

Abstract
Type 1 diabetes (T1D) is a T cell-mediated autoimmune disease that involves the slow, progressive destruction of islet β-cells and loss of insulin production, as a result of interaction with environmental factors, in genetically susceptible individuals. The gut microbiome is established very early in life. Commensal microbiota establish mutualism with the host and form an important part of the environment to which individuals are exposed in the gut, providing nutrients and shaping immune responses. Here, we studied the impact of targeting most Gram-negative bacteria in the gut of Non obese diabetic (NOD) mice at different time points in their life, using a combination of three antibiotics - neomycin, polymyxin B and streptomycin (NPS), on diabetes development. We found that the prenatal period is a critical time for shaping the immune tolerance in the progeny, influencing development of autoimmune diabetes. Prenatal NPS treatment protected NOD mice from diabetes development through alterations in the gut microbiota, as well as induction of tolerogenic antigen-presenting cells (APCs), which led to reduced activation of diabetogenic CD8 T cells. Most importantly, we found that the protective effect was age-dependent and the most profound protection was found when the mice were treated before birth. This indicates the importance of the prenatal environment and early exposure to commensal bacteria in shaping the host immune system and health.
Introduction
Type 1 diabetes (T1D), caused by a T cell-mediated destruction of islet beta cells, is results from a complex interaction between genetic susceptibility and environmental factors (1-3). The sharp rise of T1D incidence that we have seen in recent years, especially in young children (4, 5), is likely to be due to environmental influences. The gut microbiota are an important element of the environment and may play an important role in the development of T1D. We and others have provided direct evidence to support this notion (6-12). More broadly, changes in gut microbiota composition are associated with the development of a number of proinflammatory disorders (13-17).
Since the discovery of penicillin, whilst antibiotics have saved millions of human lives, these potentially life-saving drugs can alter homeostasis of the gut microbiome. Increasing evidence suggests that disturbances in the gut microbiome may contribute to a number of different health problems including autoimmunity (18-20).
Bacteria are classified into Gram positive (G+) and Gram negative (G−), according to their cell wall composition (21). Most G+ and G− bacteria belong to the Firmicutes and Bacteroidetes phyla. Vancomycin specifically inhibits G+ bacteria (22) and a recent study showed that vancomycin treatment disturbed G+ bacteria and protected from diabetes development in NOD mice (23). It was not clear, however, what the effect of elimination of G− bacteria on diabetes development would be. In this study, we used a combination of neomycin, polymyxin B and streptomycin (NPS) to target most of the G− bacteria in the gut of NOD mice and study the impact on T1D development and the possible mechanism(s). NPS treatment protected NOD mice from diabetes development, through alterations in the gut microbiota, as well as affecting the function of antigen-presenting cells (APCs). More importantly, we found that the protection was age-dependent with the most profound protection occurring when the mice were treated before birth, by administering the antibiotics to the mothers during gestation. Thus, early exposure to commensal bacteria is very important in shaping the host immune system and health.
Materials and Methods
Mice
Female NOD/Caj mice have been maintained at Yale University for many years. BDC2.5NOD and NY8.3 transgenic mice were purchased from the Jackson Laboratory. The mice used in this study were kept in specific pathogen–free conditions in a 12-hour dark/light cycle, in individually-ventilated filter cages with autoclaved food at the Yale University animal facility. The use of the animals in this study was approved by the Yale University Institutional Animal Care and Use Committee.
Antibiotic treatment
The antibiotics neomycin, polymyxin B and streptomycin (NPS) (Sigma) were added to the drinking water at a final concentration of 1mg/ml for neomycin and streptomycin and 1,600U/ml for polymyxin B.
To investigate the period in early life, when mice were most susceptible to the effects of antibiotics, we treated pregnant (plugged) NOD mice with NPS (withdrawing treatment on giving birth) and observed for diabetes development in the offspring. This group was designated as NPS/preg. In a second group, newborn mice from NPS-treated mothers were “sprayed” with a gut bacterial suspension from the feces of adult untreated female NOD mice, once a week for 3 weeks, until the mice were weaned. This group was named NPS+NOD. To further investigate whether NPS could also inhibit diabetes development at later time points, we compared three groups of mice. As in our first experiement, the mice in the NPS/preg group were the offspring of pregnant mice receiving antibiotics in drinking water for 3 weeks from mating until delivery. Mice in the NPS/born group were the newborn mice that received antibiotic through mother milk (the mothers were given antibiotic water for 3 weeks from the date of pups delivery to the date of weaning). Mice in the NPS/wean group were three-week-old mice receiving antibiotic water for 3 weeks from weaning. After the 3- week treatment period, antibiotics were withdrawn from the water in all groups of mice.
Reagents
All fluorochrome-conjugated monoclonal antibodies (mAbs) were purchased from Biolegend or eBioscience. The Luminex kit for cytokine measurement was purchased from Bio-Rad, and antibody-conjugated magnetic beads for T cell and APC purification were from Qiagen and Polyscience.
Ratio of Gram-positive (G+) and Gram-negative (G−) Fecal Bacteria
Bacterial DNA from mouse feces was isolated as previously described [28]. The ratio of the DNA content of G+ and G− bacteria was quantified by qPCR (IQ5, Bio-Rad) using G+ and G− specific forward primers (G+F and G−F) and a universal bacterial reverse primer (sequences in Supplementary table). Total 16S rRNA was used as a positive control and the results were analyzed by the delta-delta CT method after normalization with 16S rRNA. Each sample was analyzed in triplicate and the experiments were repeated twice.
16S rRNA sequencing analysis
Fecal samples were collected from offspring of 4-5 independent breeders and DNA extraction was performed. 16S rRNA V4 and V5 regions of fecal bacteria were amplified from DNA samples (barcoded primer sequences in Supplementary table). PCR products were purified by Qiagen gel extraction kit and DNA concentration was determined with a Nanodrop spectrophotometer and gel electrophoresis. Following dilution to 1×109 molecules/μl, equal volumes (10μl) were mixed together to prepare the library, followed by a final dilution to 1×106 molecules/μl. 20μl of the library was sequenced using GS Junior Titanium Series 454 sequencing system (Roche).
Sequencing results were analyzed with the QIIME software package (http://qiime.org). Taxonomic assignment was performed using representative sequences of each picked operational taxonomic unit (OTU). Beta-diversity was calculated to compare differences between microbial communities and shown as Principal Coordinate Analysis (PCoA).
