

Abstract
Background
In frontotemporal dementia (FTD) there is a critical lack in the understanding of biological and molecular mechanisms involved in disease pathogenesis. The heterogeneous genetic features associated with FTD suggest that multiple disease-mechanisms are likely to contribute to the development of this neurodegenerative condition.
We here present a systems biology approach with the scope of i) shedding light on the biological processes potentially implicated in the pathogenesis of FTD and ii) identifying novel potential risk factors for FTD. We performed a gene co-expression network analysis of microarray expression data from 101 individuals without neurodegenerative diseases to explore regional-specific co-expression patterns in the frontal and temporal cortices for 12 genes (MAPT, GRN, CHMP2B, CTSC, HLA-DRA, TMEM106B, C9orf72, VCP, UBQLN2, OPTN, TARDBP and FUS) associated with FTD and we then carried out gene set enrichment and pathway analyses, and investigated known protein-protein interactors (PPIs) of FTD-genes products.
Results
Gene co-expression networks revealed that several FTD-genes (such as MAPT and GRN, CTSC and HLA-DRA, TMEM106B, and C9orf72, VCP, UBQLN2 and OPTN) were clustering in modules of relevance in the frontal and temporal cortices. Functional annotation and pathway analyses of such modules indicated enrichment for: i) DNA metabolism, i.e. transcription regulation, DNA protection and chromatin remodelling (MAPT and GRN modules); ii) immune and lysosomal processes (CTSC and HLA-DRA modules), and; iii) protein meta/catabolism (C9orf72, VCP, UBQLN2 and OPTN, and TMEM106B modules). PPI analysis supported the results of the functional annotation and pathway analyses.
Conclusions
This work further characterizes known FTD-genes and elaborates on their biological relevance to disease: not only do we indicate likely impacted regional-specific biological processes driven by FTD-genes containing modules, but also do we suggest novel potential risk factors among the FTD-genes interactors as targets for further mechanistic characterization in hypothesis driven cell biology work.
Electronic supplementary material
The online version of this article (doi:10.1186/s13024-016-0085-4) contains supplementary material, which is available to authorized users.
Keywords: Frontotemporal dementia, Gene expression, Co-expression, Network, Systems biology, DNA protection, Immune system, Protein catabolism
Background
Frontotemporal dementia (FTD) is the second most common early onset form of dementia after Alzheimer’s disease (AD) [1]. Its main clinical presentations (behavioural or language variants) directly reflect atrophy patterns in the frontal or temporal lobes [2]. Sub-cortical regions are also implicated [3–5] as damage to white matter (uncinated fasciculus, cingulum bundle and corpus callosum) [6, 7] and deep grey matter structures (putamen, insula, thalamus and hippocampus) have been recently reported [8, 9].
The majority of cases show either tau (FTLD-tau) or ubiquitin/TDP-43 (FTLD-TDP) inclusions (≤40–50 %), whereas a minority has FUS (≤10 %; FTLD-FUS) or ubiquitin/p62 (≤1–2 %; FTLD-UPS) inclusions [10]. Some (but not all) FTLD-tau cases carry mutations in the microtubule associated protein tau (MAPT) [11], whilst FTLD-TDP cases are almost never associated with variability in the TAR-DNA binding protein 43 (TARDBP), rather, in progranulin (GRN) and the chromosome 9 open reading frame 72 (C9orf72) genes [3]. Furthermore, the FTLD-FUS cases do not have a clear genetic component, whilst the rare FTLD-UPS cases have been associated with variability in the charged multivesicular body protein 2B (CHMP2B) gene [3]. These features suggest that there is no unidirectional relationship between the genetics and the molecular pathology of FTD and that the observed pathological signatures result from complex molecular mechanisms.
To date a handful of genes has been associated with FTD: besides MAPT, GRN and C9orf72 (found in 2–11 %, 5–11 % and 7–20 % of cases, respectively) [11, 12], genetic variability in other genes, including CHMP2B, valosin containing protein (VCP), sequestosome 1 (SQSTM1) and ubiquilin 2 (UBQLN2), is extremely rare [3, 11]. Furthermore, pathogenic variants in TARDBP and the fused in sarcoma (FUS) genes seem nearly absent in FTD comparatively to ALS or ALS-FTD cases [13]; however, given that TARDBP and FUS are pathological hallmarks of FTLD subtypes [10], they likely hold functional relevance in the pathogenesis of FTD. Finally, recent genome wide association studies (GWAS) revealed association with FTD for the modifying factor transmembrane protein 106B (TMEM106B) [14, 15] and two further loci, one containing the RAB38, member RAS oncogene family (RAB38) and catepsin C (CTSC) genes, and one pointing to the HLA-locus [16].
In this study we used a systems biology approach based on gene co-expression network analysis of microarray expression data generated from 101 individuals without neurodegenerative diseases (UK Human Brain Expression Consortium [UKBEC]) [17] to further investigate genes and loci associated with FTD and, particularly, to: i) evaluate their co-expression patterns in brain areas known to be affected in FTD; ii) annotate and highlight biological processes potentially implicated in disease mechanisms, and; iii) identify novel potential risk factors for FTD.
Results
We grouped the FTD-genes into two categories: the pure (= mainly or exclusively associated with FTD) and the spectrum (= associated with more than one condition) genes (Table 1). We evaluated gene expression levels across different brain regions (frontal and temporal cortices as well as putamen, thalamus, hippocampus, white matter, cerebellum and medulla; see Methods section for details), assessed co-expression profiles and performed functional annotation and pathway analyses for the relevant modules (= we defined ‘relevant modules’ those modules containing one or more FTD-genes with hub status [1-q < 0.1] and/or module membership [MM] values > 0.5; see Methods section for details). Finally, we investigated whether the observed gene clustering is supported by known protein-protein interactors (PPIs) of FTD-genes products to infer the potential extent of the translation of the regional-specific co-expression patterns into the protein domain.
Table 1.
FTD-genes analysed in this study
| Genes | Type of variant | Clinical phenotype | Pathology | Type of association | Ref | |
|---|---|---|---|---|---|---|
| Pure FTD-genes | MAPT | exonic/intronic point mutations | bvFTD; FTD-17 | FTLD-Tau | mendelian | 3, 11 |
| small/large indels | ||||||
| missense | ||||||
| GRN | non-sense | bvFTD; PNFA | FTLD-TDP | 3, 11 | ||
| missense | ||||||
| frame-shift | ||||||
| large deletions | ||||||
| CHMP2B | non-sense | FTD3 | FTLD-UPS | 3, 11 | ||
| missense | ||||||
| RAB38 | unknown | bvFTD | unknown | from GWAS | 16 | |
| CTSC | unknown | bvFTD | unknown | |||
| BTNL2 | unknown | bvFTD; PNFA; SD | unknown | |||
| HLA-DRA | unknown | bvFTD; PNFA; SD | unknown | |||
| HLA-DRB5 | unknown | bvFTD; PNFA; SD | unknown | |||
| TMEM106B | unknown | FTD | FTLD-TDP | 14 | ||
| Spectrum FTD-genes | C9orf72 | expansion | ALS; ALS-FTD | FTLD-TDP | mendelian | 12, 13 |
| VCP | missense | IBMPFD; ALS; FTD | FTLD-TDP | 3, 11, 12 | ||
| SQSTM1 | non-sense | PBD; ALS; FTD | FTLD-TDP | 3, 11, 12 | ||
| missense | ||||||
| UBQLN2 | non-sense | MS; ALS; FTD | FTLD-TDP | |||
| missense | ||||||
| OPTN | non-sense | PDB; ALS-FTD | FTLD-TDP | |||
| missense | ||||||
| TDP-43 | missense | ALS; ALS-FTD | FTLD-TDP | |||
| FUS | missense | ALS; ALS-FTD | FTLD-FUS | |||
| frame-shift | ||||||
| indel |
The FTD-genes used in our analysis were divided into two groups: the pure and the spectrum genes. For each gene, the associated genetic variability, as well as the associated clinical and pathological features are summarized
bvFTD behavioural variant FTD, FTD-17 frontotemporal dementia linked to chromosome 17, PNFA progressive non-fluent aphasia, FTD3 frontotemporal dementia linked to chromosome 3, SD semantic dementia, ALS amyotrophic lateral sclerosis, IBMPFD Inclusion body myopathy with early-onset Paget disease and frontotemporal dementia, PBD Paget’s disease of bone, MS multiple sclerosis
Expression levels in brain
To gain insight into the expression profile of FTD-genes in brain we searched for expression levels and patterns online and in our own repositories (Human Brain Atlas and Braineac, respectively; see Methods section for further details). The general, and cross-supportive, outcome was that all FTD-genes are indeed expressed in brain: for the pure FTD-genes, MAPT, GRN, CHMP2B, CTSC, HLA-DRA and TMEM106B showed moderate to high expression levels across brain tissues throughout the lifespan of an individual, whilst, comparatively, expression levels for RAB38, BTNL2 and HLA-DRB5 appeared to be lower. All spectrum FTD-genes, C9orf72, VCP, SQSTM1, UBQLN2, OPTN, TARDBP and FUS had high expression levels across brain tissues during development and aging, and C9orf72, TARDBP and FUS showed exceptionally high levels in the cerebellum. A focused assessment of the expression levels and patterns in frontal and temporal cortices revealed that MAPT, UBQLN2, VCP, TMEM106B, FUS, TARDBP, OPTN, GRN and CHMP2B had high expression rates, whilst these were comparatively lower for C9orf72, HLA-DRA and CTSC (Fig. 1a-b).
