

Abstract
Gram-negative bacteria are intrinsically resistant to many antibiotics. Species that have acquired multidrug resistance and cause infections that are effectively untreatable present a serious threat to public health. The problem is broadly recognized and tackled at both the fundamental and applied levels. This paper summarizes current advances in understanding the molecular bases of the low permeability barrier of Gram-negative pathogens, which is the major obstacle in discovery and development of antibiotics effective against such pathogens. Gaps in knowledge and specific strategies to break this barrier and to achieve potent activities against difficult Gram-negative bacteria are also discussed.
Keywords: Gram-negative resistance, permeability barrier, outer membrane, multidrug efflux
Graphical Abstract
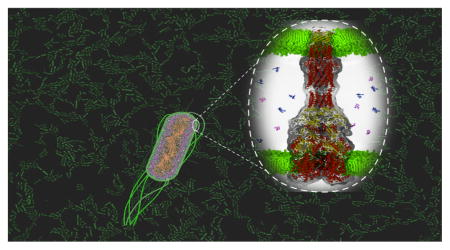
GRAM-NEGATIVE PATHOGENS AND CHALLENGES OF ANTIBIOTIC DISCOVERY
Drug resistance presents an ever-increasing threat to public health and encompasses all major microbial pathogens and antimicrobial drugs.1,2 Some pathogens have acquired resistance to multiple antibiotics and cause infections that are effectively untreatable. Among pathogenic Gram-negative Enterobacteriaceae, Acinetobacter, and Pseudomonas, species have emerged that are resistant to all good antibiotics.3 One of the most troubling event is the worldwide spread of carbapenem-resistant Klebsiella spp.4 Infections caused by these resistant variants have a mortality rate of up to 50%. By 2013, 17% of Escherichia coli infections became multidrug resistant. In some regions, fluoroquinolones are no longer on the lists of recommended treatment options.5,6 Such environmental species as Pseudomonas aeruginosa, Stenotrophomonas malto-philia, Burkholderia cepacia complex (Bcc), and Acinetobacter baumannii are intrinsically resistant to antibiotics. Among these species, P. aeruginosa is a common nosocomial pathogen, the causative agent of many life-threatening infections and the major reason for the shortened life span of people with cystic fibrosis (CF). P. aeruginosa infections can be successfully treated by only a few specific representatives of fluoroquinolones, β-lactams, or aminoglycosides. However, even these few antibiotics fail against antibiotic-resistant P. aeruginosa isolates. Thus, there is a strong need for new therapeutic options, particularly those directed against multiresistant Gram-negative bacteria.
The discovery of new antibiotics effective against Gram-negative bacteria is a major challenge, primarily because of a low hit rate during screening of compound libraries, which is up to 1000-fold lower in P. aeruginosa than against Gram-positive bacteria.7 The major reasons for such a low hit rate are the low permeability barrier of two-membrane cell envelopes of Gram-negative bacteria and insufficient chemical diversity of compound libraries to probe this barrier. Gram-negative bacteria vary significantly in their permeability to antibiotics, but one could expect that the basic principles established by extensive studies of E. coli would apply equally to such “impermeable” species as Burkholderia spp. or Pseudomonas spp. It remains unclear, however, whether permeation rules,8 in analogy with Lipinski’s rules,9 if such existed and were applied to structure–activity relationships or to filtering compound libraries, would yield compounds that permeate all Gram-negative barriers. Here, we briefly review the current state of understanding of molecular bases of low-permeability barriers of the problematic Gram-negative pathogens and current efforts to define the physicochemical properties that enable uptake of various compounds into bacterial cells.
THE TWO-MEMBRANE BARRIER OF GRAM-NEGATIVE BACTERIA
The susceptibility of Gram-negative bacteria to antibiotics is defined by two opposing fluxes across the two membranes of these species (Figure 1).10–12 The influx and uptake of antibiotics are significantly slowed by the elaborate outer membrane (OM). This membrane is an asymmetric bilayer of lipopolysaccharides (LPS) and phospholipids, into which nonspecific porins and specific uptake channels are embedded.13,14 The LPS-containing bilayers are more rigid than normal bilayers, slowing passive diffusion of hydrophobic compounds, whereas narrow pores limit by size the penetration of hydrophilic drugs. The slow influx of drugs across the OM is further opposed by active efflux mediated by multidrug efflux transporters. Multidrug efflux transporters are structurally and functionally diverse, with some transporters pumping antibiotics across the inner membrane and reducing concentrations of antibiotics in the cytoplasm, whereas others expel antibiotics from the periplasm into the external medium. The latter transporters confer resistance to antibiotics by associating with the periplasmic and OM accessory proteins to form trans-envelope complexes (Figure 1).15,16 These complexes enable conversion of the energy stored in the inner membrane into active efflux of antibiotics across the OM. Efflux of antibiotics across the inner membrane acts synergistically with the trans-envelope efflux and, as a result, inactivation of efflux pumps leads to dramatic sensitization of Gram-negative bacteria to antibiotics. The clinical relevance of efflux of multiple antibiotics has also been established. For example, in clinical isolates of E. coli and Klebsiella pneumoniae, fluoroquinolone resistance is linked to overproduction of the AcrAB-TolC efflux pump, whereas multiple efflux pumps confer antibiotic resistance in P. aeruginosa, Burkholderia spp., and A. baumannii.17–20 The interplay between uptake and efflux defines the steady-state accumulation level of antibiotics at targets.
Figure 1.

Cross-section view of a modeled P. aeruginosa cell envelope. The outer leaflet of the outer membrane is assembled of LPS (pink color) corresponding to the band A antigen,25 and the inner leaflet contains glycerophospholipids 1,2-dipalmitoyl-sn-glycero-3-phosphoe-thanolamine (DPPE). Green spheres are magnesium ions bound to O-chains and core polysaccharides of LPS. The inner membrane contains an equimolar mixture of cardiolipin, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE)b and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol (POPG). Embedded in the outer membrane are the main porin OprF (PDB 4RLC) in a yellow surface density. In the inner bilayer is localized MdfA transporter, represented in a magenta surface (PDB 4ZOW). The modeled structure of the assembled MexAB-OprM multidrug efflux pump (MexB, blue; MexA, orange; OprM, red) spans both the inner and outer membranes. The structure of MexAB-OprM is from a long (1 μs) all-atom molecular dynamics simulations with the tripartite complex embedded in 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayers mimicking inner and outer membranes (see Figure 3). The MexA component lacks the N-terminal lipid modification. The outer and inner membranes are based on separate protein-free all-atom MD simulations with the above composition. Other structures were taken from the Protein Data Bank. Ciprofloxacin molecules were added to illustrate a difference of concentrations created by slow diffusion across the outer membrane through porins and LPS-containing bilayer and the active efflux across the outer (MexAB-OprM) and inner (MdfA) membranes. (Inset) Single representative LPS molecule from the simulations. The peptidoglycan is pictorially represented by fiber-like structures underneath the outer membrane. The image was composed and posteriorly rendered using the VMD Molecular Viewer.
Composition and Properties of LPS-Containing Bilayers
Despite a similar structural organization, outer membranes of Gram-negative bacteria differ dramatically in their permeability properties. These differences are largely attributed to differences in the permeability properties of general porins12,14 (see also below), but the chemical structure and properties of the LPS-containing bilayer also play an important role (Figure 1). Typical LPS comprises a basic lipid A structure containing an N- and O-acylated diglucosamine bisphosphate backbone. Chemical variations of lipid A among species involve the number of primary acyl groups and the types of fatty acids substituting the primary and secondary acyl groups (Figure 2). Escherichia lipid A is most frequently described as a hexa-acylated molecular species, although penta-and tetra-acylated molecules are also present in various amounts.21 Most of the laboratory-adapted strains of P. aeruginosa synthesize a penta-acylated (band A, 75% of the molecules) LPS (Figure 1), with some proportion made as a hexa-acylated LPS (25% of the molecules).22,23 Growth conditions, notably magnesium levels, can affect the acylation pattern of P. aeruginosa lipid A (Figure 2). Among isolates from chronically infected CF patients, which are known to be mutants generally unable to synthesize O-antigen side chains, a hexa-acylated LPS form predominates, although a hepta-acylated lipid A has been isolated, containing an additional palmitoyl (C16:0) group linked to the primary 3-hydroxyde-canoic acid group at position 3′ of glucosamine 2.24,25 The hexa- and hepta-acylated lipid A moieties also contain cationic 4-amino-4-deoxy-L-arabinose sugars. Similarly, the major lipid A species in Burkholderia spp. (B. cepacia, B. mallei, B. pseudomallei) consists of a biphosphorylated disaccharide backbone, but it is constitutively modified with 4-amino-4-deoxy-L-arabinose and penta- or tetra-acylated.26 Lipid A of A. baumannii is often modified by phosphorylethanolamine and an unusual sugar galactosamine and hexa- and hepta-acylated27 (Figure 2).
Figure 2.

Diversity of chemical structures and modifications in LPS. The P. aeruginosa band A LPS molecule is shown for comparison. Abbreviations: Kdo, α-3-deoxy-D-manno-oct-2-ulosonic acid; Hep, heptulose; NGal, galactosamine; EPS, extracellular polysaccharide; S-type, “smooth” LPS containing O-chains; R-type, “rough” LPS lacking O-chains; Ko, D-glycero-D-talo-oct-2-ulosonic acid.