Cell purification
APCs were purified by removing CD4+ (clone GK1.5) and CD8+ T cells (clone T1B105) using mAb hybridoma supernatants, followed by magnetic bead (conjugated with goat anti-Rat IgG) separation. CD4+ T cells were purified by removing CD8+ T cells (mAb T1B105), MHC class II+ cells (mAb 10.2.16) and B cells (anti-mouse IgM and IgG (Qiagen)) using mAb hybridoma supernatants and magnetic bead separation. CD8 T cells were purified similarly, except that anti-CD4 (clone GK1.5) was used instead of anti-CD8 mAb. Individual B cells, CD11b+ cells and CD11c+ cells were purified by EasySep Mouse B cell enrichment kit, Mouse CD11b positive selection kit and Mouse CD11c positive selection kit II, respectively, from STEMCELL Technology according to manufacturer’s instructions. The purity of the cells was routinely ≥90%, analyzed by flow cytometry.
T cell proliferation assay
Spleens were harvested from mice and red blood cells were lysed to obtain splenocytes. Splenocytes were counted and plated at 1 × 105 cells/well in a volume of 150 μl in 96-well round-bottomed plates. The splenocytes were stimulated with anti-CD3 (clone 2C11 supernatant), at different dilutions, in the presence or absence of anti-CD28 (clone 37.51 supernatant, 1:100). The cells were cultured for 72 hours and the culture supernatants were collected before adding [3H]-thymidine (0.5μci/well). The plates were further incubated for 14-18 hours and the cells were harvested and cpm was counted.
Purified CD4+ BDC2.5 or CD8+ NY8.3 T cells (105/well) were co-cultured with irradiated APCs (5 × 104/well) from 12-week old NOD mice in the presence or absence of BDC2.5 mimotope peptide (RTRPLWVRME) or IGRP206-214 peptide (VYLKTNVFL), respectively. Cell proliferation was determined by stimulation index (SI), which was calculated by [3H]-thymidine incorporation (cpm) with Ag/[3H]-thymidine incorporation (cpm) without Ag.
TLR agonist stimulation
Splenocytes (1×105/well) were cultured in a volume of 150 μl in 96-well round-bottomed plates and stimulated with 0.01-1μg/ml of lipopolysaccharide (LPS) or peptidoglycan (PGN). As for the T cell proliferation assay, [3H]-thymidine was added after 72-hour stimulation and the cell peroliferation was counted in a beta-plate counter and presented as SI, as described above.
Intracellular staining
Intracellular Foxp3 was detected using a Foxp3 staining kit (eBioscience) following the manufacturer’s instructions. For intracellular cytokine (ICC) staining, 106 cells were cultured for 4 hours with 50ng/ml PMA (Sigma), 500ng/ml of ionomycin (Sigma) and 1μl/ml of Golgiplug (BD Bioscience), before staining with antibodies against surface markers. Isotype controls were used to set the gates for flow cytometric analysis. Immune cells were isolated from 12-week-old mice.
Bacterial transfer
New-born pups were “sprayed” with a freshly prepared suspension of mouse feces (from offspring of either untreated or maternally NPS-treated female NOD mice) once a week for the first two weeks. Starting from the third week, a freshly prepared fecal suspension was administered by gavage (containing ~1×106 culturable bacteria) to NOD recipients (either untreated or pre-treated with ampicillin (1mg/ml), metronidazole (1mg/ml), neomycin (1mg/ml) and vancomycin (0.5mg/ml) (AMNV), to deplete most bacteria in the gut). Recipient mice were monitored for diabetes by weekly screening glycosuria with Diastix (Bayer) and diabetes was confirmed by blood glucose measurement (≥250 mg/dl) (FreeStyle, Abbot).
Lymphocyte Adoptive Transfer
Splenocytes from NPS/preg mice were injected into NOD.scid or irradiated NOD recipient mice (107/mouse, i.v.). The recipient mice were monitored for diabetes as described above. APCs from NPS/preg or control NOD mice (3×106/mouse) plus T cells from diabetic NOD mice (3×106/mouse) were injected into 5 week old NOD.scid mice (i.v.). The recipient mice were monitored for diabetes as described above.
Insulitis scoring
Mice were dissected at the prediabetic stage (12 weeks old) and pancreata were collected. Pancreata were fixed in 10% buffered formalin and then paraffin-embedded. Tissues were sectioned and stained with H&E. Insulitis was scored under light microscopy using the following grading: 0-25, no insulitis or affecting <25% of the islet; 25-50, insulitis affecting 25–50% of the islet; 50-75, insulitis affecting 50-75% of the islet; and 75-100, >75% islet infiltration.
Statistics
Statistical analysis was performed using GraphPad Prism software. P values of < 0.05 were considered significant.
Results
Neomycin/Polymyxin/Streptomycin (NPS) treatment has a time-dependent impact on type 1 diabetes development
To investigate the effect of prenatal environment on the offspring, we treated pregnant NOD mice with NPS. We found that diabetes development in the offspring (NPS/preg) of NPS-treated parents was significantly delayed and the incidence was also significantly reduced compared with the offspring from untreated dams (p=0.01; Fig. 1A). It is interesting that by introducing normal gut bacteria soon after birth via “spraying” pups with gut bacterial suspension from non-treated adult NOD mice (NPS+NOD), the “sprayed” pups lost the maternal protective effect related to NPS treatemnt and developed a similar incidence of diabetes to the untreated control group although there was ~3-week delay in diabetes onset (p=0.01 compared to NPS/preg offspring; Fig. 1A).
Fig. 1. Effect of NPS treatment on diabetes development.