Fig. 1.
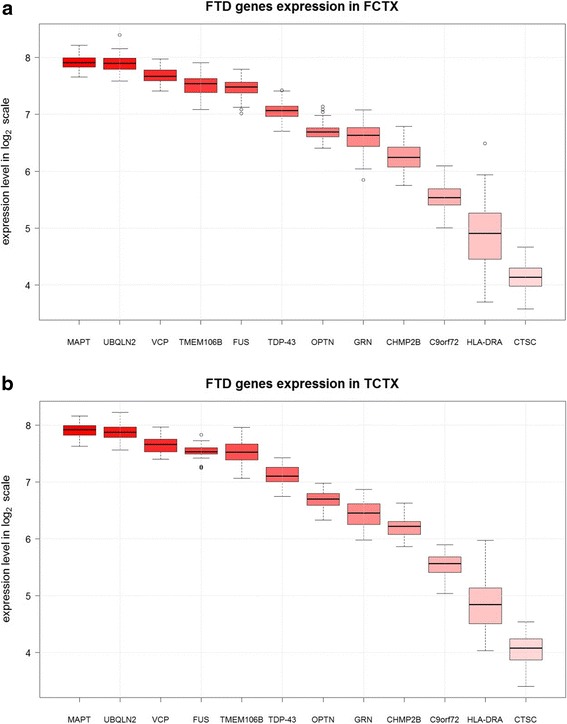
Expression levels. Expression levels of the FTD-genes considered in the network analysis in frontal cortex (a) and temporal cortex (b). FUS here was represented by Affymetrix transcript ID 3656904
For a more detailed description and visualization of all genes’ expression patterns across brain tissues see the Additional file 1 (pp 1–4) and Additional file 1: Figures S1-S16 (pp 8–23).
Weighted gene co-expression network analysis (WGCNA)
We performed the WGCNA with a primary focus on frontal and temporal cortices, the classically affected brain areas in FTD. Nevertheless, due to their recent increasing relevance to disease, we also extended the analysis to other brain regions such as putamen, thalamus, hippocampus and white matter for both the pure and spectrum genes, and the cerebellum and medulla for the spectrum genes only. Assessments on frontal and temporal cortices are presented hereafter and summarized in Table 2, whilst evaluations on any other brain region are shown in the Additional file 1 (pp 8–9).
Table 2.
Module assignments for FTD-genes in frontal and temporal cortex
| FTD genes | Affymetrix ID | type | Frontal cortex | Temporal cortex | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| module | Sz (n) | MM | 1-q | module | Sz (n) | MM | 1-q | |||
| MAPT | 3723687 | pure | black | 791 | 0.83 | 0.03 | lightyellow | 210 | 0.67 | 0.31 |
| GRN | 3722917 | 0.66 | 0.34 | cyan | 276 | 0.56 | 0.51 | |||
| CHMP2B | 2631845 | darkolivegreen | 63 | 0.76 | 0.48 | green | 1439 | 0.53 | 0.49 | |
| HLA-DRA | 2903189 | darkred ( p = 0.0046) | 141 | 0.8 | 0.23 | lightcyan ( p = 0.0139) | 250 | 0.66 | 0.51 | |
| CTSC | 3385769 | 0.73 | 0.5 | 0.69 | 0.47 | |||||
| TMEM106B | 2990342 | red | 925 | 0.78 | 0.04 | darkturquoise | 164 | 0.86 | 0.01 | |
| C9orf72 | 3202421 | spectrum | purple ( p = 0.0226) | 1559 | 0.74 | 0.1 | purple ( p = 0.0206) | 830 | 0.75 | 0.14 |
| VCP | 3204404 | 0.68 | 0.19 | 0.71 | 0.24 | |||||
| UBQLN2 | 3978999 | 0.55 | 0.45 | 0.63 | 0.45 | |||||
| OPTN | 3235726 | 0.59 | 0.38 | grey60 | 220 | 0.53 | 0.8 | |||
| FUS | 3656904 | lightcyan | 209 | 0.58 | 0.61 | midnightblue | 268 | 0.64 | 0.31 | |
| FUS | 3656950 | blue | 3329 | 0.31 | 0.69 | pink | 2979 | 0.39 | 0.58 | |
| FUS | 3656954 | 0.5 | 0.38 | magenta | 1000 | 0.44 | 0.72 | |||
| TDP-43 | 2320048 | turquoise | 4759 | 0.22 | 0.89 | 0.53 | 0.54 | |||
The FTD-genes (with corresponding Affymetrix IDs) are listed. Co-expression modules for the FTD-genes and their relevance within modules are displayed. Sz = size, i.e. number of transcripts contained in the module; MM = module membership; 1-q = 1-quantile of MM. The bolded parts highlight the fact that WGCNA indicated the following: 1) (MAPT and GRN), (HLA-DRA and CTSC), (C9orf72, VCP, UBQLN2 and OPTN) and (2 FUS) transcripts were respectively present in the same modules in frontal cortex, and; 2) (HLA-DRA and CTSC), (C9orf72, VCP and UBQLN2) and (FUS and TARDBP) transcripts were respectively present in the same modules in temporal cortex. The modules containing (HLA-DRA and CTSC) and (C9orf72, VCP, UBQLN2 [and OPTN]) were significantly enriched for FTD transcripts
We found four modules of interest in frontal cortex (Table 2): one included MAPT and GRN, where MAPT was a hub gene. Another module comprised HLA-DRA and CTSC: both genes showed high module membership (MM) values, whilst none was a hub gene. A third module displayed TMEM106B that was a hub, and the fourth included C9orf72, VCP, UBQLN2 and OPTN, where C9orf72 was a hub. The remaining genes (CHMP2B, FUS and TARDBP) had poor module assignments (Table 2).
In the temporal cortex, besides MAPT and GRN that were assigned to two distinct modules (where neither one was a hub), we observed similar co-expression patterns as in the case of the frontal cortex (Table 2): HLA-DRA and CTSC were again co-assigned to the same module with moderately high MM values (>0.65) but no hub status; TMEM106B was again a hub within its module of membership, and C9orf72, VCP and UBQLN2 were all again assigned to the same module where each had high MM values, but none was a hub. As seen in the frontal cortex, CHMP2B, OPTN, FUS and TARDBP did not hold major relevance in temporal cortex modules (Table 2).
Enrichment analysis identified the modules including HLA-DRA and CTSC, and C9orf72, VCP, UBQLN2 (and OPTN) as significantly enriched for FTD-genes (p < 0.05; Table 2). To visualize these as well as the interconnection networks of MAPT, GRN and TMEM106B we generated Figs. 2, 3, 4 and 5 for which the main nodes are listed in Additional file 2: Table S20.
Fig. 2.
