

Abstract
Understanding the function of genes mutated in hereditary forms of Parkinson's disease yields insight into disease etiology and reveals new pathways in cell biology. Although mutations or variants in many genes increase the susceptibility to Parkinson's disease, only a handful of monogenic causes of Parkinsonism have been identified. Biochemical and genetic studies reveal that the products of two genes that are mutated in autosomal recessive Parkinsonism, PINK1 and Parkin, normally work together in the same pathway to govern mitochondrial quality control, bolstering previous evidence that mitochondrial damage is involved in Parkinson's disease. PINK1 accumulates on the outer membrane of damaged mitochondria, activates Parkin's E3 ubiquitin ligase activity and recruits Parkin to the dysfunctional mitochondrion. Then, Parkin ubiquitinates outer mitochondrial membrane proteins to trigger selective autophagy. This review covers the normal functions that PINK1 and Parkin play within cells, their molecular mechanisms of action, and the pathophysiological consequences of their loss.
Introduction
Parkinson's disease (PD) affects 1-2% of the population and is the second most common neurodegenerative disease (de Rijk et al., 1995). Four cardinal signs characterize PD: rigidity, bradykinesia, postural instability, and tremor (Lang and Lozano, 1998a, b) that stem from the progressive loss of dopaminergic neurons in the substantia nigra pars compacta (SN). Motor symptoms appear when approximately 50-60% of these neurons degenerate causing a 70-80% depletion of dopamine (DA) levels in the dorsal striatum (Lang and Lozano, 1998a, b). The remaining SN neurons typically contain inclusions in the cytoplasm called Lewy bodies that immunostain for α-synuclein and other proteins (Spillantini et al., 1998). There is no cure, and the best treatment options only provide symptomatic relief with no abatement or reversal of disease progression. Although a large majority of diagnosed PD cases are idiopathic, autosomal dominant and recessive familial forms have been identified. By understanding how monogenic forms of PD lead to cell dysfunction and neuron death, researchers are identifying causes of Parkinsonism and new pathways in cell biology.
PARK2 (Parkin) and PARK6 (PINK1) Identification
In 1997, a genetic linkage analysis showed that chromosome 6q25.2-27 harbored an unidentified gene responsible for autosomal recessive juvenile Parkinsonism (AR-JP) in 13 Japanese families (Matsumine et al., 1997). One year later, the Shimizu group cloned the AR-JP gene, PARK2, and identified more Japanese AR-JP patients with either an exon 4 or a large-scale deletion between exons 3-7 (Kitada et al., 1998). Other patients of various ethnicities with early-onset PD were soon reported to also harbor PARK2 mutations with varying deletions or point mutations that cause PARK2 protein loss of function (Hattori et al., 1998a; Hattori et al., 1998b; Leroy et al., 1998; Lucking et al., 1998). PARK2 contains 12 exons that encode the 465 amino acid protein, Parkin (Kitada et al., 1998). Parkin is an E3 ubiquitin ligase with an amino-terminal ubiquitin-like (Ubl) domain and a carboxyl-terminal ubiquitin ligase domain (Hristova et al., 2009; Shimura et al., 2000).
The second gene to be identified in early-onset recessive PD cases was found in 2001 within a large Italian pedigree on chromosome 1 at the PARK6 locus (Valente et al., 2001; Valente et al., 2002b). These PARK6 mutation patients had symptoms clinically identical to those of patients with sporadic forms of PD (Bentivoglio et al., 2001; Valente et al., 2002a). The 8 exon PARK6 gene encodes the 581 amino acid protein phosphatase and tensin homolog (PTEN)-induced kinase 1 (PINK1) (Valente et al., 2004), a name stemming from its prior identification in a screen for proteins transcriptionally upregulated by exogenous PTEN overexpression (Unoki and Nakamura, 2001). The protein sequence reveals a predicted C-terminal kinase domain and a mitochondrial targeting sequence at the N-terminus suggesting that it is imported into the mitochondria, consistent with its mitochondrial localization in cells (Valente et al., 2004). This mitochondrial localization supported prior evidence of an involvement of mitochondrial dysfunction in the pathophysiology of PD.
Mitochondrial Dysfunction in Parkinson's Disease
Evidence linking mitochondrial dysfunction to PD arose in the late 1970's when accidental exposure to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), a contaminant from the synthesis of 1-methyl-4-phenyl-4-propionoxy-piperidine (MPPP) (a drug used for illicit purposes), was found to cause Parkinsonism and DA neurodegeneration (Langston et al., 1983). Follow-up studies found that MPTP oxidized to MPP+, which is selectively taken up into DA neurons causing an inhibition of complex I, a mitochondrial respiratory chain component of the oxidative phosphorylation (OXPHOS) machinery (Javitch et al., 1985; Nicklas et al., 1985; Ramsay and Singer, 1986). Complex I deficiency was also found in the brain, skeletal muscle, and platelets of sporadic PD patients (Schapira et al., 1990; Schapira et al., 1989). Pesticides and herbicides that selectively inhibit complex I, such as rotenone and paraquat, also cause Parkinsonism in animal models and possibly in man (Betarbet et al., 2000; Liou et al., 1997; Tanner et al., 2011). These early observations indicated that DA neurons appear particularly sensitive to mitochondrial dysfunction.
Pathogenic mitochondrial DNA (mtDNA) mutations are also associated with PD. MtDNA is a 16,569 base pair length genome that encodes 13 genes for subunit components of OXPHOS and its own tRNAs and rRNAs (Anderson et al., 1981). As hundreds to thousands of copies of mtDNA reside in virtually each mammalian cell, a state of heteroplasmy arises when different mtDNA genotypes, such as wild type and mutant forms, co-exist within the same cell. SN neurons from autopsies of normal aged people and PD patients harbor high levels of mutated mtDNA with large-scale deletions that cause mitochondrial dysfunction (Bender et al., 2006; Kraytsberg et al., 2006). Furthermore, mitochondrial disease patients with mutations in polymerase γ, the polymerase responsible for mtDNA replication, excessively accumulate mtDNA mutations and also have an increased risk of developing PD (Luoma et al., 2004; Reeve et al., 2013). The many links between mitochondrial dysfunction and the pathogenesis of PD stimulated interest in the role PINK1 plays in that organelle.
PINK1 and Parkin Mediate Mitophagy
Although PINK1 localizes to mitochondria and Parkin resides in the cytosol, genetic epistasis analyses in Drosophila revealed that both proteins work in the same pathway (Clark et al., 2006; Park et al., 2006) and maintain mitochondrial fidelity (Greene et al., 2003). Cell biology studies then revealed that Parkin is recruited from the cytosol to depolarized mitochondria to mediate the selective autophagic removal of the damaged organelle (mitophagy) (Narendra et al., 2008). PINK1 accumulates on dysfunctional mitochondria and its kinase activity is required for Parkin translocation to mitochondria and mitophagy linking the function of these autosomal recessive PARK gene products to the same biochemical pathway (Geisler et al., 2010a; Matsuda et al., 2010; Narendra et al., 2010b; Vives-Bauza et al., 2010). These findings have led to the unraveling of an intriguing process, by which PINK1 detects mitochondrial dysfunction and then signals Parkin to ubiquitinate specifically the damaged mitochondria to instigate their removal by autophagy. This suggests that acting together, PINK1 and Parkin constitute a mitochondrial quality control function (Fig. 1). Further supporting the model that mitochondrial quality control is the essential function of PINK1 and Parkin contributing to the prevention of Parkinsonism in man, patient mutations in both PINK1 (Geisler et al., 2010b; Narendra et al., 2010b) and Parkin (Geisler et al., 2010a; Lee et al., 2010; Matsuda et al., 2010; Narendra et al., 2010b) prevent PINK1 recruitment of Parkin to mitochondria (Fig. 2, Table 1). These results also offer further evidence that mitochondrial dysfunction may play a role in sporadic PD (Fig. 3). Recent advances have revealed the molecular mechanisms of PINK1 sensing mitochondrial damage and activation of Parkin.
Figure 1. Model of Parkin-induced Mitophagy.

Dysfunctional mitochondria (Yellow Mitochondrion) fail to import and degrade PINK1 stabilizing it on the outer mitochondrial membrane. After PINK1 accumulation, PINK1 phosphorylates ubiquitin and Parkin to activate Parkin's E3 ligase activity. Parkin ubiquitinates substrates on the outer mitochondria for two divergent processes: autophagosome recruitment and ubiquitin proteasome degradation of ubiquitinated mitochondrial substrates. Fis1 is a receptor on the outer membrane that binding two proteins TBC1D15/TBC1D17 to govern the developing LC3 isolation membrane to generate the autophagosome around the damaged mitochondria. The autophagosome is then delivered to the lysosome for degradation.
Figure 2. Cartoon Depiction of the Location of Mutations in Monogenic PINK1 and Parkin PD Patients.

* = heterozygous mutation; ins = insertion; / = compound heterozygous; fs = frame shift; dup = duplication; bp = base pairs; TM = transmembrane domain; MTS = mitochondrial targeting sequence; UBL = ubiquitin-like domain; RING = really interesting new gene domain; IBR = in between RING domain.
Table 1.
Summary of PARK2 and PARK6 Knockout Models in Comparison to Monogenic PD Patients.
| Gene Inactivated | Species | Age of Onset | DA Neurotransmission Defects | Motor Phenotypes | SN Neuron Loss | Lewy Body Formation | References |
|---|---|---|---|---|---|---|---|
| PARK2 | Human | Juvenile > 20 years Early onset 20-40 years |
DA depletion in striatum | Cardinal Signs | Progressive | Absent or present |
Matsumine et al. 1997
Kitada et al. 1998 Pramstaller et al. 2005 |
| PARK6 | Human | Juvenile > 20 years Early onset 20-40 years |
DA depletion in striatum | Cardinal Signs | Progressive | Absent or present |
Valente et al. 2001
Samaranch et al. 2010 |
| PARK2 | Drosophila | N/A | Absent | Flight wing degeneration, motor coordination deficits | Absent | Unknown | Greene et al. 2003 |
| N/A | Absent | Flight wing degeneration, motor coordination deficits | Absent | Unknown | Pesah et al. 2004 | ||
| 1 day (DA neuron loss progressive) | Unknown | Flight wing degeneration, motor coordination deficits | DL region | Unknown | Whitworth et al. 2005 | ||
| N/A | DA depletion | Flight wing degeneration, motor coordination deficits | Absent | Unknown | Cha et al. 2005 | ||
| PARK6 | Drosophila | N/A | Absent | Flight wing degeneration, motor coordination deficits | Absent | Unknown | Clark et al. 2006 |
| 30 days | Decreased TH+ Expression | Flight wing degeneration, motor coordination deficits | DM and DL region | Unknown | Park et al. 2006 | ||
| 25 days | DA depletion | Flight wing degeneration, motor coordination deficits | DM and DL region | Unknown | Yang et al. 2006 | ||
| PARK2 | Mouse | N/A | Increased extracellular DA release | Beam slip motor deficit | Absent | Unknown | Goldberg et al. 2003 |
| N/A | Decreased DA uptake | Motor coordination deficits | Absent | Unknown | Itier et al. 2003 | ||
| N/A | Absent | Startle response deficit | Absent | Unknown | Von Coelln et al. 2004 | ||
| N/A | Absent | Absent | Absent | Unknown | Perez and Palmiter 2004 | ||
| Conditional Cre-loxP | Unknown | Unknown | Yes, Lentiviral-Cre infection | Unknown | Shin et al. 2011 | ||
| PARK6 | Mouse | N/A | Evoked DA release deficit | Unknown | Absent | Unknown | Kitada et al. 2007 |
| N/A | DA depletion | Unknown | Absent | Unknown | Akundi et al. 2011 |
# = indicates that these phenotypes belong to a specific in vivo model within that subcategory.
Figure 3. The Balance Between Mitochondrial Damage and its Removal Contributes to the Pathogenesis of PD.
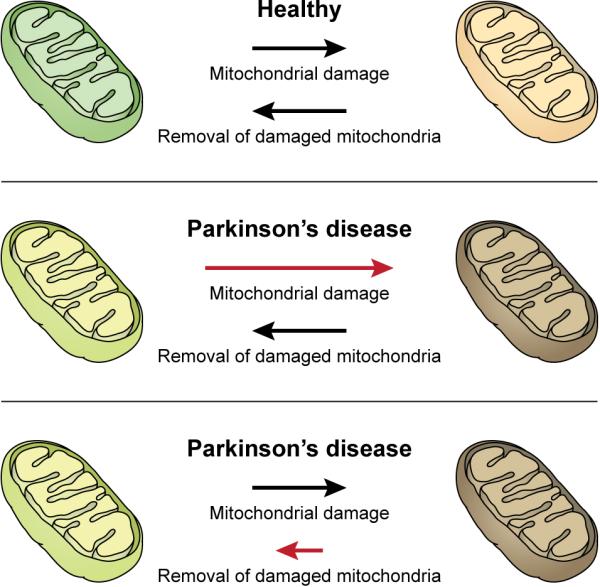
Healthy neurons efficiently remove damaged mitochondria by mitophagy as a quality control mechanism to ensure cell survival. Excess mitochondrial damage (from triggers such as MPTP exposure, paraquat, and aging-induced dysfunction) causes neuronal demise contributing to the pathogenesis of PD. Likewise, reducing the cells ability to remove damaged mitochondria (from the loss of Parkin or PINK1) may cause an accumulation of these dysfunctional organelles leading to early onset-PD.
