

Abstract
Many therapeutic monoclonal antibodies act as antagonists to receptors by targeting and blocking the natural ligand binding site (orthosteric site). In contrast, the use of antibodies to target receptors at allosteric sites (distinct from the orthosteric site) has not been extensively studied. This approach is especially important in metabolic diseases in which endogenous ligand levels are dysregulated. Herein, we review our investigations of 3 categories of human monoclonal antibodies that bind allosterically to the insulin receptor (INSR) and affect its activity: XMetA, XMetS and XMetD. XMetA directly activates the INSR either alone or in combination with insulin. XMetS, in contrast, does not directly activate the INSR but markedly enhances the receptor’s ability to bind insulin and potentiate insulin signaling. Both XMetA and XMetS are effective in controlling hyperglycemia in mouse models of diabetes. A third allosteric antibody, XMetD, is an inhibitor of INSR signaling. This antibody reverses insulin-induced hypoglycemia in a mouse model of hyperinsulinemia. These studies indicate, therefore, that allosteric antibodies to INSR can modulate its signaling and correct conditions of glucose dysregulation. These studies also raise the possibility that the use of allosteric antibodies can be expanded to other receptors for the treatment of metabolic disorders.
Keywords: monoclonal antibody, blood glucose, glycemic disorders, hyperinsulinemia, diabetes, allosteric
Recombinant monoclonal antibodies, by blocking cellular pathways that are dysregulated, are powerful targeted therapeutics for certain severe diseases.1-2 However, these cellular pathways, when functioning normally, also have important roles in normal physiology. Therefore successful treatment of these diseases may require modulation, rather than complete inhibition, of signaling pathways to restore a normal physiological state with a potentially low side-effect profile. Monoclonal antibodies also have the potential to treat disease states by either stimulating or enhancing biological signaling pathways. This class of antibodies has the potential to maintain the spatial and temporal aspects of endogenous ligands such as hormones, cytokines, and neurotransmitters.
The site on receptors at which the endogenous ligand binds is defined as the orthosteric site (Figure 1A). Sites on the receptor at which nonligand molecules bind are termed allosteric sites.1 Regulation of receptor activity by molecules binding to allosteric sites has been recognized as common mechanism for the control of enzyme activity and protein function.3 We previously described a new approach to target disease using an allosteric antibody approach based on the modulation of ligand–receptor binding kinetics.2 Both allosteric and orthosteric antibodies can induce conformational changes in receptors that may markedly activate or modulate receptor function. This approach has been previously employed to regulate receptors using small molecules.3
Figure 1.

Schematic models of allosteric modulation of insulin receptor by monoclonal antibodies. (A) Binding of insulin to the orthosteric site of the insulin receptor (INSR) with the generation of various metabolic and mitogenic signals. (B) Binding of a positive allosteric activator (PAA) to an allosteric site on the INSR with the generation of either similar or different metabolic and mitogenic signals. (C) Binding of a modulator to an allosteric site on the INSR with either positive (PAM) or negative modulation (NAM) of insulin binding and/or intracellular signaling.
Allosteric antibodies have the potential to bind and regulate receptors more selectively than orthosteric antibodies (Figure 1). This selectivity is due to lower sequence and structural homology at allosteric sites relative to orthosteric sites.4,5 This selectivity of allosteric molecules is particularly useful for targeting closely related receptors that bind to similar natural ligands. Moreover, because of their noncompetitive nature with the natural ligand, allosteric modulators allow for a more controlled and selective action on the receptor (Figures 1B). In fact, most allosteric antibodies may have little or no effect on receptor function until the active site is bound by the orthosteric endogenous ligand (Figure 1C). However, certain allosteric antibodies can directly stimulate cellular functions in the absence of the natural ligand, and can also act in concert with this ligand (Figure 2A). This type of antibody has been termed a positive allosteric activator (PAA). Moreover, certain allosteric antibodies can either enhance or inhibit the activity of the natural ligand (Figure 2B). These latter types of antibodies have been termed either positive allosteric modulators (PAM) or negative allosteric modulators (NAM).3
Figure 2.