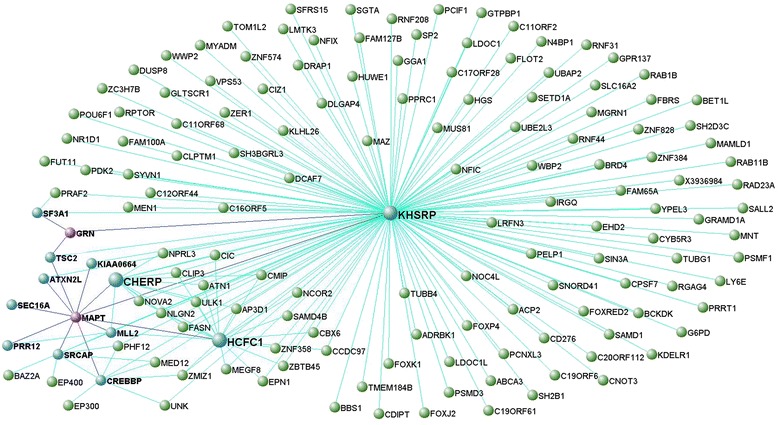
Network visualisation of genes of the black module in frontal cortex. VisANT plot shows genes (blue, gene names in bold) directly connected with a topological overlap measure TOM > 0.10 to FTD-genes, MAPT and GRN (purple, gene names in bold). All such genes are important hubs within this module whose connections are based on TOM > 0.115
Fig. 3.
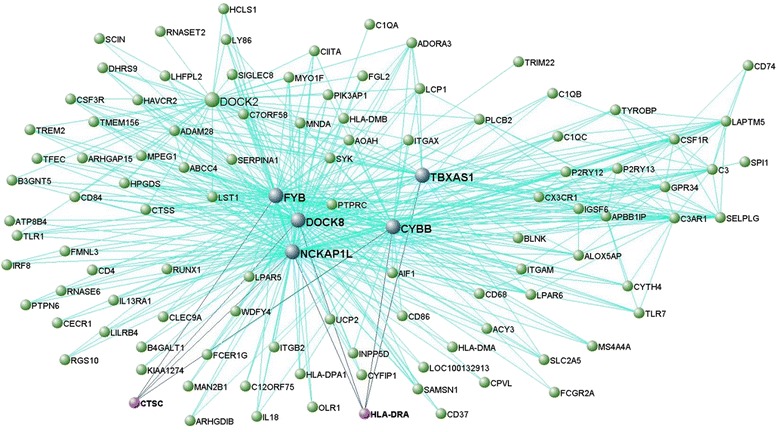
Network visualisation of genes of the darkred module in frontal cortex. VisANT plot shows genes (blue, gene names in bold) directly connected to FTD-genes, CTSC and HLA-DRA (purple, gene names in bold). All such genes are important hubs within this module and all connections shown here are based on TOM >0.165
Fig. 4.
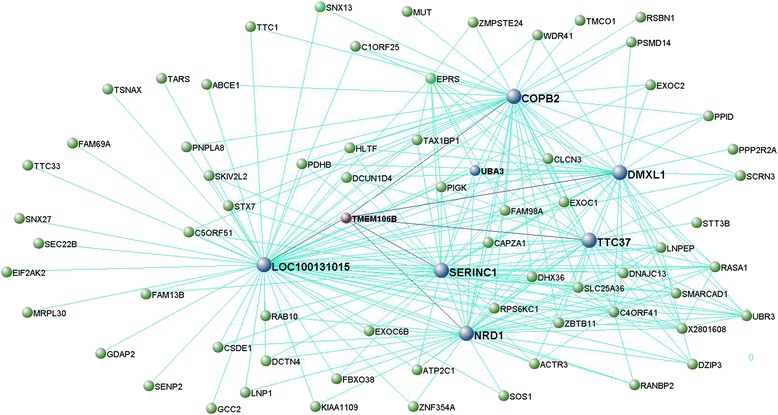
Network visualisation of genes of the red module in frontal cortex. VisANT plot shows genes (blue, gene names in bold) directly connected to FTD gene, TMEM106B (purple, gene name in bold). All such genes are important hubs within this module and all connections shown here are based on TOM > 0.1
Fig. 5.
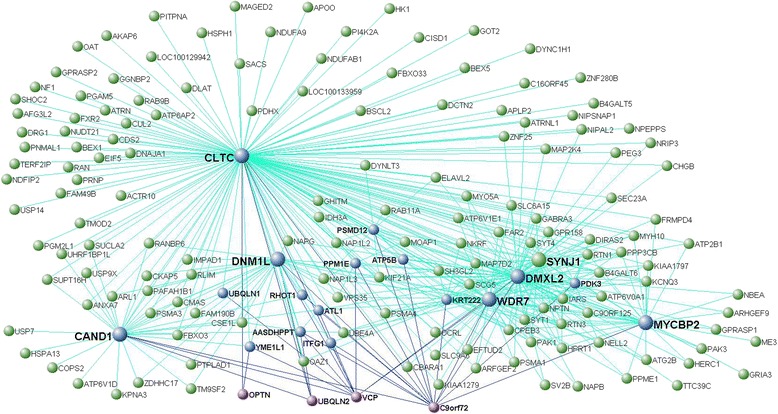
Network visualisation of genes of the purple module in frontal cortex. VisANT plot shows genes (blue, gene names in bold) directly connected with a topological overlap measure TOM > 0.08 to FTD-genes, OPTN, UBQLN2, VCP and C9orf72 (purple, gene names in bold). All such genes are important hubs within this module whose remaining connections are based on TOM >0.11
Since the FTD-genes distributed rather similarly within the modules in frontal and temporal cortex, we sought to further investigate the clustering across other brain tissues. We discovered moderate to high quantitative overlap (i.e. number of shared genes) between frontal and temporal cortex modules as shown and explained in the Additional file 1: Figure S17 (p 26). Also, we verified through composite Z-summary preservation statistics that the modules containing FTD-genes in frontal and temporal cortex were, for the most, preserved across other brain regions (Additional file 1, p 27).
Replication
In order to replicate and support our findings, we assessed the reproducibility of the frontal cortex networks as a ‘whole ‘and ‘module by module’, using an independent and well established dataset [18]. Our WGCNA TOM (topological overlap measure) was significantly reproduced in the WGCNA TOM matrix constructed from the Colauntoni dataset (p-value = 1x10-3), thus supporting robust correlation between the two datasets. Further, through the Z-summary preservation estimate we noted that 10 of our frontal cortex modules showed strong evidence of preservation (Z-summary > 10) in the Colauntoni dataset, and 30 had moderate to high preservation levels (2 < Z-summary < 10). Particularly, the Z scores for the modules of interest – based on Table 2 – were all supportive of preservation: red (TMEM106B) module (Z = 3.61), black (MAPT and GRN) module (Z = 7.15), purple (C9orf72, VCP, UBQLN2 and OPTN) module (Z = 9.28) and darkred (HLA-DRA and CTSC) module (Z = 29.73) (Fig. 6).
Fig. 6.
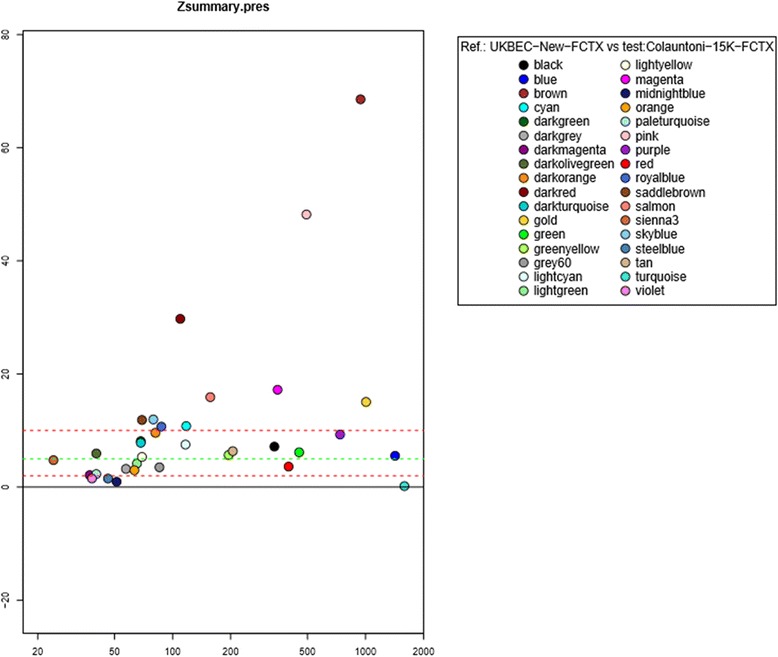
Z-summary graph. The ‘Z-summary preservation estimate’ estimates how interaction patterns of genes within modules one dataset are preserved in another dataset, i.e. it estimates how the genes which cluster together in the reference tissue maintain that clustering in the other dataset. The Z preservation summary is a mean of both estimates. The colour codes are those of reference, i.e. from our own dataset
Functional annotation and pathway analyses
To gain insight into their biological significance, we annotated genes in the network assessing gene ontology (GO) terms for biological processes (BPs), cellular components (CCs) and molecular functions (MFs) and performed pathways analyses for the relevant FTD-modules. The analyses of modules in frontal and temporal cortices (based on Table 2) are presented hereafter, whilst those of other brain tissues are summarized in the Additional file 1 (pp 8–9) and Additional file 2: Table S19.
