

Abstract
Background
The objective of this study was to increase understanding about genetic mechanisms affecting calyx persistence in Korla fragrant pear (Pyrus brestschneideri Rehd). Flowers were collected at early bloom, full bloom, and late bloom. The RNA was extracted from the flowers and then combined according to calyx type. Transcriptome and digital gene expression (DGE) profiles of flowers, ovaries, and sepals with persistent calyx (SC_hua, SC_ep, and SC_zf, respectively) were compared with those of flowers, ovaries, and sepals with deciduous calyx (TL_hua, TL_ep, and TL_zf, respectively). Temporal changes in the expression of selected genes in floral organs with either persistent or deciduous calyx were compared using real-time quantitative PCR (qRT-PCR).
Results
Comparison of the transcriptome sequences for SC_hua and TL_hua indicated 26 differentially expressed genes (DEGs) with known relationship to abscission and 10 DEGs with unknown function. We identified 98 MYB and 21 SPL genes from the assembled unigenes. From SC_zf vs TL_zf, we identified 21 DEGs with known relationship to abscission and 18 DEGs with unknown function. From SC_ep vs TL_ep, 12 DEGs with known relationship to abscission were identified along with 11 DEGs with unknown function. Ten DEGs were identified by both transcriptome sequencing and DGE sequencing.
Conclusions
More than 50 DEGs were observed that were related to calyx persistence in Korla fragrant pear. Some of the genes were related to cell wall degradation, plant hormone signal transduction, and stress response. Other DEGs were identified as zinc finger protein genes and lipid transfer protein genes. Further analysis showed that calyx persistence in Korla fragment pear was a metabolic process regulated by many genes related to cell wall degradation and plant hormones.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-2470-3) contains supplementary material, which is available to authorized users.
Keywords: Persistent calyx, Deciduous calyx, Transcriptome sequencing, DGE sequencing
Background
Korla fragrant pear is one of the most valuable fruits in China’s Xinjiang Province [1]. The calyx of Korla fragrant pear is sometimes persistent. This can negatively affect pear shape and quality. Previous studies about Korla fragrant pear have examined the relationship between calyx persistence and cultivation practice [2], tree vigor [2], pollen source [3–5], growth regulators [6–8], and plant nutrition [9]. Some studies have investigated the molecular mechanisms for calyx persistence in Korla fragrant pear. For example, Dong et al. and Wang et al. cloned a kfpMYB gene related to calyx persistence using differential display RT-PCR [10, 11]. Qi et al. used digital transcript abundance measurements to identify genes correlated with calyx abscission [12].
High-throughput sequencing has contributed greatly to the study of gene function in non-model plants. High-throughput sequencing makes it possible to understand the genome and the transcriptome of a species more comprehensively [13–15]. High-throughput sequencing of RNA (RNA-Seq) has been successfully applied in Malus domestica [16, 17], Myrica rubra [18, 19], Vaccinium section Cyanococcus [20], Litchi chinensis Sonn [21], Pyrus bretschneideri Rehd [22], Vitis vinifera cv. Shiraz [23], Musa acuminate [24, 25], Citrus sinensis [26, 27], Prunus persica [28], and Diospyros kaki [29]. The objective of this experiment was to identify candidate genes related to calyx persistence in Korla fragrant pear using both transcriptome and digital gene expression (DGE) sequencing.
Results and discussion
Transcriptome sequencing and assembly
In total, 107202492 raw reads were generated by Illumina sequencing of SC_hua vs TL_hua (Table 1). There were 103466288 clean reads after removing low-quality sequences. Assembly of the clean reads resulted in 39891341 unigenes ranging in size between 201 and 16666 bp (Fig. 1). The N50 length of the unigenes was 1579 bp and the N90 length was 289 bp.
Table 1.
Summary of the sequence analyses
| Sample | Raw Reads | Clean Reads | Clean Bases | Error (%) | Q20 (%) | Q30 (%) | GC Content (%) |
|---|---|---|---|---|---|---|---|
| SC_hua_1 | 27216916 | 26238309 | 2.62G | 0.03 | 98.51 | 94.71 | 47.18 |
| SC_hua_2 | 27216916 | 26238309 | 2.62G | 0.04 | 96.71 | 91.53 | 47.24 |
| TL_hua_1 | 26384330 | 25494835 | 2.55G | 0.03 | 98.55 | 94.82 | 46.87 |
| TL_hua_2 | 26384330 | 25494835 | 2.55G | 0.04 | 96.79 | 91.67 | 46.93 |
| Summary | 107202492 | 103466288 | 10.34G |
Sample: Sample name_1, left reads; Sample name_2, right reads. The total number of clean reads is left + right. Clean reads: The number of reads after removing low-quality sequences. The subsequent analysis is based on clean reads. Error rate: Base error rate.Q20 and Q30, the percentage of bases with Phred values >20 and >30, respectively. GC content: the GC ratio of the total base number
Fig. 1.

Length distribution of the assembled unigenes
Sequence annotation
The unigenes were aligned with seven public databases [i.e., NR (NCBI non-redundant protein sequences), NT (NCBI nucleotide sequences), KEGG (Kyoto Encyclopedia of Genes and Genomes), SwissProt (A manually annotated and reviewed protein sequence database), PFAM (Protein family), GO (Gene Ontology) and KOG/COG (Clusters of Orthologous Groups of proteins)] (Table 2). The results showed that 18605 unigenes (38.05 %) had significant matches in the NR database, 16700 unigenes (34.15 %) had significant matches in the NT database, and 17326 unigenes (35.43 %) had significant matches in the SwissProt database. In total, 26088 unigenes (53.35 %) were annotated in at least one database, with 3037 unigenes (6.21 %) being annotated in all seven databases.
Table 2.
The success rate of gene annotation
| Number of Unigenes | Percentage (%) | |
|---|---|---|
| Annotated in NR | 18605 | 38.05 |
| Annotated in NT | 16700 | 34.15 |
| Annotated in KO | 6925 | 14.16 |
| Annotated in SwissProt | 17326 | 35.43 |
| Annotated in PFAM | 16935 | 34.63 |
| Annotated in GO | 17749 | 36.3 |
| Annotated in KOG | 8891 | 18.18 |
| Annotated in all Databases | 3037 | 6.21 |
| Annotated in at least one Database | 26088 | 53.35 |
| Total Unigenes | 48894 | 100 |
Annotated in NR: The unigene number and annotation rate in the NR database. Annotated in NT: The unigene number and annotation rate in the NT database. Annotated in KO: The unigene number and annotation rate in the KO database. Annotated in SwissProt: The unigene number and annotation rate in the SwissPort database. Annotated in PFAM: The unigene number and annotation rate in the PFAM database. Annotated in GO: The unigene number and annotation rate in the GO database. Annotated in KOG: The unigene number and annotation rate in the KOG database. Annotated in all Databases: The unigene number and annotation rate in all seven databases. Annotated in at least one database: The unigene number and annotation rate in at least one database
A total of 17749 unigenes were subjected to GO analysis (Fig. 2). In the cellular component (CC) category, genes involved in ‘cell’ (6093), ‘cell part’ (6087), and ‘organelle’ (4357), were highly represented. The molecular function category (MF) mainly included genes involved in ‘binding’ (10493), ‘catalytic activity’ (8571) and ‘transporter activity’ (1176). In the biological process (BP) category, ‘cellular process’ (10437), ‘metabolic process’ (9848) and ‘single-organism process’ (5155) were highly represented.
Fig. 2.
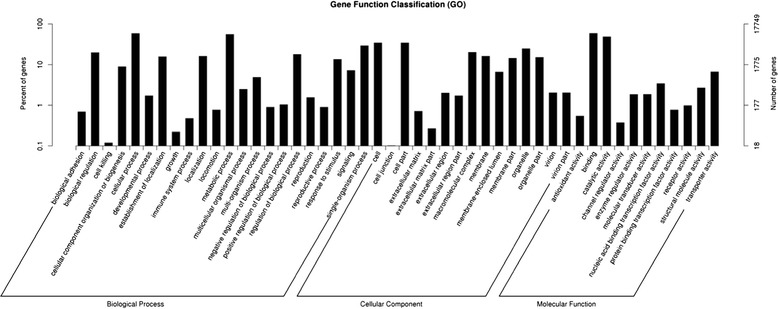
GO categorization of unigenes
The unigenes were all subjected to a search against the COG database for functional prediction and classification. In total, 8891 unigenes were assigned to COG classification and divided into 26 specific categories (Fig. 3). The largest group was ‘general function prediction only’ (1626), followed by ‘post-translational modification, protein turnover, chaperones’ (1152), ‘signal transduction mechanisms’ (800), ‘intracellular trafficking, secretion, and vesicular transport’ (535), and ‘carbohydrate transport and metabolism’ (485). Only a few unigenes were assigned to ‘extracellular structures’ (28) and ‘cell motility’ (3).
Fig. 3.

KOG annotation of putative proteins
Unigene metabolic pathway analysis was also conducted using KEGG. This process predicted a total of 258 pathways, representing 6925 unigenes (Fig. 4). The pathways involving the highest number of unique transcripts were ‘carbohydrate metabolism’ (662), followed by ‘translation’ (639) and ‘signal transduction’ (542). The above data is a very valuable genetic resource for studying calyx persistence in Korla Fragrant Pear.
Fig. 4.

KEGG annotation of putative proteins
Differential expression analysis in SC_hua vs TL_hua
Differentially expressed genes (DEGs) are defined as genes that are significantly enriched or depleted in one sample relative to another (q value < 0.005 and |log2 (foldchange)| >1). In the rest of this paper, up-regulated means that the gene expression level was greater in samples with persistent calyx than in samples with deciduous calyx. Down-regulated means that the gene expression level was less in samples with persistent calyx than in samples with deciduous calyx. There were 103 DEGs among 48894 unigenes in SC_hua vs TL_hua. Among these, 47 DEGs were up-regulated and 56 DEGs were down-regulated (Fig. 5).
Fig. 5.
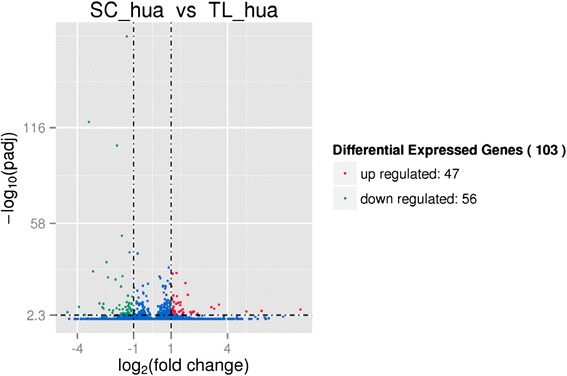
Up-regulated and down-regulated differentially expressed genes in SC_hua vs TL_hua
To further characterize the function of the DEGs, GO enrichment analysis was conducted for all of the DEGs in SC_hua vs TL_hua with the whole transcriptome as the background (Additional file 1). In the BP category, the top three enriched terms were ‘heterocycle biosynthetic process’, ‘organic cyclic compound biosynthetic process’ and ‘cellular nitrogen compound biosynthetic process’. In the CC category, ‘nuclear part’, ‘membrane-enclosed lumen’, ‘intracellular organelle lumen’, ‘organelle lumen’ and ‘nuclear lumen’ were the dominant enriched terms. In the MF category, ‘nucleic acid binding transcription factor activity’ and ‘sequence-specific DNA binding transcription factor activity’ were most highly enriched. A GO enrichment analysis was also conducted for the up-regulated DEGs (Additional file 2). In the BP category, ‘biological regulation’, ‘regulation of biological process’, and ‘regulation of cellular process’ were most highly enriched. In the CC category, ‘membrane-enclosed lumen’, ‘intracellular organelle lumen’, ‘organelle lumen’ and ‘nuclear lumen’ were the main enriched terms. In MF, the top two enriched terms were ‘nucleic acid binding transcription factor activity’ and ‘sequence-specific DNA binding transcription factor activity’.