PINK1 Detects Mitochondrial Damage via Selective Proteolysis
PINK1 accumulates specifically on damaged mitochondria flagging them for elimination. The damage-sensing mechanism stems from the rapid and constitutive degradation of PINK1 in healthy mitochondria within a cell, which is circumvented when one mitochondrion becomes impaired, allowing PINK1 to accumulate on the outer membrane of the dysfunctional mitochondrion (Jin et al., 2010; Meissner et al., 2011; Narendra et al., 2010b) (Fig. 4). Under normal steady state conditions, PINK1 is imported through the TOM complex of the outer mitochondrial membrane and into the TIM complex of the inner mitochondrial membrane, where it is cleaved initially by the mitochondrial processing peptidase (MPP) (Greene et al., 2012), as imported proteins typically are. Then PINK1 is cleaved in its hydrophobic domain spanning the inner mitochondrial membrane by the rhomboid protease, presenilin-associated rhomboid-like protein (PARL) (Jin et al., 2010; Meissner et al., 2011). The membrane-spanning domain of PINK1 is rich in Gly/Pro amino acids that can cause kinks in membrane spanning helices to target them for rhomboid protease cleavage (Urban and Freeman, 2003). PARL, an inner mitochondrial membrane protease, cleaves PINK1 between amino acids Ala103 and Phe104 generating a 52 kD, N-terminal deleted form of PINK1 (Deas et al., 2011). PARL cleavage releases the N-terminal deleted PINK1 into the cytosol where the N-terminal phenylalanine generated by the proteolysis is identified by the N-degron type-2 E3 ubiquitin ligases according to the N-end rule and degraded by the ubiquitin proteasome system (UPS) (Yamano and Youle, 2013). This explains earlier work showing that proteasome inhibition stabilizes a 52kD cleaved fragment of PINK1 (Lin and Kang, 2008). This continuous import and degradation cycle yields very low to undetectable levels of PINK1 on healthy mitochondria (Fig. 4). However, when mitochondrial import through the TIM complex is disrupted by mitochondrial depolarizing agents, OXPHOS inhibitors, genetic or environmental stresses and even unfolded proteins, PINK1 processing by PARL is prevented because import into the inner membrane where PARL and MPP reside is blocked. Instead, PINK1 accumulates uncleaved on the outer mitochondrial membrane bound to the TOM complex in a 2:1 molar ratio (Lazarou et al., 2012; Okatsu et al., 2013). Interestingly, this accumulation of PINK1 bound to TOM is completely blocked by the loss of a small subunit of the TOM complex, Tomm7 (Hasson et al., 2013). Protein import through the TOM complex into the matrix compartment is not blocked by loss of Tomm7 suggesting that the Tomm7 subunit may be devoted to releasing outer mitochondrial membrane proteins such as PINK1 laterally through the TOM complex into the outer mitochondrial membrane (Hasson et al., 2013). This model of how PINK1 senses mitochondrial damage by regulated proteolysis explains how an individually impaired mitochondrion in a milieu of healthy mitochondria may be flagged by PINK1 (Fig. 4).
Figure 4. PINK1 is Turned Over by Sequential Proteolysis.

(Left) PINK1 is continuously imported into healthy mitochondria then degraded. Mitochondrial membrane potential drives mitochondrial import through the TIM complex of the mitochondria where proteases MPP and PARL cleave PINK1's mitochondrial targeting sequence and transmembrane domain. PARL's cleavage between Ala103 and Phe104 creates a free N-terminal Phenylalanine that is identified by N-degron type 2 E3 ubiquitin ligases targeting PINK1 to the ubiquitin proteasome. (Right) Depolarization of mitochondria or blocking mitochondrial import causes PINK1 to accumulate on the outer mitochondrial membrane. Tomm7 is an accessory subunit of the TOM complex that shunts and retains PINK1 on the outer mitochondrial membrane.
Two recent papers show that the mitochondrial matrix LON protease is also involved in PINK1 stability. It had appeared that loss of mitochondrial membrane potential (Δψm), known to prevent protein import into mitochondria through the TIM complex which in turn prevents PINK1 proteolysis, was the unifying mechanism through which diverse mitochondrial stresses induced PINK1 accumulation (Youle and Narendra, 2011). However, an unfolded protein expressed in the mitochondrial matrix was found to stabilize PINK1 and activate Parkin without depolarizing the inner mitochondrial membrane (Jin and Youle, 2013). If expression of the LON protease was diminished, the levels of PINK1 increased even further (Jin and Youle, 2013). The authors concluded that loss of LON, which is known to help degrade misfolded mitochondrial matrix proteins, increased misfolded protein accumulation and thereby increased PINK1 accumulation. How misfolded proteins cause PINK1 accumulation without mitochondrial depolarization is yet unresolved, although it is possible that misfolded proteins somehow inhibit TIM complex import of PINK1 (Fig. 4). Another study showed that LON mutant flies accumulate PINK1, although a different interpretation of the mechanism was reached (Thomas et al., 2014). The Drosophila phenotype suggested that LON directly degraded PINK1 in the matrix compartment and that loss of LON prevented this degradation. However, this model is hard to reconcile with the finding that PARL processed PINK1 is degraded mainly in the cytosol by the N-end rule (Yamano and Youle, 2013). Nevertheless, in both flies and mammals it appears that upon mitochondrial damage PINK1 becomes stabilized on the outer mitochondrial membrane and from there it recruits Parkin to mitochondria and activates Parkin's E3 ubiquitin ligase activity.
PINK1 is a Ubiquitin Kinase
The substrate of PINK1 kinase activity mediating Parkin activation and recruitment to mitochondria was predicted to be in the cytosol and not a mitochondrial protein because ectopic expression of PINK1 on peroxisomes recruits Parkin to induce pexophagy (Lazarou et al., 2012). Consistent with the cytosolic substrate model, PINK1 directly phosphorylates Parkin at Ser65 within the Ubl domain of Parkin, and Ser65 phosphorylation stimulates Parkin's E3 ligase activity and recruitment to mitochondria (Kondapalli et al., 2012; Shiba-Fukushima et al., 2012). However, although Parkin mutated at Ser65 prevents phosphorylation by PINK1, Ser65Ala Parkin as well as Parkin lacking the entire Ubl domain still translocate to mitochondria in a PINK1 kinase-dependent process indicating that another cytosolic PINK1 substrate is involved in Parkin activation (Kane et al., 2014). This was initially confusing because cell free systems displayed Parkin activation simply by mixing recombinant PINK1 and recombinant Parkin, and critically, but not realized at the time, E2 enzymes and ubiquitin for assessing Parkin enzyme activity (Lazarou et al., 2013).
Three groups independently discovered that PINK1 phosphorylates ubiquitin at Ser65 and that phospho-ubiquitin activates Parkin E3 ligase activity (Kane et al., 2014; Kazlauskaite et al., 2014b; Koyano et al., 2014). This explained all the prior in-cell and cell-free results and fit seamlessly with the previous finding that PINK1 phosphorylates Parkin in its Ubl domain at Ser65, homologous to ubiquitin Ser65 (Kondapalli et al., 2012). Kane et al. used mass spectrometry to compare PINK1 knockout (KO) cells to wild type cells to reveal that endogenous PINK1 stabilized by carbonyl cyanide m-chlorophenyl hydrazine (CCCP)-induced depolarization of mitochondria produces phospho-Ser65 ubiquitin. It was found that phospho-ubiquitin activates Parkin's E3 ubiquitin ligase activity in a purified cell-free system and that Parkin binds to phosphomimetic ubiquitin Ser65Asp. In living cells, inhibition of ubiquitin phosphorylation by expression of ubiquitin Ser65Ala inhibits Parkin translocation to depolarized mitochondria. The fact that phospho-Ub peptides were isolated from purified mitochondrial OMM (outer mitochondrial membrane) peptides suggests that PINK1 phosphorylates ubiquitin chains linked to OMM proteins that recruit Parkin to mitochondria and activate it (Kane et al., 2014). Kazlauskaite et al. overexpressed PINK1 in cells treated with CCCP to identify ubiquitin phosphorylation at Ser65 by mass spectrometry. Purified recombinant PINK1 was shown to phosphorylate ubiquitin in vitro at Ser65. It was also found that Parkin mutated in the Ubl at Ser65 to mimic phosphorylation (Ser65Asp) displayed greater ubiquitin ligase activity upon incubation with phospho-ubiquitin than did wild type Parkin indicating that PINK1 phosphorylation of both Parkin and ubiquitin may be important in activation. However, both Kane et al. and Kazlauskaite et al. found that phosphomimetic mutants of ubiquitin Ser65 are sufficient for Parkin activation whereas phosphomimetic mutants of Parkin Ser65, at least in cells, are not. Koyano et al. also found ubiquitin is phosphorylated by PINK1. Like Kazlauskaite et al., Koyano et al. report that Parkin Ser65 phosphorylation in combination with ubiquitin Ser65 phosphorylation yields maximal Parkin activity. They also show that ubiquitin phosphomimetic Ser65Asp further mutated at the C-terminus to prevent ubiquitin chain formation activates Parkin indicating phospho-ubiquitin is allosteric and unrelated to thioester loading (see the following section) on Parkin. Parkin can also conjugate both phosphomimetic Ser65Asp and phospho-null Ser65Ala ubiquitin mutants to mitochondrial proteins supporting an allosteric role as phospho-ubiquitin is neither required nor excluded as a Parkin substrate. By immunoprecipitating ubiquitin chains Ordureau et al. find PINK1 increases phospho-Ser65-ubiquitin on mitochondria further indicating that chains of ubiquitin attached to proteins on the OMM become phosphorylated by PINK1 and may serve as Parkin receptors on mitochondria (Ordureau et al., 2014). Parkin binds not only to phospho-ubiquitin but also to phosphorylated ubiquitin chains. Ordureau et al. further show that phospho-ubiquitin binds to Ser65 phosphorylated Parkin with a 21x higher affinity than to Parkin, suggesting a feed forward model of Parkin activation – first Parkin is phosphorylated by PINK1 and then further activated by phospho-Ser65 ubiquitin. Interestingly, the active site Cys431 of Parkin becomes exposed to Ubiquitin-vinyl sulfone when Parkin is activated by its Ubl phosphorylation but not by activation by phospho-ubiquitin suggesting that phospho-Ser65 ubiquitin and phospho-Ser65 Parkin have different modes of activating Parkin (Ordureau et al., 2014).
Although biochemical studies indicate that Parkin phosphorylation at Ser65 is required for Parkin activation, PINK1 null and Parkin null Drosophila can be partially rescued with Ser65Ala Parkin indicating that Parkin displays Ser65 phosphorylation-independent functions in vivo (Shiba-Fukushima et al., 2014) Whether the Ser65Ala Parkin activity in vivo is due to phospho-Ser65 ubiquitin remains to be explored. A model is presented showing that a positive feedback amplification mechanism drives mitophagy to completion when the product of Parkin (ubiquitinated OMM proteins) yields substrates for PINK1 phosphorylation that in turn further activate Parkin leading to an amplification loop (Fig. 5). The recruitment of Parkin to mitochondria is likely initiated by PINK1 phosphorylating ubiquitin already attached to mitochondrial outer membrane proteins such as mitofusin 1 (Mfn1) prior to Parkin activation.
Figure 5. The Activation of Parkin Drives a Positive Feedback Loop Providing Ubiquitin as a Substrate for PINK1 to Complete Mitophagy.
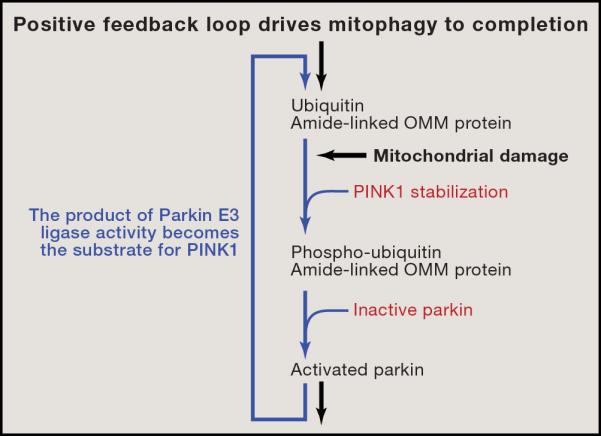
Mitochondrial dysfunction stabilizes PINK1 enabling it to phosphorylate ubiquitin and Parkin. This phosphorylation event activates Parkin, which ubiquitinates substrates on the outer membrane. These ubiquitin chains on the proteins of the outer mitochondrial membrane provide additional substrates for PINK1's kinase activity causing a strong positive feedback cycle assuring mitochondrial clearance. OMM = outer mitochondrial membrane.
In conclusion, phosphorylation of ubiquitin at Ser65 activates, or derepresses, the ubiquitin ligase activity of Parkin. Ubiquitin-phosphate is an allosteric effector of Parkin ubiquitin ligase activity. As ubiquitin is the substrate of Parkin, this resembles homotropic allosteric regulation in which an enzyme substrate binds to an allosteric site on a protein to activate enzyme activity, although in this case the allosteric site binds phospho-ubiquitin generated by PINK1 and not unmodified ubiquitin. Where phospho-ubiquitin binds to Parkin and the conformation of activated Parkin remain unknown. This interplay of ubiquitin with Parkin as both activator and substrate may relate to why a full genome siRNA screen for modifiers of Parkin translocation yielded a rich interactome of ubiquitin pathway genes that both accelerated and inhibited Parkin translocation after CCCP treatment (Hasson et al., 2013).