In frontal cortex, significant GO terms associated with the module containing MAPT and GRN indicated transcription processes and chromatin metabolism with a particular involvement of the histone methyltransferase complex as well as transcription factor and nucleic acid binding activities (Table 3a); such annotations were supported by pathways analysis that also indicated the metabolism of the chromatin. Evaluation of the list of predefined enrichment for brain terms in WGCNA (see Additional file 1, pp 28–29) revealed significant overlap with the green M10 module associated with Glutamatergic Synaptic Function in neurons identified in [19] (Table 3a). In temporal cortex, the MAPT-containing module was enriched for transcription processes (Table 3b and Additional file 1, p 5), whereas the GRN-containing module for broad biology of membrane-bounded organelle (Table 3b and Additional file 1, p 5). Here, the list of predefined enrichment for brain terms in WGCNA showed significant overlap particularly with the green M10 module associated with Glutamatergic Synaptic Function in neurons identified in [19] for both modules. The complete list of GO terms associated with the modules including MAPT and GRN in frontal and temporal cortices can be found in Additional file 2: Tables S2, S6 and S7.
Table 3.
Summary of most relevant GO terms, pathways and brain lists annotation for relevant modules in frontal cortex (a) and temporal cortex (b)
| FTD gene | MM | 1-q | type | module | sz | G:Profiler Annotation (GO terms & Pathways analyses) | WGCNA – Brain lists |
|---|---|---|---|---|---|---|---|
| a. | |||||||
| MAPT | 0.83 | 0.03 | pure | black | 791 |
BP: Transcription, DNA-templated (p=2.77x10-5); RNA biosynthetic process (p=3.17x10-5); Chromatin modification (p=9.45x10-5) CC: Nuclear lumen (p=2.81x10-13); Histone methyltransferase complex (p=3.2x10-7) MF: Transcription factor binding transcription factor activity (p=6.26x10-6); Nucleic acid binding (p=7.19x10-6)c3v1234 Pathways: Chromatin organization and Chromatin modifying enzymes (p=4.84x10-3ZX) |
Glutamatergic Synaptic Function (CTX) (p=1.36x10-7) |
| GRN | 0.66 | 0.34 | |||||
| HLA-DRA | 0.8 | 0.23 | darkred | 141 |
BP: Immune system process (p=1.39x10-31); Defense response (p=5.23x10-29); Innate immune response (p=2.51x10-19); Adaptive immune response (p=1.71x10-7); Phagocytosis (p=6.2x10-3) CC: MHC class II protein complex (p=1.17x10-7); Lysosome (p=7.69x10-5); Lytic vacuole (p=7.69x10-5) MF: Receptor activity (p=1.88x10-8); Signal transducer activity (p=6.45x10-6) Pathways: Immune system (p=4.8x10-21); Phagosome (p=6.69 x 10-12); Adaptive immune system (p=5.08 x 10-10); Cytokine signaling in immune system (p=2.63 x 10-7); Lysosome (p=4.89 x 10-2) |
Microglia (Type1) (HumanMeta) (p=1.53x10-84) Microglia (Type1) (CTX) (p=7.01x10-31) Up In Frontal Cortex (Early AD) (p=2.96x10-19) |
|
| CTSC | 0.73 | 0.5 | |||||
| TMEM106B | 0.78 | 0.04 | red | 925 |
BP: Cellular protein metabolic process (p=3.84x10-5) CC: Nuclear lumen (p=9.36x10-6); Catalytic complex (p=1.36x10-2) MF: Ligase activity (p=1.96x10-2) Pathways: Chromatin organization (p=1.1x10-2); HATs acetylate histones (p=1.3 x 10-2); HDACs deacetylate histones (p=3.2 x 10-2) |
Protein metabolism (CTX) (p=7.57x10-10) Neuron (CTX) (p=4.83x10-5) Nucleus (HumanMeta) (p=1.09x10-5) Oligodendrocyte probable (Cahoy) (p=2.42x10-5) Brown pyramidal Neurons Layer5/basolateral Amygdala (Sugino/Winden) (p=2.80x10-5) |
|
| C9orf72 | 0.74 | 0.1 | spectrum | purple | 1559 |
BP: Modification-dependent macromolecule catabolic process (p=3.9x10-7); Ubiquitin-dependent protein catabolic process (p=1.08x10-6); Proteolysis involved in cellular protein catabolic process (p=1.72x10-6) CC: Cytoplasm (p=2.95x10-14); Mitochondrion (p=2.4x10-12); Proteasome complex (p=2.81x10-4) MF: Catalytic activity (p=3.55x10-4) Pathways: Protein processing in endoplasmic reticulum (p=1.01x10-5); Mitochondrial translation (p=2.28x10-3) |
Neuron (CTX) (p=8.76x10-27) Neuron (HumanMeta) (p=9.02x10-25) Mitochondrion (p=1.63x10-19) Post Synaptic Density proteins (Bayes) (p=8.73x10-18) |
| VCP | 0.68 | 0.19 | |||||
| UBQLN2 | 0.55 | 0.45 | |||||
| OPTN | 0.59 | 0.38 | |||||
| b. | |||||||
| MAPT | 0.67 | 0.31 | pure | lightyellow | 210 |
BP: Transcription from RNA polymerase II promoter (p=1.23x10-4) CC: Nucleus (p=3.69x10-5) MF: Protein binding (p=2.93x10-4); Transcription factor binding (p=1.42x10-2) Pathways: RNA Polymerase II Transcription Elongation (p=4.21x10-2) |
Glutamatergic Synaptic Function (CTX) (p=3.79x10-5) |
| GRN | 0.56 | 0.51 | cyan | 276 |
CC: intracellular membrane-bounded organelle (p=7.56x10-3) Pathways: Signaling by Wnt (p=7.61x10-3); Lysosome (p=1.21x10-2) |
Glutamatergic Synaptic Function (CTX) (p=9.89x10-8) Mitochondria (HumanMeta) (p=1.74 x10-5) |
|
| HLA-DRA | 0.66 | 0.51 | lightcyan | 250 |
BP: Immune system process (p=1.89x10-37); Defense response (p=1.51x10-32); Phagocytosis (p=1.39x10-5) CC: MHC class II protein complex (p=3.02x10-6); Lysosome (p=5.43x10-5); Lytic vacuole (p=5.43x10-5) MF: Receptor activity (p=4.41x10-10) Pathways: Immune system (p=8x10-22); Phagosome (p=7.2x10-13); Innate immune system (p=1x10-11); Adaptive immune system (p=2.56x10-9) |
Microglia (Type1) (HumanMeta) (p=1.53x10-84) Microglia (Type1) (CTX) (p=1.08x10-30) Up In Frontal Cortex (EarlyAD) (p=7.85x10-19) |
|
| CTSC | 0.69 | 0.47 | |||||
| TMEM106B | 0.86 | 0.01 | darkturquoise | 164 |
CC: Cytoplasm (p=2.8x10-5); Nucleus (p=2.49x10-4) Pathways: Protein processing in endoplasmic reticulum (p=4.86x10-5) |
Nucleus (CTX) (p=3.67x10-9) Metabolism (CTX) (p=3.48x10-5) |
|
| C9orf72 | 0.75 | 0.14 | spectrum | purple | 830 |
BP: Proteolysis involved in cellular protein catabolic process (p=1.81x10-8); Ubiquitin-dependent protein catabolic process (p=3.04x10-8); Protein catabolic process (p=1.87x10-6) CC: Cytoplasm (p=1.28x10-12); Proteasome complex (p=1.16x10-5); Endoplasmic reticulum membrane (p=5.21x10-3) MF: Catalytic activity (p=1.08x10-4) Pathways: Protein processing in endoplasmic reticulum (p=5.25x10-6) |
Neuron (CTX) (p=9.29x10-24) Neuron (HumanMeta) (p=6.96x10-11) Post Synaptic Density proteins (Bayes) (p=4.34x10-5) |
| VCP | 0.71 | 0.24 | |||||
| UBQLN2 | 0.63 | 0.45 | |||||
Functional annotation analysis for the modules comprising HLA-DRA and CTSC indicated modulation of the immune responses via the innate and adaptive systems in both the frontal (Table 3a) and temporal cortices (Table 3b). Pathways analysis revealed implication of immune system, phagosomes, antigen processing and presentation, interferon gamma- and cytokine-signalling, and lysosomes (Table 3a and b, and Additional file 2: Table S17), and the list of predefined enrichment for brain terms in WGCNA suggested enrichment for microglia markers given significant overlap with pink M10 and purple M4 Microglia (Type1) modules identified in [17, 19, 20] (Table 3a and b). The complete list of GO terms associated with the modules containing HLA-DRA and CTSC in the frontal and temporal cortices can be found in Additional file 2: Tables S4 and S9.