Early studies showed that the permeabilities of E. coli and P. aeruginosa OM to hydrophobic steroid probes are similar, suggesting that the differences in the lipid A acylation state and the length of fatty acids do not affect significantly the OM permeability to small planar molecules.28,29 On the other hand, amphiphilic and charged molecules are likely to interact with the backbone of lipid A and LPS cores, and their permeation could be sensitive to modifications of both the lipids and polysaccharides of the LPS-containing bilayers.
LPS cores of enterobacteria typically consist of 8–12 often branched sugar units (Figure 2).30 The sugar at the reducing end is always α-3-deoxy-D-manno-oct-2-ulosonic acid (Kdo) (2→6)-linked to lipid A. At the C-4 position of Kdo, there may be one or two Kdo groupings. Three L-glycero-D-manno-heptose residues are also (1→5)-linked to the first Kdo. A heptose residue may be substituted by a phosphate, pyrophosphate, or phosphorylethanolamine group or by another sugar to make up the inner core. In Pseudomonas spp. cores, one often finds an alanyl group substituting a galactosamine residue and a carbamoyl group on heptose I.31 P. aeruginosa PAO1 serotype O5 and its two rough-type mutants share these characteristics but have, in addition, three phosphomonoester groups on their heptose.31,32 It was suggested that these groups play a role in interactions with antibiotics because their presence correlates with resistance to β-lactams but not to aminoglycosides. Some Acinetobacter and Burkholderia spp. produce LPS cores devoid of heptose. B. cepacia and A. hemolyticus synthesize and incorporate the Kdo analogue D-glycero-D-talo-oct-2-ulosonic acid (Ko).33–35
The O-chains determine the specificity of each bacterial serotype. A combination of monosaccharide diversity, the numerous possibilities of glycosidic linkage, substitution, and configuration of sugars, and the genetic capacities of the diverse organisms have all contributed to the uniqueness of the great majority of O-chain structures.
It is broadly accepted that LPS cores and lipid A of enterobacteria and P. aeruginosa are modified in response to specific growth conditions and stresses (Figure 2).36–38 These modifications are critical under the low magnesium ion conditions that destabilize the LPS leaflet and increase permeability of the OM. They also play an important role in the development of resistance against cationic antimicrobial peptides and polymyxins.36,39 However, in Acinetobacter spp. and Burkholderia spp. some of the modifications that reduce the negative-charge character of LPS are constitutive (Figure 2). How the phosphate, pyrophosphate, or phosphorylethanolamine groups on Kdo and heptoses, terminal sugars, amino acid, and other groups occurring nonstoichiometrically correlate with specific growth conditions of these bacteria remains unclear. Even less is known of how these modifications affect the packing and rigidity of the LPS layer and its interactions with proteins and small molecules in the context of the outer membrane.
Recent advances in molecular dynamics (MD) simulations of asymmetric bilayers containing LPS offer first glimpses into the packing and dynamics of such structures.40–42 The inner leaflet of the outer membrane is composed of phospholipids with a composition similar to that of the cytoplasmic membrane. Hence, the geometry of the LPS leaflet should match that of the phospholipid leaflet. On the basis of MD simulations, LPS-containing bilayers appear to be more disordered and thinner than bilayers assembled from phospholipids only (Figure 1). Magnesium ions form a layer of ionic bonds with phosphoryl moieties of lipid A and the core and stabilize LPS molecules in the bilayers. Also, LPS changes the hydration profile at membrane–water interfaces.43 The packing and rigidity of LPS bilayers are expected to affect permeation of amphiphilic and hydrophobic antibiotics. However, this picture remains incomplete because about 50% of the E. coli OM mass consists of protein and the OM resembles a LPS–protein aggregate. Changes in hydration and electrostatic profiles due to LPS may affect the environment of embedded proteins and influence protein–protein interactions.
Outer Membrane Proteins and Permeability
In E. coli, a few integral membrane proteins, such as OmpA and the general porins OmpF/C, are expressed at high levels (Table 1). Besides these, there are minor proteins whose synthesis in some cases is strongly induced when they are needed, such as specific porins (e.g., PhoE and LamB), TonB-dependent receptors (e.g., FhuA and FepA), components of several protein export systems, proteins involved in the biogenesis of flagella and pili, and enzymes (e.g., OmpT protease and phospholipase A).14,44 The permeability properties of E. coli membranes are largely defined by general porins that have an exclusion limit at about 600 Da for OmpF. Most hydrophilic and amphiphilic antibiotics reach the periplasm through porins, whereas larger antibiotics, for example, erythromycin or novobiocin, are believed to cross the outer membrane through lipid bilayer by diffusion, which is expected to be slow.14,45
Table 1.
Major Porins and Efflux Pumps of Representative Gram-Negative Bacteria
| species | relative OM permeability (%) | major general porins | major efflux pumps |
|---|---|---|---|
| E. coli | 100 | OmpF/OmpC | AcrAB-TolC |
| P. aeruginosa | 1–8 | OprF | MexAB-OprM, MexXY-OprM |
| B. cepacia | 11 | OpcP1/OpcP2 | AmrAB-OprA, BpeAB-OprB, BpeEF-OprC |
| A. baumannii | 1–5 | OmpA-AB | AdeABC and AdeIJK |
The existing paradigm that only small molecules (MW ~ 600 Da) can cross the membrane by passive diffusion has recently been challenged by studies of the CymA channel from Klebsiella oxytoca.46 CymA mediates diffusion of cyclodextrins with diameters up to 15 Å by a novel mechanism, which involves a mobile N-terminal peptide acting as a periplasmic gate. Binding of the incoming substrate displaces the N-terminal constriction and allows diffusion of the bulky molecule without compromising the permeability barrier of the OM. One could imagine that antibiotics could be modified in a way to mimic the interactions of substrates with specific porins and facilitate their own diffusion across the OM.
In addition, some leakage of large antibiotics is possible through export systems. E. coli cells lacking the TolC channel, which is involved in efflux of antibiotics and export of proteins as a part of the type I secretion pathway, are more resistant to vancomycin (2–4-fold increase in minimal inhibitory concentrations) than the wild-type cells, suggesting some penetration of this ~1200 Da molecule through TolC.47,48 Interestingly, this penetration requires the presence of active AcrAB transporter, further suggesting that vancomycin slips through TolC when it is engaged by the AcrAB pump (see also below). It is possible that permeation through other specific porins and TonB-dependent channels and the Bam complex responsible for the assembly of the outer membrane and at protein–LPS interfaces also contribute to the intracellular accumulation of antibiotics.
The overall compositions and architectures of Pseudomonas spp. and Acinetobacter spp. outer membranes are similar to those of other Gram-negative bacteria.49 About 160 various proteins are embedded into the asymmetric bilayer and play a role in generalized and specific uptake and export of various compounds and polypeptides. Unlike enterobacteria, outer membranes of Pseudomonads and Acinetobacter spp. do not contain general trimeric porins such as OmpF and OmpC of E. coli. It is estimated that the permeability of P. aeruginosa and A. baumannii outer membranes is only 1–8% of that E. coli and that nonspecific “slow” porins OprF and OmpA-AB, respectively, restrict access of molecules larger than ~200 Da, which is the size of a typical monosaccharide.49–51 However, the growth rate of P. aeruginosa as well as of A. baumannii in a rich medium is comparable to that of E. coli, and hence to sustain the high rate of metabolism, the net influx of nutrients is expected to be comparable for the three species. P. aeruginosa likely solves this conundrum of the low permeability of the outer membrane despite a high nutrient uptake in several ways: (i) by secreting degrading enzymes that convert larger molecules into monounits (pseudomonads are masters of biodegradation) and (ii) by producing a larger fraction of substrate-specific channels (such as OprB and OprD).
OprF is one of the most abundant proteins in P. aeruginosa with a copy number of 200 000 per cell.49 This protein plays a structural role by associating with LPS and peptidoglycan. P. aeruginosa mutants deficient in OprF synthesis have an almost spherical appearance, are shorter than wild-type cells, and do not grow in low-osmolarity medium. OprF also plays an important nonspecific uptake function by existing in closed and open conformations (400 open conformers of 200 000 total OprF).52,53 Much less is known about the major porin OmpA of A. baumannii. This protein has a permeability similar to that OprF and also plays both structural and uptake roles in the outer membrane, but whether or not it exists as different conformers is unclear.54
In addition to “slow” porins, P. aeruginosa and A. baumannii use specific porins for uptake of small molecules. The large number of specific porins provides an advantage in nutrient-deficient environments, but could be limiting in rich growth media.55–57 P. aeruginosa OprB, specific for glucose uptake, and OprD, specific for the diffusion of basic amino acids and peptides, are best characterized. OprD is the primary channel for the entry of carbapenems across the OM, and the reduced expression or loss of OprD has been frequently observed in carbapenem-resistant clinical isolates. A. baumannii produces an OprD homologue CarO, but whether or not this porin provides a path for carpenems remains under debate.58,59
Interestingly, although the outer membrane permeability of A. baumannii to cephalothin and cephaloridine, measured in intact cells, was found to be about 100-fold lower than that of E. coli K-12,54 these cells are more susceptible to large antibiotics such as novobiocin and erythromycin.28,60 Many Acinetobacter spp. are capable of using long-chain hydrocarbons as growth substrates, suggesting that their LPS–phospholipid bilayers of the outer membrane are more permeable for the hydrophobic molecules.60
Outer membranes of Burkholderia spp. contain a trimeric general porin Omp38 (OpcP), a homologue of E. coli OmpF.61–63 The two studies of Omp38 permeability produced contradictory results: this porin is either similar to E. coli OmpF or by 1 or 2 orders of magnitude less permeable than OmpF. Further studies are needed to analyze the properties of porins and outer membranes for challenging Gram-negative bacteria such as A. baumannii and Burkholderia spp.