Other substrates of PINK1 have been reported to facilitate mitophagy by altering mitochondrial trafficking or dynamics. PINK1 was reported to phosphorylate Miro1 at Ser156 to activate Parkin-mediated proteasomal degradation of Miro1 to arrest mitochondrial motility (Wang et al., 2011). However, while in agreement that Miro1 and Miro2 stability is genetically linked and dependent upon the PINK1/Parkin pathway, others were unable to replicate the result that PINK1 phosphorylates Ser156 on Miro1 (Kazlauskaite et al., 2014a; Liu et al., 2012). Mitofusin2 (Mfn2), a protein involved in outer mitochondrial membrane fusion, was identified to be phosphorylated at Thr111 and Ser442 by PINK1 to induce Mfn2 binding to Parkin and initiate Mfn2 ubiquitination and degradation (Chen and Dorn, 2013). However, Parkin translocates constitutively to mitochondria in Mfn1/Mfn2 knockout cells arguing that Mfn2 is not involved in Parkin translocation (Narendra et al., 2008). Alternatively, PINK1 may phosphorylate mono-ubiquitin and/or poly-ubiquitin chains covalently linked to Mfn2 to recruit Parkin to mitochondria. Mitofusins are ubiquitinated even in yeast that lack PINK1 and Parkin (Neutzner et al., 2008) supporting the idea that ubiquitinated mitofusins, among other ubiquitinated OMM proteins, may represent a starting point for the PINK1/Parkin positive feedback ubiquitin phosphorylation cycle (Fig. 5).
PINK1 may phosphorylate other mitochondrial targets, but they appear unrelated to the PINK1/Parkin mitophagic pathway. Omi/HtrA2, a serine protease, and tumor necrosis factor receptor-associated protein 1 (TRAP-1), a chaperone protein, both have been reported to be targets of PINK1 (Plun-Favreau et al., 2007; Pridgeon et al., 2007). In vitro data, however, failed to validate Omi/HtrA2 and TRAP-1 as PINK1 targets (Kondapalli et al., 2012). Furthermore, Omi/HtrA2 is not downstream of the PINK1/Parkin pathway and TRAP-1 completely rescues PINK1 deficient, but not Parkin mutant flies (Costa et al., 2013; Yun et al., 2008; Zhang et al., 2013).
Activating PINK1 kinase activity could have therapeutic applications. The ATP analog, kinetin triphosphate, rescues kinase activity of a patient mutant form of PINK1 and increases wild type PINK1 activity revealing a new way to drug the PINK1/Parkin pathway (Hertz et al., 2013).
Parkin E3 Ubiquitin Ligase Structure and Function
Parkin is an E3 ubiquitin ligase of an unusual class called ring between ring or RBR domain proteins that share three tandem zinc coordination domains (Marin, 2009). In contrast to single RING E3 ligases that juxtapose an E2 enzyme (linked via a high energy thioester bond to ubiquitin) with a protein substrate to facilitate ubiquitin transfer from the cysteine of the E2 to a lysine on the substrate, RBR proteins, Parkin (Lazarou et al., 2013; Zheng and Hunter, 2013), HOIP (Smit et al., 2012) and HHARI (Wenzel et al., 2011) all transfer ubiquitin from the E2 directly onto the RBR itself yielding another high energy thioester intermediate, similar to HECT domain ubiquitin ligases (Wenzel et al., 2011). Parkin has 4 zinc coordinating domains, RING0, RING1, IBR and RING2 (Fig. 2, Fig. 6) (Hristova et al., 2009). However, only RING1 adopts the canonical ubiquitin ligase RING domain structure that binds to E2 ubiquitin ligases and facilitates the discharge of thioester-bound ubiquitin from the E2 to the substrate. Like other RBR proteins, Parkin requires activation to allow the ubiquitin charged E2 to bind to Parkin RING1 and transfer ubiquitin to Parkin Cys431 in RING2 to form the HECT-like thioester intermediate (Fig. 6a) (Riley et al., 2013; Trempe et al., 2013; Wauer and Komander, 2013). Several E2s can transfer ubiquitin to PINK1 activated-Parkin in cell free systems including UBE2K, UBE2D1, UBE2D2, UBE2E1, UBE2L3 and UBE2C (Kazlauskaite et al., 2014a; Lazarou et al., 2013). The structure of Parkin shows that the RING0, RING1 and the IBR domains interact and a linker domain wraps around the interface of RING0 and RING1 to reach the RING2 (Riley et al., 2013; Trempe et al., 2013; Wauer and Komander, 2013) (Fig. 6b). The structure of Parkin represents the inactive or autoinhibited state because the Cys431 site of thioester formation in RING2 (yellow in Fig. 6) is occluded by the RING0 domain and the E2 binding site on RING1 is blocked by the linker region called a repressor element (blue helix in Fig. 6) (Riley et al., 2013; Trempe et al., 2013; Wauer and Komander, 2013). A substantial conformational change induced by phospho-ubiquitin and probably contributed to by phospho-S65 Parkin must shift the ubiquitin-E2 binding site on RING1 proximal to the Cys431 acceptor site on RING2. Loading of ubiquitin onto Parkin Cys431 can be activated by PINK1 (Lazarou et al., 2013; Zheng and Hunter, 2013), the PINK1 substrate phosphomimetic ubiquitin Ser65Asp (Kane et al., 2014), or the Parkin Ubl phosphomimetic Ser65Glu (Iguchi et al., 2013). Determining the structure of activated Parkin bound to phospho-ubiquitin will be illuminating.
Figure 6. Structural Representation of Parkin.

(A) Ribbon diagram of the various domains in individual colors - cyan (Ubl), grey (RING0), green (RING1), violet (IBR), blue (Rep), and bronze (RING2). The residues Ser65 (red), Arg275 (blue), and Cys431 (yellow) are highlighted in stick representation. (B) Juxtaposition of the ribbon diagram over a surface presentation of Parkin (Trempe et al., 2013).
The ubiquitin-like domain at the N-terminus of Parkin folds back and binds to the RING1 domain (Spratt et al., 2013; Trempe et al., 2013). Although cell-free experiments indicate that this Ubl domain interaction inhibits Parkin E3 ligase activity (Chaugule et al., 2011), these experiments did not take PINK1 activation of Parkin into account. Parkin lacking the Ubl domain is still activated by PINK1 indicating that phospho-ubiquitin binding to Parkin does more than displace the Parkin Ubl domain (Kazlauskaite et al., 2014b). However, phosphorylation of the Parkin Ubl domain Ser65 and mutations in the linker region between the IBR and RING2 domains near to the Ubl domain may destabilize the Ubl domain binding to RING0 and increase Parkin sensitivity to activation (Koyano et al., 2014; Trempe et al., 2013). Parkin conformational instability may also be linked to age-related or PD related decreases in Parkin solubility that could connect the PINK1/Parkin pathway to sporadic forms of PD (Meng et al., 2011; Pawlyk et al., 2003; Vandiver et al., 2013).
Parkin Ubiquitination: Targets and Consequences
Once activated, Parkin modifies many cytosolic and outer mitochondrial membrane proteins with K48- and K63- linked ubiquitin chains (Chan et al., 2011; Narendra et al., 2012; Sarraf et al., 2013). Quantification of all different chain linkages reveals that Parkin primarily forms non-canonical K6- and K11- linked chains and K48- and K63-linked chains. Mitochondrial proteins from cells displayed more K48- and K63-linked chains than K6- and K-11 whereas purified Parkin produced more non-canonical chain linkages than K48- and K-63 linked chains (Ordureau et al., 2014).
Although the role of K6-linked chains is unknown, the K63-linked chains appear to be involved in p62 recruitment (Geisler et al., 2010a; Narendra et al., 2010a; Okatsu et al., 2010; Sims et al., 2012; van Wijk et al., 2012). Although an initial report indicated that p62 is required for Parkin-mediated mitophagy (Geisler et al., 2010a); two groups using p62 KO mouse embryonic fibroblasts (MEFs) did not find that it is essential for mitophagy (Narendra et al., 2010a; Okatsu et al., 2010). However, the robust clumping of mitochondria that occurs following Parkin activation is prevented in p62 KO cells. The NBR1 ubiquitin adaptor also accumulates on mitochondria after Parkin translocation (Chan et al., 2011). However, the significance of autophagy adaptors in mediating mitophagy remains unclear.
Parkin generated K11- and K48-linked chains on mitochondria likely lead to an endoplasmic reticulum-associated degradation (ERAD)-like extraction of proteins from the outer mitochondrial membrane (OMMAD) by p97/VCP and subsequent degradation by the proteasome (Chan et al., 2011; Kim et al., 2013; Narendra et al., 2012; Yoshii et al., 2011). This removal and proteolysis of outer mitochondrial membrane proteins appears to be necessary for mitophagy (Chan et al., 2011; Tanaka et al., 2010). Modulating the levels of outer mitochondrial membrane proteins also controls mitochondrial motility and dynamics during the mitophagy process. Mfn1 and 2 are ubiquitinated and degraded by the proteasome in a Parkin-dependent manner (Gegg et al., 2010; Poole et al., 2010; Rakovic et al., 2011; Tanaka et al., 2010; Ziviani et al., 2010). Miro1, a mitochondrial Rho GTPase involved in trafficking was identified and confirmed in vitro as a Parkin substrate (Kazlauskaite et al., 2014a; Sarraf et al., 2013; Wang et al., 2011). One group reported the ability of Parkin to indirectly generate linear ubiquitin chains on mitochondria via NF-κB essential modulator (NEMO) and to elicit protection from apoptosis by stimulating the NF-κB pathway (Muller-Rischart et al., 2013). Thirty-six OMM substrates of Parkin have been identified with high confidence (Sarraf et al., 2013) suggesting that no specific substrate is required for ubiquitin signaling of mitophagy and that the chain linkage type and density on mitochondria may be the trigger.
Mitophagy Mechanisms
Inhibiting autophagic machinery genetically or pharmacologically does not disrupt PINK1/Parkin recruitment to the mitochondria, but it does prevent or delay the elimination of Parkin-ubiquitinated mitochondria (Fogel et al., 2013; Narendra et al., 2008). Different assays used to assess mitophagy have distinct advantages and drawbacks. Loss of immunostaining of mitochondrial outer membrane proteins such as Tom20 is convenient and robust, but may also reflect OMMAD, the proteasomal degradation of outer mitochondrial membrane proteins, rather than mitophagy (Yoshii et al., 2011). Thus, staining for inner mitochondrial membrane or matrix proteins or labeling with mitotracker green is a better measure of mitophagy than staining of outer membrane proteins. However, inner membrane proteins are turned over at different rates and are not necessarily precise measures of mitophagy. The fluorescent protein Keima imported into the mitochondrial matrix changes its excitation wavelength from 440 to 586 when exposed to the acidic lysosomal pH following autophagy and yields a convenient measure of mitophagy (Katayama et al., 2011). Loss of mitochondrial DNA in nucleoids is another clear way to measure loss of mitochondria. If nucleoid loss is prevented in Atg5 KO cells or by lysosomal inhibitors, such as bafilomycin, one can be confident the process is mitophagy.
Presumably, the ubiquitin chains that Parkin attaches to mitochondrial proteins initiate mitophagy, as is observed for other autophagy substrates (Kirkin et al., 2009). Mitophagy (Itakura et al., 2012), like starvation-induced autophagy (Itakura and Mizushima, 2011) involves the independent recruitment adjacent to the mitochondria of three components of the autophagosome targeting and expansion machinery; the serine/threonine kinase, UNC-51-like kinase 1 (ULK1), the membrane spanning Atg9 protein, and LC3-associated isolation membranes. Thus, ubiquitin chains attached to mitochondria seem to independently recruit different complexes to growing isolation membranes that expand alongside mitochondria. How these autophagy components are recruited to isolation membranes growing around mitochondria remains unclear except that adaptor proteins such as p62, NBR1, NDP52, Tax1BP1 and optineurin, proteins that bind both to ubiquitin chains and LC3/GABARAP family members, may be involved. On the other hand, it is becoming clear how LC3 autophagosomal membrane generation is restricted to accurately surround the mitochondria as seen in EM images of Parkin-mediated mitophagy (Fig. 7). Two mitochondrial localized RabGAPs, TBC1D15 and TBC1D17, bound to the outer mitochondrial membrane protein Fis1 govern isolation membrane engulfment of mitochondria. Knock out of either Fis1 or TBC1D15/17 leads to excessive expansion of LC3 labeled autophagosomal membranes during Parkin-mediated mitophagy (Yamano et al., 2014). Parkin activation also induces dysregulated trafficking of LC3 bound tubules in the absence of TBC1D15 that appears to be caused by excessive activation of Rab7 (Yamano et al., 2014). Fis1 also regulates autophagosomal membrane engulfment of mitochondria in vivo in C. elegans (Shen et al., 2014).
Figure 7. Electron Micrographs of Parkin-induced Mitophagy and Autophagosomal Engulfment of Mitochondria.

Mouse embryonic fibroblasts overexpressing Parkin and treated with 20mM CCCP for 12 hours. White arrowhead points to autophagosome forming around mitochondria. Scale bar = 500 nm. Images from (Yoshii et al., 2011).
There are tantalizing links between the mitochondrial fission machinery and mitophagy. In yeast the mitochondrial fission protein, Dnm1, homologous to mammalian Drp1, is required for certain forms of mitophagy (Abeliovich et al., 2013; Frank et al., 2012; Mao et al., 2013). This leads to the model that the fragmentation of mitochondria may facilitate their engulfment by autophagosomes (Buhlman et al., 2014; Tanaka et al., 2010; Twig et al., 2008) and fits with phenotypes of PINK1 and Parkin mutant flies that are reversed by promoting mitochondrial fission (Deng et al., 2008; Poole et al., 2008; Yang et al., 2008).
PINK1 and Parkin can remove small pieces of vesicles from mitochondria (McLelland et al., 2014; Yang and Yang, 2013). This may be related to the piecemeal disposal of the nuclear envelope by autophagy (Krick et al., 2008), during which membrane can be pinched off from larger cargo for degradation and may explain the selective disposal of OXPHOS complex subunits by mitophagy (Vincow et al., 2013).
