsley J.M., Davies K.E. (1996) Utrophin: a structural and functional comparison to dystrophin. Brain Pathol., 6, 37–47. [DOI] [PubMed] [Google Scholar]
- 23.Woloszynek J.C., Hu Y., Pham C.T. (2012) Cathepsin G-regulated release of formyl peptide receptor agonists modulate neutrophil effector functions. J. Biol. Chem., 287, 34101–34109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McNeil A.K., Rescher U., Gerke V., McNeil P.L. (2006) Requirement for annexin A1 in plasma membrane repair. J. Biol. Chem., 281, 35202–35207. [DOI] [PubMed] [Google Scholar]
- 25.Gonzalez-Noriega A., Michalak C., Antonio Sosa Melgarejo J. (2005) Cation-independent mannose 6-phosphate and 78 kDa receptors for lysosomal enzyme targeting are located in different cell compartments. Biochim. Biophys. Acta., 1745, 7–19. [DOI] [PubMed] [Google Scholar]
- 26.Ghosh P., Dahms N.M., Kornfeld S. (2003) Mannose 6-phosphate receptors: new twists in the tale. Nat. Rev. Mol. Cell Biol., 4, 202–212. [DOI] [PubMed] [Google Scholar]
- 27.Sano M., Wada Y., Ii K., Kominami E., Katunuma N., Tsukagoshi H. (1988) Immunolocalization of cathepsins B, H and L in skeletal muscle of X-linked muscular dystrophy (mdx) mouse. Acta. Neuropathol., 75, 217–225. [DOI] [PubMed] [Google Scholar]
- 28.Fang J., Shi G.P., Vaghy P.L. (2000) Identification of the increased expression of monocyte chemoattractant protein-1, cathepsin S, UPIX-1, and other genes in dystrophin-deficient mouse muscles by suppression subtractive hybridization. J. Cell Biochem., 79, 164–172. [DOI] [PubMed] [Google Scholar]
- 29.Granchelli J.A., Avosso D.L., Hudecki M.S., Pollina C. (1996) Cromolyn increases strength in exercised mdx mice. Res. Commun. Mol. Pathol. Pharmacol., 91, 287–296. [PubMed] [Google Scholar]
- 30.Radley H.G., Grounds M.D. (2006) Cromolyn administration (to block mast cell degranulation) reduces necrosis of dystrophic muscle in mdx mice. Neurobiol. Dis., 23, 387–397. [DOI] [PubMed] [Google Scholar]
- 31.Sawada H., Nagahiro K., Kikukawa Y., Ban S., Kakefuda R., Shiomi T., Yokosawa H. (2003) Therapeutic effect of camostat mesilate on Duchenne muscular dystrophy in mdx mice. Biol. Pharm. Bull., 26, 1025–1027. [DOI] [PubMed] [Google Scholar]
- 32.Liu J., Burkin D.J., Kaufman S.J. (2008) Increasing alpha 7 beta 1-integrin promotes muscle cell proliferation, adhesion, and resistance to apoptosis without changing gene expression. Am. J. Physiol. Cell. Physiol., 294, C627–C640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burkin D.J., Wallace G.Q., Nicol K.J., Kaufman D.J., Kaufman S.J. (2001) Enhanced expression of the alpha 7 beta 1 integrin reduces muscular dystrophy and restores viability in dystrophic mice. J. Cell. Biol., 152, 1207–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Doe J.A., Wuebbles R.D., Allred E.T., Rooney J.E., Elorza M., Burkin D.J. (2011) Transgenic overexpression of the alpha7 integrin reduces muscle pathology and improves viability in the dy(W) mouse model of merosin-deficient congenital muscular dystrophy type 1A. J. Cell Sci., 124, 2287–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gettins P.G. (2002) The F-helix of serpins plays an essential, active role in the proteinase inhibition mechanism. FEBS Lett., 523, 2–6. [DOI] [PubMed] [Google Scholar]
- 36.Hack A.A., Lam M.Y., Cordier L., Shoturma D.I., Ly C.T., Hadhazy M.A., Hadhazy M.R., Sweeney H.L., McNally E.M. (2000) Differential requirement for individual sarcoglycans and dystrophin in the assembly and function of the dystrophin-glycoprotein complex. J. Cell Sci., 113, 2535–2544. [DOI] [PubMed] [Google Scholar]
- 37.Wissing E.R., Boyer J.G., Kwong J.Q., Sargent M.A., Karch J., McNally E.M., Otsu K., Molkentin J.D. (2014) P38alpha MAPK underlies muscular dystrophy and myofiber death through a Bax-dependent mechanism. Hum. Mol. Genet., 23, 5452–5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barallobre-Barreiro J., Didangelos A., Yin X., Domenech N., Mayr M. (2013) A sequential extraction methodology for cardiac extracellular matrix prior to proteomics analysis. Methods Mol. Biol., 1005, 215–223. [DOI] [PubMed] [Google Scholar]
- 39.Kobayashi Y.M., Rader E.P., Crawford R.W., Iyengar N.K., Thedens D.R., Faulkner J.A., Parikh S.V., Weiss R.M., Chamberlain J.S., Moore S.A. et al. (2008) Sarcolemma-localized nNOS is required to maintain activity after mild exercise. Nature, 456, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Accornero F., Kanisicak O., Tjondrokoesoemo A., Attia A.C., McNally E.M., Molkentin J.D. (2014) Myofiber-specific inhibition of TGFbeta signaling protects skeletal muscle from injury and dystrophic disease in mice. Hum. Mol. Genet., 23, 6903–6915. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.