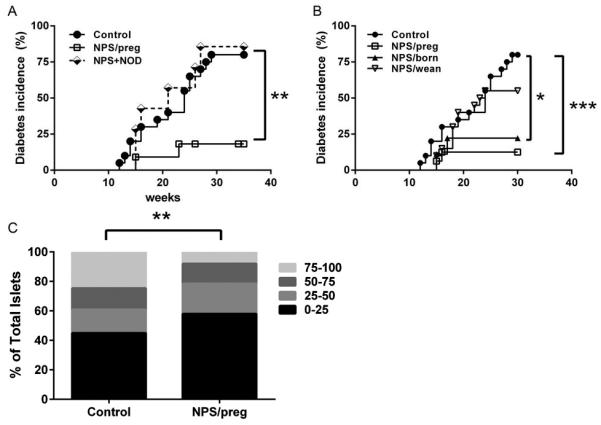
A. Diabetes incidence in NOD and NPS-treated female mice. Mice without NPS treatment (control, n=20, solid circles) or NPS treatment for 3 weeks during gestation (NPS/preg, n=11, open squares) or NPS treatment for 3 weeks during gestation, but the newborn mice received gut microbiota of non-treated adult NOD mice (NPS+NOD, n=7, diamonds). Survival curves were analyzed by log-rank test, **: p<0.01. B. Diabetes incidence in untreated NOD and NPS-treated female mice at different time points. Mice without NPS treatment (control, NOD, n=20, solid circles) or NPS treatment for 3 weeks during gestation (NPS/preg, n=16, open squares) or newborn mice (NPS/born, n=9, solid triangles) or young mice treated after weaning (NPS/wean, n=20, open reverse triangles) are shown. Survival curves were analyzed by log-rank test, *: p<0.05, ***: p<0.001. C. Insulitis in NOD and NPS/preg mice (12 weeks old), n=7-9/group, 264 islets scored. Statistical differences were analyzed by chi-square test, **: p<0.01.
We further investigated the effect of NPS treatment at different time points in the early life of female NOD mice. We compared mice from mothers treated during pregnancy (NPS/preg) with two additional groups of mice - treated just after birth for 3 weeks (NPS/born) and just after weaning for 3 weeks (NPS/wean), and observed the mice for diabetes. Confirming results shown in Fig. 1A, the NPS/preg mice had the most significant decrease of diabetes incidence while NPS/born mice also showed delayed and reduced diabetes onset (p<0.05, Figure 1B). The incidence of diabetes in NPS/wean mice was markedly higher compared to the mice treated during the prenatal or neonatal period, although the overall incidence was still lower (55%) than in the untreated control group (80%, Fig. 1B).
Furthermore, NPS treatment led to significant reduction of insulitis compared to untreated control mice (Fig. 1C).
NPS antibiotics altered gut microbiota
We next investigated the change in composition of gut microbiota induced by antibiotic treatment. As expected, NPS/preg mice had a much higher ratio of G+/G− compared to untreated mice (Fig. 2A), measured by qPCR, which indicated that most of the G− bacteria were depleted by NPS treatment and therefore more G+ bacteria colonized the gut. It is noteworthy that although the NPS/preg mice were treated with NPS in the prenatal period only, the high G+/G− ratio of gut bacteria in these mice persisted even at 6 months of age (Fig. 2A), which suggests that once established, the gut microbiome is stable.
Fig. 2. Change of gut microbiota after NPS treatment.

A. Gram positive : Gram negative bacteria (G+/G−) ratio in fecal samples from mice given NPS during gestation ( NPS/preg) and control NOD mice at different ages by qPCR using specific primers for G+ and G− bacteria. B. PCoA analysis of gut microbiota 16S rRNA sequence data from mice treated with NPS during gestation (NPS/preg, squares in upper left quadrant), NPS in the neonatal period (NPS/born, upward triangles, lower right quadrant), NPS after weaning (NPS/wean, rightward triangles, lower left quadrant) and untreated NOD mice (dots, upper right quadrant). C. Sequencing results showing the proportion of Proteobacteria, as a percentage of total bacteria. Statistical differences were analyzed by t-test, *: p<0.05. D. Comparison of two G+ families, Lachnospiraceae and Coriobacteriaceae, belonging to the Firmicutes phylum, as a percentage of total bacteria. Statistical differences were analyzed by t-test, *: p<0.05.
We further performed pyrosequencing of bacterial 16S rRNA of fecal samples collected from 12-wk old NPS-treated and untreated mice, regardless of the time of NPS treatment. Principal Coordinate Analysis (PCoA) revealed different clusters of gut microbiota from each group of mice shown in Fig. 2B. It is interesting that NOD mice treated with NPS at different time points had distinct profiles of gut microbiota from each other, compared to untreated control NOD mice (dots). Although the incidence of diabetes in mice from the NPS/preg and NPS/born groups was very similar, their gut microbial communities showed very different clustering (Fig. 2B, squares in upper left and upward triangles in lower right quadrant, respectively). Likewise, the incidence of diabetes in NPS/wean mice was similar to the untreated control NOD mice, whereas their gut microbial communities were very different (Fig. 2B, rightward triangels in lower left and dots in upper right quadrant, respectively). This indicated that antibiotic treatment not only significantly shifted the gut bacterial composition, but the timing of the treatment also made a substantial difference, although these differences were not always correlated with diabetes development. The pyrosequencing results showed that a major phylum of G− bacteria, Proteobacteria, were markedly reduced in NPS-treated mice compared to untreated control mice (Fig. 2C). In addition, there were also some changes in the G+ bacteria and we found that among the most abundant G+ Firmicutes, two families, Lachnospiraceae and Coriobacteriaceae, showed significantly higher abundance in NPS/preg mice than in control mice (Fig. 2D).
Diabetes protection induced by NPS treatment could be transferred to new hosts
To investigate if the protection was long-lasting, we adoptively transferred splenocytes from NPS/preg donors to two different recipients. NOD/scid mice have no mature T cells or B cells which makes it a good model to test the diabetogenic function of exogenous lymphocytes. The irradiated NOD mice were treated with a sub-lethal dose of X-rays to temporarily reduce most of the endogenous lymphocytes at the time of the transfer, but these endogenous lymphocytes will reconstitute after X-ray treatment. Donor splenocytes from 12-week-old NPS/preg mice and age and sex-matched untreated mice (non-diabetic) were used as controls. As shown in Figure 3, diabetes development was significantly delayed and reduced in both NOD.scid (Fig.3A) and irradiated NOD (Fig.3B) recipient mice that were injected with splenocytes from NPS/preg mice, compared to the mice injected with splenocytes from untreated NOD mice.
Fig. 3. Diabetes development by adoptive transfer.
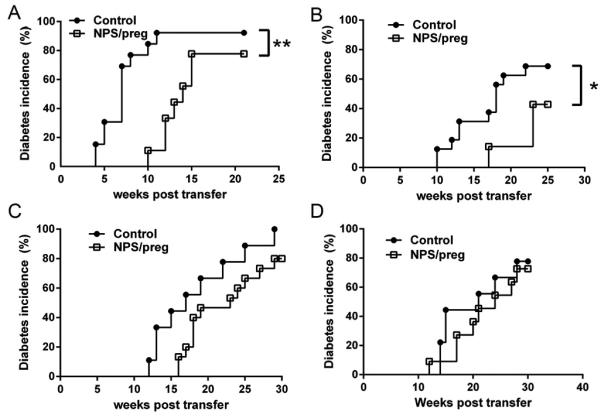
Transfer of immune cells: total splenocytes (1×107) from offspring of untreated NOD (control) or NPS/preg (treated during gestation) NOD mice were isolated and adoptively transferred to either NOD.scid (A) (Control, n=13, NPS/preg, n=9) or irradiated NOD mice (B) (Control, n=16, NPS/preg, n=7). Statistical differences were analyzed by log-rank test, *: p<0.05, **: p<0.01.