The KEGG pathway enrichment analysis for DEGs also revealed both common and tissue specific patterns of over-representation (Additional file 3). The top-four enriched pathways for DEGs in SC_hua vs TL_hua were ‘cysteine and methionine metabolism’, ‘porphyrin and chlorophyll metabolism’, ‘phenylalanine metabolism’ and ‘isoquinoline alkaloid biosynthesis’. For up-regulated DEGs (Additional file 4), ‘calcium signaling pathway’, ‘porphyrin and chlorophyll metabolism’, ‘phosphatidylinositol signaling system’ and ‘glycerolipid metabolism’ were most highly enriched. For down-regulated DEGs (Additional file 5), ‘cysteine and methionine metabolism’, ‘isoquinoline alkaloid biosynthesis’ and ‘biosynthesis of amino acids’ were the three main enriched pathways.
DGE sequencing
A DGE analysis was performed to compare gene expression in SC_ep, SC_zf, TL_ep, and TL_zf. After removing low-quality sequences, we obtained 12283115, 10084701, 9449491 and 9999449 clean reads in SC_ep, SC_zf, TL_ep, and TL_zf, respectively (Table 3). The clean data were mapped back onto the assembled transcriptome using RSEM software. The bowtie parameter mismatch was 2. Among the four DGE sequencing results, at least 91.50 % of the sequences could be mapped back to the reference sequences (Table 4).
Table 3.
Statistics of DGE sequencing
| Sample | Raw Reads | Clean Reads | Clean Bases | Error (%) | Q20 (%) | Q30 (%) | GC Content (%) |
|---|---|---|---|---|---|---|---|
| SC_ep | 12343471 | 12283115 | 0.61G | 0.01 | 99.21 | 97.67 | 46.89 |
| SC_zf | 10138431 | 10084701 | 0.5G | 0.01 | 99.2 | 97.62 | 46.98 |
| TL_ep | 9486992 | 9449491 | 0.47G | 0.01 | 99.24 | 97.74 | 46.69 |
| TL_zf | 10139423 | 9999449 | 0.5G | 0.01 | 99.22 | 97.7 | 46.79 |
Clean reads: The number of reads after removing low-quality sequences. The subsequent analysis is based on clean reads. Error rate: Base error rate. Q20 and Q30, the percentage of bases with Phred values >20 and >30, respectively. GC content: the GC ratio of the total base number
Table 4.
DGE reads mapped to the reference sequences
| Sample name | Total reads | Total mapped |
|---|---|---|
| SC_ep | 12283115 | 11280554 (91.84 %) |
| SC_zf | 10084701 | 9248894 (91.71 %) |
| TL_ep | 9449491 | 8646697 (91.50 %) |
| TL_zf | 9999449 | 9172179 (91.73 %) |
Total reads: Number of reads after removing low-quality sequences (clean data). Total mapped: Number of reads that could be mapped back to the reference sequences. Values within the parenthesis represent total mapped divided by total reads × 100 %
Comparison of four DGE databases
We obtained 64 DEGs by comparing the DGE results of SC_ep vs TL_ep. Among the DEGs, 49 were up-regulated and 15 were down-regulated. There were 95 DEGs in SC_zf vs TL_zf, with 71 DEGs being up-regulated and 24 being down-regulated. There were 48 DEGs in SC_ep vs TL_ep and 79 DEGs in SC_zf vs TL_zf. In total, SC_ep vs TL_ep and SC_zf vs TL_zf had 16 DEGs in common (Fig. 6).
Fig. 6.
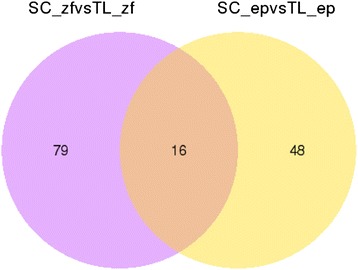
Venn diagram of DEGs from SC_ep vs TL_ep and SC_zf vs TL_zf
KEGG pathway analysis of four DGE databases
The KEGG database was used to analyze the metabolic pathways of the DEGs of SC_ep vs TL_ep and of SC_zf vs TL_zf. The results showed that 31 DEGs in SC_ep vs TL_ep were enriched in 22 KEGG pathways (Additional file 6). In comparison, 53 DEGs in SC_zf vs TL_zf were enriched in 26 KEGG pathways (Additional file 7). The top four KEGG pathways of SC_ep vs TL_ep were ‘nitrogen metabolism’, ‘cysteine and methionine metabolism’, ‘flavone and flavonol biosynthesis’ and ‘isoquinoline alkaloid biosynthesis’. Regarding up-regulated DEGs, ‘nitrogen metabolism’, ‘flavone and flavonol biosynthesis’, ‘diterpenoid biosynthesis’ and ‘selenocompound metabolism’ were most highly enriched. Regarding down-regulated DEGs, ‘isoquinoline alkaloid biosynthesis’, ‘tropane, piperidine and pyridine alkaloid biosynthesis’, ‘beta-Alanine metabolism’ and ‘carotenoid biosynthesis’ were the four main enriched KEGG pathways. Among the 26 KEGG pathways of SC_zf vs TL_zf, the most enriched pathways were ‘nitrogen metabolism’, ‘alpha-Linolenic acid metabolism’, and ‘glutathione metabolism’. Regarding the up-regulated DEGs, ‘nitrogen metabolism’, and ‘glutathione metabolism’ were mostly highly enriched. For down-regulated DEGs, ‘linoleic acid metabolism’, ‘alpha-linolenic acid metabolism’ and ‘cysteine and methionine metabolism’ were the three main enriched pathways. These results show that calyx persistence in Korla fragrant pear is regulated by a complex transcription mechanism.
We observed that 60 DEGs from SC_ep vs SC_zf, 179 DEGs from TL_ep vs TL_zf, 4 DEGs from SC_ep vs TL_ep, and 3 DEGs from SC_zf vs TL_zf were enriched in the STRING database (http://string-db.org/).
Real-time quantitative PCR
Ten DEGs were identified by both transcriptome sequencing and DGE sequencing (Table 5). These DEGs included three genes related to plant hormones [ethylene-responsive transcription factor ERF109 OS = Arabidopsis thaliana GN = ERF109 PE = 1 SV = 1 (comp36863_c0), ethylene-responsive transcription factor ERF027 OS = Arabidopsis thaliana GN = ERF027 PE = 2 SV = 1 (comp44254_c0), auxin-induced protein 5NG4 OS = Pinus taeda PE = 2 SV = 1 (comp50752_c0)]. Three genes were related to cell wall degradation [polygalacturonase inhibitor OS = Pyrus communis GN = PGIP PE = 1 SV = 1 (comp49798_c0), beta-galactosidase OS = Malus domestica PE = 1 SV = 1 (comp49925_c0), glucan endo-1,3-beta-glucosidase, acidic isoform GI9 OS = Nicotiana tabacum GN = PR2 PE = 1 SV = 1 (comp43208_c0)]. Two genes were related to stress [dehydration-responsive protein RD22 [Prunus persica] (comp44869_c0), dehydration-responsive element-binding protein, partial [Malus × domestica] (comp49899_c0)]. One gene was related to lipid transfer protein precursor [Pisum sativum] (comp36582_c0), and one gene was involved in NAC domain-containing protein 2 OS = Arabidopsis thaliana GN = NAC002 PE = 2 SV = 2 (comp41728_c0). We randomly selected five genes (comp36863_c0, comp41728_c0, com46544_c0, comp49798_c0, comp49614_c0) from the ten DEGs and all of the MYB and SPL genes. The expression levels of these five genes were measured in different floral organs at the early bloom, full bloom, and late bloom stages using qRT-PCR.
Table 5.
Genes shared by transcriptome and DGE sequencing
| Gene Id | Gene description |
|---|---|
| comp36863_c0 | Ethylene-responsive transcription factor ERF109 OS = Arabidopsis thaliana GN = ERF109 PE = 1 SV = 1 |
| comp44254_c0 | Ethylene-responsive transcription factor ERF027 OS = Arabidopsis thaliana GN = ERF027 PE = 2 SV = 1 |
| comp50752_c0 | Auxin-induced protein 5NG4 OS = Pinus taeda PE = 2 SV = 1 |
| comp49798_c0 | Polygalacturonase inhibitor OS = Pyrus communis GN = PGIP PE = 1 SV = 1 |
| comp49925_c0 | Beta-galactosidase OS = Malus domestica PE = 1 SV = 1 |
| comp43208_c0 | Glucan endo-1,3-beta-glucosidase, acidic isoform GI9 OS = Nicotiana tabacum GN = PR2 PE = 1 SV = 1 |
| comp44869_c0 | Dehydration-responsive protein RD22 OS = Arabidopsis thaliana GN = RD22 PE = 2 SV = 1 |
| comp49899_c0 | Dehydration-responsive element-binding protein 1A OS = Arabidopsis thaliana GN = DREB1A PE = 1 SV = 2 |
| comp36582_c0 | Non-specific lipid-transfer protein OS = Pyrus communis PE = 1 SV = 1 |
| comp41728_c0 | NAC domain-containing protein 2 OS = Arabidopsis thaliana GN = NAC002 PE = 2 SV = 2 |
The expression of ERF109 at the early bloom and late bloom stages was significantly (P = 0.01) greater in flowers with persistent calyx than in flowers with deciduous calyx. Regardless of whether the flower had a deciduous or a persistent calyx, ERF109 expression was significantly (P = 0.01) greater at the early bloom stage than at either the full bloom or late bloom stages (Fig. 7a). The expression of ERF109 at the late bloom stage was significantly (P = 0.01) greater in ovaries with persistent calyx than in sepals with persistent calyx (Fig. 7b). Regardless of bloom stage, the expression of ERF109 in ovaries with deciduous calyx was not significantly different than that in sepals with deciduous calyx (Fig. 7c).
Fig. 7.
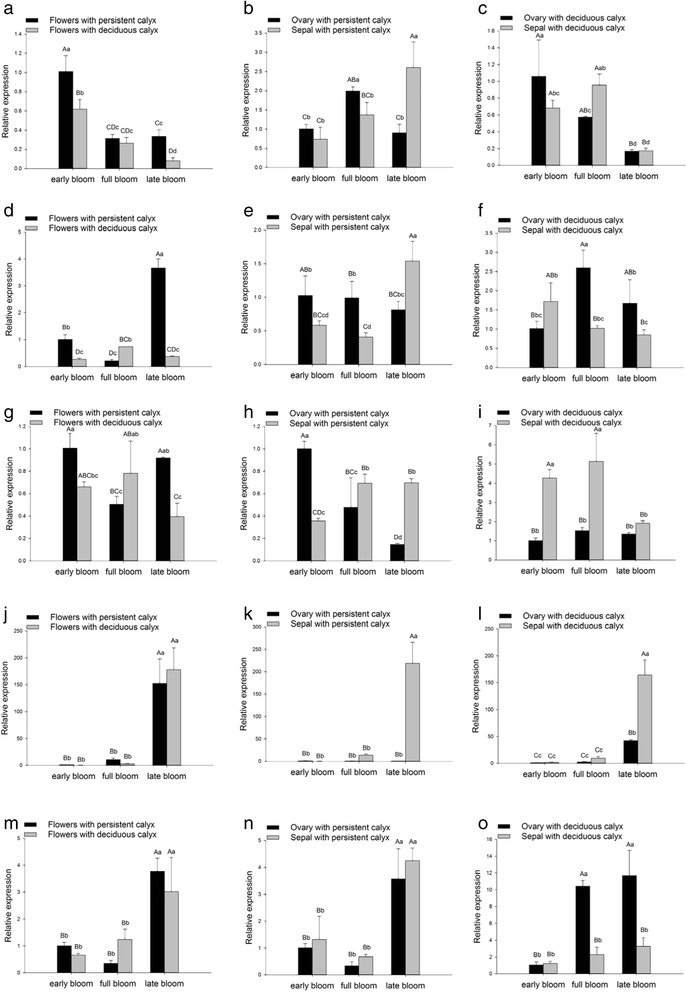
Temporal changes in the expression of selected genes in complete flowers, ovaries, and sepals. Error bars indicate SD. Different lowercase letters within a panel indicate significant differences at P = 0.05. Different uppercase letters within a panel indicate significant differences at P = 0.01
The expression of NAC002 in flowers varied significantly depending on the type of calyx and the flower stage. Specifically, NAC002 expression at early bloom and late bloom was significantly (P = 0.01) greater in flowers with persistent calyx than in flowers with deciduous calyx; however, the opposite was observed at full bloom (Fig. 7d). The NAC002 expression in flowers with a persistent calyx was significantly (P = 0.01) highest at the late bloom and early bloom stages. In contrast, NAC002 expression in flowers with a deciduous calyx was significantly (P = 0.05) greatest at the full bloom stage. The expression of NAC002 in ovaries with persistent calyx was significantly greater than that in sepals with persistent calyx at the early bloom stage (P = 0.05) and at the full bloom stage (P = 0.01) (Fig. 7e). In contrast, at the late bloom stage, NAC002 expression in ovaries with persistent calyx was significantly (P = 0.01) less than that in sepals with persistent calyx. The expression of NAC002 in ovaries with deciduous calyx was significantly greater than that in sepals with deciduous calyx at the full bloom (P = 0.01) and late bloom stages (P = 0.05) (Fig. 7f).