Modifiers of the PINK1/Parkin Pathway
RNAi screens have been used to uncover a number of genes that modify Parkin translocation and/or mitophagy. Knockdown of genes such as HK1 and HK2 (McCoy et al., 2014), BAG4 and HSPA1L (Hasson et al., 2013), IF1 (Lefebvre et al., 2013), SREBF1 and FBXW7 (Ivatt et al., 2014), and SMURF1 (Orvedahl et al., 2011) inhibits Parkin translocation, indicating that those gene products normally participate in Parkin activation. Knockdown of other genes such as SIAH3 (Hasson et al., 2013) accelerate Parkin translocation indicating the gene product normally inhibits Parkin activation. Deubiquitinases (DUBs) such as USP30 (Bingol et al., 2014) and USP15 (Cornelissen et al., 2014) antagonizes Parkin ubiquitin ligase activity by cleaving ubiquitin chains on mitochondria. Another DUB USP8 has been identified to remove K6- chains from Parkin itself to positively regulate Parkin recruitment (Durcan et al., 2014). Drugging the DUBs may be a strategy to activate the PINK/Parkin pathway and promote mitochondrial quality control. Targeted screens of E2 ubiquitin ligases that may load Parkin with ubiquitin (Fiesel et al., 2014; Geisler et al., 2014) show that UBE2D, UBE2L3, and UBE2N participate in Parkin activity.
Affinity binding studies indicate that AMBRA1, a protein mediating autophagy, binds to Parkin and promotes mitophagy (Van Humbeeck et al., 2011). Overexpression of an outer membrane mitochondrial targeted AMBRA1 also stimulates mitophagy in the presence or absence of Parkin (Strappazzon et al., 2014). Similarly, PINK1 has been reported to bind another autophagy protein, Beclin1, to promote autophagy (Michiorri et al., 2010).
Parkin-mediated mitophagy is also linked to apoptosis (Winklhofer, 2014; Zhang et al., 2014). Interestingly, anti-apoptotic Bcl-2 family proteins that localize to the outer mitochondrial membrane, Bcl-xL and Mcl-1, inhibit Parkin translocation and therefore suppress mitophagy (Hollville et al., 2014). Furthermore, translocation of the pro-apoptotic Bcl-2 family protein Bax to mitochondria, which is a key decision step in apoptosis (Wolter et al., 1997), is inhibited by Parkin, perhaps contributing to Parkin inhibition of apoptosis (Johnson et al., 2012). However, in some experimental systems Parkin promotes apoptosis by ubiquitinating and inducing proteasomal degradation of Mcl-1 (Zhang et al., 2014).
Parkin Activity Beyond Mitochondria
Numerous Parkin substrates were identified before Parkin's link to mitochondria was identified (Scarffe et al., 2014) and recently Parkin was found also to ubiquitinate the transcriptional repressor named PARIS causing it to be degraded by the proteasome thereby inducing mitochondrial biogenesis (Shin et al., 2011). In addition to mitochondria, Parkin has been found to accumulate on intracellular Mycobacterium likely to induce their autophagy (Manzanillo et al., 2013) as a host defense mechanism and consistent with genetic studies linking Parkin mutations to increased susceptibility to leprosy (Mira et al., 2004). Parkin, like other RBR proteins, needs to be activated to display ubiquitin ligase activity. A mystery for these non-mitochondrial substrates is how Parkin's autoinhibition is relieved. For mitochondrial substrates, phospho-ubiquitin produced by PINK1 kinase activity is sufficient to bind and induce Parkin activation. The kinase(s) that may activate Parkin for these proposed extra-mitochondrial substrates remains to be identified.
Physiologic Triggers of Parkin Translocation – Beyond CCCP
It is experimentally convenient in cultured cell lines, in primary neurons (Ashrafi et al., 2014) and in iPSCs differentiated into neurons (Rakovic et al., 2013; Seibler et al., 2011) to trigger PINK1/Parkin-mediated mitophagy using pharmacological reagents to depolarize mitochondrial membrane potential (Δψm) such as CCCP, oligomycin combined with antimycin A, or valinomycin, or the photosensitizer KillerRed (Wang et al., 2012; Yang and Yang, 2011). However, the pathway also proceeds constitutively in neurons without chemical perturbation. Using mitochondrial targeted Keima to assess mitophagy, Bingol and colleagues show that mitophagy occurs over a period of days in mouse primary hippocampal neurons without the addition of depolarizing drugs and that this constitutive mitophagy requires endogenous PINK1 and Parkin (Bingol et al., 2014).
Genetic models of mitochondrial dysfunction that mimic pathophysiological processes also show that Parkin recruitment is possible without pharmacological manipulation. Genetic loss of Mfn1 and Mfn2 leads to a subset of mitochondria lacking membrane potential (Chen et al., 2005) that selectively recruit Parkin (Narendra et al., 2008). Parkin is also recruited to dysfunctional mitochondrial complex I components in neurons differentiated from induced pluripotent stem cells (iPSCs) derived from MELAS (Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes) patients harboring the mtDNA mutation m.3243A>G (Hamalainen et al., 2013). Cybrid cell lines harboring mtDNA mutations display Parkin-mediated mitophagy in the presence of overexpressed Parkin or upon inhibition of mammalian target of rapamycin complex 1, mTORC1, thereby activating autophagy (Gilkerson et al., 2012; Suen et al., 2010).
A type of mitochondrial dysfunction without loss of Δψm also may trigger Parkin-mediated mitophagy. Overexpression of a mutant form of ornithine transcarbamylase (ΔOTC), an enzyme involved in the urea cycle, abnormally aggregates inside the mitochondrial matrix and triggers the mitochondrial unfolded protein response (UPR) (Zhao et al., 2002). The aggregated ΔOTC causes stabilization of PINK1, recruitment of Parkin to mitochondria and subsequent removal of ΔOTC without depolarizing mitochondria (Jin and Youle, 2013). ΔOTC expressed in Drosophila phenocopies the muscle and flight wing mitochondrial defects found in PINK1 and Parkin mutant flies and this phenotype can be rescued by the overexpression of Parkin in ΔOTC flies (Pimenta de Castro et al., 2012).
Mouse Models Manipulating Parkin or PINK1 Expression
After the discovery that autosomal recessive mutations in Parkin lead to a loss of function, many laboratories independently generated Parkin KO mice to study Parkin's function in vivo. The first two Parkin KO mouse models lacked exon 3 and exhibited only mild phenotypes such as the disruption of fine motor skills and slight abnormalities in dopamine metabolism and release, but disappointingly, no dopaminergic neuron loss (Goldberg et al., 2003; Itier et al., 2003). Another PARK2 mouse model with a deletion of exon 7 also did not cause motor behavioral phenotypes or DA neurodegeneration (von Coelln et al., 2004). An exhaustive study of a Parkin null mouse lacking exon 2 revealed no substantial progressive motor phenotype, no DA neurochemical defects in the striatum, and no DA neurodegeneration (Perez and Palmiter, 2005). A conditional Cre-loxP exon 7 Parkin KO mouse was generated that showed DA neurodegeneration after lentiviral delivery of a GFP-tagged Cre recombinase was delivered stereotactically to the midbrain of adult mice (Shin et al., 2011). This mouse model showed a loss of Parkin protein and upregulation of PARIS, which the authors’ concluded was responsible for DA neuron death (Shin et al., 2011). However, it is unclear if the DA loss in this inducible model leads to locomotor deficits. It was also untested if the conditional loss of Parkin in adults causes neurodegeneration of other neuronal populations. Most Parkin knockout mouse models fail to model the pathophysiology of PD or display the selective DA neuron loss with the ubiquitous loss of Parkin seen in man.
The lack of DA neurodegeneration and phenotypes that poorly recapitulate human PD also occurred when research groups generated PINK1 KO mouse models. The deletion in exons 4-7 of PINK1 resulted in mice displaying no signs of DA neurodegeneration or altered DA metabolism (Kitada et al., 2007). A PINK1 KO mouse with exons 4-5 deleted displayed a progressive reduction of DA in the striatum, but these results were puzzling because there was no evidence of DA neuron loss (Akundi et al., 2011).
Mitochondrial impairments have been reported in the central nervous system of Parkin and PINK1 KO mice. Aged Parkin KO brains have altered expression of mitochondrial proteins and a reduction in respiratory capacity (Palacino et al., 2004; Periquet et al., 2005; Stichel et al., 2007). Primary PINK1 KO/KD fibroblasts and neurons display reduced Δψm, impairments in respiration, Ca++ overload in mitochondria, and heightened ROS production (Gandhi et al., 2009; Gautier et al., 2012; Heeman et al., 2011). ATP levels are not changed in the striatum of PINK1 KO mice (Gautier et al., 2008), whereas PINK1 KO/KD fibroblasts and neurons in culture display impaired ATP synthesis (Heeman et al., 2011). Mitochondria isolated from the striatum or whole brain of aged PINK1 KO mice display defects in complex I (Gautier et al., 2008; Morais et al., 2009), and mitochondria derived from the brains of PINK1 KO mice have reduced calcium-buffering capacity (Akundi et al., 2011). Despite these mitochondrial dysfunctions, Parkin and PINK1 KO DA neurons remain viable and functional. Even two-year-old Parkin/PINK1/DJ-1 triple KO mouse lacked DA neurodegeneration (Kitada et al., 2009). These results suggest that mice compensate for the loss of PINK1 and Parkin in DA neurons or that central nervous system neurons in aged mice do not reach a threshold of mitochondrial dysfunction necessary to cause detrimental phenotypes. The murine resistance to PINK1 KO phenotypes does not extend to rats as PINK1 KO Long Evans rats present progressive DA neurodegeneration and motor phenotypes (Dave et al., 2014). Although neuronal health appears unaffected, PINK1 KO mice develop a progressive cardiomyopathy caused by mitochondrial dysfunction and oxidative stress (Billia et al., 2011). Consistent with this cardiac defect, Parkin KO mice display a more severe phenotype than wild type mice following experimental myocardial infarction that was attributed to an accumulation of damaged mitochondria, however, they did not display the cardiac hypertrophy reported for PINK1 KO mice (Kubli et al., 2013). Cardiac ischemic preconditioning also required Parkin activity (Huang et al., 2011).
As unchallenged Parkin KO mice lack obvious phenotypes, several groups crossed mouse models of mitochondrial dysfunction with the Parkin KO mouse to assess potential roles for Parkin in vivo in mitochondrial quality control. The phenotype caused by deletion of mitochondrial transcription factor A (TFAM) in DA neurons was no worse in a Parkin KO background (Sterky et al., 2011). This lack of synthetic phenotype indicates that endogenous Parkin does not mitigate the effects on mitochondria caused by loss of TFAM. This could be due to the essential role of TFAM in mtDNA replication that leaves no viable pool of mitochondria for a quality control pathway to enrich. However, one might anticipate Parkin to be recruited to the defective mitochondria in TFAM null cells, which was not detected (Sterky et al., 2011). In addition, there are reports that the loss of Parkin did not worsen motor phenotypes or translocate respectively in two mouse models affecting mitochondrial fission and fusion - Purkinje cell-specific Drp-1 KO (Kageyama et al., 2012) and dopaminergic neuron-specific Mfn2 KO mice (Lee et al., 2012). On the other hand, knocking out choline kinase beta in the skeletal muscle of mice leads to mitochondrial dysfunction, Parkin translocation to mitochondria, and mitophagy (Mitsuhashi et al., 2011). In a model of muscle atrophy following denervation, Parkin translocation to mitochondria is more readily detected in vivo in ATG7 knockout mice than in wild type mice (Furuya et al., 2014). Parkin translocation appears to be more apparent in vivo when mitophagy is blocked downstream and thereby delaying the elimination of the mitochondrial Parkin. Thus, Parkin translocation to mitochondria can occur in vivo, at least in skeletal muscle. However, there is no experimental evidence to date in mammals that Parkin rescues mitochondrial damage in dopaminergic neurons.
Drosophila Models Manipulating Parkin or PINK1 Expression
The absence of adequate mammalian models led to the generation of other mutant organisms to elucidate the functions of these genes. The generation of Parkin and PINK1 mutant flies was the cornerstone in understanding the genetic link between these two proteins and their relationship to mitochondrial integrity.
Parkin mutant Drosophila display a severe flight muscle defect leading to locomotive behavioral problems, male sterility, and reduced lifespan (Greene et al., 2003; Pesah et al., 2004). Parkin mutant flies are more susceptible to oxidative stress and, although not all DA neuron clusters are affected, some DA neurons display abnormal shrinkage and morphology and one group reported neuron loss (Cha et al., 2005; Greene et al., 2005; Greene et al., 2003; Pesah et al., 2004; Whitworth et al., 2005). Mitochondria are abnormal and swollen in muscle cells lacking Parkin activity and this observation became important in connecting PINK1 to Parkin mechanistically (Greene et al., 2003).
PINK1 mutant or knockdown Drosophila phenocopy Parkin mutant flies and display the same flight muscle degeneration, sensitivity to environmental stressors, and reduced lifespan (Clark et al., 2006; Park et al., 2006; Yang et al., 2006). PINK1 knockdown and mutant flies also display selective DA neuron degeneration and the same abnormal mitochondrial morphology present in Parkin mutant flies (Clark et al., 2006; Park et al., 2006; Yang et al., 2006). Such strong similarities between the two models indicated that PINK1/Parkin work in the same pathway. This was corroborated when researchers showed that PINK1/Parkin double mutant flies were no worse then single mutant flies. Overexpression of Parkin rescued the PINK1 deficient phenotype, but not visa versa indicating that PINK1 acts upstream of Parkin in the same pathway (Clark et al., 2006; Park et al., 2006; Yang et al., 2006).