Theoretical dose response profiles of different types of allosteric anti-insulin receptor antibodies on INSR activities. (A) Positive allosteric activator (PAA) directly activates the INSR without altering insulin activity. The effect of a PAA on the INSR is shown either alone or in combination with insulin. The activation of the INSR by PAA does not block further stimulation by insulin. (B) Allosteric modulation of insulin signaling by either a positive allosteric modulator (PAM) or a negative allosteric modulator (NAM) shifts the dose response of insulin to either the left or right, respectively.
It has been suggested that monoclonal antibodies to the insulin receptor (INSR) may be useful in various disorders of glucose metabolism including hyperglycemia and hypoglycemia.6 We have recently produced allosteric antibodies that regulate INSR signaling by either direct activation, or positive or negative modulation.7-10 These types of antibodies have been classified as selective INSR modulators.11 Naturally occurring antibodies have been shown to activate the INSR, including both spontaneous human autoantibodies and mouse monoclonal antibodies. In humans, autoantibodies to the INSR typically bind at the orthosteric insulin binding site. In most cases, these autoantibodies cause severe insulin resistance and diabetes despite compensatory hyperinsulinemia.12-15 Orthosteric INSR autoantibodies isolated from humans have been studied in rats, and have been shown to be weak agonists that cause hypoglycemia at low concentrations and hyperglycemia at high concentrations.16 Thus, these types of orthosteric antibodies are not likely to be candidates for the treatment of hyperlycemic diseases.
The major disease of altered glucose metabolism is type 2 diabetes mellitus (T2DM) in which there is both insulin deficiency and cellular resistance to insulin.17 The temporal sequence of insulin resistance and impaired insulin secretion in T2DM is still being debated. On one hand there is evidence that in T2DM patients, the disease is preceded by insulin resistance that is compensated for by increased insulin secretion from pancreatic beta cells.18 Initially, this compensation maintains glycemic control. However, in many patients, progressive beta cell dysfunction occurs, resulting in hyperglycemia and ultimately clinical diabetes. On the other hand, there is evidence that hyperinsulinemia is a cause of insulin resistance. In this case, the development of insulin resistance is an adaptation to prevent hypoglycemia in the presence of abundant insulin as a result of diets that promote high levels of insulin secretion.19
Insulin initiates the regulation of cellular glucose metabolism by binding to the INSR on the cell surface, causing a conformational shift that leads to activation of the receptor’s intrinsic kinase activity.20 When activated, the INSR undergoes autophosphorylation, followed by the recruitment and phosphorylation of INSR signaling molecules, including the insulin receptor substrate (IRS) proteins and subsequent activation of members of the phosphotidylinositol 3-kinase (PI3K)/Akt pathway.20,21 Activation of this pathway by insulin results in the translocation of glucose transporters to the cell surface with subsequent uptake of glucose and other antidiabetic functions.22,23 Thus, allosteric monoclonal antibodies to the INSR that either directly activate this INSR or potentiate insulin binding and action could theoretically be effective antidiabetic agents.
Another disease of altered glucose metabolism is hypoglycemia due to insulin excess from both endogenous and exogenous sources, which is not an infrequent clinical condition.24-27 Allosteric monoclonal antibodies that either directly inhibit the INSR or decrease insulin binding and signaling of this receptor, would theoretically be effective antihypoglycemic agents.
We have recently described allosteric modulating antibodies targeting the INSR. These antibodies were identified by panning naïve human antibody phage display libraries using the recombinant extracellular domain of the INSR complexed to insulin.7,9,10 This approach was employed to minimize obtaining antibodies to the INSR orthosteric site. Three allosteric antibodies will be described here; XMetA and XMetS, antibodies that activate or positively modulate the INSR and XMetD, an antibody that negatively modulates the INSR. These antibodies regulate the human INSR as well as the INSRs of other mammalian species.