Efflux Pumps
Efflux across the Outer Membrane
All Gram-negative bacteria examined so far contain at least one multidrug efflux transporter responsible for protection against a variety of antimicrobial agents.10,11 AcrAB-TolC of E. coli is the best characterized efflux pump, and its homologues are broadly represented in enterobacteria and other Gram-negative species.16,64–66 In this three-component complex, AcrB is a proton-motive force driven transporter from the resistance–nodulation–division (RND) superfamily of proteins, TolC is an outer membrane channel, and AcrA is a periplasmic membrane fusion protein (MFP). The three proteins form a trans-envelope complex that expels multiple antibiotics from E. coli cells (Figure 3).67,68 Extensive structural and functional analyses, including our own studies, showed that AcrB captures its substrates from the membrane and from the periplasm and expels them through the TolC channel into the external medium with the help of AcrA protein.69–71 At least two substrate binding sites in AcrB, the proximal and the distal, are both located in the periplasmic domains of the protein. Thus, unlike other typical secondary transporters, AcrB with the help of AcrA and TolC expels substrates across the outer membrane while driven by the proton transfer across the cytoplasmic membrane. All RND-type multidrug efflux pumps are likely to share this mechanism, but it remains unclear whether some of these protein complexes can actually pump substrates not only across the outer but also across the inner membrane, thus performing the trans-envelope efflux of antibiotics from the cytoplasm directly into the external medium.
Figure 3.
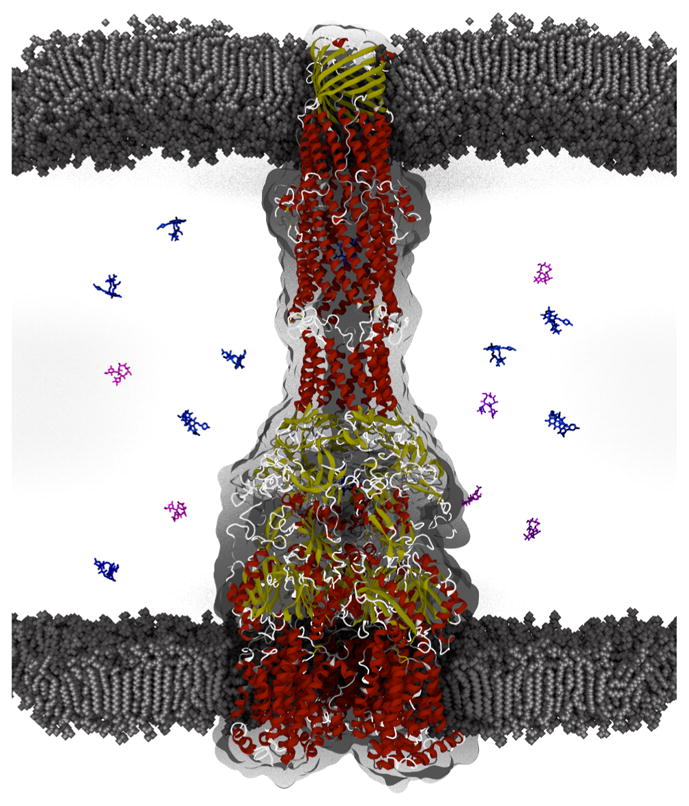
Model of the assembled MexB-MexA-OprM multidrug efflux pump from P. aeruginosa. MexA and other MFPs are proposed to translate the conformational changes in MexB transporter driven by a proton-motive force into opening of the outer membrane channel OprM that enables efflux across the outer membrane. The tripartite complex shown is a snapshot of a 1 μs MD simulation of the complex embedded in a bilayer composed of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC). Proteins are colored according to secondary structure. For visual clarity water molecules are not shown. Accompanying drugs are shown for illustration purposes only (blue, rifampicin; pink, erythromycin). The gray shape corresponds to the AcrAB-TolC density obtained by the cryo-EM imaging. The equilibrated MexAB-OprM complex in the bacterial envelope maintains the expected distance of the P. aeruginosa periplasm. Image was initially exported from VMD Molecular Viewer in STL format and posteriorly rendered in Blender (https://www.blender.org). The gray shape corresponds to the AcrAB-TolC density obtained by the cryo-EM imaging.124
Among AcrAB-TolC substrates are organic solvents, antibiotics, detergents, dyes, and even hormones. Examination of chemical structures, as well as cocrystallization and molecular dynamics studies, significantly advanced the understanding of the molecular mechanism of multidrug recognition by AcrB.72,73 However, physicochemical properties of compounds that would distinguish AcrB substrates from nonsubstrates remain elusive. The current consensus is that AcrAB-TolC is the most effective against amphiphilic compounds that diffuse slowly across the OM. Recent attempts to determine the affinity of AcrAB-TolC toward its substrates demonstrated convincingly that minimal inhibitory concentrations of antibiotics is a very poor, if at all, measure of whether an antibiotic is a good or a bad substrate of an efflux pump.74 Several uptake assays were developed to analyze drug efflux, with most of them informative in both E. coli and P. aeruginosa. However, further characterization of the kinetic behavior of efflux pumps in the context of two-membrane envelopes of Gram-negative bacteria is needed.
P. aeruginosa MexAB-OprM is a close homologue of AcrAB-TolC and the major “house-keeping” efflux pump (Figure 3), but it is not the only pump that is expressed and contributes to antibiotic resistance in this species. The constitutive expression of MexAB-OprM confers intrinsic antibiotic resistance,17–19 whereas elevated expression of MexXY-OprM is the major cause of aminoglycoside resistance in the absence of modifying enzymes.75 In addition, efflux pumps MexCD-OprJ and MexEF-OprN are often overproduced in multiresistant clinical isolates. MexEF-OprN and MexGHI-OprD play important roles in P. aeruginosa physiology and are involved in the secretion of quorum-sensing signals and biofilm formation.76–78 Furthermore, P. aeruginosa lacking either MexAB or MexHI is attenuated in animal models.78,79
As in the case of P. aeruginosa, genomes of Burkholderia spp. and Acinetobacter spp. contain multiple operons encoding RND-type efflux pumps. Some of these pumps are constitutively expressed and confer intrinsic antibiotic resistance, whereas others are inducible and expressed under specific physiological conditions. AmrAB-OprA B. pseudomallei and its homologues in other Burkholderia spp. are largely responsible for intrinsic antibiotic resistance.80 AmrAB-OprA is closely related to MexXY-OprM of P. aeruginosa and, in addition to various antibiotics, confers resistance to aminoglycosides. In the most comprehensive genetic analysis, deletion of the 16 putative RND operons from B. cenocepacia strain J2315 showed that these pumps play differential roles in the drug resistance of sessile (biofilm) and planktonic cells. These studies revealed that (1) RND-3 (a homologue of AmrAB-OprA) and RND-4 play important roles in resistance to various antibiotics, including fluoroquinolones and aminoglycosides, in planktonic populations; (2) RND-3, RND-8, and RND-9 protect against the antimicrobial effects of tobramycin in biofilm cells; and (3) RND-8 and RND-9 do not play a role in ciprofloxacin resistance.81
The A. baumannii genome encodes seven RND-type efflux pumps. AdeIJK confers intrinsic resistance to various antibiotics including β-lactams, fluoroquinolones, tetracyclines, lincosamides, and chloramphenicol. In addition, AdeABC and AdeFGH are overproduced in clinical multidrug resistant isolates.82 Surprisingly, overproduction of AdeABC and AdeIJK could also alter bacterial membrane composition, resulting in decreased biofilm formation but not motility.83 When expressed in E. coli at levels comparable to those of AcrAB-TolC, AdeIJK but not AdeABC was more effective in the removal of lipophilic β-lactams, novobiocin, and ethidium bromide.83
Efflux across the Cytoplasmic Membrane
Highly abundant in genomes of various Gram-negative bacteria, single-component transporters extruding drugs from the cytoplasm into periplasm remain understudied in clinical settings, in part because their drug efflux activities are important only in the context of intact OM and in the presence of active efflux across the OM. Inactivation of single-component transporters that pump antibiotics across the inner membrane usually does not lead to significant changes in susceptibilities to antibiotics.84 However, inactivation of multiple such pumps in E. coli is able to negate even the activity of AcrAB-TolC.85
Drug-specific transporters, such as TetA, are also spread by plasmids, and their contribution could be either additive or multiplicative depending on the combination of transporters expressed in host cells.86 Experimental data and kinetic modeling showed that the coexpression of one- and three-component transporters leads to a synergistic loss of susceptibility to shared substrates.86,87
One-component transporters can belong to one of the three protein families: small multidrug resistance (SMR), major facilitator superfamily (MFS), or multidrug and toxic extrusion (MATE). E. coli SMR transporter EmrE, MFS MdfA, and MATE transporter NorE are the best characterized representatives.88–90 MdfA (Figure 1) and NorE are commonly expressed in fluoroquinolone-resistant isolates and contribute to resistance against these antibiotics.18 The properties and mechanisms of these proteins are well-characterized, but very little is known about their homologues in “difficult” Gram-negative pathogens.