The expression of MYB5 was significantly greater in flowers with persistent calyx than in flowers with deciduous calyx at the early bloom (P = 0.05) and late bloom (P = 0.01) stages (Fig. 7g). In contrast, at the full bloom stage, MYB5 expression was significantly (P = 0.05) less in flowers with persistent calyx than in flowers with deciduous calyx. The expression of MYB5 in sepals with persistent calyx was significantly greater than that in ovaries with persistent calyx at the full bloom (P = 0.05) and late bloom (P = 0.01) stages (Fig. 7h). In contrast, MYB5 expression at the early bloom stage was significantly (P = 0.01) less in sepals with persistent calyx than in ovaries with persistent calyx. The expression of MYB5 in sepals with deciduous calyx was significantly greater than that in ovaries with deciduous calyx at early bloom and full bloom (Fig. 7i, P = 0.01).
Regardless of whether the flower had a deciduous or a persistent calyx, PGIG expression was significantly (P = 0.01) greater at the late bloom stage than at either the early bloom or full bloom stages (Fig. 7j). There was no significant difference in PGIG expression between flowers with persistent calyx and flowers with deciduous calyx. Regardless of whether the calyx was persistent or deciduous, the expression of PGIG in sepals was significantly greater than that in ovaries at the late bloom stage (Fig. 7k and l, P = 0.01).
The expression of SPL9 at the early bloom and late bloom stages was greater in flowers with persistent calyx than in flowers with deciduous calyx; however the opposite was true at the full bloom stage. The expression of SPL9 in flowers with deciduous calyx was not significantly different from that in flowers with deciduous calyx. Regardless of whether the flower had a deciduous or a persistent calyx, SPL9 expression was significantly (P = 0.01) greater at the late bloom stage than at either the early bloom or full bloom stages (Fig. 7m). There was no significant difference in SPL9 expression between ovaries with persistent calyx and sepals with deciduous calyx (Fig. 7n). The expression of MYB5 in ovaries with deciduous calyx was significantly greater than that in sepals at the full bloom and late bloom stages (Fig. 7o, P = 0.01).
The total expression pattern of the three genes ((ERF109 (comp36863_c0), NAC002 (comp41728_c0), and PGIG (comp49798_c0)) obtained with qRT-PCR was consistent with the RNA-seq data. This confirmed the validity of our results.
Plant hormone and organ abscission
Many hormones, especially IAA and ethylene, regulate organ abscission [30–35]. From 103 DEGs in SC_hua vs TL_hua, 11 genes were identified that were related to plant hormone metabolism. Five of these genes were related to ethylene-responsive transcription factor, two genes were related to auxin-induced protein, one gene was related to gibberellin-regulated protein, one gene was related to EREBP-like factor, one gene was related to the auxin responsive GH3 gene family, and one gene was related to brassinosteroid-regulated protein. From 64 DEGs in SC_ep vs TL_ep, seven genes were identified that were involved in plant hormone metabolism. Four of these genes were related to ethylene-responsive transcription factor, one gene was related to gibberellin 2-beta-dioxygenase 1, one gene was related to auxin-induced protein, and one gene was related to abscisic acid 8'-hydroxylase 4. We also identified five genes related to ethylene-responsive transcription factor from 95 DEGs in SC_zf vs TL_zf (Table 6).
Table 6.
Genes related to plant hormones
| Gene Id | Gene description |
|---|---|
| SC-hua vs TL-hua | |
| comp33730_c0 | Gibberellin-regulated protein 14 OS = Arabidopsis thaliana GN = GASA14 PE = 1 SV = 1 |
| comp36863_c0 | Ethylene-responsive transcription factor ERF109 OS = Arabidopsis thaliana GN = ERF109 PE = 1 SV = 1 |
| comp43830_c0 | Auxin-induced protein 5NG4 OS = Pinus taeda PE = 2 SV = 1 |
| comp44254_c0 | Ethylene-responsive transcription factor ERF027 OS = Arabidopsis thaliana GN = ERF027 PE = 2 SV = 1 |
| comp44440_c0 | Ethylene-responsive transcription factor ERF109 OS = Arabidopsis thaliana GN = ERF109 PE = 1 SV = 1 |
| comp47703_c0 | Ethylene-responsive transcription factor CRF4 OS = Arabidopsis thaliana GN = CRF4 PE = 1 SV = 2 |
| comp50752_c0 | Auxin-induced protein 5NG4 OS = Pinus taeda PE = 2 SV = 1 |
| comp54623_c0 | Ethylene-responsive transcription factor ERF109 OS = Arabidopsis thaliana GN = ERF109 PE = 1 SV = 1 |
| comp39099_c0 | EREBP-like factor |
| comp50238_c0 | auxin responsive GH3 gene family |
| comp49181_c2 | Brassinosteroid-regulated protein BRU1 OS = Glycine max PE = 2 SV = 1 |
| SC_ep vs TL_ep | |
| comp33823_c0 | Gibberellin 2-beta-dioxygenase 1 OS = Pisum sativum GN = GA2OX1 PE = 1 SV = 1 |
| comp36863_c0 | Ethylene-responsive transcription factor ERF109 OS = Arabidopsis thaliana GN = ERF109 PE = 1 SV = 1 |
| comp43552_c0 | Ethylene-responsive transcription factor RAP2-4 OS = Arabidopsis thaliana GN = RAP2-4 PE = 1 SV = 1 |
| comp44254_c0 | Ethylene-responsive transcription factor ERF027 OS = Arabidopsis thaliana GN = ERF027 PE = 2 SV = 1 |
| comp50299_c0 | Abscisic acid 8’-hydroxylase 4 OS = Arabidopsis thaliana GN = CYP707A4 PE = 2 SV = 2 |
| comp50752_c0 | Auxin-induced protein 5NG4 OS = Pinus taeda PE = 2 SV = 1 |
| comp48588_c0 | Ethylene-responsive transcription factor 1A OS = Arabidopsis thaliana GN = ERF1A PE = 1 SV = 2 |
| SC_zf vs TL_zf | |
| comp36863_c0 | Ethylene-responsive transcription factor ERF109 OS = Arabidopsis thaliana GN = ERF109 PE = 1 SV = 1 |
| comp40246_c0 | Ethylene-responsive transcription factor ERF023 OS = Arabidopsis thaliana GN = ERF023 PE = 2 SV = 1 |
| comp41236_c0 | Ethylene-responsive transcription factor ERF019 OS = Arabidopsis thaliana GN = ERF019 PE = 2 SV = 1 |
| comp43552_c0 | Ethylene-responsive transcription factor RAP2-4 OS = Arabidopsis thaliana GN = RAP2-4 PE = 1 SV = 1 |
| comp47393_c0 | Ethylene-responsive transcription factor ERF105 OS = Arabidopsis thaliana GN = ERF105 PE = 2 SV = 1 |
Genes related to cell wall degradation and organ abscission
The dissolution of the middle lamella is related to abscission, especially the loss of adhesion by separation layer cells due to the effects of cell wall degrading enzymes such as polygalacturonases. Several researchers have reported that cell wall modifying proteins such as expansin [36] and pectinesterase [37] have a role in abscission. Other researchers have observed that polygalacturonases have important function in the abscission process in oil palm [38], tomato [39], oilseed rape and Arabidopsis [40]. Beta-galactosidase [41], xyloglucan endotransglucosylase/hydrolase [42], and glucanase [43] genes have also been shown to be related to abscission. We obtained eight genes related to cell wall degradation from DEGs in SC_hua vs TL_hua. These eight genes included one gene related to polygalacturonase, one gene related to polygalacturonase inhibition, one gene related to beta-galactosidase, one gene related to glucan endo-1,3-beta-glucosidase, one gene related to lignin catabolic process, one gene related to tissue regeneration, and two genes related to xyloglucan endotransglucosylase. One expansin gene was obtained from DEGs in SC_ep vs TL_ep. From DEGs in SC_zf vs TL_zf, we obtained genes related to glucan endo-1,3-beta-glucosidase, beta-galactosidase, polygalacturonase inhibition, xyloglucan endotransglucosylase, and pectinesterase (Table 7).
Table 7.
Genes related to cell wall metabolism
| Gene Id | Gene description |
|---|---|
| SC-hua vs TL-hua | |
| comp47965_c0 | Probable polygalacturonase OS = Vitis vinifera GN = GSVIVT00026920001 PE = 1 SV = 1 |
| comp49798_c0 | Polygalacturonase inhibitor OS = Pyrus communis GN = PGIP PE = 1 SV = 1 |
| comp49925_c0 | Beta-galactosidase OS = Malus domestica PE = 1 SV = 1 |
| comp43208_c0 | Glucan endo-1,3-beta-glucosidase, acidic isoform GI9 OS = Nicotiana tabacum GN = PR2 PE = 1 SV = 1 |
| comp40498_c0 | lignin catabolic process//oxidation-reduction process |
| comp45343_c2 | tissue regeneration//cell adhesion//regulation of transcription, DNA-dependent |
| comp49181_c2 | Malus x domestica xyloglucan endotransglucosylase/hydrolase 7 mRNA, complete cds |
| comp38937_c0 | Probable xyloglucan endotransglucosylase/hydrolase protein 23 OS = Arabidopsis thaliana GN = XTH23 PE = 2 SV = 1 |
| SC_ep vs TL_ep | |
| comp43526_c0 | Expansin-A8 OS = Arabidopsis thaliana GN = EXPA8 PE = 2 SV = 1 |
| SC_zf vs TL_zf | |
| comp45273_c0 | Glucan endo-1,3-beta-glucosidase 8 OS = Arabidopsis thaliana GN = At1g64760 PE = 1 SV = 2 |
| comp49925_c0 | Beta-galactosidase OS = Malus domestica PE = 1 SV = 1 |
| comp43208_c0 | Glucan endo-1,3-beta-glucosidase, acidic isoform GI9 OS = Nicotiana tabacum GN = PR2 PE = 1 SV = 1 |
| comp49798_c0 | Polygalacturonase inhibitor OS = Pyrus communis GN = PGIP PE = 1 SV = 1 |
| comp38937_c0 | Probable xyloglucan endotransglucosylase/hydrolase protein 23 OS = Arabidopsis thaliana GN = XTH23 PE = 2 SV = 1 |
| comp51877_c0 | Putative pectinesterase/pectinesterase inhibitor 28 OS = Arabidopsis thaliana GN = PME28 PE = 2 SV = 1 |
Function of SPL and MYB genes in organ abscission
The SPL genes play an important role in the growth process of plants, including morphogenesis, the transition between developmental stages, sporogenesis, floral and fruit development, stress response, and plant hormone signal transduction [44]. In addition, SPL genes are induced during cell senescence leading to cell death [45, 46]. The MYB genes participate in plant secondary metabolism [47] as well as the plant’s response to hormones and environmental factors [48–50]. The MYB genes also regulate cellular differentiation, the cell life cycle [51, 52], and the morphogenesis of organs such as leaves [53–55]. The MYB genes are also involved in abscission [11, 56, 46]. We obtained 98 MYB and 21 SPL genes from the 48894 annotated unigenes (Table 8).