Allosteric Antibodes to the INSR
XMetA: A Positive Allosteric Activator of the INSR (PAA)
XMetA is high affinity antibody that specifically binds and activates CHO and other cells expressing the human INSR and INSRs of other species.7 XMetA did not alter the affinity of insulin binding to the human INSR (Figure 3A). Moreover, XMetA partially activated the INSR as demonstrated by enhanced INSR autophosphorylation (Figure 3B). This activation did not block insulin action.7 Similarly, XMetA partially stimulated the phosphorylation of Akt, a major intracellular mediator of INSR signaling (Figure 3C). This activation of pAkt allowed XMetA to fully stimulate 2-deoxy-D-glucose uptake (Figure 3D). Although XMetA activated pAkt, in contrast to insulin it did not significantly activate pERK.7 While XMetA had a major effect on the INSR, it had no effect on the closely related IGF-1R receptor.7
Figure 3.

XMetA is a positive allosteric activator (PAA) of the INSR. Studies were performed in cultured CHO cells engineered to express the human INSR. (A) XMetA did not influence insulin binding to INSR. (B) XMetA induced INSR autophosphorylatio, but to a lower magnitude than insulin. (C) XMetA induced pAkt phosphorylation, but to a lower magnitude than insulin. (D) XMetA induced cellular glucose uptake to a degree similar to that induced by insulin.
To evaluate the in vivo activity of XMetA, we utilized an animal model of insulinopenic, insulin-resistant diabetes, the multi-low-dose streptozotocin, high-fat diet (MLDS/HFD) mouse.28 In this diabetic animal, fasting blood glucose levels were elevated to nearly 500 mg/dL compared to normal animals whose fasting blood glucose levels were approximately 150 mg/dL. Treatment with XMetA lowered fasting glucose levels to near normal levels (Figure 4). It also reduced nonfasted glucose levels and improved other metabolic indices in the diabetic animals, including plasma non-HDL cholesterol, free fatty acids and beta hydroxy butyrate.7 Consistent with the decrease in glucose levels, XMetA had a major effect to decrease hemoglobin A1c.7 In studies using an animal model of only insulin resistance, the diabetes induced obesity mouse (DIO), XMetA also reversed hyperglycemia.8
Figure 4.
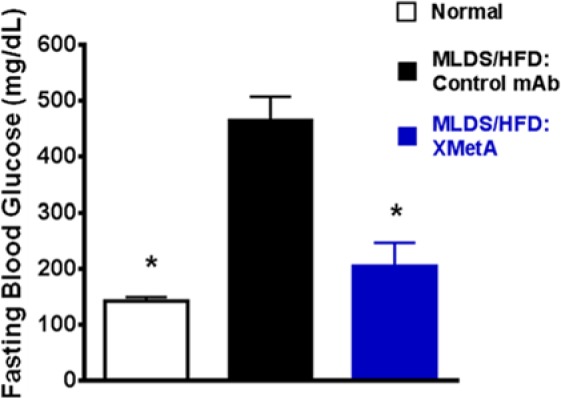
XMetA improves fasting blood glucose in diabetic mice. Studies were performed in mice rendered diabetic by streptozotocin and high fat diet. Animals were treated with XMetA intraperitoneally twice weekly (10 mg/kg) for 6 weeks and fasting blood glucose measured at the end of the study. Open bar, untreated normal mice; black, diabetic mice treated with a control antibody; blue, diabetic mice treated with XMetA.
XMetS: A Positive Allosteric Modulator (PAM) of the INSR
XMetS is another allosteric INSR antibody that binds CHO and other cells expressing the human and mouse INSRs.10 In contrast to XMetA, XMetS did not have any major effects on human INSR signaling in the absence of insulin. However, XMetS enhanced insulin binding affinity approximately 18-fold (Figure 5A) due mainly to a decrease in the insulin dissociation rate.10 This increase in affinity resulted in 14-fold increase in INSR autophosphorylation (Figure 5B). Moreover, XMetS induced a greater than 30-fold increase in the sensitivity of insulin-stimulated Akt phosphorylation (Figure 5C). These observations suggested that XMetS had a major effect on insulin binding, but also had a small additional effect on insulin signaling. As a result of these effects, XMetS markedly enhanced insulin-stimulated 2-deoxy-D-glucose (2DG) uptake (Figure 5D).