Transfer of gut microbiota: The recipients are the offspring of untreated or AMNV treated (during pregnancy) mothers as described in Materials and Methods. Total gut microbiota from either non-diabetic 12 week old control NOD or NPS/preg mice were transferred to offspring of AMNV treated mothers (C) (Control, n=9, NPS/preg, n=15, p=0.07) or offspring of untreated NOD mothers (D) (Control, n=9, NPS/preg, n=11). Statistical differences were analyzed by log-rank test.
We also tested the protective effect of gut microbiota by fecal transplant experiments. To avoid the effects of existing gut commensals, we first treated the pregnant NOD mice with AMNV until delivery, to mimic a relatively commensal-free prenatal environment (more than 90% of the gut bacteria were depleted by AMNV treatment in the dams, comfirmed by CFU analysis of feces, data not shown). We also set up an untreated pregnant NOD control group. Soon after birth, the offspring from each group were given gut microbiota from diabetes-free 12-week old NPS/preg mice. Gut microbiota from unmanipulated non-diabetic 12-week old NOD mice were used as a control. Diabetes development in the offspring treated with gut microbiota from NPS/preg mice was delayed and decreased, compared to the controls (the offspring that received gut microbiota from normal adult NOD mice). This phenomenon was more clearly seen in the group of recipient mice whose mothers had been treated by AMNV in pregnancy (Fig. 3C, p=0.06), compared with the group of recipient mice born to untreated mothers (Fig. 3D). These results indicated that diabetes protection was transferable by altered gut microbiota if endogenous microbiota of the recipients was limited.
Effects of NPS on T cell function
To investigate which immune cells contribute to this protection from diabetes in addition to the altered gut microbiota, we isolated the lymphocytes from spleen, MLN, PLN and Peyer’s patches (PP) to study the expression of inflammatory cytokines. There were lower frequencies of both CD4 and CD8 MLN T cells expressing intracellular IFNγ in NPS/preg mice (Figure 4A). Consistent with the reduced frequency of IFNγ producing cells, the frequency of cells expressing T-bet, a “master” transcription factor for IFNγ production, was also significantly lower in MLN T cells from NPS/preg mice compared with untreated control mice. Similar results were seen in PLN (Fig. 4B) and splenic T cells. Moreover, we found a reduction in the frequency of intracellular IL-17 producing CD4+ and CD8+ T cells from NPS/preg mice (data not shown). The frequency of inflammatory cytokine-producing lymphocytes in PP of NPS mice was also reduced (data not shown).
Fig. 4. Functional characterization of T cells from NPS/preg mice.

A. Percentage of CD4+ and CD8+ IFNg-expressing T cells in MLN from NOD and NPS/preg mice. Statistical differences were analyzed by t-test *: p<0.05, **: p<0.01. B. Percentage of CD4+ and CD8+ T-bet-expressing T cells in PLN from NOD and NPS/preg groups. Statistical differences were analyzed by t-test, *: p<0.05, ****:p<0.0001. C. T cell proliferation in response to anti-CD3 stimulation. Splenocytes from NPS/preg mice and untreated control NOD mice were stimulated with anti-CD3 at different concentrations with or without anti-CD28. T cell proliferation was determined by 3H-thymidine incorporation and calculated as stimulation index (CPM in the presence of anti-CD3 + anti-CD28/CPM in the absence of antibodies). D. IL-17 and IFN-g secretion from the culture supernatants in C. Statistical differences were analyzed by t-test, *: p<0.05, ****: p<0.0001. E. Representative flow cytometric plots of regulatory T cells from splenocytes of untreated NOD (Control) or NPS/preg mice. The total number of CD25+FoxP3+ cells in gated CD4+ T cells from splenocytes, MLN and PLN cells is shown. N=4/group. Statistical differences were analyzed by t-test, *: p<0.05, **: p<0.01.
We then tested the effect of the antibiotic treatment on T cell function following stimulation in vitro, and found that although prenatal NPS treatment did not alter T cell proliferation (Fig. 4C), the T cells secreted less inflammatory cytokines, IFNγ and IL-17 (Fig. 4D).
To investigate whether regulatory T cells (Tregs) contributed to this protection, we analyzed Foxp3+ Tregs in 12-week old NPS/preg mice. We found that the frequency of Foxp3+ Treg cells was significantly increased in all the lymphoid tissues examined (spleen, MLN and PLN). More importantly, the absolute number of Foxp3+ Treg cells was also significantly increased in spleen, MLN and PLN of NPS/preg mice (Fig. 4E, representing FACS plot showing the frequency of Foxp3+CD25+ Treg cells in splenocytes).
Effect of NPS treatment on APCs
We next investigated the APCs from 12-week old mice. The percentages of IFN-γ-producing splenic CD11c+ and IL-12 and IL-17-producing CD11b+ cells were reduced in NPS/preg mice (Fig. 5A-D). Furthermore, the CD11b+ cells expressing the immunoregulatory cytokine IL-10 were increased, with a significantly higher frequency in the NPS/preg group (Fig. 5E). This strongly suggested that APCs may contribute to diabetes protection by NPS treatment.
Fig. 5. Cytokine expression profiles of antigen-presenting cells.

A: Representative FACS plot of IFNγ+ cells gated on CD11c+ cells from control NOD or NPS/preg mice. B: The percentage of IFN-g+ CD11c+ cells in splenocytes from control NOD or NPS/preg mice (n=4/group). Statistical analysis was performed by t-test, *: P<0.05. C-E: The percentage of CD11b+IL-12+ (C), CD11b+IL-17+ (D) and CD11b+IL-10+ (E) cells between control NOD and NPS/preg (n=4/group). Statistical analysis was performed by t-test, *: P<0.05, **: p<0.01, ***: p<0.001.