Table 8.
The MYB and SPL genes
| Gene ID | Gene description |
|---|---|
| SPL | |
| comp40233_c0 | Squamosa promoter-binding-like protein 8 OS = Arabidopsis thaliana GN = SPL8 PE = 1 SV = 2 |
| comp36894_c0 | Squamosa promoter-binding-like protein 13B OS = Arabidopsis thaliana GN = SPL13B PE = 3 SV = 1 |
| comp54049_c0 | Squamosa promoter-binding-like protein 14 OS = Arabidopsis thaliana GN = SPL14 PE = 2 SV = 3 |
| comp15760_c0 | Squamosa promoter-binding-like protein 5 OS = Arabidopsis thaliana GN = SPL5 PE = 2 SV = 1 |
| comp53959_c0 | Squamosa promoter-binding-like protein 1 OS = Arabidopsis thaliana GN = SPL1 PE = 1 SV = 2 |
| comp48948_c0 | Squamosa promoter-binding-like protein 1 OS = Arabidopsis thaliana GN = SPL1 PE = 1 SV = 2 |
| comp51995_c0 | Squamosa promoter-binding-like protein 6 OS = Arabidopsis thaliana GN = SPL6 PE = 2 SV = 2 |
| comp43799_c2 | Squamosa promoter-binding-like protein 12 OS = Arabidopsis thaliana GN = SPL12 PE = 1 SV = 1 |
| comp33051_c0 | Putative squamosa promoter-binding-like protein 19 OS = Oryza sativa subsp. japonica GN = SPL19 PE = 3 SV = 2 |
| comp43799_c1 | Squamosa promoter-binding-like protein 1 OS = Arabidopsis thaliana GN = SPL1 PE = 1 SV = 2 |
| comp19424_c0 | Squamosa promoter-binding-like protein 16 OS = Arabidopsis thaliana GN = SPL16 PE = 2 SV = 2 |
| comp43328_c0 | Squamosa promoter-binding-like protein 4 OS = Arabidopsis thaliana GN = SPL4 PE = 1 SV = 1 |
| comp34651_c0 | Malus x domestica SPL domain class transcription factor (SPL3) mRNA, complete cds |
| comp48364_c1 | Squamosa promoter-binding-like protein 12 OS = Oryza sativa subsp. indica GN = SPL12 PE = 2 SV = 1 |
| comp30499_c0 | Malus x domestica SPL domain class transcription factor (SPL2) mRNA, complete cds |
| comp46477_c1 | Squamosa promoter-binding-like protein 7 OS = Oryza sativa subsp. japonica GN = SPL7 PE = 2 SV = 2 |
| comp49614_c0 | Squamosa promoter-binding-like protein 9 OS = Arabidopsis thaliana GN = SPL9 PE = 2 SV = 2 |
| comp53802_c0 | Squamosa promoter-binding-like protein 7 OS = Arabidopsis thaliana GN = SPL7 PE = 1 SV = 2 |
| comp47538_c0 | Squamosa promoter-binding-like protein 6 OS = Arabidopsis thaliana GN = SPL6 PE = 2 SV = 2 |
| comp48561_c0 | Squamosa promoter-binding-like protein 8 OS = Arabidopsis thaliana GN = SPL8 PE = 1 SV = 2 |
| comp17109_c0 | Malus x domestica SPL domain class transcription factor (SPL3) mRNA, complete cds |
| MYB | |
| comp491996_c0 | putative MYB transcription factor [Rosa rugosa] |
| comp47342_c0 | Myb-related protein 308 OS = Antirrhinum majus GN = MYB308 PE = 2 SV = 1 |
| comp47241_c0 | Myb-related protein 306 OS = Antirrhinum majus GN = MYB306 PE = 2 SV = 1 |
| comp45253_c0 | Anthocyanin regulatory C1 protein OS = Zea mays GN = C1 PE = 2 SV = 1 |
| comp44151_c0 | Protein ODORANT1 OS = Petunia hybrida GN = ODO1 PE = 2 SV = 1 |
| comp31710_c0 | Transcription factor MYB39 OS = Arabidopsis thaliana GN = MYB39 PE = 2 SV = 1 |
| comp42545_c0 | Transcription factor RAX3 OS = Arabidopsis thaliana GN = RAX3 PE = 2 SV = 1 |
| comp41210_c0 | Myb-related protein 3R-1 OS = Arabidopsis thaliana GN = MYB3R-1 PE = 2 SV = 1 |
| comp2739_c0 | Myb-related protein Myb4 OS = Oryza sativa subsp. japonica GN = MYB4 PE = 2 SV = 2 |
| comp23664_c0 | Myb-related protein 306 OS = Antirrhinum majus GN = MYB306 PE = 2 SV = 1 |
| comp49924_c0 | Transcription factor MYB1R1 OS = Solanum tuberosum PE = 2 SV = 1 |
| comp47011_c1 | Transcription factor MYB86 OS = Arabidopsis thaliana GN = MYB86 PE = 2 SV = 1 |
| comp45831_c0 | Transcription repressor MYB6 OS = Arabidopsis thaliana GN = MYB6 PE = 1 SV = 1 |
| comp259366_c0 | Malus x domestica MYBR domain class transcription factor (MYBR14) mRNA, complete cds |
| comp25899_c0 | Malus x domestica MYB domain class transcription factor (MYB31) mRNA, complete cds |
| comp51661_c2 | Malus x domestica MYB domain class transcription factor (MYB88) mRNA, complete cds |
| comp38641_c1 | Anthocyanin regulatory C1 protein OS = Zea mays GN = C1 PE = 2 SV = 1 |
| comp36088_c0 | Transcription factor MYB113 OS = Arabidopsis thaliana GN = MYB113 PE = 1 SV = 1 |
| comp33026_c0 | Transcription factor MYB3 OS = Arabidopsis thaliana GN = MYB3 PE = 1 SV = 1 |
| comp44651_c0 | Myb-related protein 306 OS = Antirrhinum majus GN = MYB306 PE = 2 SV = 1 |
| comp41277_c0 | Malus x domestica MYB92 mRNA, complete cds |
| comp42019_c0 | Transcription factor MYB21 OS = Arabidopsis thaliana GN = MYB21 PE = 1 SV = 1 |
| comp42660_c0 | Transcription factor MYB39 OS = Arabidopsis thaliana GN = MYB39 PE = 2 SV = 1 |
| comp617_c1 | Pyrus communis R2R3 MYB transcription factor 10 (MYB10) gene, promoter region and partial cds |
| comp266782_c0 | MYB11 [Malus x domestica] |
| comp5228_c0 | Transcription factor MYB82 OS = Arabidopsis thaliana GN = MYB82 PE = 1 SV = 1 |
| comp40270_c0 | Transcription repressor MYB4 OS = Arabidopsis thaliana GN = MYB4 PE = 1 SV = 1 |
| comp41339_c0 | Malus x domestica MYB7 mRNA, complete cds |
| comp40714_c1 | MYB92 [Malus x domestica] |
| comp44744_c0 | Transcription factor MYB44 OS = Arabidopsis thaliana GN = MYB44 PE = 2 SV = 1 |
| comp38255_c0 | Malus x domestica cultivar Royal Gala MYB10 (MYB10) gene, promoter region and complete cds |
| comp33193_c0 | Malus x domestica MYB domain class transcription factor (MYB33) mRNA, complete cds |
| comp411_c0 | PREDICTED: Cicer arietinum transcription factor MYB12-like (LOC101507446), Mrna |
| comp33184_c0 | Malus x domestica MYB domain class transcription factor (MYB33) mRNA, complete cds |
| comp38919_c0 | MYB24 [Malus x domestica] |
| comp9080_c0 | PREDICTED: Fragaria vesca subsp. vesca transcription factor MYB32-like (LOC101307403), mRNA |
| comp37971_c0 | Myb-related protein 305 OS = Antirrhinum majus GN = MYB305 PE = 2 SV = 1 |
| comp8954_c0 | Malus x domestica MYB domain class transcription factor (MYB36) mRNA, complete cds |
| comp52545_c0 | Malus x domestica MYBR domain class transcription factor (MYBR8) mRNA, complete cds |
| comp51661_c0 | Myb-related protein B OS = Xenopus laevis GN = mybl2 PE = 2 SV = 2 |
| comp28973_c0 | Malus x domestica MYB domain class transcription factor (MYB1) mRNA, complete cds |
| comp404278_c0 | Transcription factor MYB39 OS = Arabidopsis thaliana GN = MYB39 PE = 2 SV = 1 |
| comp44434_c0 | Transcription factor MYB12 OS = Arabidopsis thaliana GN = MYB12 PE = 2 SV = 1 |
| comp40714_c0 | MYB92 [Malus x domestica] |
| comp620621_c0 | Transcription factor MYB23 OS = Arabidopsis thaliana GN = MYB23 PE = 1 SV = 1 |
| comp23111_c0 | Myb-related protein Myb4 OS = Oryza sativa subsp. japonica GN = MYB4 PE = 2 SV = 2 |
| comp43823_c0 | Myb-related protein Myb4 OS = Oryza sativa subsp. japonica GN = MYB4 PE = 2 SV = 2 |
| comp44151_c1 | MYB19 [Malus x domestica] > gi|189339113|dbj|BAG48172.1| myb-related transcription factor [Malus x domestica] |
| comp49161_c0 | Transcription factor MYB44 OS = Arabidopsis thaliana GN = MYB44 PE = 2 SV = 1 |
| comp49501_c0 | Transcription factor MYB1R1 OS = Solanum tuberosum PE = 2 SV = 1 |
| comp48408_c0 | Transcription factor MYB44 OS = Arabidopsis thaliana GN = MYB44 PE = 2 SV = 1 |
| comp35657_c0 | Transcription factor MYB48 OS = Arabidopsis thaliana GN = MYB48 PE = 2 SV = 1 |
| comp46544_c0 | Transcription repressor MYB5 OS = Arabidopsis thaliana GN = MYB5 PE = 1 SV = 1 |
| comp7072_c1 | Myb-related protein 306 OS = Antirrhinum majus GN = MYB306 PE = 2 SV = 1 |
| comp46515_c0 | Transcription factor MYB86 OS = Arabidopsis thaliana GN = MYB86 PE = 2 SV = 1 |
| comp30457_c0 | Malus x domestica MYB domain class transcription factor (MYB25) mRNA, complete cds |
| comp49893_c0 | Transcription factor MYB44 OS = Arabidopsis thaliana GN = MYB44 PE = 2 SV = 1 |
| comp46778_c0 | Myb-related protein 305 OS = Antirrhinum majus GN = MYB305 PE = 2 SV = 1 |
| comp89753_c0 | Transcription factor MYB12 OS = Arabidopsis thaliana GN = MYB12 PE = 2 SV = 1 |
| comp44726_c0 | Transcription factor AS1 OS = Arabidopsis thaliana GN = AS1 PE = 1 SV = 1 |