For the modules including TMEM106B, we noted GO terms indicating protein metabolic processes exerted through catalytic complexes in frontal cortex (Table 3a), whereas terms were rather general in temporal cortex (Table 3b). Pathway analysis pointed to chromatin metabolism for the frontal cortex (Table 3a) and protein processing in endoplasmic reticulum (ER) for the temporal cortex (Table 3b). The list of predefined enrichment for brain terms in WGCNA indicated that both the TMEM106B-containing modules in frontal and temporal cortex held overlapping features with the yellow M18 (enriched for protein metabolism) and the blue M16 Neuron modules identified in [19] (Table 3a). In addition, there was further overlap – for the frontal cortex module only – with the turquoise M14 Nucleus, Oligodendrocyte probable and brown pyramidal Neurons Layer5/basolateral Amygdala modules found in [20–23] (Table 3a), indicating that the TMEM106B protein most probably takes part in different biological processes in diverse brain areas. The complete list of GO terms associated with the modules including TMEM106B in frontal and temporal cortices can be found in Additional file 2: Tables S5 and S10.
Functional annotation analysis for the module containing C9orf72, VCP, UBQLN2 (and OPTN) in the frontal and temporal cortices pointed to ubiquitin-mediated protein catabolic process entailing proteasome and proteolysis activities, and to ER-associated ubiquitin-dependent protein catabolic process and endosomal transport (Table 3a and b). Pathways analysis hinted to protein processing in ER and also supported the ubiquitin-dependent degradation of proteins and the proteasome biology (Table 3a and b, and Additional file 2: Table S17). In addition, there was significant overlap with numerous WGCNA lists particularly pointing to the blue M16 Neuron module identified in [19] (Table 3a and b). The complete list of GO terms associated with the modules including C9orf72, VCP, UBQLN2 (and OPTN) in the frontal and temporal cortices can be found in Additional file 2: Tables S11 and S14.
For completeness, we also performed functional annotation and pathway analysis for the genes with poor module assignments (CHMP2B, FUS and TARDBP; Table 2): results are briefly detailed in the Additional file 1, pp 4–8 and in Additional file 2: Table S19.
Protein-protein interactors (PPIs) of FTD-genes
We sought to verify whether genes clustering together with FTD-genes in the relevant FTD-modules are found to interact at the protein level with the FTD-genes products. We searched for known PPIs of MAPT, GRN, HLA-DRA, CTSC, TMEM106B, C9orf72, VCP, UBQLN2 and OPTN (Additional file 2: Table S21a and b), and assessed any nominal overlap with genes co-clustering in modules containing the FTD-genes in the frontal and temporal cortices. Only for TMEM106B no nominal overlap between PPIs and related transcripts was seen.
Among the MAPT-PPIs, MARK2 (MAP/microtubule affinity-regulating kinase 2), MARK4 (MAP/microtubule affinity-regulating kinase 4) and EP300 (E1A binding protein p300) were assigned to the module containing MAPT in frontal cortex. Here, MARK2 was a hub, whilst MARK4 and EP300 were among the top ~15 % interactive genes (Additional file 2: Table S1a). MARK2 and MARK4 encode kinases that target proteins involved in stabilizing the microtubules, while EP300 encodes an acetyltransferase; of note, MARK2 and EP300 co-clustered with MAPT within GO terms pointing to the nucleus and indicated features such as DNA-, RNA- and protein-binding (MARK2), and transferase/catalitic complexes and nuclear chromatin (EP300) in frontal cortex (Additional file 2: Table S2). Conversely, MARK2, EP300 and AKT1 (v-akt murine thymoma viral oncogene homolog 1) were assigned to the MAPT-containing module in temporal cortex (Additional file 2: Table S6), where MARK2 was a hub (Additional file 2: Table S1b). The protein kinase AKT1 is a critical mediator of growth factor-induced neuronal survival in the developing nervous system. These 3 genes were found in GO terms pointing to the nucleus, together with MAPT, and transcription or RNA metabolic processes (Additional file 2: Table S6).
Among the GRN-PPIs, ATN1 (atrophin 1), SGTA (small glutamine-rich tetratricopeptide repeat (TPR)-containing, alpha), CRKL (v-crk avian sarcoma virus CT10 oncogene homolog-like) and TLE3 (transducin-like enhancer of split 3) were assigned to the module containing GRN in frontal cortex. Here, ATN1, that encodes atrophin 1, a conserved transcriptional co-repressor [24] and whose expansions have been associated with neurodegeneration [25], was a hub (Additional file 2: Table S1a). SGTA encodes a small glutamine-rich tetratricopeptide repeat (TPR)-containing, alpha that might be involved in neuronal apoptotic processes [26]. CRKL encodes an oncogene and seems pleiotropic in physiologic signalling [27], whilst TLE3 is a transcriptional co-repressor. ATN1 and TLE3 were found in gene lists indicating GO terms pointing to the nucleus and transcription related processes (Additional file 2: Table S2). No relevant functional annotations were available for SGTA, whereas CRKL appeared to be involved in DNA- and RNA-binding (with GRN; Additional file 2: Table S2). Conversely, TLE3 and CRKL were assigned to the GRN-containing module in temporal cortex and annotation analysis indicated GO terms involving the biology of membrane-bounded organelle (with GRN; Additional file 2: Table S7).
There was nominal overlap between HLA-DRA-PPIs and relative genes (Additional file 2: Table S21a), whereas there was none for CTSC. HLA-DMB (major histocompatibility complex, class II, DM beta), HLA-DMA (major histocompatibility complex, class II, DM alpha) and CD74 (CD74 molecule, major histocompatibility complex, class II invariant chain) were present in both modules containing HLA-DRA in the frontal (Additional file 2: Table S4) and temporal cortex (Additional file 2: Table S9). HLA-DMA and HLA-DMB encode the major histocompatibility complex, class II, DM alpha and beta, which is anchored in the membrane of intracellular vesicles and plays a central role in the peptide loading of MHC class II molecules [28]. CD74 encodes a chaperone that regulates antigen presentation during immune response. HLA-DMB, HLA-DMA and CD74 were included within all or most of the GO terms indicated by our functional annotation analysis together with HLA-DRA and/or CTSC (Additional file 2: Tables S4 and S9).