Figure 5.

XMetS is a positive allosteric modulator (PAM) of the INSR. Studies were performed in cultured CHO cells engineered to express the human INSR. (A) XMetS positively enhanced insulin binding to the INSR. (B) XMetS enhanced insulin-induced INSR autophosphorylation. (C) XMetS enhanced insulin-induced pAkt phosphorylation. (D) XMetS enhanced insulin-induced glucose uptake.
To evaluate the in vivo activity of XMetS, we studied its effect using the insulin resistant DIO mouse model of diabetes. XMetS improved glucose tolerance in these animals (Figure 6A). Moreover in these animals, XMetS may have potential beta cell sparing activity as treated mice had decreased insulin (Figure 6B) and C-peptide levels. In addition, we studied the effects of XMetS in MLDS/HFD diabetic mice. In these animals, fasting hyperglycemia was markedly reduced and the blood glucose response to exogenous insulin was improved.10
Figure 6.

XMetS improves glucose metabolism in diabetic mice. Studies were performed in DIO mice. Animals were treated twice weekly with either XMetS (10 mg/kg) or a control antibody given intraperitoneally. (A) XMetS improved glucose tolerance. After 1 week of treatment and following a 14-hour overnight fast, a glucose bolus was administered intraperitoneally (1 g/kg) and blood glucose levels were measured for 120 minutes. (B) XMetS decreased fasting insulin levels in DIO mice.
XMetD: A Negative Allosteric Modulator (NAM) of the INSR
We next investigated XMetD, a negative modulator of the INSR.9 In CHO cells expressing the human INSR, XMetD negatively shifted the EC50 by ~ 3-fold (Figure 7A). However, the interaction of XMetD with the INSR resulted in a greater than 40-fold decrease in the sensitivity of insulin-stimulated INSR autophosphorylation (Figure 7B). The much greater inhibition by XMetD on insulin-mediated INSR autophosphorylation relative to its inhibitory effect on insulin binding to the INSR suggested that the antagonistic effects of the antibody were mediated both by its negative modulation of insulin binding and by its negative modulation of INSR activation. To investigate this negative modulation, the effects of XMetD on INSR efficacy were further characterized. XMetD induced a greater than 100-fold decrease in the sensitivity of insulin dependent Akt phosphorylation (Figure 7C). We also assessed whether XMetD inhibited the IGF-1 receptor (IGF-1R); XMetD did not antagonize IGF-1 mediated activation of Akt, indicating that the inhibitory effect of XMetD was specific to the INSR.9 XMetD antagonized insulin stimulation of glucose uptake (Figure 7D) by 70-fold. Thus, these studies indicated that inhibition of insulin stimulated INSR activation by XMetD resulted in greatly diminished metabolic signal activation.
Figure 7.

XMetD is a negative allosteric modulator (NAM) of the INSR. Studies were performed in cultured CHO cells engineered to express the human INSR. (A) XMetD decreased insulin binding to the INSR. (B) XMetD markedly decreased insulin-induced INSR autophosphorylation. (C) XMetD markedly decreased insulin-induced pAkt phosphorylation. (D) XMetS decreased insulin-induced glucose uptake.
To explore whether XMetD would antagonize hypoglycemia induced by insulin excess, we created a mouse model of hyperinsulinemic hypoglycemia by the insertion of slow release insulin implants into normal mice. In this model, the implants continually release insulin for over 2 weeks providing a sustained exposure to high concentrations of the hormone. Three days post implantation; the mice were hypoglycemic with blood glucose levels in the range of 50 mg/dL (Figure 8). In fasted mice with implants, following a 1-week treatment with control IgG, blood glucose levels remained in the range of 50 mg/dL. In contrast, following a 1-week treatment with XMetD, fasted mice with implants were no longer hypoglycemic (Figure 8).9
Figure 8.