APPROACHES TO BYPASS OR BREAK THE PERMEABILITY BARRIER
In general, several approaches are envisioned as to how antibiotic penetration across the permeability barrier of Gram-negative bacteria could be improved, but all of these approaches suffer from intrinsic limitations that need to be further addressed for new therapeutics to emerge.
Inhibition of New Accessible Targets
The most traditional approach would be to identify new accessible targets on cell surfaces or in the periplasm that could be interrogated in drug discovery efforts. The major bottleneck here is that such targets are optimized over millions of years of evolution to be resistant to antibiotics. On the other hand, recent breakthroughs in understanding the molecular mechanisms of the cell envelope assembly promise that such a goal is attainable, albeit through a more target-oriented approach. LPS and its biosynthesis pathway have been considered highly attractive targets in Gram-negative bacteria for decades.7,91 Polymyxins, cationic cyclic lipopeptides, bind to LPS and permeabilize OM. These peptides recently re-emerged in clinics to treat multidrug-resistant Gram-negative infections, and their less toxic and “improved” variants attract significant attention.92,93 Burkholderia spp. are intrinsically resistant to cationic peptides, but they have high efficacy against susceptible enterobacteria, P. aeruginosa and A. baumannii. There are two drug candidates in clinical trials that target the LPS pathway in P. aeruginosa.94 ACHN-975 (Achaogen) is a synthetic molecule that inhibits LpxC deacylase, and POL7080 (Polyphor) is a synthetic peptide targeting the outer membrane protein LptD, involved in exporting LPS molecules across the periplasm. In addition to targeting essential enzymes, these inhibitors are expected to be synergistic with other antibiotics by enabling their permeation across the cell envelope.
Identification of Uptake Pathways and the “Trojan Horse” Approach
Another approach to bypass the permeability problem is to achieve fast or facilitated uptake of an antibiotic. Such efficient uptake would negate the impact of efflux and increase the concentration of an antibiotic at the target. As discussed above, P. aeruginosa and other “impermeable” Gram-negative species rely on specific porins and an active uptake system to fulfill their metabolic demands and to maximize their growth rates.55,57 These pathways could be potentially exploited for delivery of antibiotics to their targets through the characterization of specificity determinants and modification of antibiotics to enable their uptake through such specific systems. In the recent study of carbapenem penetration into P. aeruginosa, molecular metadynamics simulations were used to delineate the lowest energy path of native substrates and carbapenem antibiotics through OccD1(OprD) and OccD3 channels and to identify molecular features in antibiotics that enable diffusion through specific channels.95 Subsequent synthesis of carbapenem analogues to chemically probe some of the required features of permeation while maintaining the inherent activity led to an analogue of meropenem, for which a simple substitution on the side chain resulted in diminished dependence on OccD1 for translocation. This study is the first success story and demonstrates that a rational design of compounds with enhanced OM permeability is feasible. The major limitation of such an approach is a high frequency of resistance because the pathways are redundant and nonessential. However, identification and exploitation of uptake systems that are essential during establishment and proliferation of infections could facilitate the development of new therapeutics.
The “Trojan Horse” approach, which is largely derived from siderophore-conjugated antibiotics, is an example of such a strategy. Siderophore-conjugated antibiotics can be actively transported into the periplasm and cytoplasm by various iron-uptake systems.96 A common pathway of bacterial siderophore transport systems in Gram-negative bacteria has been identified.97 An outer membrane transporter binds the Fe3+–siderophore complex with an affinity in the range of 1 nM and translocates this complex across the outer membrane with the help of TonB protein anchored in the cytoplasmic membrane. This process is driven by the cytoplasmic membrane potential. The Fe3+–siderophore in the periplasm is bound by a binding protein that delivers its cargo via the cognate ABC transporter into the cytoplasm. Alternatively, the reduced iron is unloaded in the periplasm, as in the case of yersiniabactin and P. aeruginosa pyoverdines.98 Hence, siderophore-conjugated antibiotics could be delivered by such uptake pathways either into the periplasm or all the way into the cytoplasm.99 BAL30072 (Basilea) is the only candidate currently in clinical trials.100 This compound is a monocyclic β-lactam conjugated to a dihydropyridone siderophore moiety, which has a potent activity against multidrug resistant Gram-negative pathogens including “impermeable” species.
Studies of natural peptide antibiotics suggested additional pathways that could be exploited for bypassing the “impermeable” cell envelopes. Pacidamycins are uridyl peptide antibiotics, which specifically kill strains against P. aeruginosa.101–103 These antibiotics inhibit MraY translocase by catalyzing the first step in peptidoglycan synthesis. With MW > 800 Da, pacidamycins cannot penetrate the OM but exploit a specific uptake system to reach the target. The ABC transporter NppABCD belonging to the PepT family of transporters is implicated in the uptake of pacidamycin and other peptidyl nucleoside antibiotics, such as blastidin S, albomycin, and microcin C, across the inner membrane.104 These antibiotics are likely to cross the OM through the TonB-dependent receptors involved in the uptake of siderophores.
Rules of Permeation
It is likely that permeation of different classes of compounds is affected by the outer membrane barrier and by the active efflux to different degrees. At present no rules exist to predict whether increasing uptake or reducing efflux would be the most efficient way to increase the potency of a specific class of compounds. It is believed that these rules will emerge when we address a critical gap in knowledge about what physicochemical properties and specific functional groups define the permeation of compounds across cell walls of Gram-negative pathogens.8 Having such rules to guide medicinal chemistry efforts could potentially facilitate the discovery of novel anti-Gram negative drugs.105 Although the task is complex, recent success in the development of a predictive model for drug accumulation in Caenorhabditis elegans106 is inspiring. Several investigators107,108 led the efforts to develop experimental approaches and to generate representative data sets of compound penetration into difficult Gram-negative pathogens. These studies complement the activity-based approaches to identify physicochemical properties that facilitate efflux of compounds.109
In addition, the first bacterial membrane models reflecting the biological complexity are emerging and could be used to address a wide range of questions about protein–lipid packing and dynamics that are not accessible by experiments110 (see also Figure 1). With these models and methods it is now possible to study interactions of antimicrobial peptides and antibiotics with membranes and to identify their preferable penetration routes.111 The “ideal” rules of permeation will also have to account for genetic and cellular processes that change the expression of efflux pumps and influx porins or modify the permeability of the OM in response to the uptake of antibiotics.
Efflux Pump Inhibitors (EPIs)
The unquestionably significant impact of multidrug efflux pumps on bacterial physiology and the resistance to antibiotics in clinical settings makes them attractive targets for inhibition. Several classes of EPIs have been reported in the literature and are being pursued in drug development programs.112–116 Phenylalanyl-arginyl-β-naphthylamide and analogous arylamines are broad-spectrum EPIs with activities against Gram-negative bacteria;113,117,118 arylpiperidines119 and more recently pyrinopyridine120 are inhibitors of AcrAB-TolC from E. coli. Likewise, pyridopyrimidines are specific inhibitors of P. aeruginosa MexB and E. coli AcrB.121,122 Importantly, broad-spectrum EPIs that target multiple efflux pumps have been reported not only to restore activities of antibiotics but also to reduce frequencies of antibiotic resistance.123 As with many combination therapies, the task of EPI development is not easy, and multiple hurdles must be overcome, starting with the choice of an antibiotic for potentiation and all the way to matching the pharmacological properties of an EPI/antibiotic pair.115 At the same time, some efflux pumps are critical for virulence and biofilm formation, for example, MexGHI-OpmH and MexEF-OprN from P. aeruginosa. EPIs effective against such transporters, if available, could be useful alternative therapeutics on their own.
CONCLUSIONS
The emerging multidrug-resistant Gram-negative pathogens present a significant challenge in clinics that should be addressed in a timely manner. Several decades of antibiotic discovery experience suggested that the permeability barrier is the major hurdle in the development of new therapeutics against these pathogens. Significant efforts are currently directed both at understanding at the molecular level the permeability properties of the OM from different bacterial species and at finding correlations between physicochemical properties of compounds and their permeation across the two membrane cell envelopes in the presence of efflux. Ultimately, the development of effective therapeutic modalities against multidrug-resistant Gram-negative pathogens will be strongly facilitated by a system-level approach that integrates the rules of permeation at the molecular level with supramolecular genetic and cellular processes of the organism. The task is complex and multifaceted but could be achieved by combined efforts at the government, industry, and academic levels.
Acknowledgments
Studies in H.I.Z.’s laboratory are sponsored by the Department of the Defense, Defense Threat Reduction Agency and by the National Institute of Health (Grant AI052293). S.G. is supported by DOE/LANL-DR project on drug resistance. C.A.L. is supported by a fellowship from the Center of Nonlinear Studies at LANL. The content of this paper does not necessarily reflect the position or the policy of the federal government, and no official endorsement should be inferred.
Footnotes
Notes
The authors declare no competing financial interest.