A large number of PPIs of VCP, UBQLN2 and OPTN nominally overlapped with the genes co-expressed with C9orf72, VCP, UBQLN2 (and OPTN) in the relative modules in the frontal and temporal cortices; conversely, this was the case only for a few of the C9orf72-PPIs (Additional file 2: Table S21b). We found up to 10 PPIs of VCP that were also hubs in frontal and/or temporal cortex (Additional file 2: Table S1a and b): CUL2 (cullin 2), UBQLN1 (ubiquilin 1), NF1 (neurofibromin 1), NIPSNAP1 (nipsnap homolog 1), BTRC (beta-transducin repeat containing E3 ubiquitin protein ligase), ARFGEF2 (ADP ribosylation factor guanine nucleotide exchange factor 2), COPS3 (COP9 signalosome subunit 3), PLAA (phospholipase A2-activating protein), CLTA (clathrin, light chain A) and ANXA7 (annexin A7); all such proteins are involved, mainly, in either protein catabolic or intracellular vesicle transport processes (Table 4). For UBQLN2 we found 5 PPIs that were also hubs in the frontal and/or temporal cortex (Additional file 2: Table S1a and b): UBQLN1, SEC23A (Sec23 homolog A, COPII coat complex component), USP9X (ubiquitin specific peptidase 9, X-linked), STAM (signal transducing adaptor molecule) and HSPA13 (heat shock protein family A (Hsp70) member 13); these are, overall, involved in protein degradation and ER-Golgi protein transport processes (Table 4). In the case of OPTN we counted two PPIs that were also hubs (Additional file 2: Table S1a and b): RAB11A (RAB11A, member RAS oncogene family) and RTN3 (reticulon 3), involved in protein transport and modulation of ß-amyloid production, respectively (Table 4). Finally, for C9orf72 we found only three PPIs (none of which was a hub): APP (amyloid beta (A4) precursor protein), ELAVL1 (ELAV like RNA binding protein 1) and EIF2B2 (eukaryotic translation initiation factor 2B subunit beta), linking C9orf72, provided further evidence, to the ß-amyloid production, RNA metabolism and protein synthesis, respectively (Table 4). Interestingly, we noticed that all genes mentioned above (and PPI interactors of VCP, UBQLN2, OPTN or C9orf72) were included together with the spectrum FTD-genes in the gene lists supporting GO terms indicating functions pointing to protein catabolism pathways and cytoplasmic protein transport and/or vesicle trafficking as highlighted in Additional file 2: Tables S11 and/or S14.
Table 4.
Novel potential risk factors in FTD
| Novel potential risk factor | Interactor of | Function | Topography | Evidence |
|---|---|---|---|---|
| KHSRP | MAPT, GRN | involved in alternative pre-mRNA splicing and mRNA localization | FCTX | WGCNA + VisANT |
| SF3A1 | GRN | |||
| CREBBP | MAPT | acetyltransferase involved in chromatin remodelling and transcriptional activation/regulation | ||
| MLL2 | methyl-transferase involved in chromatin remodelling | |||
| SRCAP | involved in transcriptional activation/regulation | |||
| HCFC1 | ||||
| MARK2 | MAPT | kinase involved in stabilizing the microtubules and tau’s phosphorylation | FCTX, TCXT | WGCNA + PPI |
| MARK4 | FCTX | |||
| EP300 | acetyltransferase involved in tau’s acetylation | FCTX, TCXT | ||
| AKT1 | kinase involved in growth factor-induced neuronal survival in the developing nervous system | TCTX | ||
| ATN1 | GRN | transcriptional co-repressor factor | FCTX | |
| SGTA | involved in neuronal apoptotic processes | |||
| CRKL | oncogene pleiotropic in physiologic signalling | FCTX, TCXT | ||
| TLE3 | transcriptional co-repressor factor | |||
| CYBB | HLA-DRA, CTSC | critical component of the oxidase system of phagocytes | FCTX | WGCNA + VisANT |
| DOCK8 | involved in neuronal development and immune cells shaping | |||
| HLA-DMA | HLA-DRA | transmembrane protein of intracellular vesicles involved in peptide loading of MHC class II molecules | FCTX, TCXT | WGCNA + PPI |
| HLA-DMB | ||||
| CD74 | chaperone involved in antigen presentation during immune response | |||
| COPB2 | TMEM106B | involved in Golgi budding and vesicular trafficking | FCTX | WGCNA + VisANT |
| SERINC1 | involved in lipid biosynthesis in neurons at the ER level | |||
| NRD1 | metalloprotease with potential neuropathogenic role | |||
| TTC37 | protein-protein interactor with chaperone activity | |||
| CAND1 | C9orf72, VCP | involved in ubiquitin ligase network | FCTX | WGCNA + VisANT |
| PSMD12 | C9orf72 | subunit of a multi-catalytic proteinase complex | ||
| MYCBP2 | C9orf72 | E3 ubiquitin protein ligase (alias) | ||
| ATL1 | C9orf72, VCP | involved in axonal maintenance | ||
| UBQLN1 | VCP | ubiquitin-like protein which links the ubiquitination and proteasome machineries | ||
| APP | C9orf72 | cell surface receptor and transmembrane precursor protein cleaved by secretases into different peptides: some of these can bind to the acetyltransferase complex (APBB1/TIP60) to promote transcriptional activation; others form the protein basis of the amyloid plaques | FCTX, TCTX | WGCNA + PPI |
| ELAVL1 | RNA-binding protein that contain several RNA recognition motifs, and selectively bind AU-rich elements (AREs) found in the 3' untranslated regions of mRNAs. AREs signal degradation of mRNAs as a means to regulate gene expression; the ELAVL family stabilizes ARE-containing mRNAs | |||
| EIF2B2 | beta subunit of eukaryotic initiation factor-2B (EIF2B). EIF2B is involved in protein synthesis and exchanges GDP and GTP for its activation and deactivation | FCTX | ||
| CUL2 | VCP | Cullins are a family of NEDD8 targets important in the stabilization and degradation of proteins | FCTX, TCTX | |
| UBQLN1 | part of the ubiquitination machinery of the proteasome to affect in vivo protein degradation | |||
| NF1 | negative regulator of the ras signal transduction pathway (control such processes as actin cytoskeletal integrity, proliferation, differentiation, cell adhesion, apoptosis and cell migration) | |||
| NIPSNAP1 | family of proteins involved in vesicular transport | FCTX | ||
| BTRC | constitutes one of the four subunits of ubiquitin protein ligase complex called SCFs (SKP1-cullin-F-box) that function in phosphorylation-dependent ubiquitination | FCTX, TCTX | ||
| ARFGEF2 | plays an important role in intracellular vesicular trafficking; involved in Golgi transport | FCTX | ||
| COPS3 | kinase activity that phosphorylates regulators involved in signal transduction | |||
| PLAA | activation of protein kinase C (PKC) and PKC-dependent responses (in response to inflammatory mediators and release during apoptosis) | |||
| CLTA | part of structural component of the lattice-type cytoplasmic face of coated pits and vesicles which entrap specific macromolecules during receptor-mediated endocytosis with regulatory function | |||
| ANXA7 | a membrane binding protein with diverse properties (voltage-sensitive calcium channel activity, ion selectivity and membrane fusion) | |||
| UBQLN1 | UBQLN2 | part of the ubiquitination machinery of the proteasome to affect in vivo protein degradation | FCTX, TCTX | |
| SEC23A | suggested to play a role in the ER-Golgi protein trafficking | |||
| USP9X | protein similar to ubiquitin-specific proteases | |||
| STAM | mediates downstream signaling of cytokine receptors and also play a role in ER to Golgi trafficking | |||
| HSPA13 | member of the heat shock protein 70 family and is found associated with microsomes. Members of this protein family play a role in the processing of cytosolic and secretory proteins, as well as in the removal of denatured or incorrectly-folded proteins | |||
| RAB11A | OPTN | involvedin constitutive, regulated secretory pathways and protein transport | FCTX | |
| RTN3 | expressed in neuroendocrine tissues: interacts with and modulates the activity of beta-amyloid converting enzyme 1 (BACE1), and the production of amyloid-beta |
Each novel potential risk factor is listed along with the interacting FTD-gene(s). The evidence of interaction is primarily defined by our WGCNA data that assigned each transcript to a module containing one (or more) FTD-gene(s) in frontal and/or temporal cortex. The nomenclature (WGCNA + VisANT) indicates that the novel potential risk factor is chosen because of its hub status and its interaction with FTD-gene(s) based on topological overlap measure (TOM) > 0.10 (see also Figs. 2, 3, 4 and 5). The nomenclature (WGCNA + PPI) indicates that the novel potential risk factor is chosen based on nominal overlap between interactive transcript(s) and protein(s). The potential risk factors belonging to the latter category are bolded as an indication that the WGCNA + PPI combination could be a strong indicator for regional-specific impacted functional networks. The main known function(s) of each novel potential genetic and/or functional risk factor is included in the central column. FCTX = frontal cortex; TCTX = temporal cortex
Taken all together, the module assignments in the frontal and temporal cortices as well as the functional annotation, pathways and PPIs analyses provide not only a more comprehensive picture of the potential biological and cellular mechanisms involved in the development of FTD but also an enlarged domain of novel potential genetic or functional risk factors associated with FTD (that are comprehensively summarized in Table 4).