XMetD reversed insulin-induced hypoglycemia in mice. Normal male C57BL/6 mice were given insulin implants and fasting glucose levels were measured after either no treatment, treatment with 10 mg/kg XMetD intraperitoneally, or treatment with control IgG. Open bar, untreated normal mice; black, untreated mice with insulin implants; yellow, implanted mice treated with control IgG; red, implanted mice treated with XMetD.
Discussion
We have described 3 different classes of allosteric antibodies to the INSR. These antibodies include XMetA, a PAA; XMetS, a PAM; and XMetD, a NAM. Each of these classes of antibodies may have clinical efficacy in diseases of altered glucose metabolism.
In the treatment of diabetes, novel and improved therapeutic modalities for those individuals with impaired insulin secretory function would be helpful. Currently, long-acting basal insulins are given daily, and are often associated with poor compliance, hypoglycemia, and weight gain.29 To more effectively control basal hyperglycemia, either very long acting new insulin analogues or other novel agents that stimulate the INSR could be of benefit. Fully human antibodies typically have a half-life greater than 1 week and relatively low immunogenicity in humans.30 Thus, a fully human monoclonal antibody that activates the INSR has the potential for dosing in humans at once weekly intervals or less.
XMetA, a PAA, both activated the INSR and allowed the binding of insulin and its subsequent signaling. Moreover, XMetA activated the INSR in a manner that was different than insulin, an effect that could be the result of a distinct structural state of the INSR induced by the binding of XMetA. In animal models of T2D, XMetA reversed hyperglycemia and improved the metabolic profiles.7,8 Moreover, the blood glucose response to exogenous insulin was improved.7,8
These observations suggested the interesting possibility that antibodies such as XMetA, by activating INSR, both in vitro and in vivo, have the potential to improve glycemic control in a model of insulinopenic diabetes without causing hypoglycemia or promoting weight gain. To our knowledge, XMetA is the first fully human monoclonal antibody to the INSR reported to correct hyperglycemia and improve diabetes in vivo. These observations suggest that antibodies like XMetA, either alone or in combination with short acting insulins/oral agents, may represent a novel strategy for controlling blood glucose levels in insulinopenic diabetes.
XMetS markedly enhanced the affinity of insulin for the INSR. XMetS slowed the rate of dissociation of insulin from the INSR. Thus, XMetS was a PAM of the INSR. XMetS also markedly enhanced INSR autophosphorylation, pAkt activation, and glucose uptake several-fold. XMetS slowed the rate of dissociation of insulin from the INSR.10 In humans, insulin may promote tumor progression via the INSR, while insulin analogs with INSR dissociation rates slower than insulin may have greater mitogenic activity.31 Thus we studied the effect of XMetS on insulin-stimulated proliferation in MCF-7 breast cancer cells and Saos-2 osteosarcoma cells, 2 cancer cells lines that are commonly used to determine the mitogenic effects of insulin analogs.32 We observed that XMetS did not enhance the effect of either insulin or IGF-1 on the proliferation of these cell lines.10 One potential explanation for the lack of effect of XMetS on ligand induced proliferation of cancer cells may be related to the relatively limited effect of XMetS on insulin-dependent ERK activation.10 Another explanation is that XMetS does not activate hybrids of INSR and IGF-1R, which are commonly formed in these cancers cell lines.33-35
In the DIO mouse, a model of obese insulin resistance and hyperglycemia, XMetS reduced fasting blood glucose levels and normalized glucose tolerance. Improved glycemic control in these animal models was accompanied by reduced insulin and C-peptide levels, suggesting that XMetS increased insulin sensitivity and lowered the requirement for insulin. These observations suggest that XMetS could improve glycemic control, prevent compensatory hyperinsulinemia, and, in early ti midstage T2DM pateinets, possibly delay or obviate the need for exogenous insulin therapy by preserving beta cell function in early to midstage T2DM patients.