References
- 1.Levy SB, Marshall B. Antibacterial resistance worldwide: causes, challenges and responses. Nat Med. 2004;10:S122. doi: 10.1038/nm1145. [DOI] [PubMed] [Google Scholar]
- 2.Sievert DMP, Ricks PP, Edwards JRMS, Schneider AMPH, Patel JP, Srinivasan AMD, Kallen AMD, Limbago BP, Fridkin SMD. For the National Healthcare Safety Network, T., and Participating, N. F. Antimicrobial-Resistant Pathogens Associated with Healthcare-Associated Infections: Summary of Data Reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2009–2010. Infect Control Hospital Epidemiol. 2013;34:1–14. doi: 10.1086/668770. [DOI] [PubMed] [Google Scholar]
- 3.Livermore DM. The need for new antibiotics. Clin Microbiol Infect. 2004;10(Suppl 4):1–9. doi: 10.1111/j.1465-0691.2004.1004.x. [DOI] [PubMed] [Google Scholar]
- 4.Munoz-Price LS, Poirel L, Bonomo RA, Schwaber MJ, Daikos GL, Cormican M, Cornaglia G, Garau J, Gniadkowski M, Hayden MK, Kumarasamy K, Livermore DM, Maya JJ, Nordmann P, Patel JB, Paterson DL, Pitout J, Villegas MV, Wang H, Woodford N, Quinn JP. Clinical epidemiology of the global expansion of Klebsiella pneumoniae carbapenemases. Lancet Infect Dis. 2013;13:785–796. doi: 10.1016/S1473-3099(13)70190-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rice LB. Mechanisms of Resistance and Clinical Relevance of Resistance to β-Lactams, Glycopeptides, and Fluoroquinolones. Mayo Clin Proc. 2012;87:198–208. doi: 10.1016/j.mayocp.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vasoo S, Barreto JN, Tosh PK. Emerging Issues in Gram-Negative Bacterial Resistance: An Update for the Practicing Clinician. Mayo Clin Proc. 2015;90:395–403. doi: 10.1016/j.mayocp.2014.12.002. [DOI] [PubMed] [Google Scholar]
- 7.Silver LL. Challenges of Antibacterial Discovery. Clin Microbiol Rev. 2011;24:71–109. doi: 10.1128/CMR.00030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lewis K. Antibiotics: Recover the lost art of drug discovery. Nature. 2012;485:439–440. doi: 10.1038/485439a. [DOI] [PubMed] [Google Scholar]
- 9.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Delivery Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 10.Zgurskaya HI. Mechanisms of drug efflux and strategies to combat them. Preface Biochim Biophys Acta, Proteins Proteomics. 2009;1794:723–724. doi: 10.1016/j.bbapap.2009.03.017. [DOI] [PubMed] [Google Scholar]
- 11.Nikaido H. Multidrug Resistance in Bacteria. Annu Rev Biochem. 2009;78:119–146. doi: 10.1146/annurev.biochem.78.082907.145923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li XZ, Plésiat P, Nikaido H. The Challenge of Efflux-Mediated Antibiotic Resistance in Gram-Negative Bacteria. Clin Microbiol Rev. 2015;28:337–418. doi: 10.1128/CMR.00117-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delcour AH. Outer membrane permeability and antibiotic resistance. Biochim Biophys Acta, Proteins Proteomics. 2009;1794:808–816. doi: 10.1016/j.bbapap.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nikaido H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev: MMBR. 2003;67:593–656. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lomovskaya O, Zgurskaya HI, Totrov M, Watkins WJ. Waltzing transporters and ‘the dance macabre’ between humans and bacteria. Nat Rev Drug Discovery. 2007;6:56–65. doi: 10.1038/nrd2200. [DOI] [PubMed] [Google Scholar]
- 16.Zgurskaya HI. Multicomponent drug efflux complexes: architecture and mechanism of assembly. Future Microbiol. 2009;4:919–932. doi: 10.2217/fmb.09.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ziha-Zarifi I, Llanes C, Kohler T, Pechere JC, Plesiat P. In vivo emergence of multidrug-resistant mutants of Pseudomonas aeruginosa overexpressing the active efflux system MexA-MexB-OprM. Antimicrob Agents Chemother. 1999;43:287–291. doi: 10.1128/aac.43.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Swick MC, Morgan-Linnell SK, Carlson KM, Zechiedrich L. Expression of multidrug efflux pump genes acrAB-tolC, mdfA, and norE in Escherichia coli clinical isolates as a function of fluoroquinolone and multidrug resistance. Antimicrob Agents Chemother. 2011;55:921–924. doi: 10.1128/AAC.00996-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Padilla E, Llobet E, Doménech-Sánchez A, Martínez-Martínez L, Bengoechea JA, Albertí S. Klebsiella pneumoniae AcrAB Efflux Pump Contributes to Antimicrobial Resistance and Virulence. Antimicrob Agents Chemother. 2010;54:177–183. doi: 10.1128/AAC.00715-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chiu CH, Lee HY, Tseng LY, Chen CL, Chia JH, Su LH, Liu SY. Mechanisms of resistance to ciprofloxacin, ampicillin/sulbactam and imipenem in Acinetobacter baumannii clinical isolates in Taiwan. Int J Antimicrob Agents. 2010;35:382–386. doi: 10.1016/j.ijantimicag.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 21.Caroff M, Karibian D. Structure of bacterial lipopolysaccharides. Carbohydr Res. 2003;338:2431–2447. doi: 10.1016/j.carres.2003.07.010. [DOI] [PubMed] [Google Scholar]
- 22.Kocincova D, Lam JS. Structural diversity of the core oligosaccharide domain of Pseudomonas aeruginosa lipopolysaccharide. Biochemistry. 2011;76:755–760. doi: 10.1134/S0006297911070054. [DOI] [PubMed] [Google Scholar]
- 23.Pier GB. Pseudomonas aeruginosa lipopolysaccharide: a major virulence factor, initiator of inflammation and target for effective immunity. Int J Med Microbiol. 2007;297:277–295. doi: 10.1016/j.ijmm.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ernst RK, Moskowitz SM, Emerson JC, Kraig GM, Adams KN, Harvey MD, Ramsey B, Speert DP, Burns JL, Miller SI. Unique Lipid A Modifications in Pseudomonas aeruginosa Isolated from the Airways of Patients with Cystic Fibrosis. J Infect Dis. 2007;196:1088–1092. doi: 10.1086/521367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ernst RK, Hajjar AM, Tsai JH, Moskowitz SM, Wilson CB, Miller SI. Pseudomonas aeruginosa lipid A diversity and its recognition by Toll-like receptor 4. J Endotoxin Res. 2003;9:395–400. doi: 10.1179/096805103225002764. [DOI] [PubMed] [Google Scholar]
- 26.De Soyza A, Silipo A, Lanzetta R, Govan JR, Molinaro A. Review: Chemical and biological features of Burkholderia cepacia complex lipopolysaccharides. Innate Immun. 2008;14:127–144. doi: 10.1177/1753425908093984. [DOI] [PubMed] [Google Scholar]
- 27.Beceiro A, Llobet E, Aranda J, Bengoechea JA, Doumith M, Hornsey M, Dhanji H, Chart H, Bou G, Livermore DM, Woodford N. Phosphoethanolamine Modification of Lipid A in Colistin-Resistant Variants of Acinetobacter baumannii Mediated by the pmrAB Two-Component Regulatory System. Antimicrob Agents Chemother. 2011;55:3370–3379. doi: 10.1128/AAC.00079-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Plésiat P, Nikaido H. Outer membranes of Gram-negative bacteria are permeable to steroid probes. Mol Microbiol. 1992;6:1323–1333. doi: 10.1111/j.1365-2958.1992.tb00853.x. [DOI] [PubMed] [Google Scholar]
- 29.Plesiat P, Aires JR, Godard C, Köhler T. Use of steroids to monitor alterations in the outer membrane of Pseudomonas aeruginosa. J Bacteriol. 1997;179:7004–7010. doi: 10.1128/jb.179.22.7004-7010.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Caroff M, Karibian D. Structure of bacterial lipopolysaccharides. Carbohydr Res. 2003;338:2431–2447. doi: 10.1016/j.carres.2003.07.010. [DOI] [PubMed] [Google Scholar]
- 31.Knirel YA, Bystrova OV, Kocharova NA, Zahringer U, Pier GB. Conserved and variable structural features in the lipopolysaccharide of Pseudomonas aeruginosa. J Endotoxin Res. 2006;12:324–336. doi: 10.1179/096805106X118906. [DOI] [PubMed] [Google Scholar]
- 32.Bystrova OV, Lindner B, Moll H, Kocharova NA, Knirel YA, Zahringer U, Pier GB. Full structure of the lipopolysaccharide of Pseudomonas aeruginosa immunotype 5. Biochemistry (Moscow) 2004;69:170–175. doi: 10.1023/b:biry.0000018947.60328.8d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Isshiki Y, Zähringer U, Kawahara K. Structure of the core-oligosaccharide with a characteristic D-glycero-α-D-talo-oct-2-ulosylonate-(2→4)-3-deoxy-D-manno-oct-2-ulosonate [α-Ko-(2→4)-Kdo] disaccharide in the lipopolysaccharide from Burkholderia cepacia. Carbohydr Res. 2003;338:2659–2666. doi: 10.1016/j.carres.2003.07.005. [DOI] [PubMed] [Google Scholar]