Discussion
In this study we used a systems biology approach based on gene co-expression network analysis of microarray expression data to investigate genes known to be associated with FTD. After a general assessment of their expression levels in different brain regions, we particularly evaluated co-expression patterns in brain areas known to be affected in FTD with a major focus on the frontal and temporal cortices and inferred biological processes potentially implicated in the pathogenesis of FTD. We also sought to identify novel potential risk factors for FTD.
Prior to putting our results into context, a number of limitations that apply to this work need to be acknowledged: i) although the prevalence of FTD is almost equal among males and females [3], data available for this study were generated mainly in male individuals (78/101); ii) the disease generally manifests between mid 50s to early 60s years of age and, although expression data were corrected for individual effects including sex and age at death, the study cohort had a mean age of 50 years (ranging from 16 to 83 years); iii) besides that there is no golden standard approach or pipeline in systems biology studies, it is important to note that data supporting functional annotation and enrichment analyses presented here are based on the current literature (i.e. focused on a restricted number of targets or pathways) and that data mining has been manually curated or generated by semantic associations, thus some or novel interactions might be overlooked.
The data on expression levels across brain tissues indicated that MAPT, GRN, CHMP2B, CTSC, HLA-DRA, TMEM106B, C9orf72, VCP, SQSTM1, UBQLN2, OPTN, TARDBP and FUS are all robustly expressed in (thus clearly hold relevance for the biology of) the aging brain. Comparatively, by showing rather lower expression rates, it is a possibility that RAB38, BTNL2 and HLA-DRB5 might rather represent sensitive cellular markers and sudden changes in their expression levels might impact cellular homeostasis.
The current work revealed that MAPT–GRN, HLA-DRA–CTSC, TMEM106B and C9orf72–VCP–UBQLN2–OPTN clustered in interesting modules in the frontal and/or temporal cortex and that, particularly on the basis of functional annotation and pathway analyses, three main biological processes with a direct relation to FTD-genes hold relevance to the pathogenesis of FTD: i) DNA & chromatin biology; ii) immune & lysosomal processes, and; iii) protein meta/catabolism.
The first biological process, i.e. DNA and chromatin implication (including transcription [i.e. RNA biosynthesis and gene expression] and chromatin remodelling), was defined by modules containing MAPT (and GRN), and appeared to be specific to neurons in the frontotemporal cortices, and the putamen (Fig. 7). This is, overall, a relatively novel concept in FTD, and it is noteworthy to appreciate that genes responsible for alternative pre-mRNA splicing and mRNA localization (KHSRP [KH-type splicing regulatory protein] and SF3A1 [splicing factor 3a, subunit 1]), chromatin remodelling such as acetyl- (CREBBP [CREB binding protein]) and methyl-transferases (MLL2 [lysine (K)-specific methyltransferase]), and transcriptional activation/regulation (CREBBP, SRCAP [Snf2-related CREBBP activator protein] and HCFC1 [host cell factor C1]) were strong hubs and highly co-expressed and interconnected with MAPT and/or GRN (Additional file 2: Table S1a and S20). Even more importantly, we noted that genes such as MARK2, MARK4, EP300 and AKT1, and ATN1, SGTA, CRKL and TLE3 not only were co-expressed with MAPT and GRN, respectively, in frontal cortex and/or temporal cortex, but also that they are PPIs of either tau or GRN proteins: these could be novel functional risk factors to be further investigated in the functional environment. Particularly, MARK2, MARK4, AKT1 and EP300 influence Tau’s phosphorylation and acetylation [29–32], whilst ATN1 and TLE3 regulate transcription, and CRKL is involved in DNA-binding processes. If the protein interactors of GRN support transcription related processes, those of MAPT appear to directly influence tau’s activities and function, fostering some intriguing considerations. Tau is classically known for binding and stabilizing microtubules in neuronal soma and axons [33], but it also localizes in their nucleus [34]. Recent data showed that when DNA damage occurs, tau is dephosphorylated and imported into the nucleus to exert protective effect in stressed neurons [35, 36]. Conversely, acetylation was suggested to affect tau’s phosphorylation patterns leading to early events of tau pathology [29]. All this suggests that MAPT-phosphorylation dynamics are highly sensitive and critical in regulating not only i) microtubules homeostasis and ii) aberrant cytoplasmic accumulation of hyper-phosphorylated tau, but also iii) tau’s shuttling between the cytoplasm and the nucleus. Impairment of the latter might have detrimental downstream effect on DNA protection. Our data support tau’s involvement in DNA binding and given evidence for its involvement in DNA protection [35], it follows that tau must hold relevance in protecting neurons from apoptosis and/or aberrant transcription events, thus supporting neuronal longevity. Finally, our data also indicate that tau’s RNA might be target of genes regulating mRNA splicing and gene expression and determining which tau isoform is being expressed; this supports the hypothesis that aberrant regulation of tau splicing/expression might contribute to tau pathology [31]. With this work we are now highlighting potential effectors of tau’s splicing/expression regulation such as the above mentioned KHSRP and SF3A1 (which are splicing regulatory protein), and CREBBP, SRCAP and HCFC1 (which are transcriptional activators/regulators); these factors should be further investigated in the cell-biology setting.
Fig. 7.
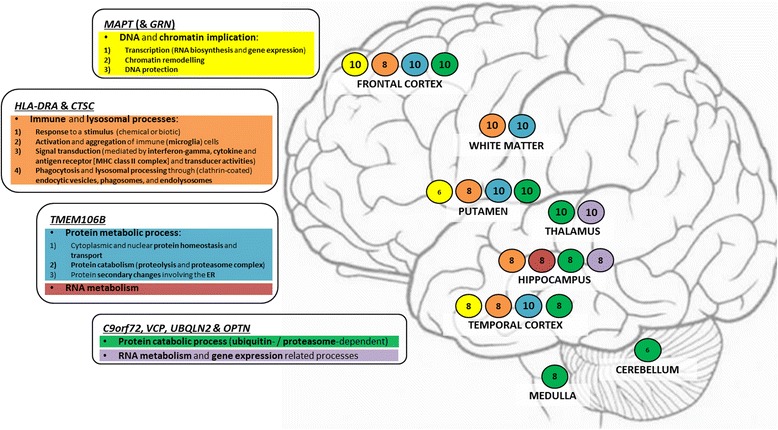
Scheme of the regional distribution of the FTD-genes and associated annotated biological processes or functions. Rectangles contain FTD-gene(s) and their main associated regional-specific biological process. Each colour indicates a particular biological process. Font size differs based on statistics associated with the transcript(s) within their assigned modules. Font 10 = hub (1-q < 0.1) + MM > 0.7; Font 8 = 1-q > 0.1 + MM > 0.6, and; Font 6 = 1-q > 0.1 + MM ≤ 0.6. If multiple transcripts belong to a module, at least 1 has values that justify the font size
The second biological process was defined by HLA-DRA and CTSC that clustered together in all assessed brain regions (thalamus, cerebellum and medulla excluded; Fig. 7) within preserved modules indicating immune- and lysosomal-related processes in microglia. Our data support the idea of a synergistic interplay between immune system and degradation processes relying on the activation of immune responses, phagocytosis and lysis in the lysosomes, particularly through: i) response to a stimulus (stress, chemical [interferon-gamma or cytokine] or biotic stimulus); ii) activation and aggregation of immune (microglial) cells; iii) signal transduction (mediated by interferon-gamma, cytokine and antigen receptor [MHC class II protein complexes] and transducer activities), and; iv) phagocytosis and lysosomal processing through endocytic vesicles, phagosomes, and endolysosomes. Interestingly, a number of transcripts highly interconnected with both the HLA-DRA and CTSC also support these processes, and might be novel functional markers to be further investigated in the cell-biology setting, such as: CYBB (cytochrome b-245, beta polypeptide) encodes a critical component of the oxidase system of phagocytes [37] and DOCK8 (dedicator of cytokinesis 8) encodes a protein involved in neuronal development [38] and immune cells shaping [39]. Furthermore, and probably more importantly, we identified overlap between our transcripts data and protein interactors of HLA-DRA such as HLA-DMB, HLA-DMA and CD74 to be considered for further investigation in the functional environment. These not only appeared to support immune cell (microglia) activation and maturation, cell-cell adhesion and MHC class II complex activity, but also the biology of extracellular vesicles such as exosomes, and that of lysosomes and lytic vacuoles. These data support the results of our GWAS [16] and the idea that immune processes and lysosomal biology are important and likely common elements across different neurodegenerative diseases [17, 40, 41].