XMetD, a NAM, markedly antagonized insulin-dependent INSR autophosphorylation and downstream metabolic effects, including Akt phosphorylation and glucose transport. XMetD was effective at blocking hypoglycemia induced by insulin excess in mice that were made hyperinsulinemic by the insertion of sustained-release insulin implants. This observation raises the possibility XMetD may be useful for the long term treatment of hypoglycemia caused by insulin. There are several conditions of sustained endogenous hyperinsulinemia where current therapies are not always adequate. These conditions include insulinoma,26,27,36 excess secretion of IGF-II37 and congenital hyperinsulinemia (CHI).38-39 In the latter condition, particularly in those patients with defects of the ATP-regulated potassium channel, control of hypoglycemia can be extremely difficult.39,40 Preliminary studies in the SUR-1 knockout mouse (an animal model of CHI) indicate that XMetD treatment prevents fasting hypoglycemia in these animals.41
Not infrequently, diabetic patients treated with either insulin or insulin-releasing agents develop symptomatic hypoglycemia. On occasion, this hypoglycemia can be severe and sustained, requiring either visits to the emergency department and or prolonged hospitalization.26,42 Thus it is possible that antagonist allosteric INSR antibodies could be a potential new treatment for patients with hypoglycemia due to exogenous hyperinsulinism. In this case, a short acting version of XmetD with a faster clearance rate may be needed. Additional studies with this class of antibodies will be required to understand their optimum use in the clinic.
Conclusion
In this review, we have focused on allosteric modulation of the INSR both in vitro and in vivo by 3 types of monoclonal antibodies. XMetA, a PAA, directly activated the INSR and was effective in ameliorating diabetes in mouse models. Such an antibody may be used either alone or in combination with other agents for insulinopenic diabetes. XMetS, a PAM, enhanced insulin binding to the INSR and was also effective in ameliorating diabetes in mouse models. Such an antibody may be useful in the earlier stages of diabetes in which insulin secretion is still occurring. Finally, we studied XMetD, a NAM that inhibited both insulin binding and insulin-induced INSR signaling. Antibodies such as XMetD may prove useful to treat condition of hyperinsulinemic hypoglycemia.
These data with allosteric antibodies to the INSR suggest that monoclonal antibodies to receptors can be used in conditions of metabolic dysfunction. There are other metabolic conditions in which altered regulation of receptor signaling occurs.43 It is possible, therefore, that allosteric monoclonal antibodies to receptors may have broad clinical utility.
Acknowledgments
We thank Marina Roell, Toshi Takeushi, and Diane Wilcock for critical reviews of the manuscript and insightful comments.
Footnotes
Abbreviations: INSR, insulin receptor; NAM, negative allosteric modulator; PAA, positive allosteric activator; PAM, positive allosteric modulator; T2DM, type 2 diabetes mellitus.
Declaration of Conflicting Interests: The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: HI, DHB, JAC, IDG, VB, MLW, PR and PJS have been or are employees of XOMA Corporation.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
- 1. Nussinov R, Tsai CJ. Allostery in disease and in drug discovery. Cell. 2013;153:293-305. [DOI] [PubMed] [Google Scholar]
- 2. Roell MK, Issafras H, Bauer RJ, et al. Kinetic approach to pathway attenuation using XOMA 052, a regulatory therapeutic antibody that modulates interleukin-1beta activity. J Biol Chem. 2010;285:20607-20614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Christopoulos A, Kenakin T. G protein-coupled receptor allosterism and complexing. Pharmacol Rev. 2002;54:323-374. [DOI] [PubMed] [Google Scholar]