The swollen mitochondrial phenotype in both PINK1 and Parkin mutant flies led to the investigation of proteins that control mitochondrial morphology. Genetic loss of dynamin-related protein 1 (Drp1), a mitochondrial fission protein, caused synthetic lethality in PINK1 and Parkin mutants (Deng et al., 2008; Poole et al., 2008). The relationship with mitochondrial morphogenesis was extended to Marf (a homologue of human Mfn) and optical atrophy 1 (OPA1), two proteins required for mitochondrial fusion (Deng et al., 2008; Poole et al., 2008). The silencing of Marf or OPA1 or the overexpression of Drp1 reverted the mitochondrial morphology and biochemical deficits exhibited in PINK1 and Parkin mutant flies (Deng et al., 2008; Liu et al., 2011; Park et al., 2009; Poole et al., 2008; Yang et al., 2008). The link between mitochondrial morphology and PINK1/ Parkin is not so clear in cultured mammalian cells as mitochondria appear grossly normal in Parkin and PINK1 null cells. However, as mitophagy has been linked to mitochondrial fission in yeast and mammals, perhaps the fission activity of PINK1 and Parkin in flies reflects mitochondrial division to segregate debris for autophagic elimination (Twig and Shirihai, 2011; Youle and van der Bliek, 2012).
Ubiquitous or neuronal overexpression of Parkin boosts mitochondrial number and increases the lifespan of flies (Rana et al., 2013). PINK1 deficient flies have deficits in complex I, decreased Δψm, and reduced ATP levels (Liu et al., 2011; Morais et al., 2009). Cardiomyocyte-specific Parkin KO flies accumulate depolarized mitochondria that produce excess ROS (Bhandari et al., 2014). Increasing mitochondrial function by facilitating electron transport or overexpressing the yeast equivalent of complex I rescues the PINK1 KO phenotypes but not Parkin KO phenotypes in Drosophila (Vilain et al., 2012; Vos et al., 2012). Constitutively active Ret, the receptor for glia cell-lined derived neurotrophic factor (GDNF), also recues only PINK1 KO flies restoring the mitochondrial deficits seen in this model (Klein et al., 2014). These data suggest PINK1 has functions independent of Parkin that may stem from PINK1 phosphorylation of ubiquitin. Interestingly, an E3 ubiquitin ligase located on the outer mitochondrial membrane named MULAN can compensate for Parkin activity and appears to work in a parallel pathway to ubiquitinate mitofusin (Yun et al., 2014).
Loss-of-function mutations in Parkin and PINK1 in Drosophila lead to the accumulation of dysfunctional mitochondria in DA neurons due to defective mitophagy (Burman et al., 2012). Mass spectrometry analysis of fly heads enriched in neurons shows that the turnover rates of different subunits of mitochondrial OXPHOS complexes differ despite occurring via mitophagy (Vincow et al., 2013). This indicates that segregation of mitochondrial proteins occurs, possibly of properly folded and functional proteins versus misfolded or aggregated proteins. The molecular basis of such protein segregation mechanisms leading to mitophagy remains to be found.
PINK1 and Parkin Patients Compared to Sporadic PD Patients
Patients with PINK1 and Parkin recessive mutations have PD that is clinically indistinguishable from that of sporadic cases, although they have an earlier onset of the disease (Houlden and Singleton, 2012; Matsumine et al., 1997; Valente et al., 2001). However, it is controversial whether or not Parkin and PINK1 patients have a different neuropathological presentation of Lewy body formation as compared to sporadic cases (Houlden and Singleton, 2012; Kitada et al., 1998; Pramstaller et al., 2005; Samaranch et al., 2010). As α-synuclein mutations are linked to PD, and α-synuclein is a major component of Lewy bodies, its aggregation is a leading candidate for the etiology of PD. Are mutations in α-synuclein and PINK1/Parkin unrelated causes of PD or do they share common downstream targets? Mouse models and cell culture models have attempted to elucidate whether there is an interaction between the PINK1/Parkin pathway and mutant α-synuclein expression; however, to date it is unclear if this is the case (Choubey et al., 2011; Oliveras-Salva et al., 2014; Stichel et al., 2007; von Coelln et al., 2006). α-synuclein aggregates can localize to mitochondria and may cause mitochondrial dysfunction (Devi et al., 2008; Dryanovski et al., 2013). Macroautophagy is impaired by α-synuclein (Winslow et al., 2010) and blocking autophagy causes the accumulation of damaged mitochondria in vivo (Takamura et al., 2011). Whether mitochondrial dysfunction can lead to α-synuclein aggregation is unknown. Analyses of potential mechanistic links between these and other PD disease genes are active areas of investigation.
Concluding Remarks
Elucidating the functions of genes mutated in monogenic forms of PD has helped researchers better grasp an understanding of mitochondrial cell biology pathways and how these may go awry in sporadic cases and contribute to disease (Fig. 3). Augmenting mitophagy by activating the PINK1/Parkin pathway is an attractive target for therapeutic intervention for PD and a variety of maternally inherited mitochondrial diseases. The new understanding of mechanism and the importance of mitochondrial quality control may also extend to other common neurodegenerative diseases associated with mitochondrial dysfunction such as Alzheimer's disease and Amyotrophic lateral sclerosis.
Acknowledgements
The National Institutes of Health NINDS intramural program (RJY, AMP), and the Postdoctoral Research Associate Program (PRAT) at the National Institute of General Medical Sciences (AMP) supported this work. We thank Soojay Banerjee for assistance with artwork. We also thank Katherine Roche for thoughtful reading of the manuscript and members of the Youle lab for comments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abeliovich H, Zarei M, Rigbolt KT, Youle RJ, Dengjel J. Involvement of mitochondrial dynamics in the segregation of mitochondrial matrix proteins during stationary phase mitophagy. Nature Commun. 2013;4:2789. doi: 10.1038/ncomms3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akundi RS, Huang Z, Eason J, Pandya JD, Zhi L, Cass WA, Sullivan PG, Bueler H. Increased mitochondrial calcium sensitivity and abnormal expression of innate immunity genes precede dopaminergic defects in Pink1-deficient mice. PloS one. 2011;6:e16038. doi: 10.1371/journal.pone.0016038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- Ashrafi G, Schlehe JS, LaVoie MJ, Schwarz TL. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J Cell Biol. 2014;206:655–670. doi: 10.1083/jcb.201401070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. 2006;38:515–517. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- Bentivoglio AR, Cortelli P, Valente EM, Ialongo T, Ferraris A, Elia A, Montagna P, Albanese A. Phenotypic characterisation of autosomal recessive PARK6-linked parkinsonism in three unrelated Italian families. Mov Disord. 2001;16:999–1006. doi: 10.1002/mds.10034. [DOI] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- Bhandari P, Song M, Chen Y, Burelle Y, Dorn GW., 2nd Mitochondrial contagion induced by Parkin deficiency in Drosophila hearts and its containment by suppressing mitofusin. Circ Res. 2014;114:257–265. doi: 10.1161/CIRCRESAHA.114.302734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billia F, Hauck L, Konecny F, Rao V, Shen J, Mak TW. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. P Natl Acad Sci USA. 2011;108:9572–9577. doi: 10.1073/pnas.1106291108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingol B, Tea JS, Phu L, Reichelt M, Bakalarski CE, Song Q, Foreman O, Kirkpatrick DS, Sheng M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature. 2014;510:370–375. doi: 10.1038/nature13418. [DOI] [PubMed] [Google Scholar]
- Buhlman L, Damiano M, Bertolin G, Ferrando-Miguel R, Lombes A, Brice A, Corti O. Functional interplay between Parkin and Drp1 in mitochondrial fission and clearance. Biochim Biophys Acta. 2014;1843:2012–2026. doi: 10.1016/j.bbamcr.2014.05.012. [DOI] [PubMed] [Google Scholar]
- Burman JL, Yu S, Poole AC, Decal RB, Pallanck L. Analysis of neural subtypes reveals selective mitochondrial dysfunction in dopaminergic neurons from parkin mutants. P Natl Acad Sci USA. 2012;109:10438–10443. doi: 10.1073/pnas.1120688109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha GH, Kim S, Park J, Lee E, Kim M, Lee SB, Kim JM, Chung J, Cho KS. Parkin negatively regulates JNK pathway in the dopaminergic neurons of Drosophila. P Natl Acad Sci USA. 2005;102:10345–10350. doi: 10.1073/pnas.0500346102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S, Chan DC. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet. 2011;20:1726–1737. doi: 10.1093/hmg/ddr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaugule VK, Burchell L, Barber KR, Sidhu A, Leslie SJ, Shaw GS, Walden H. Autoregulation of Parkin activity through its ubiquitin-like domain. EMBO J. 2011;30:2853–2867. doi: 10.1038/emboj.2011.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem. 2005;280:26185–26192. doi: 10.1074/jbc.M503062200. [DOI] [PubMed] [Google Scholar]
- Chen Y, Dorn GW., 2nd PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. 2013;340:471–475. doi: 10.1126/science.1231031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choubey V, Safiulina D, Vaarmann A, Cagalinec M, Wareski P, Kuum M, Zharkovsky A, Kaasik A. Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy. J Biol Chem. 2011;286:10814–10824. doi: 10.1074/jbc.M110.132514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- Cornelissen T, Haddad D, Wauters F, Van Humbeeck C, Mandemakers W, Koentjoro B, Sue C, Gevaert K, De Strooper B, Verstreken P, Vandenberghe W. The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Hum Mol Genet. 2014;23:5227–5242. doi: 10.1093/hmg/ddu244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa AC, Loh SH, Martins LM. Drosophila Trap1 protects against mitochondrial dysfunction in a PINK1/parkin model of Parkinson's disease. Cell Death Dis. 2013;4:e467. doi: 10.1038/cddis.2012.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dave KD, De Silva S, Sheth NP, Ramboz S, Beck MJ, Quang C, Switzer RC, 3rd, Ahmad SO, Sunkin SM, Walker D, et al. Phenotypic characterization of recessive gene knockout rat models of Parkinson's disease. Neurobiol Dis. 2014;70:190–203. doi: 10.1016/j.nbd.2014.06.009. [DOI] [PubMed] [Google Scholar]
- de Rijk MC, Breteler MM, Graveland GA, Ott A, Grobbee DE, van der Meche FG, Hofman A. Prevalence of Parkinson's disease in the elderly: the Rotterdam Study. Neurology. 1995;45:2143–2146. doi: 10.1212/wnl.45.12.2143. [DOI] [PubMed] [Google Scholar]
- Deas E, Plun-Favreau H, Gandhi S, Desmond H, Kjaer S, Loh SH, Renton AE, Harvey RJ, Whitworth AJ, Martins LM, et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum Mol Genet. 2011;20:867–879. doi: 10.1093/hmg/ddq526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H, Dodson MW, Huang H, Guo M. The Parkinson's disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. P Natl Acad Sci USA. 2008;105:14503–14508. doi: 10.1073/pnas.0803998105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem. 2008;283:9089–9100. doi: 10.1074/jbc.M710012200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryanovski DI, Guzman JN, Xie Z, Galteri DJ, Volpicelli-Daley LA, Lee VM, Miller RJ, Schumacker PT, Surmeier DJ. Calcium entry and alpha-synuclein inclusions elevate dendritic mitochondrial oxidant stress in dopaminergic neurons. J Neurosci. 2013;33:10154–10164. doi: 10.1523/JNEUROSCI.5311-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durcan TM, Tang MY, Perusse JR, Dashti EA, Aguileta MA, McLelland GL, Gros P, Shaler TA, Faubert D, Coulombe B, Fon EA. USP8 regulates mitophagy by removing K6-linked ubiquitin conjugates from parkin. EMBO J. 2014 doi: 10.15252/embj.201489729. 2014/09/14, 10.15252/embj.201489729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiesel FC, Moussaud-Lamodiere EL, Ando M, Springer W. Select E2 enzymes differentially regulate parkin activation and mitophagy. J Cell Sci. 2014;127:3488–3504. doi: 10.1242/jcs.147520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogel AI, Dlouhy BJ, Wang C, Ryu SW, Neutzner A, Hasson SA, Sideris DP, Abeliovich H, Youle RJ. Role of membrane association and Atg14-dependent phosphorylation in beclin-1-mediated autophagy. Mol Cell Biol. 2013;33:3675–3688. doi: 10.1128/MCB.00079-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank M, Duvezin-Caubet S, Koob S, Occhipinti A, Jagasia R, Petcherski A, Ruonala MO, Priault M, Salin B, Reichert AS. Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim Biophys Acta. 2012;1823:2297–2310. doi: 10.1016/j.bbamcr.2012.08.007. [DOI] [PubMed] [Google Scholar]