It is known that TLRs are expressed mostly on antigen presenting cells. Therefore, to test the effects of NPS treatment on the function of APCs, we stimulated splenocytes from NPS/preg mice with lipopolysaccharide (LPS), which is derived from the cell walls of G− bacteria, and peptidoglycan (PGN), the major component of G+ bacteria. Our results showed that APCs from NPS/preg mice had reduced responses to LPS but mildly enhanced responses to PGN stimulation (Fig. 6A+B). We further tested antigen-presenting function, using irradiated splenocytes as APCs to present antigen and stimulate the proliferation of BDC2.5 CD4+ and NY8.3 CD8+ diabetogenic T cells. It is interesting that NPS treatment did not affect antigen presentation and stimulation of CD4+ T cells (Figure 6C), but antigen presentation and stimulation of CD8+ T cells was significantly impaired (Figure 6D).
Fig. 6. Functional study of APC.
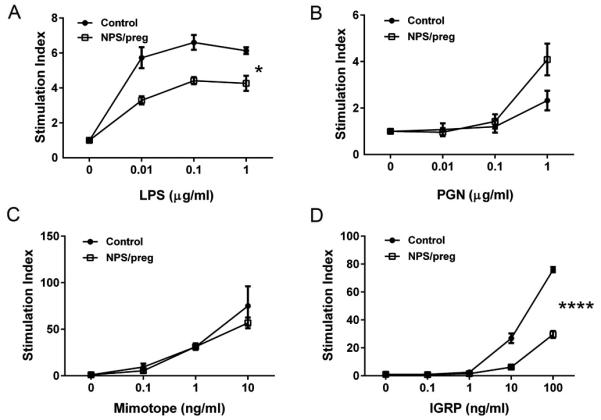
A. LPS stimulation: splenocytes from control NOD or NPS/preg mice were cultured in the presence of LPS at different concentrations for 3 days. The proliferation was determined by 3H-Thymidine incorporation and presented as stimulation index (n=4/group). B. PGN stimulation: The same proliferation assay was performed in the presence of PGN (n=4/group). C & D. Islet autoantigen presentation assays. Purified APCs (5 ×104/well) from splenocytes of control NOD (n=7) or NPS/preg (n=6) NOD mothers were cultured with purified BDC2.5 CD4+ T cells (1×105/well) in the presence of absence of BDC2.5 mimotope peptide for 72 hours. Antigen-presenting function was determined by activation and proliferation of BDC2.5 T cells (C). In a separate set of experiments, purified NY8.3 CD8+ T cells (1×105/well) were tested in the presence or absence of IGRP206-214 peptide. CD8 T cell proliferation to IGRP206-214 peptide presented by APCs from control NOD or NPS/preg mice (D). Statistical differences were analyzed by two-way ANOVA, *: p<0.05, ****: p<0.0001.
To further dissect which APC subset was responsible for the reduced antigen presentation to CD8+ T cells, we purified B cells, dendritic cells (DCs) and macrophages (Mφs) from the spleens of NPS/preg and control NOD mice and tested their antigen-presenting function to NY8.3 CD8+ T cells. NPS treatment suppressed the antigen-presenting function of all three major APCs (Fig.7A-C). This alteration in APC function could be mediated by the reduced production of IL-12, IFN-γ and IL-17 but increased production of IL-10 from the APCs, shown earlier.
Fig. 7.
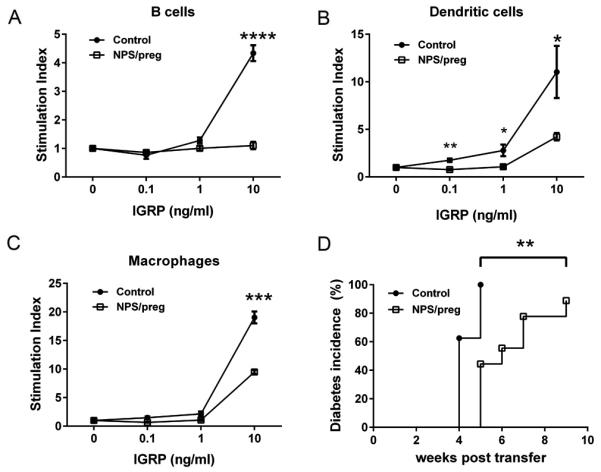
Tolerogenic function of APC in vitro and in vivo. In vitro: splenic B cells (A), dendritic cells (B) and macrophages (C) were purified from control NOD or NPS/preg mice (N=4/group). Irradiated B cells (5×104), dendritic cells (5×103), and macrophages (5×103) were incubated with purified CD8+ T cells (1×105) from NY8.3 NOD mice, together with different concentrations of IGRP206-214 peptide as described in Fig.6. Statistical differences were analyzed by t-test - *: p<0.05, **: p<0.01, ***: p<0.001, ****, p<0.0001. (D) In vivo: Purified total splenic APCs were isolated either from control NOD or NPS/preg mice, and purified total splenic T cells were isolated from diabetic NOD mice. Three million APCs and two million T cells were mixed and transferred to 5-week-old NOD.scid mice (n=9/group). Diabetes development was monitored twice a week by glycosuria and confirmed by blood glucose ≥ 250mg/dl. Statistical differences were analyzed by log-rank test, **: p<0.01.
APC function in vivo
To confirm that the protection from T1D development by NPS treatment was mediated by APCs, we isolated total splenic APCs (T cell-depleted splenocytes) either from NPS/preg or control NOD mice and co-transferred these APCs with diabetogenic T cells (from splenocytes of diabetic NOD mice) into NOD.scid recipients. Diabetes development in the NOD.scid mice was significantly delayed and reduced in the recipients that received APC from NPS/preg donors, compared to the recipients that received APC from control NOD mice (Fig.7D).
Discussion
The most important finding of our study is the critical role of prenatal environment in establishing long-term immune tolerance, mediated by gut microbiota and antigen presenting cells, to islet beta cell autoimmunity in the offspring. We have treated NOD mice with antibiotics at different times, for a defined 3-wk period, in early life and found that when mice were treated during pregnancy, the offspring were significantly protected from diabetes development. Similar protection was also seen when treatment was given soon after birth, but there was no effect when the antibiotics were given after the pups were weaned (21-day old). The protected mice had better glucose tolerance and immunologically, inflammatory cytokines were reduced in both T cells and APCs but accompanied with increased Foxp3+ Treg cells. Moreover, the protective effect was transferable, not only by the gut microbiota, but also by APCs from the protected mice.