- 4. May LT, Leach K, Sexton PM, Christopoulos A. Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2007;47:1-51. [DOI] [PubMed] [Google Scholar]
- 5. Capra JA, Laskowski RA, Thornton JM, Singh M, Funkhouser TA. Predicting protein ligand binding sites by combining evolutionary sequence conservation and 3D structure. PLOS Comput Biol. 2009;5:e1000585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ussar S, Vienberg SG, Kahn CR. Receptor antibodies as novel therapeutics for diabetes. Sci Transl Med. 2011;3:113ps147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bhaskar V, Goldfine ID, Bedinger DH, et al. A fully human, allosteric monoclonal antibody that activates the insulin receptor and improves glycemic control. Diabetes. 2012;61:1263-1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bhaskar V, Lau A, Goldfine ID, et al. XMetA, an allosteric monoclonal antibody to the insulin receptor, improves glycemic control in mice with diet-induced obesity. Diabetes Obes Metab. 2013;15:272-275. [DOI] [PubMed] [Google Scholar]
- 9. Corbin JA BV, Goldfine ID, Issafras H, et al. Inhibition of insulin receptor function by a human, allosteric monoclonal antibody: a potential new approach for the treatment of hyperinsulinemic hypoglycemia. mAbs. 2014;6:15-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Corbin JA BV, Goldfine ID, Bedinger DH, et al. A novel allosteric monoclonal antibody that improves glucose metabolism in vitro and in vivo by increasing insulin binding affinity to the insulin receptor. PLOS ONE. 2014;9(2):e88684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vigneri R, Squatrito S, Frittitta L. Selective insulin receptor modulators (SIRM): a new class of antidiabetes drugs? Diabetes. 2012;61:984-985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lupsa BC, Chong AY, Cochran EK, et al. Autoimmune forms of hypoglycemia. Medicine (Baltimore). 2009;88:141-153. [DOI] [PubMed] [Google Scholar]
- 13. Le Marchand-Brustel Y, Gorden P, Flier JS, Kahn CR, Freychet P. Anti-insulin receptor antibodies inhibit insulin binding and stimulate glucose metabolism in skeletal muscle. Diabetologia. 1978;14:311-317. [DOI] [PubMed] [Google Scholar]
- 14. De Pirro R, Roth RA, Rossetti L, Goldfine ID. Characterization of the serum from a patient with insulin resistance and hypoglycemia. Evidence for multiple populations of insulin receptor antibodies with different receptor binding and insulin-mimicking activities. Diabetes. 1984;33:301-304. [DOI] [PubMed] [Google Scholar]
- 15. Arioglu E, Andewelt A, Diabo C, et al. Clinical course of the syndrome of autoantibodies to the insulin receptor (type B insulin resistance): a 28-year perspective. Medicine (Baltimore). 2002;81:87-100. [DOI] [PubMed] [Google Scholar]
- 16. Dons RF, Havlik R, Taylor SI, et al. Clinical disorders associated with autoantibodies to the insulin receptor. Simulation by passive transfer of immunoglobulins to rats. J Clin Invest. 1983;72:1072-1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988;37:1595-1607. [DOI] [PubMed] [Google Scholar]
- 18. Reaven GM, Olefsky JM. The role of insulin resistance in the pathogenesis of diabetes mellitus. Adv Metab Disord. 1978;9:313-331. [DOI] [PubMed] [Google Scholar]
- 19. Corkey BE. Banting lecture 2011: hyperinsulinemia: cause or consequence? Diabetes. 2012;61:4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7:85-96. [DOI] [PubMed] [Google Scholar]
- 21. Asano T, Fujishiro M, Kushiyama A, et al. Role of phosphatidylinositol 3-kinase activation on insulin action and its alteration in diabetic conditions. Biol Pharm Bull. 2007;30:1610-1616. [DOI] [PubMed] [Google Scholar]
- 22. Chang L, Chiang SH, Saltiel AR. Insulin signaling and the regulation of glucose transport. Mol Med. 2004;10:65-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Simpson IA, Cushman SW. Hormonal regulation of mammalian glucose transport. Annu Rev Biochem. 1986;55:1059-1089. [DOI] [PubMed] [Google Scholar]