- Furuya N, Ikeda S, Sato S, Soma S, Ezaki J, Oliva Trejo JA, Takeda-Ezaki M, Fujimura T, Arikawa-Hirasawa E, Tada N, et al. PARK2/Parkin-mediated mitochondrial clearance contributes to proteasome activation during slow-twitch muscle atrophy via NFE2L1 nuclear translocation. Autophagy. 2014;10:631–641. doi: 10.4161/auto.27785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi S, Wood-Kaczmar A, Yao Z, Plun-Favreau H, Deas E, Klupsch K, Downward J, Latchman DS, Tabrizi SJ, Wood NW, et al. PINK1-associated Parkinson's disease is caused by neuronal vulnerability to calcium-induced cell death. Mol Cell. 2009;33:627–638. doi: 10.1016/j.molcel.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier CA, Giaime E, Caballero E, Nunez L, Song Z, Chan D, Villalobos C, Shen J. Regulation of mitochondrial permeability transition pore by PINK1. Molecular neurodegeneration. 2012;7:22. doi: 10.1186/1750-1326-7-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier CA, Kitada T, Shen J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. P Natl Acad Sci USA. 2008;105:11364–11369. doi: 10.1073/pnas.0802076105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet. 2010;19:4861–4870. doi: 10.1093/hmg/ddq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010a;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- Geisler S, Holmstrom KM, Treis A, Skujat D, Weber SS, Fiesel FC, Kahle PJ, Springer W. The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy. 2010b;6:871–878. doi: 10.4161/auto.6.7.13286. [DOI] [PubMed] [Google Scholar]
- Geisler S, Vollmer S, Golombek S, Kahle PJ. UBE2N, UBE2L3 and UBE2D2/3 ubiquitin-conjugating enzymes are essential for parkin-dependent mitophagy. J Cell Sci. 2014:127. doi: 10.1242/jcs.146035. [DOI] [PubMed] [Google Scholar]
- Gilkerson RW, De Vries RL, Lebot P, Wikstrom JD, Torgyekes E, Shirihai OS, Przedborski S, Schon EA. Mitochondrial autophagy in cells with mtDNA mutations results from synergistic loss of transmembrane potential and mTORC1 inhibition. Hum Mol Genet. 2012;21:978–990. doi: 10.1093/hmg/ddr529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A, Meloni EG, Wu NP, Ackerson LC, Klapstein GJ, et al. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem. 2003;278:43628–43635. doi: 10.1074/jbc.M308947200. [DOI] [PubMed] [Google Scholar]
- Greene AW, Grenier K, Aguileta MA, Muise S, Farazifard R, Haque ME, McBride HM, Park DS, Fon EA. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012;13:378–385. doi: 10.1038/embor.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene JC, Whitworth AJ, Andrews LA, Parker TJ, Pallanck LJ. Genetic and genomic studies of Drosophila parkin mutants implicate oxidative stress and innate immune responses in pathogenesis. Hum Mol Genet. 2005;14:799–811. doi: 10.1093/hmg/ddi074. [DOI] [PubMed] [Google Scholar]
- Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. P Natl Acad Sci USA. 2003;100:4078–4083. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamalainen RH, Manninen T, Koivumaki H, Kislin M, Otonkoski T, Suomalainen A. Tissue- and cell-type-specific manifestations of heteroplasmic mtDNA 3243A>G mutation in human induced pluripotent stem cell-derived disease model. P Natl Acad Sci USA. 2013;110:E3622–3630. doi: 10.1073/pnas.1311660110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasson SA, Kane LA, Yamano K, Huang CH, Sliter DA, Buehler E, Wang C, Heman-Ackah SM, Hessa T, Guha R, et al. High-content genome-wide RNAi screens identify regulators of parkin upstream of mitophagy. Nature. 2013;504:291–295. doi: 10.1038/nature12748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori N, Kitada T, Matsumine H, Asakawa S, Yamamura Y, Yoshino H, Kobayashi T, Yokochi M, Wang M, Yoritaka A, et al. Molecular genetic analysis of a novel Parkin gene in Japanese families with autosomal recessive juvenile parkinsonism: Evidence for variable homozygous deletions in the Parkin gene in affected individuals. Ann Neurol. 1998a;44:935–941. doi: 10.1002/ana.410440612. [DOI] [PubMed] [Google Scholar]
- Hattori N, Matsumine H, Asakawa S, Kitada T, Yoshino H, Elibol B, Brookes AJ, Yamamura Y, Kobayashi T, Wang M, et al. Point mutations (Thr240Arg and GIn311Stop) in the Parkin gene (vol 249, pg 754, 1998). Biochem Biophys Res Commun. 1998b;251:666–666. doi: 10.1006/bbrc.1998.9134. [DOI] [PubMed] [Google Scholar]
- Heeman B, Van den Haute C, Aelvoet SA, Valsecchi F, Rodenburg RJ, Reumers V, Debyser Z, Callewaert G, Koopman WJ, Willems PH, Baekelandt V. Depletion of PINK1 affects mitochondrial metabolism, calcium homeostasis and energy maintenance. J Cell Sci. 2011;124:1115–1125. doi: 10.1242/jcs.078303. [DOI] [PubMed] [Google Scholar]
- Hertz NT, Berthet A, Sos ML, Thorn KS, Burlingame AL, Nakamura K, Shokat KM. A neo-substrate that amplifies catalytic activity of parkinson's-disease-related kinase PINK1. Cell. 2013;154:737–747. doi: 10.1016/j.cell.2013.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollville E, Carroll RG, Cullen SP, Martin SJ. Bcl-2 Family Proteins Participate in Mitochondrial Quality Control by Regulating Parkin/PINK1-Dependent Mitophagy. Mol Cell. 2014 doi: 10.1016/j.molcel.2014.06.001. 2014/07/08, 10.1016/j.molcel.2014.06.001. [DOI] [PubMed] [Google Scholar]
- Houlden H, Singleton AB. The genetics and neuropathology of Parkinson's disease. Acta neuropathologica. 2012;124:325–338. doi: 10.1007/s00401-012-1013-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hristova VA, Beasley SA, Rylett RJ, Shaw GS. Identification of a Novel Zn2+-binding Domain in the Autosomal Recessive Juvenile Parkinson-related E3 Ligase Parkin. J Biol Chem. 2009;284:14978–14986. doi: 10.1074/jbc.M808700200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Andres AM, Ratliff EP, Hernandez G, Lee P, Gottlieb RA. Preconditioning involves selective mitophagy mediated by Parkin and p62/SQSTM1. PloS one. 2011;6:e20975. doi: 10.1371/journal.pone.0020975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iguchi M, Kujuro Y, Okatsu K, Koyano F, Kosako H, Kimura M, Suzuki N, Uchiyama S, Tanaka K, Matsuda N. Parkin-catalyzed ubiquitin-ester transfer is triggered by PINK1-dependent phosphorylation. J Biol Chem. 2013;288:22019–22032. doi: 10.1074/jbc.M113.467530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itakura E, Kishi-Itakura C, Koyama-Honda I, Mizushima N. Structures containing Atg9A and the ULK1 complex independently target depolarized mitochondria at initial stages of Parkin-mediated mitophagy. J Cell Sci. 2012;125:1488–1499. doi: 10.1242/jcs.094110. [DOI] [PubMed] [Google Scholar]
- Itakura E, Mizushima N. p62 Targeting to the autophagosome formation site requires self-oligomerization but not LC3 binding. J Cell Biol. 2011;192:17–27. doi: 10.1083/jcb.201009067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itier JM, Ibanez P, Mena MA, Abbas N, Cohen-Salmon C, Bohme GA, Laville M, Pratt J, Corti O, Pradier L, et al. Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse. Hum Mol Genet. 2003;12:2277–2291. doi: 10.1093/hmg/ddg239. [DOI] [PubMed] [Google Scholar]
- Ivatt RM, Sanchez-Martinez A, Godena VK, Brown S, Ziviani E, Whitworth AJ. Genome-wide RNAi screen identifies the Parkinson disease GWAS risk locus SREBF1 as a regulator of mitophagy. P Natl Acad Sci USA. 2014;111:8494–8499. doi: 10.1073/pnas.1321207111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javitch JA, D'Amato RJ, Strittmatter SM, Snyder SH. Parkinsonism-inducing neurotoxin, N-methyl-4-phenyl-1,2,3,6 - tetrahydropyridine: uptake of the metabolite N-methyl-4-phenylpyridine by dopamine neurons explains selective toxicity. P Natl Acad Sci USA. 1985;82:2173–2177. doi: 10.1073/pnas.82.7.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol. 2010;191:933–942. doi: 10.1083/jcb.201008084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SM, Youle RJ. The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin-mediated mitophagy of polarized mitochondria. Autophagy. 2013;9:1750–1757. doi: 10.4161/auto.26122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BN, Berger AK, Cortese GP, Lavoie MJ. The ubiquitin E3 ligase parkin regulates the proapoptotic function of Bax. P Natl Acad Sci USA. 2012;109:6283–6288. doi: 10.1073/pnas.1113248109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kageyama Y, Zhang Z, Roda R, Fukaya M, Wakabayashi J, Wakabayashi N, Kensler TW, Reddy PH, Iijima M, Sesaki H. Mitochondrial division ensures the survival of postmitotic neurons by suppressing oxidative damage. J Cell Biol. 2012;197:535–551. doi: 10.1083/jcb.201110034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol. 2014;205:143–153. doi: 10.1083/jcb.201402104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama H, Kogure T, Mizushima N, Yoshimori T, Miyawaki A. A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem Biol. 2011;18:1042–1052. doi: 10.1016/j.chembiol.2011.05.013. [DOI] [PubMed] [Google Scholar]
- Kazlauskaite A, Kelly V, Johnson C, Baillie C, Hastie CJ, Peggie M, Macartney T, Woodroof HI, Alessi DR, Pedrioli PG, Muqit MM. Phosphorylation of Parkin at Serine65 is essential for activation: elaboration of a Miro1 substrate-based assay of Parkin E3 ligase activity. Open Biol. 2014a;4:130213. doi: 10.1098/rsob.130213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K, Alessi DR, Knebel A, Trost M, Muqit MM. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem J. 2014b;460:127–139. doi: 10.1042/BJ20140334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim NC, Tresse E, Kolaitis RM, Molliex A, Thomas RE, Alami NH, Wang B, Joshi A, Smith RB, Ritson GP, et al. VCP is essential for mitochondrial quality control by PINK1/Parkin and this function is impaired by VCP mutations. Neuron. 2013;78:65–80. doi: 10.1016/j.neuron.2013.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell. 2009;34:259–269. doi: 10.1016/j.molcel.2009.04.026. [DOI] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Kitada T, Pisani A, Porter DR, Yamaguchi H, Tscherter A, Martella G, Bonsi P, Zhang C, Pothos EN, Shen J. Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. P Natl Acad Sci USA. 2007;104:11441–11446. doi: 10.1073/pnas.0702717104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada T, Tong Y, Gautier CA, Shen J. Absence of nigral degeneration in aged parkin/DJ-1/PINK1 triple knockout mice. J Neurochem. 2009;111:696–702. doi: 10.1111/j.1471-4159.2009.06350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein P, Muller-Rischart AK, Motori E, Schonbauer C, Schnorrer F, Winklhofer KF, Klein R. Ret rescues mitochondrial morphology and muscle degeneration of Drosophila Pink1 mutants. EMBO J. 2014;33:341–355. doi: 10.1002/embj.201284290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, Burchell L, Walden H, Macartney TJ, Deak M, et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012;2:120080. doi: 10.1098/rsob.120080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014;510:162–166. doi: 10.1038/nature13392. [DOI] [PubMed] [Google Scholar]
- Kraytsberg Y, Kudryavtseva E, McKee AC, Geula C, Kowall NW, Khrapko K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet. 2006;38:518–520. doi: 10.1038/ng1778. [DOI] [PubMed] [Google Scholar]
- Krick R, Muehe Y, Prick T, Bremer S, Schlotterhose P, Eskelinen EL, Millen J, Goldfarb DS, Thumm M. Piecemeal microautophagy of the nucleus requires the core macroautophagy genes. Mol Biol Cell. 2008;19:4492–4505. doi: 10.1091/mbc.E08-04-0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubli DA, Zhang X, Lee Y, Hanna RA, Quinsay MN, Nguyen CK, Jimenez R, Petrosyan S, Murphy AN, Gustafsson AB. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem. 2013;288:915–926. doi: 10.1074/jbc.M112.411363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang AE, Lozano AM. Parkinson's disease. First of two parts. N Engl J Med. 1998a;339:1044–1053. doi: 10.1056/NEJM199810083391506. [DOI] [PubMed] [Google Scholar]
- Lang AE, Lozano AM. Parkinson's disease. Second of two parts. N Engl J Med. 1998b;339:1130–1143. doi: 10.1056/NEJM199810153391607. [DOI] [PubMed] [Google Scholar]
- Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219:979–980. doi: 10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
- Lazarou M, Jin SM, Kane LA, Youle RJ. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell. 2012;22:320–333. doi: 10.1016/j.devcel.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarou M, Narendra DP, Jin SM, Tekle E, Banerjee S, Youle RJ. PINK1 drives Parkin self-association and HECT-like E3 activity upstream of mitochondrial binding. J Cell Biol. 2013;200:163–172. doi: 10.1083/jcb.201210111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Nagano Y, Taylor JP, Lim KL, Yao TP. Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J Cell Biol. 2010;189:671–679. doi: 10.1083/jcb.201001039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Sterky FH, Mourier A, Terzioglu M, Cullheim S, Olson L, Larsson NG. Mitofusin 2 is necessary for striatal axonal projections of midbrain dopamine neurons. Hum Mol Genet. 2012;21:4827–4835. doi: 10.1093/hmg/dds352. [DOI] [PubMed] [Google Scholar]
- Lefebvre V, Du Q, Baird S, Ng AC, Nascimento M, Campanella M, McBride HM, Screaton RA. Genome-wide RNAi screen identifies ATPase inhibitory factor 1 (ATPIF1) as essential for PARK2 recruitment and mitophagy. Autophagy. 2013;9:1770–1779. doi: 10.4161/auto.25413. [DOI] [PubMed] [Google Scholar]
- Leroy E, Anastasopoulos D, Konitsiotis S, Lavedan C, Polymeropoulos MH. Deletions in the Parkin gene and genetic heterogeneity in a Greek family with early onset Parkinson's disease. Hum Genet. 1998;103:424–427. doi: 10.1007/s004390050845. [DOI] [PubMed] [Google Scholar]
- Lin W, Kang UJ. Characterization of PINK1 processing, stability, and subcellular localization. J Neurochem. 2008;106:464–474. doi: 10.1111/j.1471-4159.2008.05398.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou HH, Tsai MC, Chen CJ, Jeng JS, Chang YC, Chen SY, Chen RC. Environmental risk factors and Parkinson's disease: a case-control study in Taiwan. Neurology. 1997;48:1583–1588. doi: 10.1212/wnl.48.6.1583. [DOI] [PubMed] [Google Scholar]