Is the type of antibiotic important? Recent studies have demonstrated that colonization with specific bacteria in the gut can protect mice from developing T1D. These bacteria include SFB (Segmented Filamentous Bacteria) (24), Lactobacillus johnsonii N6.2 (25), as well as Streptococcal extracts (26), and glycoprotein extracts from Klebsiella pneumoniae (27). Most of these are G+ bacteria. Concordant with these findings, in our laboratory, NOD mice treated with vancomycin, which depletes most G+ bacteria, have accelerated T1D development (unpublished). However, a recent study reported that vancomycin treatment propagated G− Akkermansia muciniphila and reduced diabetes development in NOD mice (23). Previous studies have not shown the influence of G− bacteria on diabetes development. Therefore, to investigate the effects of altering the balance of G+ and G− bacteria in the gut, we treated the NOD mice with a cocktail of antibiotics to target mainly G− bacteria. Polymyxin B is selectively toxic to G− bacteria due to specificity for the LPS that exists within many G− outer membranes (28). Streptomycin and neomycin are protein synthesis inhibitors which are antibiotics with broader spectrum but have excellent activity against G− bacteria, and partial activity against G+ bacteria (29, 30). Our current study supported our hypothesis that G+/G- balance is very important to diabetes development. NPS cocktail treatment would skew the gut microbial composition away from “diabetogenic” bacteria and NPS resistant bacteria in the gut may have a modulatory effect on immune cells including APCs and Tregs.
Is the timing of antibiotic administration important? In human study, researchers have found distinguishing differences in the gene expression patterns in utero in infants born in contrasting standards of living and hygiene(31). This suggests the environmental factors can shape the immunity from neonatal stage and play a role in the susceptibility or protection towards autoimmune disease. Brugman and colleagues found that BB rats treated from the time of weaning, with sulphamethoxazole, trimethoprim and colistin sulphate, that decrease both G− and G+ bacteria, had reduced insulitis and delayed diabetes onset (32). Our study suggests that timing of exposure to antibiotics is very important. In our model system, the strongest effects were seen when antibiotics were administered in the prenatal period. It is interesting to note that diabetes protection declined when treatment was started later and suggests that the critical time window is during prenatal and neonatal periods. Our results also support the study by Hansen and colleagues, using different antibiotics, who found a significant reduction in diabetes development when NOD mice were treated with Vancomycin from birth until weaning. However, when 8-week old mice were treated, diabetes reduction became less obvious (23). In focusing on the prenatal period, Tormo-Badia and colleagues also treated pregnant female mice with a cocktail of metronidazole, neomycin and polymyxin and found an increased incidence of diabetes in the offspring, although in their study, the treatment did not affect the time of diabetes onset (33). Unfortunately, the authors did not investigate other time point(s) and thus, the effect of the treatment cannot be compared with diabetes incidence at other times in their colony (33). Another recent study in pregnant females, showed that a gluten-free diet reduced the incidence of diabetes in the offspring (34) – but again this was not directly compared with effects of treatment later in life (34). In our study, we sought to systematically test treatment in the 3 early stages of life, during pregnancy, from birth to weaning and after weaning. Our results showed that increasing the G+ to G− ratio reduced diabetes incidence when antibiotics were given during pregnancy and in the neonatal period, and the bacterial composition was altered. Furthermore, this pattern of bacterial colonization was persistent, confirming the importance of bacterial flora establishment in early life, as has been shown in humans (35). The treatment increased the G+ Firmicutes, Lachnospiraceae and Coriobacteriaceae, and the bacterial change associated with protection from diabetes concords with the observation of a decrease in Firmicutes to Bacteroidetes ratio found in children who developed diabetes compared to healthy controls (36, 37). That the bacterial composition played a role in the reduction of diabetes was evident when bacterial transfer induced a delay and reduction in the onset of diabetes in the recipient mice.
How does the alteration in bacteria induce protection from diabetes? Lau and colleagues reported that introducing Lactobacillus johnsonii strain N6.2 to BBDP rats resulted in diabetes protection, mediated by enhanced Th17 cells (25). We found that changing the composition of gut microbiota affected the immune system at multiple levels. These included induction of tolerogenic APCs, with reduced antigen presentation to pathogenic CD8 T cells, an increase in Tregs and an overall reduction of the inflammatory cytokine milieu. This was seen not only in the gut-associated lymphoid tissue but also in other lymphoid organs. These changes are likely to have been effected in the period when the immune system is more sensitive to environmental changes. It emphasizes the importance of early events in shaping of immune responses and determining onset of autoimmunity. Importantly, the immuno-tolerance induced by NPS treatment was transferrable to naïve hosts and intriguingly, APCs were the major contributor to this transferable protection.
These results have important implications for considerations of the environmental effects on the development of autoimmunity. Our study emphasizes the importance of the composition of gut microbiota that is established from birth. There is an increase in diabetes in those born by Caesarean section (38). The composition of gut bacteria is dependent on the mode of delivery, with gut microbiota in babies born by vaginal delivery more closely resembling vaginal flora but in those born by Cesarean section the microbiota are similar to skin flora (39). Our study highlights another way that gut microbiota established at birth may be altered to a more protective type of flora and that changing the G+ to G− bacterial balance early in life was beneficial in protecting mice from diabetes. It is likely that although individual strains may play important roles, because of heterogeneity and the gut microbe ecosystem, no single strain will be identified as being universally protective. However, discovering any such bacteria will allow further study, to increase understanding of gut microbiota interaction with the host immune system. Furthermore, identifying how bacterial metabolites may affect differentiation of the immune cells, especially APCs, and contribute to maintenance of homeostasis and self tolerance will be particularly important.
In summary, we have made three significant findings. Firstly, targeting most of the G− bacteria and some G+ bacteria by NPS treatment early in life significantly protected the offspring from diabetes, which indicates that early-life antibiotic usage has a long-lasting effect. Secondly, the antibiotic effect is time dependent, with better protection from diabetes development, if gut microbiota is altered from birth. Lastly, we showed that changing the composition of gut mictobiota induced tolerogenic APCs, which not only failed to activate diabetogenic CD8 T cells but also transferred diabetes protection to naïve NOD.scid and irradiated NOD recipients. It is not yet clear how removal of G− gut bacteria induced tolerogenic APCs; however, this finding opens a new area of research which may also benefit other autoimmune diseases. Ultimately, we hope to develop probiotic treatment to provide simple and effective measures of promoting symbiosis of the gut microbiome with the host and maintaining a healthy “inner” environment.