| comp25436_c0 | Transcription factor MYB44 OS = Arabidopsis thaliana GN = MYB44 PE = 2 SV = 1 |
| comp27942_c0 | Transcription factor MYB44 OS = Arabidopsis thaliana GN = MYB44 PE = 2 SV = 1 |
| comp46739_c0 | Transcription factor MYB44 OS = Arabidopsis thaliana GN = MYB44 PE = 2 SV = 1 |
| comp40636_c1 | Transcription factor MYB113 OS = Arabidopsis thaliana GN = MYB113 PE = 1 SV = 1 |
| comp38343_c0 | Transcription factor MYB3 OS = Arabidopsis thaliana GN = MYB3 PE = 1 SV = 1 |
| comp41103_c0 | Transcription repressor MYB5 OS = Arabidopsis thaliana GN = MYB5 PE = 1 SV = 1 |
| comp33109_c1 | Rosa rugosa mRNA for putative MYB transcription factor (myb9 gene) |
| comp209723_c0 | putative MYB transcription factor [Rosa hybrid cultivar] |
| comp44561_c0 | Myb-related protein Myb4 OS = Oryza sativa subsp. japonica GN = MYB4 PE = 2 SV = 2 |
| comp51083_c0 | putative MYB transcription factor [Rosa hybrid cultivar] |
| comp31372_c0 | MYB domain class transcription factor [Malus x domestica] |
| comp38343_c1 | Myb-related protein Myb4 OS = Oryza sativa subsp. japonica GN = MYB4 PE = 2 SV = 2 |
| comp52029_c2 | Malus x domestica cultivar Jiangsu Fuji MYB transcription factor (MYB53) mRNA, partial cds |
| comp45889_c1 | Transcription factor MYB39 OS = Arabidopsis thaliana GN = MYB39 PE = 2 SV = 1 |
| comp37277_c1 | Transcription factor MYB59 OS = Arabidopsis thaliana GN = MYB59 PE = 2 SV = 2 |
| comp46917_c0 | Myb-related protein 330 OS = Antirrhinum majus GN = MYB330 PE = 2 SV = 1 |
| comp7908_c0 | Transcription factor MYB86 OS = Arabidopsis thaliana GN = MYB86 PE = 2 SV = 1 |
| comp7072_c0 | Transcription factor MYB39 OS = Arabidopsis thaliana GN = MYB39 PE = 2 SV = 1 |
| comp47011_c0 | Myb-related protein Hv33 OS = Hordeum vulgare GN = MYB2 PE = 2 SV = 3 |
| comp159049_c0 | Transcription factor MYB46 OS = Arabidopsis thaliana GN = MYB46 PE = 2 SV = 1 |
| comp27400_c0 | Transcription factor MYB46 OS = Arabidopsis thaliana GN = MYB46 PE = 2 SV = 1 |
| comp308054_c0 | Malus x domestica MYB domain class transcription factor (MYB18) mRNA, complete cds |
| comp125091_c0 | Malus x domestica cultivar Royal Gala MYB9 mRNA, complete cds |
| comp48140_c0 | Rosa hybrid cultivar mRNA for putative MYB transcription factor (myb1 gene), cultivar Yellow Island |
| comp8463_c0 | Transcription factor MYB44 OS = Arabidopsis thaliana GN = MYB44 PE = 2 SV = 1 |
| comp26540_c0 | Malus x domestica cultivar Royal Gala MYB9 mRNA, complete cds |
| comp28178_c0 | Malus x domestica MYB2 mRNA, complete cds |
| comp188108_c0 | Transcription factor MYB113 OS = Arabidopsis thaliana GN = MYB113 PE = 1 SV = 1 |
| comp29648_c0 | Myb-related protein 305 OS = Antirrhinum majus GN = MYB305 PE = 2 SV = 1 |
| comp611736_c0 | Malus x domestica MYB domain class transcription factor (MYB17) mRNA, complete cds |
| comp49971_c0 | Transcription factor MYB86 OS = Arabidopsis thaliana GN = MYB86 PE = 2 SV = 1 |
| comp42161_c1 | Lupinus albus LaMYB27 mRNA for R2R3-MYB transcription factor, partial cds |
| comp43170_c0 | Transcription factor MYB21 OS = Arabidopsis thaliana GN = MYB21 PE = 1 SV = 1 |
| comp37565_c0 | Trifolium repens tannin-related R2R3 MYB transcription factor (Myb14) gene, Myb14-3 allele, partial cds |
| comp49430_c0 | Myb-related protein 3R-1 OS = Arabidopsis thaliana GN = MYB3R-1 PE = 2 SV = 1 |
| comp43202_c0 | Myb-related protein 305 OS = Antirrhinum majus GN = MYB305 PE = 2 SV = 1 |
| comp38641_c0 | Malus x domestica MYB11 mRNA, complete cds |
| comp50379_c0 | Myb-related protein 305 OS = Antirrhinum majus GN = MYB305 PE = 2 SV = 1 |
Stress response genes and abscission
The sequencing results showed that many genes related to stress response exhibited differential expression. There was one heat shock factor protein, two dehydration-responsive element-binding proteins, one dehydration-responsive protein, two NAC transcription factor proteins, one NAC domain-containing protein [57, 58], and one cysteine synthase-like gene [59] among the DEGs in SC_hua vs TL_hua. There were also genes related to the NAC domain-containing protein, the pathogenesis-related protein Bet v I family, the senescence-related protein gene, dehydration-responsive protein, and dehydration-responsive element-binding protein from DEGs in SC_ep vs TL_ep. From the DEGs in SC_zf vs TL_zf, we obtained genes related to disease resistance response protein 206, dehydration-responsive protein, defensin-like protein, and senescence-related protein (Table 9).
Table 9.
Genes related to stress
| Gene Id | Gene description |
|---|---|
| SC-hua vs TL-hua | |
| comp43473_c0 | Heat shock factor protein HSF24 OS = Solanum peruvianum GN = HSF24 PE = 2 SV = 1 |
| comp44869_c0 | Dehydration-responsive protein RD22 OS = Arabidopsis thaliana GN = RD22 PE = 2 SV = 1 |
| comp49899_c0 | Dehydration-responsive element-binding protein 1A OS = Arabidopsis thaliana GN = DREB1A PE = 1 SV = 2 |
| comp39099_c0 | Dehydration-responsive element-binding protein 3 OS = Arabidopsis thaliana GN = DREB3 PE = 2 SV = 1 |
| comp45992_c0 | NAC transcription factor NAM-B2 OS = Triticum durum GN = NAM-B2 PE = 2 SV = 1 |
| comp49969_c0 | NAC transcription factor 25 OS = Arabidopsis thaliana GN = NAC025 PE = 2 SV = 1 |
| comp41728_c0 | NAC domain-containing protein 2 OS = Arabidopsis thaliana GN = NAC002 PE = 2 SV = 2 |
| comp48683_c0 | Cysteine synthase OS = Citrullus lanatus PE = 1 SV = 1 |
| SC_ep vs TL_ep | |
| comp34503_c0 | Pathogenesis-related protein Bet v I family |
| comp43933_c0 | senescence-related protein [Camellia sinensis] |
| comp44869_c0 | Dehydration-responsive protein RD22 OS = Arabidopsis thaliana GN = RD22 PE = 2 SV = 1 |
| comp41728_c0 | NAC domain-containing protein 2 OS = Arabidopsis thaliana GN = NAC002 PE = 2 SV = 2 |
| comp49899_c0 | Dehydration-responsive element-binding protein 1A OS = Arabidopsis thaliana GN = DREB1A PE = 1 SV = 2 |
| SC_zf vs TL_zf | |
| comp41222_c0 | Disease resistance response protein 206 OS = Pisum sativum GN = PI206 PE = 2 SV = 2 |
| comp44869_c0 | Dehydration-responsive protein RD22 OS = Arabidopsis thaliana GN = RD22 PE = 2 SV = 1 |
| comp51764_c0 | Defensin-like protein 2 OS = Arabidopsis thaliana GN = PDF2.2 PE = 2 SV = 1 |
| comp43933_c0 | senescence-related protein [Camellia sinensis] |
Other genes and abscission
Several researchers have reported that zinc finger protein [60] and lipid-transfer protein [61, 62] are involved in calyx abscission. We obtained one gene related to lipid-transfer protein from DEGs in SC_hua vs TL_hua. One gene related to lipid-transfer protein as well as five zinc finger genes were obtained from DEGs in SC_zf vs TL_zf (Table 10).
Table 10.
Additional genes related to abscission
| Gene Id | Gene description |
|---|---|
| SC-hua vs TL-hua | |
| comp36582_c0 | Non-specific lipid-transfer protein OS = Pyrus communis PE = 1 SV = 1 |
| SC_zf vs TL_zf | |
| comp33569_c0 | zinc finger protein, putative [Ricinus communis] > gi|223538542|gb|EEF40147.1| zinc finger protein, putative [Ricinus communis] |
| comp41672_c0 | Zinc finger, C3HC4 type (RING finger)//Ring finger domain//Anaphase-promoting complex subunit 11 RING-H2 finger//zinc-RING finger domain//RING-H2 zinc finger |
| comp43820_c0 | MYM-type Zinc finger with FCS sequence motif |
| comp46839_c0 | Putative zinc finger protein At1g68190 OS = Arabidopsis thaliana GN = At1g68190 PE = 2 SV = 1 |
| comp53961_c0 | RING finger and CHY zinc finger domain-containing protein 1 OS = Homo sapiens GN = RCHY1 PE = 1 SV = 1 |
| comp36582_c0 | Non-specific lipid-transfer protein OS = Pyrus communis PE = 1 SV = 1 |
Putative genes related to abscission
Other genes in this study showed high-level differential expression. However, the function of these genes is unknown. We defined these genes as putative genes related to abscission. There were ten putative genes among DEGs in SC_hua vs TL_hua, eleven putative genes among DEGs in SC_ep vs TL_ep, and eighteen putative genes among DEGs in SC_zf vs TL_zf (Table 11).
Table 11.