- Liu S, Sawada T, Lee S, Yu W, Silverio G, Alapatt P, Millan I, Shen A, Saxton W, Kanao T, et al. Parkinson's disease-associated kinase PINK1 regulates Miro protein level and axonal transport of mitochondria. PLoS Genet. 2012;8:e1002537. doi: 10.1371/journal.pgen.1002537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Acin-Perez R, Geghman KD, Manfredi G, Lu B, Li C. Pink1 regulates the oxidative phosphorylation machinery via mitochondrial fission. P Natl Acad Sci USA. 2011;108:12920–12924. doi: 10.1073/pnas.1107332108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucking CB, Abbas N, Durr A, Bonifati V, Bonnet AM, de Broucker T, De Michele G, Wood NW, Agid Y, Brice A, et al. Homozygous deletions in parkin gene in European and North African families with autosomal recessive juvenile parkinsonism. Lancet. 1998;352:1355–1356. doi: 10.1016/s0140-6736(05)60746-5. [DOI] [PubMed] [Google Scholar]
- Luoma P, Melberg A, Rinne JO, Kaukonen JA, Nupponen NN, Chalmers RM, Oldfors A, Rautakorpi I, Peltonen L, Majamaa K, et al. Parkinsonism, premature menopause, and mitochondrial DNA polymerase gamma mutations: clinical and molecular genetic study. Lancet. 2004;364:875–882. doi: 10.1016/S0140-6736(04)16983-3. [DOI] [PubMed] [Google Scholar]
- Manzanillo PS, Ayres JS, Watson RO, Collins AC, Souza G, Rae CS, Schneider DS, Nakamura K, Shiloh MU, Cox JS. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature. 2013;501:512–516. doi: 10.1038/nature12566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao K, Wang K, Liu X, Klionsky DJ. The scaffold protein Atg11 recruits fission machinery to drive selective mitochondria degradation by autophagy. Dev Cell. 2013;26:9–18. doi: 10.1016/j.devcel.2013.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin I. RBR ubiquitin ligases: Diversification and streamlining in animal lineages. J Mol Evol. 2009;69:54–64. doi: 10.1007/s00239-009-9252-3. [DOI] [PubMed] [Google Scholar]
- Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189:211–221. doi: 10.1083/jcb.200910140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumine H, Saito M, Shimoda-Matsubayashi S, Tanaka H, Ishikawa A, Nakagawa-Hattori Y, Yokochi M, Kobayashi T, Igarashi S, Takano H, et al. Localization of a gene for an autosomal recessive form of juvenile Parkinsonism to chromosome 6q25.2-27. Am J Hum Genet. 1997;60:588–596. [PMC free article] [PubMed] [Google Scholar]
- McCoy MK, Kaganovich A, Rudenko IN, Ding J, Cookson MR. Hexokinase activity is required for recruitment of parkin to depolarized mitochondria. Hum Mol Genet. 2014;23:145–156. doi: 10.1093/hmg/ddt407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLelland GL, Soubannier V, Chen CX, McBride HM, Fon EA. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 2014;33:282–295. doi: 10.1002/embj.201385902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner C, Lorenz H, Weihofen A, Selkoe DJ, Lemberg MK. The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J Neurochem. 2011;117:856–867. doi: 10.1111/j.1471-4159.2011.07253.x. [DOI] [PubMed] [Google Scholar]
- Meng F, Yao D, Shi Y, Kabakoff J, Wu W, Reicher J, Ma Y, Moosmann B, Masliah E, Lipton SA, Gu Z. Oxidation of the cysteine-rich regions of parkin perturbs its E3 ligase activity and contributes to protein aggregation. Molecular neurodegeneration. 2011;6:34. doi: 10.1186/1750-1326-6-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michiorri S, Gelmetti V, Giarda E, Lombardi F, Romano F, Marongiu R, Nerini-Molteni S, Sale P, Vago R, Arena G, et al. The Parkinson-associated protein PINK1 interacts with Beclin1 and promotes autophagy. Cell Death Differ. 2010;17:962–974. doi: 10.1038/cdd.2009.200. [DOI] [PubMed] [Google Scholar]
- Mira MT, Alcais A, Nguyen VT, Moraes MO, Di Flumeri C, Vu HT, Mai CP, Nguyen TH, Nguyen NB, Pham XK, et al. Susceptibility to leprosy is associated with PARK2 and PACRG. Nature. 2004;427:636–640. doi: 10.1038/nature02326. [DOI] [PubMed] [Google Scholar]
- Mitsuhashi S, Hatakeyama H, Karahashi M, Koumura T, Nonaka I, Hayashi YK, Noguchi S, Sher RB, Nakagawa Y, Manfredi G, et al. Muscle choline kinase beta defect causes mitochondrial dysfunction and increased mitophagy. Hum Mol Genet. 2011;20:3841–3851. doi: 10.1093/hmg/ddr305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morais VA, Verstreken P, Roethig A, Smet J, Snellinx A, Vanbrabant M, Haddad D, Frezza C, Mandemakers W, Vogt-Weisenhorn D, et al. Parkinson's disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO molecular medicine. 2009;1:99–111. doi: 10.1002/emmm.200900006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller-Rischart AK, Pilsl A, Beaudette P, Patra M, Hadian K, Funke M, Peis R, Deinlein A, Schweimer C, Kuhn PH, et al. The E3 ligase parkin maintains mitochondrial integrity by increasing linear ubiquitination of NEMO. Mol Cell. 2013;49:908–921. doi: 10.1016/j.molcel.2013.01.036. [DOI] [PubMed] [Google Scholar]
- Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy. 2010a;6:1090–1106. doi: 10.4161/auto.6.8.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Walker JE, Youle R. Mitochondrial quality control mediated by PINK1 and Parkin: links to parkinsonism. Cold Spring Harb Perspect Biol. 2012:4. doi: 10.1101/cshperspect.a011338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010b;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neutzner A, Benard G, Youle RJ, Karbowski M. Role of the ubiquitin conjugation system in the maintenance of mitochondrial homeostasis. Ann N Y Acad Sci. 2008;1147:242–253. doi: 10.1196/annals.1427.012. [DOI] [PubMed] [Google Scholar]
- Nicklas WJ, Vyas I, Heikkila RE. Inhibition of NADH-linked oxidation in brain mitochondria by 1-methyl-4-phenyl-pyridine, a metabolite of the neurotoxin, 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. Life Sci. 1985;36:2503–2508. doi: 10.1016/0024-3205(85)90146-8. [DOI] [PubMed] [Google Scholar]
- Okatsu K, Saisho K, Shimanuki M, Nakada K, Shitara H, Sou YS, Kimura M, Sato S, Hattori N, Komatsu M, et al. p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells. 2010;15:887–900. doi: 10.1111/j.1365-2443.2010.01426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okatsu K, Uno M, Koyano F, Go E, Kimura M, Oka T, Tanaka K, Matsuda N. A dimeric PINK1-containing complex on depolarized mitochondria stimulates Parkin recruitment. J Biol Chem. 2013;288:36372–36384. doi: 10.1074/jbc.M113.509653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveras-Salva M, Macchi F, Coessens V, Deleersnijder A, Gerard M, Van der Perren A, Van den Haute C, Baekelandt V. Alpha-synuclein-induced neurodegeneration is exacerbated in PINK1 knockout mice. Neurobiol Aging. 2014;35:2625–2636. doi: 10.1016/j.neurobiolaging.2014.04.032. [DOI] [PubMed] [Google Scholar]
- Ordureau A, Sarraf SA, Duda DM, Heo JM, Jedrychowski MP, Sviderskiy VO, Olszewski JL, Koerber JT, Xie T, Beausoleil SA, et al. Quantitative Proteomics Reveal a Feedforward Mechanism for Mitochondrial PARKIN Translocation and Ubiquitin Chain Synthesis. Mol Cell. 2014 doi: 10.1016/j.molcel.2014.09.007. 2014/10/07, 10.1016/j.molcel.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orvedahl A, Sumpter R, Jr., Xiao G, Ng A, Zou Z, Tang Y, Narimatsu M, Gilpin C, Sun Q, Roth M, et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature. 2011;480:113–117. doi: 10.1038/nature10546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, Klose J, Shen J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem. 2004;279:18614–18622. doi: 10.1074/jbc.M401135200. [DOI] [PubMed] [Google Scholar]
- Park J, Lee G, Chung J. The PINK1-Parkin pathway is involved in the regulation of mitochondrial remodeling process. Biochem Biophys Res Commun. 2009;378:518–523. doi: 10.1016/j.bbrc.2008.11.086. [DOI] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, Chung J. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- Pawlyk AC, Giasson BI, Sampathu DM, Perez FA, Lim KL, Dawson VL, Dawson TM, Palmiter RD, Trojanowski JQ, Lee VM. Novel monoclonal antibodies demonstrate biochemical variation of brain parkin with age. J Biol Chem. 2003;278:48120–48128. doi: 10.1074/jbc.M306889200. [DOI] [PubMed] [Google Scholar]
- Perez FA, Palmiter RD. Parkin-deficient mice are not a robust model of parkinsonism. P Natl Acad Sci USA. 2005;102:2174–2179. doi: 10.1073/pnas.0409598102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Periquet M, Corti O, Jacquier S, Brice A. Proteomic analysis of parkin knockout mice: alterations in energy metabolism, protein handling and synaptic function. J Neurochem. 2005;95:1259–1276. doi: 10.1111/j.1471-4159.2005.03442.x. [DOI] [PubMed] [Google Scholar]
- Pesah Y, Pham T, Burgess H, Middlebrooks B, Verstreken P, Zhou Y, Harding M, Bellen H, Mardon G. Drosophila parkin mutants have decreased mass and cell size and increased sensitivity to oxygen radical stress. Development. 2004;131:2183–2194. doi: 10.1242/dev.01095. [DOI] [PubMed] [Google Scholar]
- Pimenta de Castro I, Costa AC, Lam D, Tufi R, Fedele V, Moisoi N, Dinsdale D, Deas E, Loh SH, Martins LM. Genetic analysis of mitochondrial protein misfolding in Drosophila melanogaster. Cell Death Differ. 2012;19:1308–1316. doi: 10.1038/cdd.2012.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plun-Favreau H, Klupsch K, Moisoi N, Gandhi S, Kjaer S, Frith D, Harvey K, Deas E, Harvey RJ, McDonald N, et al. The mitochondrial protease HtrA2 is regulated by Parkinson's disease-associated kinase PINK1. Nat Cell Biol. 2007;9:1243–1252. doi: 10.1038/ncb1644. [DOI] [PubMed] [Google Scholar]
- Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway regulates mitochondrial morphology. P Natl Acad Sci USA. 2008;105:1638–1643. doi: 10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole AC, Thomas RE, Yu S, Vincow ES, Pallanck L. The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PloS one. 2010;5:e10054. doi: 10.1371/journal.pone.0010054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pramstaller PP, Schlossmacher MG, Jacques TS, Scaravilli F, Eskelson C, Pepivani I, Hedrich K, Adel S, Gonzales-McNeal M, Hilker R, et al. Lewy body Parkinson's disease in a large pedigree with 77 Parkin mutation carriers. Ann Neurol. 2005;58:411–422. doi: 10.1002/ana.20587. [DOI] [PubMed] [Google Scholar]
- Pridgeon JW, Olzmann JA, Chin LS, Li L. PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol. 2007;5:e172. doi: 10.1371/journal.pbio.0050172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakovic A, Grunewald A, Kottwitz J, Bruggemann N, Pramstaller PP, Lohmann K, Klein C. Mutations in PINK1 and Parkin impair ubiquitination of Mitofusins in human fibroblasts. PloS one. 2011;6:e16746. doi: 10.1371/journal.pone.0016746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakovic A, Shurkewitsch K, Seibler P, Grunewald A, Zanon A, Hagenah J, Krainc D, Klein C. Phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1)-dependent ubiquitination of endogenous Parkin attenuates mitophagy: study in human primary fibroblasts and induced pluripotent stem cell-derived neurons. J Biol Chem. 2013;288:2223–2237. doi: 10.1074/jbc.M112.391680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsay RR, Singer TP. Energy-dependent uptake of N-methyl-4-phenylpyridinium, the neurotoxic metabolite of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, by mitochondria. J Biol Chem. 1986;261:7585–7587. [PubMed] [Google Scholar]
- Rana A, Rera M, Walker DW. Parkin overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics, and extends lifespan. P Natl Acad Sci USA. 2013;110:8638–8643. doi: 10.1073/pnas.1216197110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve A, Meagher M, Lax N, Simcox E, Hepplewhite P, Jaros E, Turnbull D. The impact of pathogenic mitochondrial DNA mutations on substantia nigra neurons. J Neurosci. 2013;33:10790–10801. doi: 10.1523/JNEUROSCI.3525-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley BE, Lougheed JC, Callaway K, Velasquez M, Brecht E, Nguyen L, Shaler T, Walker D, Yang Y, Regnstrom K, et al. Structure and function of Parkin E3 ubiquitin ligase reveals aspects of RING and HECT ligases. Nature Commun. 2013;4:1982. doi: 10.1038/ncomms2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaranch L, Lorenzo-Betancor O, Arbelo JM, Ferrer I, Lorenzo E, Irigoyen J, Pastor MA, Marrero C, Isla C, Herrera-Henriquez J, Pastor P. PINK1-linked parkinsonism is associated with Lewy body pathology. Brain. 2010;133:1128–1142. doi: 10.1093/brain/awq051. [DOI] [PubMed] [Google Scholar]
- Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, Harper JW. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature. 2013;496:372–376. doi: 10.1038/nature12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarffe LA, Stevens DA, Dawson VL, Dawson TM. Parkin and PINK1: much more than mitophagy. Trends Neurosci. 2014;37:315–324. doi: 10.1016/j.tins.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD. Mitochondrial complex I deficiency in Parkinson's disease. J Neurochem. 1990;54:823–827. doi: 10.1111/j.1471-4159.1990.tb02325.x. [DOI] [PubMed] [Google Scholar]
- Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD. Mitochondrial complex I deficiency in Parkinson's disease. Lancet. 1989;1:1269. doi: 10.1016/s0140-6736(89)92366-0. [DOI] [PubMed] [Google Scholar]