- 34.Vinogradov EV, Müller-Loennies S, Petersen BO, Meshkov S, Thomas-Oates JE, Holst O, Brade H. Structural Investigation of the Lipopolysaccharide from Acinetobacter haemolyticus Strain NCTC 10305 (ATCC 17906, DNA Group 4) Eur J Biochem. 1997;247:82–90. doi: 10.1111/j.1432-1033.1997.00082.x. [DOI] [PubMed] [Google Scholar]
- 35.Vinion-Dubiel AD, Goldberg JB. Review: Lipopolysaccharide of Burkholderia cepacia complex. J Endotoxin Res. 2003;9:201–213. doi: 10.1179/096805103225001404. [DOI] [PubMed] [Google Scholar]
- 36.Murata T, Tseng W, Guina T, Miller SI, Nikaido H. PhoPQ-Mediated Regulation Produces a More Robust Permeability Barrier in the Outer Membrane of Salmonella enterica Serovar Typhimurium. J Bacteriol. 2007;189:7213–7222. doi: 10.1128/JB.00973-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raetz CRH, Reynolds CM, Trent MS, Bishop RE. Lipid A Modification Systems in Gram-Negative Bacteria. Annu Rev Biochem. 2007;76:295–329. doi: 10.1146/annurev.biochem.76.010307.145803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo L, Lim KB, Gunn JS, Bainbridge B, Darveau RP, Hackett M, Miller SI. Regulation of Lipid A Modifications by Salmonella typhimurium Virulence Genes phoP-phoQ. Science. 1997;276:250–253. doi: 10.1126/science.276.5310.250. [DOI] [PubMed] [Google Scholar]
- 39.Olaitan AO, Morand S, Rolain JM. Mechanisms of polymyxin resistance: acquired and intrinsic resistance in bacteria. Front Microbiol. 2014;5:643. doi: 10.3389/fmicb.2014.00643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jo S, Wu E, Stuhlsatz D, Klauda J, MacKerell A, Jr, Widmalm G, Im W. Lipopolysaccharide membrane building and simulation. In: Lütteke T, Frank M, editors. Glycoinformatics. Springer; New York: 2015. pp. 391–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khalid S, Berglund NA, Holdbrook DA, Leung YM, Parkin J. The membranes of Gram-negative bacteria: progress in molecular modelling and simulation. Biochem Soc Trans. 2015;43:162–167. doi: 10.1042/BST20140262. [DOI] [PubMed] [Google Scholar]
- 42.Dias RP, Li L, Soares TA, Alexov E. Modeling the electrostatic potential of asymmetric lipopolysaccharide membranes: the MEMPOT algorithm implemented in DelPhi. J Comput Chem. 2014;35:1418–1429. doi: 10.1002/jcc.23632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nascimento A, Pontes FJS, Lins RD, Soares TA. Hydration, ionic valence and cross-linking propensities of cations determine the stability of lipopolysaccharide (LPS) membranes. Chem Commun. 2014;50:231–233. doi: 10.1039/c3cc46918b. [DOI] [PubMed] [Google Scholar]
- 44.Nikaido H, Pages JM. Broad-specificity efflux pumps and their role in multidrug resistance of Gram-negative bacteria. FEMS Microbiol Rev. 2012;36:340–363. doi: 10.1111/j.1574-6976.2011.00290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pages JM, James CE, Winterhalter M. The porin and the permeating antibiotic: a selective diffusion barrier in Gram-negative bacteria. Nat Rev Microbiol. 2008;6:893–903. doi: 10.1038/nrmicro1994. [DOI] [PubMed] [Google Scholar]
- 46.Pletzer D, Braun Y, Dubiley S, Lafon C, Köhler T, Page MGP, Mourez M, Severinov K, Weingart H. The Pseudomonas aeruginosa PA14 ABC Transporter NppA1A2BCD Is Required for Uptake of Peptidyl Nucleoside Antibiotics. J Bacteriol. 2015;197:2217–2228. doi: 10.1128/JB.00234-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Krishnamoorthy G, Tikhonova EB, Dhamdhere G, Zgurskaya HI. On the role of TolC in multidrug efflux: the function and assembly of AcrAB-TolC tolerate significant depletion of intracellular TolC protein. Mol Microbiol. 2013;87:982–997. doi: 10.1111/mmi.12143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weeks JW, Celaya-Kolb T, Pecora S, Misra R. AcrA suppressor alterations reverse the drug hypersensitivity phenotype of a TolC mutant by inducing TolC aperture opening. Mol Microbiol. 2010;75:1468–1483. doi: 10.1111/j.1365-2958.2010.07068.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tamber S, Hancock RE. The outer membranes of Pseudomonads. In: Ramos J-L, editor. Pseudomonas. Kluwer Academic/Plenum Publishers; New York: 2004. pp. 575–601. [Google Scholar]
- 50.Sugawara E, Nikaido H. OmpA Is the Principal Nonspecific Slow Porin of Acinetobacter baumannii. J Bacteriol. 2012;194:4089–4096. doi: 10.1128/JB.00435-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vila J, Martí S, Sánchez-Céspedes J. Porins, efflux pumps and multidrug resistance in Acinetobacter baumannii. J Antimicrob Chemother. 2007;59:1210–1215. doi: 10.1093/jac/dkl509. [DOI] [PubMed] [Google Scholar]
- 52.Sugawara E, Nagano K, Nikaido H. Alternative folding pathways of the major porin OprF of Pseudomonas aeruginosa. FEBS J. 2012;279:910–918. doi: 10.1111/j.1742-4658.2012.08481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sugawara E, Nestorovich EM, Bezrukov SM, Nikaido H. Pseudomonas aeruginosa Porin OprF Exists in Two Different Conformations. J Biol Chem. 2006;281:16220–16229. doi: 10.1074/jbc.M600680200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sugawara E, Nikaido H. OmpA is the principal nonspecific slow porin of Acinetobacter baumannii. J Bacteriol. 2012;194:4089–4096. doi: 10.1128/JB.00435-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eren E, Parkin J, Adelanwa A, Cheneke B, Movileanu L, Khalid S, van den Berg B. Toward Understanding the Outer Membrane Uptake of Small Molecules by Pseudomonas aeruginosa. J Biol Chem. 2013;288:12042–12053. doi: 10.1074/jbc.M113.463570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tamber S, Ochs MM, Hancock REW. Role of the Novel OprD Family of Porins in Nutrient Uptake in Pseudomonas aeruginosa. J Bacteriol. 2006;188:45–54. doi: 10.1128/JB.188.1.45-54.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van den Berg B. Structural Basis for Outer Membrane Sugar Uptake in Pseudomonads. J Biol Chem. 2012;287:41044–41052. doi: 10.1074/jbc.M112.408518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zahn M, D’Agostino T, Eren E, Baslé A, Ceccarelli M, van den Berg B. Small-Molecule Transport by CarO, an Abundant Eight-Stranded β-Barrel Outer Membrane Protein from Acinetobacter baumannii. J Mol Biol. 2015;427:2329–2339. doi: 10.1016/j.jmb.2015.03.016. [DOI] [PubMed] [Google Scholar]
- 59.Mussi MA, Limansky AS, Viale AM. Acquisition of Resistance to Carbapenems in Multidrug-Resistant Clinical Strains of Acinetobacter baumannii: Natural Insertional Inactivation of a Gene Encoding a Member of a Novel Family of β-Barrel Outer Membrane Proteins. Antimicrob Agents Chemother. 2005;49:1432–1440. doi: 10.1128/AAC.49.4.1432-1440.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Borneleit P, Kleber H-P. The outer membrane of Acinetobacter: structure-function relationships. In: Towner KJ, Bergogne-Berezin E, Fewson CA, editors. The Biology of Acinetobacter. Plenum Press; New York: 1991. pp. 259–272. [Google Scholar]
- 61.Siritapetawee J, Prinz H, Krittanai C, Suginta W. Expression and refolding of Omp38 from Burkholderia pseudomallei and Burkholderia thailandensis, and its function as a diffusion porin. Biochem J. 2004;384:609–617. doi: 10.1042/BJ20041102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Suginta W, Mahendran KR, Chumjan W, Hajjar E, Schulte A, Winterhalter M, Weingart H. Molecular analysis of antimicrobial agent translocation through the membrane porin BpsOmp38 from an ultraresistant Burkholderia pseudomallei strain. Biochim Biophys Acta, Biomembr. 2011;1808:1552–1559. doi: 10.1016/j.bbamem.2010.10.018. [DOI] [PubMed] [Google Scholar]