Putative genes related to abscission
| Gene Id | Gene description |
|---|---|
| SC-hua vs TL-hua | |
| comp54231_c0 | Polyphenol oxidase, chloroplastic OS = Malus domestica PE = 2 SV = 1 |
| comp52712_c0 | Asparagine synthetase [glutamine-hydrolyzing] OS = Asparagus officinalis PE = 2 SV = 2 |
| comp48325_c1 | NADP-dependent D-sorbitol-6-phosphate dehydrogenase OS = Malus domestica GN = S6PDH PE = 2 SV = 1 |
| comp33824_c0 | 11S globulin seed storage protein 2 OS = Sesamum indicum PE = 2 SV = 1 |
| comp42796_c0 | CASP-like protein RCOM_0679870 OS = Ricinus communis GN = RCOM_0679870 PE = 2 SV = 1 |
| comp44393_c0 | UDP-glucose 4-epimerase 1 OS = Arabidopsis thaliana GN = At1g12780 PE = 1 SV = 2 |
| comp32282_c0 | Alpha-aminoadipic semialdehyde synthase OS = Arabidopsis thaliana GN = LKR/SDH PE = 1 SV = 1 |
| comp44627_c0 | Protein ASPARTIC PROTEASE IN GUARD CELL 1 OS = Arabidopsis thaliana GN = ASPG1 PE = 1 SV = 1 |
| comp53838_c1 | Synaptotagmin-3 OS = Arabidopsis thaliana GN = SYT3 PE = 2 SV = 1 |
| comp46366_c0 | Tonoplast dicarboxylate transporter OS = Arabidopsis thaliana GN = TDT PE = 2 SV = 2 |
| SC_ep vs TL_ep | |
| comp47776_c0 | Bidirectional sugar transporter NEC1 OS = Petunia hybrida GN = NEC1 PE = 2 SV = 1 |
| comp43067_c0 | Miraculin OS = Richadella dulcifica PE = 1 SV = 3 |
| comp44995_c0 | Taxadien-5-alpha-ol O-acetyltransferase OS = Taxus wallichiana var. chinensis PE = 2 SV = 1 |
| comp43847_c0 | Uncharacterized protein At3g61260 OS = Arabidopsis thaliana GN = At3g61260 PE = 1 SV = 1 |
| comp47017_c0 | PREDICTED: Fragaria vesca subsp. vesca uncharacterized LOC101305493 (LOC101305493), mRNA |
| comp43990_c0 | Heavy metal-associated isoprenylated plant protein 26 OS = Arabidopsis thaliana GN = HIPP26 PE = 1 SV = 1 |
| comp47206_c0 | Copper methylamine oxidase OS = Arthrobacter sp. (strain P1) GN = maoII PE = 1 SV = 1 |
| comp51202_c0 | Diacylglycerol kinase 1 OS = Arabidopsis thaliana GN = DGK1 PE = 1 SV = 2 |
| comp50351_c0 | Uncharacterized membrane protein C2G11.09 OS = Schizosaccharomyces pombe (strain 972/ATCC 24843) GN = SPAC2G11.09 PE = 2 SV = 2 |
| comp44384_c1 | LOB domain-containing protein 41 OS = Arabidopsis thaliana GN = LBD41 PE = 2 SV = 1 |
| comp46169_c0 | -- |
| SC_zf vs TL_zf | |
| comp40401_c0 | Jasmonate O-methyltransferase OS = Brassica rapa subsp. pekinensis GN = JMT PE = 1 SV = 1 |
| comp42909_c0 | Ornithine decarboxylase OS = Datura stramonium PE = 2 SV = 1 |
| comp40364_c1 | Bifunctional monodehydroascorbate reductase and carbonic anhydrase nectarin-3 OS = Nicotiana langsdorffii x Nicotiana sanderae GN = NEC3 PE = 1 SV = 1 |
| comp49118_c1 | Polyphenol oxidase, chloroplastic OS = Malus domestica PE = 2 SV = 1 |
| comp48325_c1 | NADP-dependent D-sorbitol-6-phosphate dehydrogenase OS = Malus domestica GN = S6PDH PE = 2 SV = 1 |
| comp48520_c0 | Beta-D-xylosidase 1 OS = Arabidopsis thaliana GN = BXL1 PE = 1 SV = 1 |
| comp36479_c0 | -- |
| comp44627_c0 | Protein ASPARTIC PROTEASE IN GUARD CELL 1 OS = Arabidopsis thaliana GN = ASPG1 PE = 1 SV = 1 |
| comp54257_c0 | Beta-fructofuranosidase, insoluble isoenzyme 1 OS = Daucus carota GN = INV1 PE = 1 SV = 1 |
| comp26144_c0 | (RS)-norcoclaurine 6-O-methyltransferase OS = Coptis japonica PE = 1 SV = 1 |
| comp47734_c0 | (R)-mandelonitrile lyase 3 OS = Prunus serotina GN = MDL3 PE = 2 SV = 1 |
| comp45349_c1 | Snakin-2 OS = Solanum tuberosum GN = SN2 PE = 1 SV = 1 |
| comp47553_c0 | Ferredoxin--nitrite reductase, chloroplastic OS = Betula pendula GN = NIR1 PE = 2 SV = 1 |
| comp42868_c0 | L-aspartate oxidase 1 OS = Ralstonia solanacearum (strain GMI1000) GN = nadB1 PE = 3 SV = 1 |
| comp44384_c1 | LOB domain-containing protein 41 OS = Arabidopsis thaliana GN = LBD41 PE = 2 SV = 1 |
| comp43977_c0 | -- |
| comp43311_c0 | predicted protein [Arabidopsis lyrata subsp. lyrata] > gi|297331837|gb|EFH62256.1| predicted protein [Arabidopsis lyrata subsp. lyrata] |
| comp47017_c0 | uncharacterized protein LOC100814873 [Glycine max] > gi|255637360|gb|ACU19009.1| unknown [Glycine max] |
The DEGs from transcriptome and DGE sequencing were subjected to a search against GO and KEGG databases. The results showed that many of the DEGs were involved in metabolic processes related to chlorophyll, plant hormone metabolism, carbohydrate metabolism, signal transduction and cell wall construction. The results were consistent with Qi’s (2013) [12], and suggest that calyx persistence in Korla fragrant pear is regulated by many genes.
Conclusion
More than 50 DEGs were obtained through transcriptome and DGE sequencing. These DEGS were related to cell wall metabolism, plant hormone metabolism, stress response, zinc finger protein, and lipid-transfer protein. Analysis of the functions and metabolic pathways of the DEGs indicated that calyx abscission in Korla fragrant pear was a metabolic process induced by a variety of genes related to cell wall metabolism and regulated by multiple plant hormones. Our laboratory is currently researching the protein function of the DEGs in Korla fragrant pear.
Methods
Plant material
Three trees with high vigor and three trees with low vigor were selected in spring 2013 at the Shayidong Horticulture Field, Korla, Xinjiang Province. Flowers were collected from each tree at the early bloom, full bloom, and late bloom stages. The first flower to open in clusters on trees with high vigor has a persistent calyx (Fig. 8a, b). The fourth flower to open in clusters from trees with low vigor has a deciduous calyx (Fig. 8c, d). The flowers were immediately frozen in liquid N and stored at −80 °C.
Fig. 8.

Flowers with persistent and deciduous calyx of Korla fragrant pear. The a and b indicate flowers with persistent calyx. The c and d indicate flowers with deciduous calyx
Transcriptome sequencing
Solexa/Illumina sequencing was carried out by Novogene, Beijing, China. Total RNA was extracted from the flower samples using RNAout 1.0 (Tianenze, Beijing, China). The RNA degradation and contamination was monitored on 1 % agarose gels. The purity of the RNA was checked with a NanoPhotometer® (IMPLEN, CA, USA). The RNA concentration was measured using a Qubit®RNA Assay Kit and a Qubit®2.0 Fluorometer (Life Technologies, CA, USA). The RNA integrity was assessed using an RNA Nano 6000 Assay Kit and an Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA). After quality inspection, the RNA from flowers at the early, full, and late bloom stages were combined by calyx type. The combined RNA sample from flowers with a persistent calyx will be referred to as SC_hua. The combined RNA sample from flowers with a deciduous calyx will be referred to as TL_hua. These RNA samples were used for transcriptome sequencing. Three biological replicates were used.
The RNA preparations used 3 μg RNA per sample. Sequencing libraries were generated using NEBNext®Ultra™ RNA Library Prep Kit for Illumina® (NEB, USA) following the manufacturer’s recommendations. Index codes were added to attribute sequences in each sample. Briefly, mRNA was purified from total RNA using poly-T oligo-attached magnetic beads. Fragmentation was carried out using divalent cations under elevated temperature in NEBNext First Strand Synthesis Reaction Buffer (5x). First strand cDNA was synthesized using random hexamer primer and M-MuLV Reverse Transcriptase (RNase H−). Second strand cDNA synthesis was subsequently performed using DNA Polymerase I and RNase H. Remaining overhangs were converted into blunt ends via exonuclease/polymerase activities. After adenylation of the 3’ ends, the DNA fragments were ligated with the NEBNext Adaptor with hairpin loop structure. The library fragments were purified with AMPure XP (Beckman Coulter, Beverly, USA) in order to select cDNA fragments with lengths of 150 ~ 200 bp. The size-selected, adaptor-ligated cDNA was mixed with 3 μl USER Enzyme (NEB, USA) at 37 °C for 15 min followed by 5 min at 95 °C before PCR. The PCR was performed with Phusion High-Fidelity DNA polymerase, universal PCR primers and Index (X) Primer. The PCR products were purified (AMPure XP system) and the library quality was assessed using an Agilent Bioanalyzer 2100.
The clustering of the index-coded samples was performed on a cBot Cluster Generation System using TruSeq PE Cluster Kit v3-cBot-HS (Illumia) according to the manufacturer’s instructions. After cluster generation, the library preparations were sequenced on an Illumina Hiseq 2000 platform and paired-end reads were generated.
Data analysis of transcriptome sequencing
Raw data (raw reads) in fastq format were first processed through in-house Perl scripts. Clean data (clean reads) were obtained by removing reads containing adapter sequences, reads containing poly-N, and low quality reads. The Q20, Q30, GC-content, and sequence duplication level of the clean data were calculated. All downstream analyses were based on clean data with high quality.
The left files (read1 files) from all libraries/samples were pooled into one large left.fq file. The right files (read2 files) were pooled into one large right.fq file. Transcriptome assembly was accomplished based on the left.fq and right.fq files using Trinity [63]. The min_kmer_cov was set at 2 and all other parameters were set at default. Gene function was annotated based on the following databases: NR (NCBI non-redundant protein sequences); NT (NCBI non-redundant nucleotide sequences); PFAM (Protein family); KOG/COG (Clusters of Orthologous Groups of proteins); SwissProt (A manually annotated and reviewed protein sequence database); KO (KEGG Ortholog database); GO (Gene Ontology).
DGE sequencing
The RNA was extracted from sepals and ovaries at the early, full, and late bloom stages. The RNA was combined by calyx type. The combined RNA sample from sepals with a persistent calyx will be referred to as SC_ep. The combined RNA sample from sepals with a deciduous calyx will be referred to as TL _ep. The combined RNA sample from ovaries with a persistent calyx will be referred to as SC_zf. The combined RNA sample from ovaries with a deciduous calyx will be referred to as TL _zf. The methods of RNA extraction, RNA quantification, RNA qualification, clustering, and sequencing were the same as those described above for transcriptome sequencing.
Differential expression analysis
Samples with biological replicates
Differential expression analysis of two conditions/groups was performed using the DESeqR package (1.10.1). The DESeq provides statistical routines for determining differential expression in digital gene expression data using a model based on negative binomial distribution. The resulting P values were adjusted using Benjamini and Hochberg’s approach for controlling the false discovery rate. Genes were considered to be differentially expressed if DESeq found the adjusted P-value to be <0.05.
Samples without biological replicates
Prior to differential gene expression analysis, the read counts for each sequenced library were adjusted using edgeR software through one scaling normalized factor. Differential expression analysis of two samples was performed using DEGseq R package (2010). The P value was adjusted using the q value [64]. The q value < 0.005&|log2 (fold change)| > 1 was set as the threshold for significantly differential expression.
GO enrichment analysis
Gene Ontology (GO) enrichment analysis of the differentially expressed genes (DEGs) was implemented by GOseq R packages based on Wallenius non-central hyper-geometric distribution [65] which can be adjusted for gene length bias in DEGs.
KEGG pathway enrichment analysis
KEGG [66] is a database resource for understanding high-level functions and utilities of biological systems (e.g., cell, organism, and ecosystem), from molecular-level information, especially large-scale molecular datasets generated by genome sequencing and other high-throughput experimental technologies (http://www.genome.jp/kegg/). We used KOBAS [67] software to test the statistical enrichment of differentially expressed genes in KEGG pathways.
Protein Protein Interaction (PPI)
The sequences of the DEGs were BLASTx against the genome of a related species (the PPI of which exists in the STRING database: http://string-db.org/) to get the predicted PPIs of these DEGs. The PPIs were visualized in Cytoscape [68].
Real-time quantitative PCR
The expression of five genes (Gene ID: comp36863_c0, comp41728_c0, comp46544_c0, comp49798_c0, and comp49614_c0) that might be associated with calyx persistence in Korla Fragrant Pear were analyzed by qRT-PCR. Total RNA was separately extracted from the full flowers, sepals and ovaries using RNAout 1.0 (Tianenze, Beijing, China) at the early bloom, full bloom, and late bloom stages. The RNA samples were from (i) sepals with persistent calyx, (ii) ovaries with persistent calyx, (iii) sepals with deciduous calyx, (iv) ovaries with deciduous calyx, (v) full flowers with deciduous calyx, and (vi) full flowers with persistent calyx. Each group had three biological replications. Gene-specific primers were designed according to the reference unigene sequences using Primer Premier 5.0 (Table 12). The synthesis of cDNA was performed using a Reverse Transcriptase M-MLV kit (TaKaRa, Dalian, China). Real-time quantification was performed using a CFX manager (Bio-Rad, USA) and the SYBR Green Real-time PCR Master Mix (Toyobo, Osaka, Japan). The protocol of real-time PCR was as follows: initiation with a 30 s pre-denaturation at 95 °C followed by 40 cycles of amplification with 5 s of denaturation at 95 °C, 10 s of annealing at 56 °C, 15 s of extension at 72 °C and reading the plate for fluorescence data collection at 65 °C. A melting curve was performed from 65 to 95 °C to check the specificity to the amplified product. Each reaction was repeated three times. Korla fragrant pear actin gene (forward: 5’-CCATCCAGGCTGTTCTCTC-3’, and reverse: 5’-GCAAGGTCCAGACGAAGG -3’) was used as a normalizer.