- Seibler P, Graziotto J, Jeong H, Simunovic F, Klein C, Krainc D. Mitochondrial Parkin recruitment is impaired in neurons derived from mutant PINK1 induced pluripotent stem cells. J Neurosci. 2011;31:5970–5976. doi: 10.1523/JNEUROSCI.4441-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Q, Yamano K, Head BP, Kawajiri S, Cheung JT, Wang C, Cho JH, Hattori N, Youle RJ, van der Bliek AM. Mutations in Fis1 disrupt orderly disposal of defective mitochondria. Mol Biol Cell. 2014;25:145–159. doi: 10.1091/mbc.E13-09-0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba-Fukushima K, Imai Y, Yoshida S, Ishihama Y, Kanao T, Sato S, Hattori N. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci Rep. 2012;2:1002. doi: 10.1038/srep01002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba-Fukushima K, Inoshita T, Hattori N, Imai Y. PINK1-mediated phosphorylation of Parkin boosts Parkin activity in Drosophila. PLoS Genet. 2014;10:e1004391. doi: 10.1371/journal.pgen.1004391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25:302–305. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O, Troconso JC, Dawson VL, Dawson TM. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson's disease. Cell. 2011;144:689–702. doi: 10.1016/j.cell.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims JJ, Scavone F, Cooper EM, Kane LA, Youle RJ, Boeke JD, Cohen RE. Polyubiquitin-sensor proteins reveal localization and linkage-type dependence of cellular ubiquitin signaling. Nat Methods. 2012;9:303–309. doi: 10.1038/nmeth.1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit JJ, Monteferrario D, Noordermeer SM, van Dijk WJ, van der Reijden BA, Sixma TK. The E3 ligase HOIP specifies linear ubiquitin chain assembly through its RING-IBR-RING domain and the unique LDD extension. EMBO J. 2012;31:3833–3844. doi: 10.1038/emboj.2012.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with lewy bodies. P Natl Acad Sci USA. 1998;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spratt DE, Martinez-Torres RJ, Noh YJ, Mercier P, Manczyk N, Barber KR, Aguirre JD, Burchell L, Purkiss A, Walden H, Shaw GS. A molecular explanation for the recessive nature of parkin-linked Parkinson's disease. Nature Commun. 2013;4:1983. doi: 10.1038/ncomms2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterky FH, Lee S, Wibom R, Olson L, Larsson NG. Impaired mitochondrial transport and Parkin-independent degeneration of respiratory chain-deficient dopamine neurons in vivo. P Natl Acad Sci USA. 2011;108:12937–12942. doi: 10.1073/pnas.1103295108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stichel CC, Zhu XR, Bader V, Linnartz B, Schmidt S, Lubbert H. Mono- and double-mutant mouse models of Parkinson's disease display severe mitochondrial damage. Hum Mol Genet. 2007;16:2377–2393. doi: 10.1093/hmg/ddm083. [DOI] [PubMed] [Google Scholar]
- Strappazzon F, Nazio F, Corrado M, Cianfanelli V, Romagnoli A, Fimia GM, Campello S, Nardacci R, Piacentini M, Campanella M, Cecconi F. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 2014 doi: 10.1038/cdd.2014.139. 2014/09/13, 10.1038/cdd.2014.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suen DF, Narendra DP, Tanaka A, Manfredi G, Youle RJ. Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. P Natl Acad Sci USA. 2010;107:11835–11840. doi: 10.1073/pnas.0914569107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25:795–800. doi: 10.1101/gad.2016211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010;191:1367–1380. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner CM, Kamel F, Ross GW, Hoppin JA, Goldman SM, Korell M, Marras C, Bhudhikanok GS, Kasten M, Chade AR, et al. Rotenone, paraquat, and Parkinson's disease. Environ Health Perspect. 2011;119:866–872. doi: 10.1289/ehp.1002839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas RE, Andrews LA, Burman JL, Lin WY, Pallanck LJ. PINK1-Parkin pathway activity is regulated by degradation of PINK1 in the mitochondrial matrix. PLoS Genet. 2014;10:e1004279. doi: 10.1371/journal.pgen.1004279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trempe JF, Sauve V, Grenier K, Seirafi M, Tang MY, Menade M, Al-Abdul-Wahid S, Krett J, Wong K, Kozlov G, et al. Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science. 2013;340:1451–1455. doi: 10.1126/science.1237908. [DOI] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Shirihai OS. The interplay between mitochondrial dynamics and mitophagy. Antioxid Redox Signal. 2011;14:1939–1951. doi: 10.1089/ars.2010.3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unoki M, Nakamura Y. Growth-suppressive effects of BPOZ and EGR2, two genes involved in the PTEN signaling pathway. Oncogene. 2001;20:4457–4465. doi: 10.1038/sj.onc.1204608. [DOI] [PubMed] [Google Scholar]
- Urban S, Freeman M. Substrate specificity of rhomboid intramembrane proteases is governed by helix-breaking residues in the substrate transmembrane domain. Mol Cell. 2003;11:1425–1434. doi: 10.1016/s1097-2765(03)00181-3. [DOI] [PubMed] [Google Scholar]
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, et al. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- Valente EM, Bentivoglio AR, Dixon PH, Ferraris A, Ialongo T, Frontali M, Albanese A, Wood NW. Localization of a novel locus for autosomal recessive early-onset parkinsonism, PARK6, on human chromosome 1p35-p36. American journal of human genetics. 2001;68:895–900. doi: 10.1086/319522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente EM, Brancati F, Caputo V, Graham EA, Davis MB, Ferraris A, Breteler MMB, Gasser T, Bonifati V, Bentivoglio AR, et al. PARK6 is a common cause of familial parkinsonism. Neurol Sci. 2002a;23:S117–S118. doi: 10.1007/s100720200097. [DOI] [PubMed] [Google Scholar]
- Valente EM, Brancati F, Ferraris A, Graham EA, Davis MB, Breteler MMB, Gasser T, Bonifati V, Bentivoglio AR, De Michele G, et al. PARK6-linked parkinsonism occurs in several European families. Ann Neurol. 2002b;51:14–18. [PubMed] [Google Scholar]
- Van Humbeeck C, Cornelissen T, Hofkens H, Mandemakers W, Gevaert K, De Strooper B, Vandenberghe W. Parkin interacts with Ambra1 to induce mitophagy. J Neurosci. 2011;31:10249–10261. doi: 10.1523/JNEUROSCI.1917-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wijk SJ, Fiskin E, Putyrski M, Pampaloni F, Hou J, Wild P, Kensche T, Grecco HE, Bastiaens P, Dikic I. Fluorescence-based sensors to monitor localization and functions of linear and K63-linked ubiquitin chains in cells. Mol Cell. 2012;47:797–809. doi: 10.1016/j.molcel.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandiver MS, Paul BD, Xu R, Karuppagounder S, Rao F, Snowman AM, Ko HS, Lee YI, Dawson VL, Dawson TM, et al. Sulfhydration mediates neuroprotective actions of parkin. Nature Commun. 2013;4:1626. doi: 10.1038/ncomms2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilain S, Esposito G, Haddad D, Schaap O, Dobreva MP, Vos M, Van Meensel S, Morais VA, De Strooper B, Verstreken P. The yeast complex I equivalent NADH dehydrogenase rescues pink1 mutants. PLoS Genet. 2012;8:e1002456. doi: 10.1371/journal.pgen.1002456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincow ES, Merrihew G, Thomas RE, Shulman NJ, Beyer RP, MacCoss MJ, Pallanck LJ. The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. P Natl Acad Sci USA. 2013;110:6400–6405. doi: 10.1073/pnas.1221132110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. P Natl Acad Sci USA. 2010;107:378–383. doi: 10.1073/pnas.0911187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Coelln R, Thomas B, Andrabi SA, Lim KL, Savitt JM, Saffary R, Stirling W, Bruno K, Hess EJ, Lee MK, et al. Inclusion body formation and neurodegeneration are parkin independent in a mouse model of alpha-synucleinopathy. J Neurosci. 2006;26:3685–3696. doi: 10.1523/JNEUROSCI.0414-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Coelln R, Thomas B, Savitt JM, Lim KL, Sasaki M, Hess EJ, Dawson VL, Dawson TM. Loss of locus coeruleus neurons and reduced startle in parkin null mice. P Natl Acad Sci USA. 2004;101:10744–10749. doi: 10.1073/pnas.0401297101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos M, Esposito G, Edirisinghe JN, Vilain S, Haddad DM, Slabbaert JR, Van Meensel S, Schaap O, De Strooper B, Meganathan R, et al. Vitamin K2 is a mitochondrial electron carrier that rescues pink1 deficiency. Science. 2012;336:1306–1310. doi: 10.1126/science.1218632. [DOI] [PubMed] [Google Scholar]
- Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. 2011;147:893–906. doi: 10.1016/j.cell.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Nartiss Y, Steipe B, McQuibban GA, Kim PK. ROS-induced mitochondrial depolarization initiates PARK2/PARKIN-dependent mitochondrial degradation by autophagy. Autophagy. 2012;8:1462–1476. doi: 10.4161/auto.21211. [DOI] [PubMed] [Google Scholar]
- Wauer T, Komander D. Structure of the human Parkin ligase domain in an autoinhibited state. EMBO J. 2013;32:2099–2112. doi: 10.1038/emboj.2013.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel DM, Lissounov A, Brzovic PS, Klevit RE. UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature. 2011;474:105–108. doi: 10.1038/nature09966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitworth AJ, Theodore DA, Greene JC, Benes H, Wes PD, Pallanck LJ. Increased glutathione S-transferase activity rescues dopaminergic neuron loss in a Drosophila model of Parkinson's disease. P Natl Acad Sci USA. 2005;102:8024–8029. doi: 10.1073/pnas.0501078102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winklhofer KF. Parkin and mitochondrial quality control: toward assembling the puzzle. Trends Cell Biol. 2014;24:332–341. doi: 10.1016/j.tcb.2014.01.001. [DOI] [PubMed] [Google Scholar]
- Winslow AR, Chen CW, Corrochano S, Acevedo-Arozena A, Gordon DE, Peden AA, Lichtenberg M, Menzies FM, Ravikumar B, Imarisio S, et al. alpha-Synuclein impairs macroautophagy: implications for Parkinson's disease. J Cell Biol. 2010;190:1023–1037. doi: 10.1083/jcb.201003122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolter KG, Hsu YT, Smith CL, Nechushtan A, Xi XG, Youle RJ. Movement of Bax from the cytosol to mitochondria during apoptosis. J Cell Biol. 1997;139:1281–1292. doi: 10.1083/jcb.139.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamano K, Fogel AI, Wang C, van der Bliek AM, Youle RJ. Mitochondrial Rab GAPs govern autophagosome biogenesis during mitophagy. eLife. 2014;3:e01612. doi: 10.7554/eLife.01612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamano K, Youle RJ. PINK1 is degraded through the N-end rule pathway. Autophagy. 2013;9:1758–1769. doi: 10.4161/auto.24633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JY, Yang WY. Spatiotemporally controlled initiation of Parkin-mediated mitophagy within single cells. Autophagy. 2011;7:1230–1238. doi: 10.4161/auto.7.10.16626. [DOI] [PubMed] [Google Scholar]
- Yang JY, Yang WY. Bit-by-bit autophagic removal of parkin-labelled mitochondria. Nature Commun. 2013;4:2428. doi: 10.1038/ncomms3428. [DOI] [PubMed] [Google Scholar]
- Yang Y, Gehrke S, Imai Y, Huang Z, Ouyang Y, Wang JW, Yang L, Beal MF, Vogel H, Lu B. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. P Natl Acad Sci USA. 2006;103:10793–10798. doi: 10.1073/pnas.0602493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Ouyang Y, Yang L, Beal MF, McQuibban A, Vogel H, Lu B. Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. P Natl Acad Sci USA. 2008;105:7070–7075. doi: 10.1073/pnas.0711845105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshii SR, Kishi C, Ishihara N, Mizushima N. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J Biol Chem. 2011;286:19630–19640. doi: 10.1074/jbc.M110.209338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun J, Cao JH, Dodson MW, Clark IE, Kapahi P, Chowdhury RB, Guo M. Loss-of-function analysis suggests that Omi/HtrA2 is not an essential component of the PINK1/PARKIN pathway in vivo. J Neurosci. 2008;28:14500–14510. doi: 10.1523/JNEUROSCI.5141-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun J, Puri R, Yang H, Lizzio MA, Wu C, Sheng ZH, Guo M. MUL1 acts in parallel to the PINK1/parkin pathway in regulating mitofusin and compensates for loss of PINK1/parkin. eLife. 2014;3:e01958. doi: 10.7554/eLife.01958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Lee S, Peng Y, Bunker E, Giaime E, Shen J, Zhou Z, Liu X. PINK1 Triggers Autocatalytic Activation of Parkin to Specify Cell Fate Decisions. Curr Biol. 2014;24:854–865. doi: 10.1016/j.cub.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Karsten P, Hamm S, Pogson JH, Muller-Rischart AK, Exner N, Haass C, Whitworth AJ, Winklhofer KF, Schulz JB, Voigt A. TRAP1 rescues PINK1 loss-of-function phenotypes. Hum Mol Genet. 2013;22:2829–2841. doi: 10.1093/hmg/ddt132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002;21:4411–4419. doi: 10.1093/emboj/cdf445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Hunter T. Parkin mitochondrial translocation is achieved through a novel catalytic activity coupled mechanism. Cell Res. 2013;23:886–897. doi: 10.1038/cr.2013.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziviani E, Tao RN, Whitworth AJ. Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. P Natl Acad Sci USA. 2010;107:5018–5023. doi: 10.1073/pnas.0913485107. [DOI] [PMC free article] [PubMed] [Google Scholar]