- 24. Arnoux JB, Verkarre V, Saint-Martin C, et al. Congenital hyperinsulinism: current trends in diagnosis and therapy. Orphanet J Rare Dis. 2011;6:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Batcher E, Madaj P, Gianoukakis AG. Pancreatic neuroendocrine tumors. Endocr Res. 2011;36:35-43. [DOI] [PubMed] [Google Scholar]
- 26. Service FJ. Hypoglycemic disorders. N Engl J Med. 1995;332:1144-1152. [DOI] [PubMed] [Google Scholar]
- 27. Cryer PE, Axelrod L, Grossman AB, et al. Evaluation and management of adult hypoglycemic disorders: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2009;94:709-728. [DOI] [PubMed] [Google Scholar]
- 28. Arulmozhi DK, Kurian R, Bodhankar SL, Veeranjaneyulu A. Metabolic effects of various antidiabetic and hypolipidaemic agents on a high-fat diet and multiple low-dose streptozocin (MLDS) mouse model of diabetes. J Pharm Pharmacol. 2008;60:1167-1173. [DOI] [PubMed] [Google Scholar]
- 29. Niswender KD. Basal insulin: physiology, pharmacology, and clinical implications. Postgrad Med. 2011;123:17-26. [DOI] [PubMed] [Google Scholar]
- 30. Lonberg N. Fully human antibodies from transgenic mouse and phage display platforms. Curr Opin Immunol. 2008;20:450-459. [DOI] [PubMed] [Google Scholar]
- 31. Kurtzhals P, Schaffer L, Sorensen A, et al. Correlations of receptor binding and metabolic and mitogenic potencies of insulin analogs designed for clinical use. Diabetes. 2000;49:999-1005. [DOI] [PubMed] [Google Scholar]
- 32. Sciacca L, Le Moli R, Vigneri R. Insulin analogs and cancer. Front Endocrinol (Lausanne). 2012;3:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Belfiore A, Frasca F, Pandini G, Sciacca L, Vigneri R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr Rev. 2009;30:586-623. [DOI] [PubMed] [Google Scholar]
- 34. Frasca F, Pandini G, Vigneri R, Goldfine ID. Insulin and hybrid insulin/IGF receptors are major regulators of breast cancer cells. Breast Dis. 2003;17:73-89. [DOI] [PubMed] [Google Scholar]
- 35. Malaguarnera R, Sacco A, Voci C, et al. Proinsulin binds with high affinity the insulin receptor isoform A and predominantly activates the mitogenic pathway. Endocrinology. 2012;153:2152-2163. [DOI] [PubMed] [Google Scholar]
- 36. Starke A, Saddig C, Mansfeld L, et al. Malignant metastatic insulinoma-postoperative treatment and follow-up. World J Surg. 2005;29:789-793. [DOI] [PubMed] [Google Scholar]
- 37. Arnoux JB, de Lonlay P, Ribeiro MJ, et al. Congenital hyperinsulinism. Early Hum Dev. 2010;86:287-294. [DOI] [PubMed] [Google Scholar]
- 38. Hussain K. Congenital hyperinsulinism. Semin Fetal Neonatal Med. 2005;10:369-376. [DOI] [PubMed] [Google Scholar]
- 39. Palladino AA, Bennett MJ, Stanley CA. Hyperinsulinism in infancy and childhood: when an insulin level is not always enough. Clin Chem. 2008;54:256-263. [DOI] [PubMed] [Google Scholar]
- 40. Henwood MJ, Kelly A, Macmullen C, et al. Genotype-phenotype correlations in children with congenital hyperinsulinism due to recessive mutations of the adenosine triphosphate-sensitive potassium channel genes. J Clin Endocrinol Metab. 2005;90:789-794. [DOI] [PubMed] [Google Scholar]
- 41. Patel P, Corbin JA, Goldfine ID, Rubin P, De Leon DD. A unique allosteric insulin receptor monoclonal antibody that prevents hypoglycemia in the SUR-1-/- mouse model of KATP hyperinsulinism [abstract]. Diabetes. 2013;62(suppl 1):A-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Budnitz DS, Lovegrove MC, Shehab N, Richards CL. Emergency hospitalizations for adverse drug events in older Americans. N Engl J Med. 2011;365:2002-2012. [DOI] [PubMed] [Google Scholar]
- 43. Ahima RS, Osei SY. Adipokines in obesity. Front Horm Res. 2008;36:182-197. [DOI] [PubMed] [Google Scholar]