Table 12.
Primer for qRT-PCR
| Primer ID | Primer sequences (5’ to 3’) |
|---|---|
| comp36863_c0 | AACTACTTCTCGCCATCGT |
| TGTTCTTGCTCTTCCTCGT | |
| comp41728_c0 | GCGTGGAGGTAGGAGAAC |
| CAAGAAGGGCAGCATAGA | |
| comp46544_c0 | GAGGAGGAAATGAAGAGG |
| ATCAATCAAACAGGTGGC | |
| comp49798_c0 | AATAAACTGCCCAAACGA |
| ACAAGCAACCCAATCTCA | |
| comp49614_c0 | CACGAAGTGGTCGGAAAG |
| GGAGAATGCGTCACAGTAG |
Availability of supporting data
Illumina sequencing data from ‘Korla Fragrant Pear’ SC_hua, TL_hua, SC_zf, TL_zf, SC_ep, and TL_ep were deposited in the NCBI SRA database under accession number SRP066513, bioProject accession: PRJNA303067 (http://www.ncbi.nlm.nih.gov/bioproject/303067). The release time is 2016-11-21 00:00:00. All the supporting data have been provided as Additional files (1, 2, 3, 4, 5, 6 and 7).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31360474), the Specialized Research Fund for the Doctoral Program of Higher Education (2013651810002), and the International Scientific and Technological Cooperation Projects (2009YD32).
Abbreviations
- SC_hua
flowers with persistent calyx
- SC_ep
sepals of flowers with persistent calyx
- SC_ zf
ovaries of flowers with persistent calyx
- TL_hua
flowers of flowers with deciduous calyx
- TL_ep
sepals of flowers with deciduous calyx
- TL_zf
ovaries with deciduous calyx
- DEGs
differentially expressed genes
- qRT-PCR
real-time quantitative PCR
- DGE
digital gene expression
- RNA-Seq
high-throughput sequencing of RNA
- NR
NCBI non-redundant protein sequences
- NT
NCBI nucleotide sequences
- KEGG
Kyoto encyclopedia of genes and genomes
- SwissProt
a manually annotated and reviewed protein sequence database
- PFAM
protein family
- GO
gene ontology
- KOG/COG
clusters of orthologous groups of proteins
- CC
the cellular component category
- BP
the biological process category
- MF
the molecular function category
Additional files
GO enrichment of DEGs in SC_hua vs TL_hua. GO_accession: The ID of nodal point. Description: GO description. Term_type: The category of GO. Over_represented_pValue: The pValue of enrichment. Corrected_pValue: The pValue is after correction. DEG_item: The DEGs related to this GO. DEG_list: All the DEGs annotated in GO. Bg_item: The background genes related to this GO. Bg_list: The background genes annoted in this GO. Gene_names: The DEGs’ ID related to this GO. (XLS 130 kb)
GO enrichment of up-regulated DEGs in SC_hua vs TL_hua. GO_accession: The ID of nodal point. Description: GO description. Term_type: The category of GO. Over_represented_pValue: The pValue of enrichment. Corrected_pValue: The pValue is after correction. DEG_item: The DEGs related to this GO. DEG_list: All the DEGs annotated in GO. Bg_item: The background genes related to this GO. Bg_list: The background genes annoted in this GO. Gene_names: The DEGs’ ID related to this GO. (XLS 82 kb)
The top 20 KEGG pathways enrichment of DEGs in SC_hua vs TL_hua. (XLS 1 kb)
The top 20 KEGG pathways enrichment of up DEGs in SC_hua vs TL_hua. (XLS 512 bytes)
The top 20 KEGG pathways enrichment of down DEGs in SC_hua vs TL_hua. (XLS 977 bytes)
The top 20 KEGG pathways enrichment of DEGs in SC_ep vs TL_ep. (XLS 1 kb)
The top 20 KEGG pathways enrichment of DEGs in SC_zf vs TL_zf. (XLS 1 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JXN led and coordinated the project, JXN and MSP designed the study, MSP, CJL, FJC and SWQ collected the plant materials and isolated the RNA. MSP and SWQ conducted the real-time quantitative PCR. MSP conducted the bioinformatics analysis and wrote the paper. All authors have read and agree with the final manuscript. JXN is the corresponding author and is responsible for all contact and correspondence. All authors read and approved the final manuscript.
Contributor Information
Maosong Pei, Email: 1185248506@qq.com.
Jianxin Niu, Email: njx105@163.com.
Chenjing Li, Email: 1429994796@qq.com.
Fujun Cao, Email: 528296410@qq.com.
Shaowen Quan, Email: 731982806@qq.com.
References
- 1.Gao QM, Li J, Li Y. Literature review of researches on ‘Kuerle Sweet Pear’. Nonwood Forest Res. 2005;23:79–82. [Google Scholar]
- 2.Liang SW, Wang WL, Zhang JH. The reasons and strategies of calyx persistence in Dangshan pear. Agricultural Tech Ser. 2009;6:88. [Google Scholar]
- 3.Liu N, Tao ST, Zhang SL, Cao YF, Wu HQ, Wang JZ, Wu T. Effect of different pollinizer varieties on calyx retention and quality for ‘Kuerlexiangli’ fruit. J Nanjing Agri Univ. 2011;34:43–7.
- 4.Zhu ML, Qiao JC, Xu JF. Effect of pollinating varieties and pollen amount on persistent calyx of Ya pear. J Agri Univ Hebei. 2006;29:38–40. [Google Scholar]
- 5.Xu YL, Gao ZH, Zhang JY, Yi XK, Shu B. Effects of pollination with various pollination varieties and pollen amount on fruit calyx of Dangshan Suli pear. J Anhui Agric Univ. 2009;36:1–6. [Google Scholar]
- 6.Ren YY, Li J, Qin WM, Guo QZ. Preliminary studies on characteristic and control of calyx leaving and persistent from fruit of Korla Fragrant Pear. J Xinjiang Agri Univ. 2007;30:25–9. [Google Scholar]
- 7.Heng W, Chen J, Ye ZF, Jia B, Zhang SM, Sun J, Zhu LW. Development of calyx and its controlling techniques of young fruit of Dangshansu pear. J Anhui Agri Univ. 2010;37:238–43.
- 8.Jia B, Zhu LW, Zhang SL. Effects of the growth regulators on the ratio of the fruit without calyx, fruit quality and shoot growth in Pyrus bretschneideri ‘Dangshansuli’. J Nanjing Agri Univ. 2012;35:26–32. [Google Scholar]
- 9.Jia B, Zhu LW, Zhang SL. Preliminary study on the action of the mineral element Fe in fruit calyx development of ‘Dangshan Suli’ Pear. Acta Horticulturae Sinica. 2012;39:159–67. [Google Scholar]
- 10.Dong FY, Zhang F, Wang YT, Niu JX. Differential expression anaiysis of calyx failing off Korla Fragrant Pears and their persistent related genes. Xinjiang Agri Sci. 2013;50:57–64. [Google Scholar]
- 11.Wang BH, Sun XX, Dong FY, Zhang F, Niu JX. Cloning and expression analysis of an MYB gene associated with calyx persistence in Korla fragrant pear. Plant Cell Rep. 2014;33:1333–41. doi: 10.1007/s00299-014-1619-2. [DOI] [PubMed] [Google Scholar]
- 12.Qi XX, Wu J, Wang LF, Li LT, Gao YF, Tian LM, Dong XG, Zhang SL. Identifying the candidate genes involved in the calyx abscission process of ‘Kuerlexiangli’ (Pyrus sinkiangensis Yu) by digital transcript abundance measurements. BMC Genomics. 2013;14:727. [DOI] [PMC free article] [PubMed]
- 13.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anisimov SV. Serial Analysis of Gene Expression (SAGE): 13 years of application in research. Curr Pharm Biotechnol. 2008;9:338–50. doi: 10.2174/138920108785915148. [DOI] [PubMed] [Google Scholar]
- 15.Surget-Groba Y, Montoya-Burgos JI. Optimization of de novo transcriptome assembly from next-generation sequencing data. Genome Res. 2010;20:1432–40. doi: 10.1101/gr.103846.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang YG, Zhu J, Dai HY. Characterization of transcriptional differences between columnar and standard apple trees using RNA-Seq. Plant Mol Biol Report. 2012;30:957–65. doi: 10.1007/s11105-011-0396-0. [DOI] [Google Scholar]
- 17.Gleave AP, Ampomah-Dwamena C, Berthold S, Dejnoprat S, Karunairetnam S, Nain B, Wang YY, Crowhurst RN, MacDiarmid RM. Identification and characterisation of primary microRNAs from apple (Malus domestica cv. Royal Gala) expressed sequence tags. Tree Genet Genomes. 2008;4:343–58.