- 63.Siritapetawee J, Prinz H, Samosornsuk W, Ashley RH, Suginta W. Functional reconstitution, gene isolation and topology modelling of porins from Burkholderia pseudomallei and Burkholderia thailandensis. Biochem J. 2004;377:579–587. doi: 10.1042/BJ20031118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tikhonova EB, Dastidar V, Rybenkov VV, Zgurskaya HI. Kinetic control of TolC recruitment by multidrug efflux complexes. Proc Natl Acad Sci U S A. 2009;106:16416–16421. doi: 10.1073/pnas.0906601106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tikhonova EB, Yamada Y, Zgurskaya HI. Sequential mechanism of assembly of multidrug efflux pump AcrAB-TolC. Chem Biol. 2011;18:454–463. doi: 10.1016/j.chembiol.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tikhonova EB, Zgurskaya HI. AcrA, AcrB, and TolC of Escherichia coli Form a Stable Intermembrane Multidrug Efflux Complex. J Biol Chem. 2004;279:32116–32124. doi: 10.1074/jbc.M402230200. [DOI] [PubMed] [Google Scholar]
- 67.Nikaido H, Zgurskaya HI. Antibiotic efflux mechanisms. Curr Opin Infect Dis. 1999;12:529–536. doi: 10.1097/00001432-199912000-00001. [DOI] [PubMed] [Google Scholar]
- 68.Phillips JL, Gnanakaran S. A data-driven approach to modeling the tripartite structure of multidrug resistance efflux pumps. Proteins: Struct, Funct, Genet. 2015;83:46–65. doi: 10.1002/prot.24632. [DOI] [PubMed] [Google Scholar]
- 69.Zgurskaya HI, Nikaido H. Bypassing the periplasm: reconstitution of the AcrAB multidrug efflux pump of Escherichia coli. Proc Natl Acad Sci U S A. 1999;96:7190–7195. doi: 10.1073/pnas.96.13.7190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yu EW, McDermott G, Zgurskaya HI, Nikaido H, Koshland DE., Jr Structural basis of multiple drug-binding capacity of the AcrB multidrug efflux pump. Science. 2003;300:976–980. doi: 10.1126/science.1083137. [DOI] [PubMed] [Google Scholar]
- 71.Murakami S, Nakashima R, Yamashita E, Matsumoto T, Yamaguchi A. Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. Nature. 2006;443:173–179. doi: 10.1038/nature05076. [DOI] [PubMed] [Google Scholar]
- 72.Vargiu AV, Nikaido H. Multidrug binding properties of the AcrB efflux pump characterized by molecular dynamics simulations. Proc Natl Acad Sci U S A. 2012;109:20637–20642. doi: 10.1073/pnas.1218348109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nakashima R, Sakurai K, Yamasaki S, Nishino K, Yamaguchi A. Structures of the multidrug exporter AcrB reveal a proximal multisite drug-binding pocket. Nature. 2011;480:565–569. doi: 10.1038/nature10641. [DOI] [PubMed] [Google Scholar]
- 74.Nagano K, Nikaido H. Kinetic behavior of the major multidrug efflux pump AcrB of Escherichia coli. Proc Natl Acad Sci U S A. 2009;106:5854–5858. doi: 10.1073/pnas.0901695106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Poole K, Srikumar R. Multidrug efflux in Pseudomonas aeruginosa: components, mechanisms and clinical significance. Curr Top Med Chem. 2001;1:59–71. doi: 10.2174/1568026013395605. [DOI] [PubMed] [Google Scholar]
- 76.Pumbwe L, Piddock LJ. Two efflux systems expressed simultaneously in multidrug-resistant Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2000;44:2861–2864. doi: 10.1128/aac.44.10.2861-2864.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Poole K. Efflux-mediated multiresistance in Gram-negative bacteria. Clin Microbiol Infect. 2004;10:12–26. doi: 10.1111/j.1469-0691.2004.00763.x. [DOI] [PubMed] [Google Scholar]
- 78.Aendekerk S, Diggle SP, Song Z, Hoiby N, Cornelis P, Williams P, Camara M. The MexGHI-OpmD multidrug efflux pump controls growth, antibiotic susceptibility and virulence in Pseudomonas aeruginosa via 4-quinolone-dependent cell-to-cell communication. Microbiology. 2005;151:1113–1125. doi: 10.1099/mic.0.27631-0. [DOI] [PubMed] [Google Scholar]
- 79.Hirakata Y, Srikumar R, Poole K, Gotoh N, Suematsu T, Kohno S, Kamihira S, Hancock RE, Speert DP. Multidrug efflux systems play an important role in the invasiveness of Pseudomonas aeruginosa. J Exp Med. 2002;196:109–118. doi: 10.1084/jem.20020005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Podnecky NL, Rhodes KA, Schweizer HP. Efflux Pump-mediated Drug Resistance in Burkholderia. Front Microbiol. 2015;6:305. doi: 10.3389/fmicb.2015.00305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Buroni S, Matthijs N, Spadaro F, Van Acker H, Scoffone VC, Pasca MR, Riccardi G, Coenye T. Differential Roles of RND Efflux Pumps in Antimicrobial Drug Resistance of Sessile and Planktonic Burkholderia cenocepacia Cells. Antimicrob Agents Chemother. 2014;58:7424–7429. doi: 10.1128/AAC.03800-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Coyne S, Courvalin P, Périchon B. Efflux-Mediated Antibiotic Resistance in Acinetobacter spp. Antimicrob Agents Chemother. 2011;55:947–953. doi: 10.1128/AAC.01388-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yoon EJ, Nait Chabane Y, Goussard S, Snesrud E, Courvalin P, Dé E, Grillot-Courvalin C. Contribution of Resistance-Nodulation-Cell Division Efflux Systems to Antibiotic Resistance and Biofilm Formation in Acinetobacter baumannii. mBio. 2015;6:e00309–15. doi: 10.1128/mBio.00309-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sulavik MC, Houseweart C, Cramer C, Jiwani N, Murgolo N, Greene J, DiDomenico B, Shaw KJ, Miller GH, Hare R, Shimer G. Antibiotic Susceptibility Profiles of Escherichia coli Strains Lacking Multidrug Efflux Pump Genes. Antimicrob Agents Chemother. 2001;45:1126–1136. doi: 10.1128/AAC.45.4.1126-1136.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tal N, Schuldiner S. A coordinated network of transporters with overlapping specificities provides a robust survival strategy. Proc Natl Acad Sci U S A. 2009;106:9051–9056. doi: 10.1073/pnas.0902400106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lee A, Mao W, Warren MS, Mistry A, Hoshino K, Okumura R, Ishida H, Lomovskaya O. Interplay between efflux pumps may provide either additive or multiplicative effects on drug resistance. J Bacteriol. 2000;182:3142–3150. doi: 10.1128/jb.182.11.3142-3150.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zgurskaya HI, Nikaido H. Mechanistic parallels in bacterial and human multidrug efflux transporters. Curr Protein Pept Sci. 2002;3:531–540. doi: 10.2174/1389203023380512. [DOI] [PubMed] [Google Scholar]
- 88.Fluman N, Bibi E. Bacterial multidrug transport through the lens of the major facilitator superfamily. Biochim Biophys Acta, Proteins Proteomics. 2009;1794:738–747. doi: 10.1016/j.bbapap.2008.11.020. [DOI] [PubMed] [Google Scholar]
- 89.Schuldiner S. EmrE, a model for studying evolution and mechanism of ion-coupled transporters. Biochim Biophys Acta, Proteins Proteomics. 2009;1794:748–762. doi: 10.1016/j.bbapap.2008.12.018. [DOI] [PubMed] [Google Scholar]
- 90.Kuroda T, Tsuchiya T. Multidrug efflux transporters in the MATE family. Biochim Biophys Acta, Proteins Proteomics. 2009;1794:763–768. doi: 10.1016/j.bbapap.2008.11.012. [DOI] [PubMed] [Google Scholar]
- 91.Walsh CT, Wencewicz TA. Prospects for new antibiotics: a molecule-centered perspective. J Antibiot. 2014;67:7–22. doi: 10.1038/ja.2013.49. [DOI] [PubMed] [Google Scholar]
- 92.Vaara M. Polymyxins and their novel derivatives. Curr Opin Microbiol. 2010;13:574–581. doi: 10.1016/j.mib.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 93.Velkov T, Roberts KD, Nation RL, Wang J, Thompson PE, Li J. Teaching ‘Old’ Polymyxins New Tricks: New-Generation Lipopeptides Targeting Gram-Negative ‘Superbugs’. ACS Chem Biol. 2014;9:1172–1177. doi: 10.1021/cb500080r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pucci MJ, Bush K. Investigational Antimicrobial Agents of 2013. Clin Microbiol Rev. 2013;26:792–821. doi: 10.1128/CMR.00033-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Isabella VM, Campbell AJ, Manchester J, Sylvester M, Nayar AS, Ferguson KE, Tommasi R, Miller AA. Toward the Rational Design of Carbapenem Uptake in Pseudomonas aeruginosa. Chem Biol. 2015;22:535–547. doi: 10.1016/j.chembiol.2015.03.018. [DOI] [PubMed] [Google Scholar]
- 96.Schalk IJ, Mislin GLA, Brillet K. Chapter 2. Structure, function and binding selectivity and stereoselectivity of siderophore–iron outer membrane transporters. In: José MA, Svetlana L, editors. Current Topics in Membranes. Academic Press; San Diego, CA, USA: 2012. pp. 37–66. [DOI] [PubMed] [Google Scholar]