Supplementary Material
Acknowledgments
This project was funded by a JDRF innovative research grant (5-2010-664), NIH (RC1DK-087699), NIH (RO1DK088181), NIH (RO1DK092882) and the Yale Diabetes Research Center (P30-DK-45735).
References
- 1.Redondo MJ, Yu L, Hawa M, Mackenzie T, Pyke DA, Eisenbarth GS, Leslie RD. Heterogeneity of type I diabetes: analysis of monozygotic twins in Great Britain and the United States. Diabetologia. 2001;44:354–362. doi: 10.1007/s001250051626. [DOI] [PubMed] [Google Scholar]
- 2.Metcalfe KA, Hitman GA, Rowe RE, Hawa M, Huang X, Stewart T, Leslie RD. Concordance for type 1 diabetes in identical twins is affected by insulin genotype. Diabetes Care. 2001;24:838–842. doi: 10.2337/diacare.24.5.838. [DOI] [PubMed] [Google Scholar]
- 3.Babaya N, Nakayama M, Eisenbarth GS. The stages of type 1A diabetes. Ann N Y Acad Sci. 2005;1051:194–204. doi: 10.1196/annals.1361.061. [DOI] [PubMed] [Google Scholar]
- 4.Marcovecchio ML, Tossavainen PH, Dunger DB. Prevention and treatment of microvascular disease in childhood type 1 diabetes. Br Med Bull. 2010;94:145–164. doi: 10.1093/bmb/ldp053. [DOI] [PubMed] [Google Scholar]
- 5.Patterson CC, Gyurus E, Rosenbauer J, Cinek O, Neu A, Schober E, Parslow RC, Joner G, Svensson J, Castell C, Bingley PJ, Schoenle E, Jarosz-Chobot P, Urbonaite B, Rothe U, Krzisnik C, Ionescu-Tirgoviste C, Weets I, Kocova M, Stipancic G, Samardzic M, de Beaufort CE, Green A, Dahlquist GG, Soltesz G. Trends in childhood type 1 diabetes incidence in Europe during 1989-2008: evidence of non-uniformity over time in rates of increase. Diabetologia. 2012;55:2142–2147. doi: 10.1007/s00125-012-2571-8. [DOI] [PubMed] [Google Scholar]
- 6.Brown CT, Davis-Richardson AG, Giongo A, Gano KA, Crabb DB, Mukherjee N, Casella G, Drew JC, Ilonen J, Knip M, Hyoty H, Veijola R, Simell T, Simell O, Neu J, Wasserfall CH, Schatz D, Atkinson MA, Triplett EW. Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PLoS One. 2011;6:e25792. doi: 10.1371/journal.pone.0025792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cerf-Bensussan N, Gaboriau-Routhiau V. The immune system and the gut microbiota: friends or foes? Nat Rev Immunol. 2010;10:735–744. doi: 10.1038/nri2850. [DOI] [PubMed] [Google Scholar]
- 8.King C, Sarvetnick N. The incidence of type-1 diabetes in NOD mice is modulated by restricted flora not germ-free conditions. PLoS One. 2011;6:e17049. doi: 10.1371/journal.pone.0017049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Manirarora JN, Parnell SA, Hu YH, Kosiewicz MM, Alard P. NOD dendritic cells stimulated with Lactobacilli preferentially produce IL-10 versus IL-12 and decrease diabetes incidence. Clin Dev Immunol. 2011;2011:630187. doi: 10.1155/2011/630187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9:313–323. doi: 10.1038/nri2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Valladares R, Sankar D, Li N, Williams E, Lai KK, Abdelgeliel AS, Gonzalez CF, Wasserfall CH, Larkin J, Schatz D, Atkinson MA, Triplett EW, Neu J, Lorca GL. Lactobacillus johnsonii N6.2 mitigates the development of type 1 diabetes in BB-DP rats. PLoS One. 2010;5:e10507. doi: 10.1371/journal.pone.0010507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, Hu C, Wong FS, Szot GL, Bluestone JA, Gordon JI, Chervonsky AV. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature. 2008;455:1109–1113. doi: 10.1038/nature07336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Atkinson MA, Chervonsky A. Does the gut microbiota have a role in type 1 diabetes? Early evidence from humans and animal models of the disease. Diabetologia. 2012;55:2868–2877. doi: 10.1007/s00125-012-2672-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Couturier-Maillard A, Secher T, Rehman A, Normand S, De Arcangelis A, Haesler R, Huot L, Grandjean T, Bressenot A, Delanoye-Crespin A, Gaillot O, Schreiber S, Lemoine Y, Ryffel B, Hot D, Nunez G, Chen G, Rosenstiel P, Chamaillard M. NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J Clin Invest. 2013;123:700–711. doi: 10.1172/JCI62236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duca FA, Sakar Y, Lepage P, Devime F, Langelier B, Dore J, Covasa M. Replication of obesity and associated signaling pathways through transfer of microbiota from obese-prone rats. Diabetes. 2014;63:1624–1636. doi: 10.2337/db13-1526. [DOI] [PubMed] [Google Scholar]
- 16.Hara N, Alkanani AK, Ir D, Robertson CE, Wagner BD, Frank DN, Zipris D. The role of the intestinal microbiota in type 1 diabetes. Clin Immunol. 2013;146:112–119. doi: 10.1016/j.clim.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 17.Pachikian BD, Neyrinck AM, Deldicque L, De Backer FC, Catry E, Dewulf EM, Sohet FM, Bindels LB, Everard A, Francaux M, Guiot Y, Cani PD, Delzenne NM. Changes in intestinal bifidobacteria levels are associated with the inflammatory response in magnesium-deficient mice. J Nutr. 2010;140:509–514. doi: 10.3945/jn.109.117374. [DOI] [PubMed] [Google Scholar]
- 18.Boerner BP, Sarvetnick NE. Type 1 diabetes: role of intestinal microbiome in humans and mice. Ann N Y Acad Sci. 2011;1243:103–118. doi: 10.1111/j.1749-6632.2011.06340.x. [DOI] [PubMed] [Google Scholar]
- 19.Cenit MC, Matzaraki V, Tigchelaar EF, Zhernakova A. Rapidly expanding knowledge on the role of the gut microbiome in health and disease. Biochim Biophys Acta. 2014;1842:1981–1992. doi: 10.1016/j.bbadis.2014.05.023. [DOI] [PubMed] [Google Scholar]