- 18.Feng C, Chen M, Xu CJ, Bai L, Yin XR, Li X, Allan AC, Ferguson IB, Chen KS. Transcriptomic analysis of Chinese bayberry (Myrica rubra) fruit development and ripening using RNA-Seq. BMC Genomics. 2012;13:19. [DOI] [PMC free article] [PubMed]
- 19.Zhang SY, Li X, Feng C, Zhu CQ, Grierson D, Xu CJ, Chen KS. Development and characterization of 109 polymorphic EST-SSRs derived from Chinese bayberry (Myrica rubra) transcriptome. J Am Botany. 2012;99:501–7. [DOI] [PubMed]
- 20.Rowland LJ, Alkharouf N, Darwish O, Ogden EL, Polashock JJ, Bassil NV, Main D. Generation and analysis of blueberry transcriptome sequences from leaves, developing fruit, and flower buds from cold acclimation through deacclimation. BMC Plant Biol. 2012;12:46. [DOI] [PMC free article] [PubMed]
- 21.Li CQ, Wang Y, Huang XM, Li J, Wang HC, Li JG. De novo assembly and characterization of fruit transcriptome in Litchi chinensis Sonn and analysis of differentially regulated genes in fruit in response to shading. BMC Genomics. 2013;14:552. doi: 10.1186/1471-2164-14-552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xie M, Huang Y, Zhang YP, Wang X, Yang H, Yu O, Dai WH, Fang CB. Transcriptome profiling of fruit development and maturation in Chinese white pear (Pyrus bretschneideri Rehd). BMC Genomics. 2013;14:823. [DOI] [PMC free article] [PubMed]
- 23.Sweetman C, Wong DC, Ford CM, Drew DP. Transcriptome analysis at four developmental stages of grape berry (Vitis vinifera cv. Shiraz) provides insights into regulated and coordinated gene expression. BMC Genomics. 2012;13:691. doi: 10.1186/1471-2164-13-691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.D’Hont A, Denoeud F, Aury JM, Baurens FC, Carreel F, Garsmeur O, Noel B, Bocs S, Droc G, Rouard M, Da Silva C, Jabbari K, Cardi C, Poulain J, Souquet M, Labadie K, Jourda C, Lengellé J, Rodier-Goud M, Alberti A, Bernard M, Corr ea M, Ayyampalayam S, Mckain MR, Leebens-Mack J, Burgess D, Freeling M, Mbéguié-A-Mbéguié D, Chabannes M, Wi cker T, et al. The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature. 2012;488:213–7. [DOI] [PubMed]
- 25.Li CY, Deng GM, Yang J, Viljoen A, Jin Y, Kuang RB, Zuo CW, Lv ZC, Yang QS, Sheng O, Wei YR, Hu CH, Dong T, Yi GJ. Transcriptome profiling of resistant and susceptible Cavendish banana roots following inoculation with Fusarium oxysporum f. sp. cubense tropical race 4. BMC Genomics. 2012;13:374. [DOI] [PMC free article] [PubMed]
- 26.Yu KQ, Xu Q, Da XL, Guo F, Ding YD, Deng XX. Transcriptome changes during fruit development and ripening of sweet orange (Citrus sinensis) BMC Genomics. 2012;13:10. doi: 10.1186/1471-2164-13-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu Q, Liu YL, Zhu AD, Wu XM, Ye JL, Yu KQ, Guo WW, Deng XX. Discovery and comparative profiling of microRNAs in a sweet orange red-flesh mutant and its wild type. BMC Genomics. 2010;11:246. [DOI] [PMC free article] [PubMed]
- 28.Zhu H, Xia R, Zhao BY, An YQ, Dardick CD, Callahan AM, Liu ZG. Unique expression, processing regulation, and regulatory network of peach (Prunus persica) miRNAs. BMC Plant Biol. 2012;12:149. [DOI] [PMC free article] [PubMed]
- 29.Sablok G, Luo C, Lee WS, Rahman F, Tatarinova TV, Harikrishna JA, Luo ZR. Bioinformatic analysis of fruit-specific expressed sequence tag libraries of Diospyros kaki Thunb.: view at the transcriptome at different developmental stages. Biotech. 2011;1:35–45. [DOI] [PMC free article] [PubMed]
- 30.Dal Cin V, Velasco R, Ramina A. Dominance induction of fruitlet shedding in Malus × domestica (L. Borkh): molecular changes associated with polar auxin transport. BMC Plant Biol. 2009;9:139. doi: 10.1186/1471-2229-9-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li JG, Yuan RC. NAA and Ethylene regulate expression of genes related to ethylene biosynthesis, perception, and cell wall degradation during fruit abscission and ripening in ‘Delicious’ apples. J Plant Growth Regul. 2008;27:283–95. doi: 10.1007/s00344-008-9055-6. [DOI] [Google Scholar]
- 32.Corbacho J, Romojaro F, Pech JC, Latché A, Gomez-Jimenez MC. Transcriptomic events involved in melon mature-fruit abscission comprise the sequential induction of cell-wall degrading genes coupled to a stimulation of endo and exocytosis. PLoS ONE. 2013 doi: 10.1371/journal.pone.0058363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Agustí J, Merelo P, Cercós M, Tadeo FR, Talón M. Comparative tran-scriptional survey between laser-microdissected cells from laminar abscission zone and petiolar cortical tissue during ethylene-promoted abscission in citrus leaves. BMC Plant Biology. 2009;9:127. doi: 10.1186/1471-2229-9-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu H, Dardick CD, Beers EP, Callanhan AM, Xia R, Yuan RC. Transcriptomics of shading-induced and NAA-induced abscission in apple (Malus domestica) reveals a shared pathway involving reduced photosynthesis, alterations in carbohydrate transport and signaling and hormone crosstalk. BMC Plant Biol. 2011;11:138. doi: 10.1186/1471-2229-11-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parra-Lobato MC, Gomez-Jimenez MC. Polyamine-induced modulation of genes involved in ethylene biosynthesis and signaling pathways and nitric oxide production during olive mature fruit abscission. Exp Bot. 2011;62:4447–65. doi: 10.1093/jxb/err124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sane AP, Tripathi SK, Nath P. Petal abscission in rose (Rosa bourboniana var Gruss an Teplitz) is associated with the enhanced expression of an alpha expansin gene, RbEXPA1. Plant Sci. 2007;172:481–7. doi: 10.1016/j.plantsci.2006.10.005. [DOI] [Google Scholar]
- 37.Tucker ML, Burke A, Murphy CA, Thai VK, Ehrenfried ML. Gene expression profiles for cell wall-modifying proteins associated with soybean cyst nematode infection, petiole abscission, root tips, flowers, apical buds, and leaves. J Exp Bot. 2007;58:3395–406. doi: 10.1093/jxb/erm188. [DOI] [PubMed] [Google Scholar]
- 38.Roongsattham P, Morcillo F, Jantasuriyarat C, Pizot M, Moussu S, Jayaweera D, Collin M, Gonzalez-Carranza ZH, Amblard P, Tregear JW, Tragoonrung S, Verdeil JL, Tranbarger TJ. Temporal and spatial expression of polygalacturonase gene family members reveals divergent regulation during fleshy fruit ripening and abscission in the monocot species oil palm. BMC Plant Biol. 2012;12:150. [DOI] [PMC free article] [PubMed]
- 39.Hong SB, Sexton R, Tucker ML. Analysis of gene promoters for two tomato polygalacturonases expressed in abscission zones and the stigma. Plant Physiol. 2000;123:869–81. doi: 10.1104/pp.123.3.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Imoto K, Yokoyama R, Nishitani K. Comprehensive approach to genes involved in cell wall modifications in Arabidopsis thaliana. Plant Mol Biol. 2005;58:177–92. doi: 10.1007/s11103-005-5344-7. [DOI] [PubMed] [Google Scholar]
- 41.Wu ZC, Burns JK. A beta-galactosidase gene is expressed during mature fruit abscission of ‘Valencia’ orange (Citrus sinensis) J Exp Bot. 2004;55:1483–90. doi: 10.1093/jxb/erh163. [DOI] [PubMed] [Google Scholar]
- 42.Singh AP, Tripathi SK, Nath P, Sane AP. Petal abscission in rose is associated with the differential expression of two ethylene-responsive xyloglucan endotransglucosylase/hydrolase genes, RbXTH1 and RbXTH2. J Exp Bot. 2011;62:5091–103. doi: 10.1093/jxb/err209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taylor JE, Coupe SA, Picton S, Roberts JA. Characterization and accumulation pattern of an mRNA encoding an abscission-related β-1,4-glucanase from leaflets of Sambucus nigra. Plant Mol Biol. 1994;24:961–4. doi: 10.1007/BF00014449. [DOI] [PubMed] [Google Scholar]
- 44.Gou JY, Felippes FF, Liu CJ, Weigel D, Wang JW. Negative regulation of anthocyanin biosynthesis in Arabidopsis by a miR156-targeted SPL transcription factor. Plant Cell. 2011;23:1512–22. doi: 10.1105/tpc.111.084525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Niu Y, Chen KL, Wang JZ, Liu X, Qin HJ, Zhang AM, Wang DW. Molecular and functional characterization of sphingosine-1-phosphate lyase homolog from higher plants. Plant Biol. 2007;49:323–35.
- 46.Reiss U, Oskouian B, Zhou JH, Gupta V, Sooriyakumaran P, Kelly S, Wang E, Merrill AH, Saba JD. Sphingosine-phosphate lyase enhances stress-induced ceramide generation and apoptosis. J Biol Chem. 2003;279:1281–90. [DOI] [PubMed]
- 47.Uimari A, Strommer J. Myb26: a MYB-like protein of pea flowers with affinity for promoters of phenylpropanoid genes. Plant J. 1997;12:1273–84. doi: 10.1046/j.1365-313x.1997.12061273.x. [DOI] [PubMed] [Google Scholar]
- 48.Chen SC, Peng SQ, Huang GX, Wu KX, Fu XH, Chen ZQ. Association of decreased expression of a Myb transcription factor with the TPD (tapping panel dryness) syndrome in Hevea brasiliensis. Plant Mol Biol. 2003;51:51–8. doi: 10.1023/A:1020719420867. [DOI] [PubMed] [Google Scholar]
- 49.Hoeren FU, Dolferus R, Wu Y, Peacock WJ, Dennis ES. Evidence for a role for AtMYB2 in the induction of the Arabidopsis alcohol dehydrogenase gene (ADH1) by low oxygen. Genetics. 1998;149:479–90. doi: 10.1093/genetics/149.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lea US, Slimestad R, Smedvig P, Lillo C. Nitrogen deficiency enhances expression of specific MYB and bHLH transcription factors and accumulation of end products in the flavonoid pathway. Planta. 2007;225:1245–53. doi: 10.1007/s00425-006-0414-x. [DOI] [PubMed] [Google Scholar]
- 51.Payne CT, Zhang F, Lloyd AM. GL3 encodes a bHLH protein that regulates trichome development in Arabidopsis through interaction with GL1 and TTG1. Genetics. 2000;156:1349–62. doi: 10.1093/genetics/156.3.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Suo JF, Liang XO, Pu L, Zhang YS, Xue YB. Identification of GhMYB109 encoding a R2R3 MYB transcription factor that expressed specifically in fiber initials and elongating fibers of cotton (Gossypium hirsutum L.) Biochim Biophys Acta. 2003;1630:25–34. doi: 10.1016/j.bbaexp.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 53.Lee MM, Schiefelbein J. Cell pattern in the Arabidopsis root epidermis determined by lateral inhibition with feedback. The Plant Cell. 2002;14:611–8. doi: 10.1105/tpc.010434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Legay S, Lacombe E, Goicoechea M, Brière C, Séguin A, Mackay J, Grima-Pettenati J. Molecular characterization of EgMYB1, a putative transcriptional repressor of the lignin biosynthetic pathway. Plant Sci. 2007;173:542–9.
- 55.Yang XY, Li JG, Pei M, Gu H, Chen ZL, Qu LJ. Over-expression of a flower-specific transcription factor gene AtMYB24 causes aberrant anther development. Plant Cell Rep. 2007;26:219–28. doi: 10.1007/s00299-006-0229-z. [DOI] [PubMed] [Google Scholar]
- 56.Gubert CM, Christy ME, Ward DL, Groner WD, Liljegren SJ. ASYMMETRIC LEAVES1 regulates abscission zone placement in Arabidopsis flowers. BMC Plant Biol. 2014;14:195. doi: 10.1186/s12870-014-0195-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhong H, Guo QQ, Chen L, Ren F, Wang QQ, Zheng Y, Li XB. Two Brassica napus genes encoding NAC transcription factors are involved in response to high-salinity stress. Plant Cell Rep. 2012;31:1991–2003. [DOI] [PubMed]
- 58.Zhu MK, Chen GP, Zhang JL, Zhang YJ, Xie QL, Zhao ZP, Pan Y, Hu ZL. The abiotic stress-responsive NAC-type transcription factor SlNAC4 regulates salt and drought tolerance and stress-related genes in tomato (Solanum lycopersicum). Plant Cell Rep. 2014;33:1851–63. [DOI] [PubMed]
- 59.Wrzaczek M, Brosché M, Salojärvi J, Kangasjärvi S, Idänheimo N, Mersmann S, Robatzek S, Karpiński S, Karpińska B, Kangasjärvi J. Transcriptional regulation of the CRK/DUF26 group of receptor-like protein kinases by ozone and plant hormones in Arabidopsis. BMC Plant Biol. 2010;10:95. [DOI] [PMC free article] [PubMed]
- 60.Cai SQ, Lashbrook CC. Stamen abscission zone transcriptome profiling reveals new candidates for abscission control: enhanced retention of floral organs in transgenic plants overexpressing Arabidopsis ZINC FINGER PROTEIN2. Plant Physiol. 2008;146:1305–21. doi: 10.1104/pp.107.110908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu ZC, Burns JK. Isolation and characterization of a cDNA encoding a lipid transfer protein expressed in ‘Valencia’ orange during abscission. J Exp Bot. 2003;54:1183–91. doi: 10.1093/jxb/erg112. [DOI] [PubMed] [Google Scholar]
- 62.Cheng YQ, Liu JF, Yang XD, Ma R, Liu CM, Liu Q. RNA-seq analysis reveals ethylene-mediated reproductive organ development and abscission in soybean (Glycine max L. Merr.) Plant Mol Biol Report. 2013;31:607–19. doi: 10.1007/s11105-012-0533-4. [DOI] [Google Scholar]
- 63.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–52. [DOI] [PMC free article] [PubMed]
- 64.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci. 2003;100:9440–5. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010 doi: 10.1186/gb-2010-11-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T, Yamanishi Y. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36:D480–4. [DOI] [PMC free article] [PubMed]
- 67.Mao X, Cai T, Olyarchuk JG, Wei L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics. 2005;21:3787–93. doi: 10.1093/bioinformatics/bti430. [DOI] [PubMed] [Google Scholar]
- 68.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504. [DOI] [PMC free article] [PubMed]