- 97.Braun V, Hantke K. Recent insights into iron import by bacteria. Curr Opin Chem Biol. 2011;15:328–334. doi: 10.1016/j.cbpa.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 98.Cunrath O, Geoffroy VA, Schalk IJ. Metallome of Pseudomonas aeruginosa: a role for siderophores. Environ Microbiol. 2015 doi: 10.1111/1462-2920.12971. n/a–n/a. [DOI] [PubMed] [Google Scholar]
- 99.Page MGP. Siderophore conjugates. Ann N Y Acad Sci. 2013;1277:115–126. doi: 10.1111/nyas.12024. [DOI] [PubMed] [Google Scholar]
- 100.Page MGP, Dantier C, Desarbre E. In Vitro Properties of BAL30072, a Novel Siderophore Sulfactam with Activity against Multiresistant Gram-Negative Bacilli. Antimicrob Agents Chemother. 2010;54:2291–2302. doi: 10.1128/AAC.01525-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chen RH, Buko AM, Whittern DN, McAlpine JB. Pacidamycins, a novel series of antibiotics with anti-Pseudomonas aeruginosa activity. II Isolation and structural elucidation. J Antibiot. 1989;42:512–520. doi: 10.7164/antibiotics.42.512. [DOI] [PubMed] [Google Scholar]
- 102.Fernandes PB, Swanson RN, Hardy DJ, Hanson CW, Coen L, Rasmussen RR, Chen RH. Pacidamycins, a novel series of antibiotics with anti-Pseudomonas aeruginosa activity. III Microbiologic profile. J Antibiot. 1989;42:521–526. doi: 10.7164/antibiotics.42.521. [DOI] [PubMed] [Google Scholar]
- 103.Karwowski JP, Jackson M, Theriault RJ, Chen RH, Barlow GJ, Maus ML. Pacidamycins, a novel series of antibiotics with anti-Pseudomonas aeruginosa activity. I Taxonomy of the producing organism and fermentation. J Antibiot. 1989;42:506–511. doi: 10.7164/antibiotics.42.506. [DOI] [PubMed] [Google Scholar]
- 104.Mistry A, Warren MS, Cusick JK, Karkhoff-Schweizer RR, Lomovskaya O, Schweizer HP. High-Level Pacidamycin Resistance in Pseudomonas aeruginosa Is Mediated by an Opp Oligopeptide Permease Encoded by the opp-fabI Operon. Antimicrob Agents Chemother. 2013;57:5565–5571. doi: 10.1128/AAC.01198-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.O’Shea R, Moser HE. Physicochemical properties of antibacterial compounds: implications for drug discovery. J Med Chem. 2008;51:2871–2878. doi: 10.1021/jm700967e. [DOI] [PubMed] [Google Scholar]
- 106.Burns AR, Wallace IM, Wildenhain J, Tyers M, Giaever G, Bader GD, Nislow C, Cutler SR, Roy PJ. A predictive model for drug bioaccumulation and bioactivity in Caenorhabditis elegans. Nat Chem Biol. 2010;6:549–557. doi: 10.1038/nchembio.380. [DOI] [PubMed] [Google Scholar]
- 107.Davis TD, Gerry CJ, Tan DS. General Platform for Systematic Quantitative Evaluation of Small-Molecule Permeability in Bacteria. ACS Chem Biol. 2014;9:2535–2544. doi: 10.1021/cb5003015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhou Y, Joubran C, Miller-Vedam L, Isabella V, Nayar A, Tentarelli S, Miller A. Thinking Outside the “Bug”: A Unique Assay To Measure Intracellular Drug Penetration in Gram-Negative Bacteria. Anal Chem. 2015;87:3579–3584. doi: 10.1021/ac504880r. [DOI] [PubMed] [Google Scholar]
- 109.Manchester JI, Buurman ET, Bisacchi GS, McLaughlin RE. Molecular determinants of AcrB-mediated bacterial efflux implications for drug discovery. J Med Chem. 2012;55:2532–2537. doi: 10.1021/jm201275d. [DOI] [PubMed] [Google Scholar]
- 110.Parkin J, Chavent M, Khalid S. Molecular Simulations of Gram-Negative Bacterial Membranes: A Vignette of Some Recent Successes. Biophys J. 2015;109:461–468. doi: 10.1016/j.bpj.2015.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Berglund NA, Piggot TJ, Jefferies D, Sessions RB, Bond PJ, Khalid S. Interaction of the antimicrobial peptide polymyxin B1 with both membranes of E. coli: a molecular dynamics study. PLoS Comput Biol. 2015;11:e1004180. doi: 10.1371/journal.pcbi.1004180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bohnert JA, Kern WV. Selected arylpiperazines are capable of reversing multidrug resistance in Escherichia coli overexpressing RND efflux pumps. Antimicrob Agents Chemother. 2005;49:849–852. doi: 10.1128/AAC.49.2.849-852.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Renau TE, Leger R, Filonova L, Flamme EM, Wang M, Yen R, Madsen D, Griffith D, Chamberland S, Dudley MN, Lee VJ, Lomovskaya O, Watkins WJ, Ohta T, Nakayama K, Ishida Y. Conformationally-restricted analogues of efflux pump inhibitors that potentiate the activity of levofloxacin in Pseudomonas aeruginosa. Bioorg Med Chem Lett. 2003;13:2755–2758. doi: 10.1016/s0960-894x(03)00556-0. [DOI] [PubMed] [Google Scholar]
- 114.Watkins WJ, Landaverry Y, Leger R, Litman R, Renau TE, Williams N, Yen R, Zhang JZ, Chamberland S, Madsen D, Griffith D, Tembe V, Huie K, Dudley MN. The relationship between physicochemical properties, in vitro activity and pharmacokinetic profiles of analogues of diamine-containing efflux pump inhibitors. Bioorg Med Chem Lett. 2003;13:4241–4244. doi: 10.1016/j.bmcl.2003.07.030. [DOI] [PubMed] [Google Scholar]
- 115.Opperman TJ, Nguyen ST. Recent advances toward a molecular mechanism of efflux pump inhibition. Front Microbiol. 2015;6:e00421. doi: 10.3389/fmicb.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lomovskaya O, Zgurskaya HI, Bostian KA. Bacterial multidrug transporters: molecular and clinical aspects. In: Ecker GF, Chiba P, editors. Transporters as Drug Carriers. Wiley and Sons; Hoboken, NJ, USA: 2008. [Google Scholar]
- 117.Lomovskaya O, Warren MS, Lee A, Galazzo J, Fronko R, Lee M, Blais J, Cho D, Chamberland S, Renau T, Leger R, Hecker S, Watkins W, Hoshino K, Ishida H, Lee VJ. Identification and characterization of inhibitors of multidrug resistance efflux pumps in Pseudomonas aeruginosa: novel agents for combination therapy. Antimicrob Agents Chemother. 2001;45:105–116. doi: 10.1128/AAC.45.1.105-116.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Watkins WJ, Chong L, Cho A, Hilgenkamp R, Ludwikow M, Garizi N, Iqbal N, Barnard J, Singh R, Madsen D, Lolans K, Lomovskaya O, Oza U, Kumaraswamy P, Blecken A, Bai S, Loury DJ, Griffith DC, Dudley MN. Quinazolinone fungal efflux pump inhibitors. Part 3: (N-methyl)piperazine variants and pharmacokinetic optimization. Bioorg Med Chem Lett. 2007;17:2802–2806. doi: 10.1016/j.bmcl.2007.02.047. [DOI] [PubMed] [Google Scholar]
- 119.Thorarensen A, Presley-Bodnar AL, Marotti KR, Boyle TP, Heckaman CL, Bohanon MJ, Tomich PK, Zurenko GE, Sweeney MT, Yagi BH. 3-Arylpiperidines as potentiators of existing antibacterial agents. Bioorg Med Chem Lett. 2001;11:1903–1906. doi: 10.1016/s0960-894x(01)00330-4. [DOI] [PubMed] [Google Scholar]
- 120.Opperman TJ, Kwasny SM, Kim HS, Nguyen ST, Houseweart C, D’Souza S, Walker GC, Peet NP, Nikaido H, Bowlin TL. Characterization of a Novel Pyranopyridine Inhibitor of the AcrAB Efflux Pump of Escherichia coli. Antimicrob Agents Chemother. 2014;58:722–733. doi: 10.1128/AAC.01866-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nakayama K, Ishida Y, Ohtsuka M, Kawato H, Yoshida K-i, Yokomizo Y, Hosono S, Ohta T, Hoshino K, Ishida H, Yoshida K, Renau TE, Léger R, Zhang JZ, Lee VJ, Watkins WJ. MexAB-OprM-Specific efflux pump inhibitors in Pseudomonas aeruginosa Part 1: Discovery and early strategies for lead optimization. Bioorg Med Chem Lett. 2003;13:4201–4204. doi: 10.1016/j.bmcl.2003.07.024. [DOI] [PubMed] [Google Scholar]
- 122.Nakashima R, Sakurai K, Yamasaki S, Hayashi K, Nagata C, Hoshino K, Onodera Y, Nishino K, Yamaguchi A. Structural basis for the inhibition of bacterial multidrug exporters. Nature. 2013;500:102–106. doi: 10.1038/nature12300. [DOI] [PubMed] [Google Scholar]
- 123.Lomovskaya O, et al. Use of a genetic approach to evaluate the consequences of inhibition of efflux pumps in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 1999;43:1340–1346. doi: 10.1128/aac.43.6.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Du D, Wang Z, James NR, Voss JE, Klimont E, Ohene-Agyei T, Venter H, Chiu W, Luisi BF. Structure of the AcrAB-TolC multidrug efflux pump. Nature. 2014;509:512–515. doi: 10.1038/nature13205. [DOI] [PMC free article] [PubMed] [Google Scholar]