- 20.Myles IA, Fontecilla NM, Janelsins BM, Vithayathil PJ, Segre JA, Datta SK. Parental dietary fat intake alters offspring microbiome and immunity. J Immunol. 2013;191:3200–3209. doi: 10.4049/jimmunol.1301057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Woese CR. Bacterial evolution. Microbiol Rev. 1987;51:221–271. doi: 10.1128/mr.51.2.221-271.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Levine DP. Vancomycin: a history. Clin Infect Dis. 2006;42(Suppl 1):S5–12. doi: 10.1086/491709. [DOI] [PubMed] [Google Scholar]
- 23.Hansen CH, Krych L, Nielsen DS, Vogensen FK, Hansen LH, Sorensen SJ, Buschard K, Hansen AK. Early life treatment with vancomycin propagates Akkermansia muciniphila and reduces diabetes incidence in the NOD mouse. Diabetologia. 2012;55:2285–2294. doi: 10.1007/s00125-012-2564-7. [DOI] [PubMed] [Google Scholar]
- 24.Kriegel MA, Sefik E, Hill JA, Wu HJ, Benoist C, Mathis D. Naturally transmitted segmented filamentous bacteria segregate with diabetes protection in nonobese diabetic mice. Proc Natl Acad Sci U S A. 2011;108:11548–11553. doi: 10.1073/pnas.1108924108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lau K, Benitez P, Ardissone A, Wilson TD, Collins EL, Lorca G, Li N, Sankar D, Wasserfall C, Neu J, Atkinson MA, Shatz D, Triplett EW, Larkin J., 3rd Inhibition of type 1 diabetes correlated to a Lactobacillus johnsonii N6.2-mediated Th17 bias. J Immunol. 2011;186:3538–3546. doi: 10.4049/jimmunol.1001864. [DOI] [PubMed] [Google Scholar]
- 26.Satoh J, Shintani S, Oya K, Tanaka S, Nobunaga T, Toyota T, Goto Y. Treatment with streptococcal preparation (OK-432) suppresses anti-islet autoimmunity and prevents diabetes in BB rats. Diabetes. 1988;37:1188–1194. doi: 10.2337/diab.37.9.1188. [DOI] [PubMed] [Google Scholar]
- 27.Sai P, Rivereau AS. Prevention of diabetes in the nonobese diabetic mouse by oral immunological treatments. Comparative efficiency of human insulin and two bacterial antigens, lipopolysacharide from Escherichia coli and glycoprotein extract from Klebsiella pneumoniae. Diabetes Metab. 1996;22:341–348. [PubMed] [Google Scholar]
- 28.Dixon RA, Chopra I. Polymyxin B and polymyxin B nonapeptide alter cytoplasmic membrane permeability in Escherichia coli. J Antimicrob Chemother. 1986;18:557–563. doi: 10.1093/jac/18.5.557. [DOI] [PubMed] [Google Scholar]
- 29.Singh B, Mitchison DA. Bactericidal activity of streptomycin and isoniazid against tubercle bacilli. Br Med J. 1954;1:130–132. doi: 10.1136/bmj.1.4854.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Obojska K, Drabikowska AK. Studies on the mechanism of resistance of Pseudomonas aeruginosa to neomycin. II. Correlation between neomycin resistance and hemoprotein concentration. Acta Microbiol Pol. 1981;30:123–131. [PubMed] [Google Scholar]
- 31.Kallionpaa H, Laajala E, Oling V, Harkonen T, Tillmann V, Dorshakova NV, Ilonen J, Lahdesmaki H, Knip M, Lahesmaa R. Standard of hygiene and immune adaptation in newborn infants. Clin Immunol. 2014;155:136–147. doi: 10.1016/j.clim.2014.09.009. [DOI] [PubMed] [Google Scholar]
- 32.Brugman S, Klatter FA, Visser JT, Wildeboer-Veloo AC, Harmsen HJ, Rozing J, Bos NA. Antibiotic treatment partially protects against type 1 diabetes in the Bio-Breeding diabetes-prone rat. Is the gut flora involved in the development of type 1 diabetes? Diabetologia. 2006;49:2105–2108. doi: 10.1007/s00125-006-0334-0. [DOI] [PubMed] [Google Scholar]
- 33.Tormo-Badia N, Hakansson A, Vasudevan K, Molin G, Ahrne S, Cilio CM. Antibiotic treatment of pregnant non-obese diabetic (NOD) mice leads to altered gut microbiota and intestinal immunological changes in the offspring. Scand J Immunol. 2014;80:250–260. doi: 10.1111/sji.12205. [DOI] [PubMed] [Google Scholar]
- 34.Hansen CH, Krych L, Buschard K, Metzdorff SB, Nellemann C, Hansen LH, Nielsen DS, Frokiaer H, Skov S, Hansen AK. A maternal gluten-free diet reduces inflammation and diabetes incidence in the offspring of NOD mice. Diabetes. 2014;63:2821–2832. doi: 10.2337/db13-1612. [DOI] [PubMed] [Google Scholar]
- 35.Salminen S, Gibson GR, McCartney AL, Isolauri E. Influence of mode of delivery on gut microbiota composition in seven year old children. Gut. 2004;53:1388–1389. doi: 10.1136/gut.2004.041640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de Goffau MC, Luopajarvi K, Knip M, Ilonen J, Ruohtula T, Harkonen T, Orivuori L, Hakala S, Welling GW, Harmsen HJ, Vaarala O. Fecal microbiota composition differs between children with beta-cell autoimmunity and those without. Diabetes. 2013;62:1238–1244. doi: 10.2337/db12-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murri M, Leiva I, Gomez-Zumaquero JM, Tinahones FJ, Cardona F, Soriguer F, Queipo-Ortuno MI. Gut microbiota in children with type 1 diabetes differs from that in healthy children: a case-control study. BMC Med. 2013;11:46. doi: 10.1186/1741-7015-11-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cardwell CR, Stene LC, Joner G, Cinek O, Svensson J, Goldacre MJ, Parslow RC, Pozzilli P, Brigis G, Stoyanov D, Urbonaite B, Sipetic S, Schober E, Ionescu-Tirgoviste C, Devoti G, de Beaufort CE, Buschard K, Patterson CC. Caesarean section is associated with an increased risk of childhood-onset type 1 diabetes mellitus: a meta-analysis of observational studies. Diabetologia. 2008;51:726–735. doi: 10.1007/s00125-008-0941-z. [DOI] [PubMed] [Google Scholar]
- 39.Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, Knight R. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A. 2010;107:11971–11975. doi: 10.1073/pnas.1002601107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.