The third biological process was defined by TMEM106B, C9orf72, VCP, UBQLN2 and OPTN. TMEM106B was ubiquitous across all assessed tissues (thalamus, cerebellum and medulla excluded; Fig. 7) in various brain cell types (neurons, oligodendrocytes, pyramidal cells) and associated, from a functional perspective, with various protein metabolism processes. Particularly, we found that TMEM106B appears to play a key role in processes supporting protein homeostasis and transport, catabolism and protein secondary changes involving the ER. Interestingly, our data confirm the importance of these processes in neurodegeneration and indicate potential interactors or targets of TMEM106B among the transcripts highly interconnected with TMEM106B that should be considered for further investigation in the cell-biology setting, including: COPB2 (coatomer protein complex, subunit beta 2, an essential protein for Golgi budding and vesicular trafficking), SERINC1 (serine incorporator 1, a carrier protein involved in lipid biosynthesis in neurons at the ER level) [42], NRD1 (nardilysin [N-arginine dibasic convertase], a metalloprotease with potential neuropathogenic role) [43] and TTC37 (tetratricopeptide repeat domain 37, a protein-protein interactor with chaperone activity).
C9orf72, VCP, UBQLN2 and OPTN were relevant in both the frontal and temporal cortices where they clustered within modules indicating primarily protein catabolic processes; this was also evident for the putamen, thalamus, hippocampus, cerebellum and medulla (Fig. 7). It is relevant to note not only that PPIs data strongly pointed towards ‘protein catabolism’ pathways (mainly driven by VCP, UBQLN2, OPTN and their associated PPIs), but also that our transcripts interconnectivity data indicated further novel potential factors, involved in catabolic processes and interactors of C9orf72, VCP, UBQLN2 and OPTN, such as: CAND1 (cullin-associated and neddylation-dissociated 1), a factor involved in ubiquitin ligase network, critical for substrate degradation [44]; PSMD12 (proteasome 26S subunit, non-ATPase 12) that encodes a subunit of a multi-catalytic proteinase complex, and; MYCBP2 (MYC binding protein 2, E3 ubiquitin protein ligase) that functions as a E3 ubiquitin protein ligase. All these factors, such as CAND1, PSMD12 and MYCBP2 (among the interconnected transcripts) as well as ELAVL1, EIF2B2, CUL2, UBQLN1, NF1, NIPSNAP1, BTRC, ARFGEF2, COPS3, PLAA, CLTA, ANXA7, SEC23A, USP9X, STAM, HSPA13, RAB11A and RTN3 (co-expressed transcripts and PPIs of C9orf72, VCP, UBQLN2 and OPTN; see Table 4 for detailed associations with the spectrum FTD-genes) should be considered for further investigation in the functional environment.
Finally, CHMP2B, FUS and TARDBP not only did not hold major relevance in their respective modules in the frontal and/or temporal cortex, but also such modules either contained very small or very large numbers of co-expressed genes (see Table 2); nevertheless, we assessed their potential biological meaning through functional annotation and pathways analyses (see Additional file 1 pp 4-5 and 7) we found very general (thus negligible) processes for CHMP2B, whilst we noted processes mainly associated with the RNA metabolism for FUS and TARDBP, confirming their involvement in the modulation of RNA processing [45].
Conclusion
In summary, with this study we further characterize known FTD-genes by providing insight into their regional-specific functional networks and associated biological processes that might be implicated in the pathogenesis of FTD (Fig. 7). Particularly, we: i) show the probable involvement of transcription regulation, chromatin remodelling and DNA protection through the networks of MAPT and GRN; ii) further support the likely involvement of immune and lysosomal processes through the networks of CTSC and HLA-DRA, and; iii) confirm implication of protein meta/catabolism through the networks of C9orf72, VCP, UBQLN2 and OPTN, and TMEM106B.
In addition, we also highlight novel potential genetic and/or functional risk factors (Table 4) to be further explored in focused and extended hypothesis driven cell biology work.
As a final remark, our work overall suggests that we are at a point in time in which there is a critical need for a shift in the study of complex traits and diseases from a reductionist ‘gene → pathology’ type of approach to a more holistic ‘gene ↔ networks ↔ pathways’ strategy.
Methods
Expression during development and aging across brain tissues
We assessed expression for FTD-genes in different brain areas by means of the Human Brain Atlas (HBA; [46]) and Braineac [47, 48]. Data from HBA allow to assess expression levels in the cerebellar cortex, mediodorsal nucleus of the thalamus, striatum, amygdala, hippocampus, and 11 areas of the neocortex; data from Braineac allow to assess expression levels in ten distinct brain areas such as the frontal cortex, temporal cortex, putamen, thalamus, hippocampus, white matter, cerebellum, medulla, substantia nigra and occipital cortex. We also extracted expression quantitative trait loci (eQTL) data from Braineac for further analysis and characterization of the newly identified potential risk factors for FTD.
Ethics
Expression data analysed in this study were generated from 101 control individuals was collected by the Medical Research Council Sudden Death Brain and Tissue Bank, Edinburgh, UK [48, 49] and are available in NCBI’s GEO through accession number GSE46706. All samples had fully informed consent for retrieval and were authorized for ethically approved scientific investigation (Research Ethics Committee [REC] number 10/H0716/3).
Weighted gene co-expression network analysis (WGCNA)
Networks were generated by weighted gene co-expression network analysis (WGCNA) (see [50, 51]) and modules of highly correlated genes were determined in an unsupervised manner based on co-expression patterns in the ten distinct brain areas: frontal cortex, temporal cortex, putamen, thalamus, hippocampus, white matter, cerebellum, medulla, substantia nigra and occipital cortex. Comparatively to previous work (see details in [17, 52]) we generated networks using 19,152 transcripts. We assessed 14 transcripts corresponding to 12 FTD-genes (MAPT, GRN, CHMP2B, CTSC, HLA-DRA, TMEM106B, C9orf72, VCP, UBQLN2, OPTN, TARDBP and FUS; Table 2). HLA-DRB5 was not present in the Braineac dataset; RAB38, SQSTM1, and BTNL2 were excluded before network analysis due to preliminary inclusion/exclusion criteria (see [17]), and; FUS had three transcripts (ID3656904, 3656950, and 3656954). We particularly focused on frontal and temporal cortex, the classically affected brain areas in FTD; we also analysed other brain regions such as the putamen, thalamus, hippocampus and white matter (pure and spectrum genes), and the cerebellum and medulla (spectrum genes only), where the FTD-genes were highly co-expressed or were hubs. To identify highly interconnected genes within each module (hubs) we used the measure of module membership (MM), a Pearson correlation between gene-expression level and module-eigengene. An elevated MM (>0.6) suggests strong inter-correlations between genes in a module. We used the 1-quantile measure of the MMs to define inter-modular hubs as any top 10 % gene (i.e. genes with a 1-quantile <0.10). We evaluated whether modules (containing a minimum of two FTD-transcripts) were significantly enriched with FTD-genes using a hypergeometric distribution (with Bonferroni correction for multiple testing) and, to validate modules’ consistency and preservation across tissues, we calculated a composite Z-summary statistic that aggregates different module preservation statistics (Z-summary > 10 = module preservation; Z-summary < 2 = no preservation) [53].
As transcriptome organization in a given biological system is highly reproducible [19], we compared the modules containing FTD-genes to previous WGCNA studies in human brain, gene-markers for cell types, and region-enriched or disease-specific genes using the WGCNA function userListEnrichment [54]. Such lists are referenced in the Additional file 1 and significant overlap (Bonferroni corrected) with each FTD-module is reported in the results section.
Mantel test and construction of replication network
To assess the reproducibility, and thus replicate, the frontal cortex networks we used the Colantuoni dataset [18] with GEO accession number GSE30272 (Illumina Human 49K Oligo array, HEEBO-7 Set). This comprises 269 prefrontal cortex human samples but we restricted our analysis to 175 (i.e. those with age > 16). We used the pre-processed/normalized data version of such dataset. In order to validate the frontal cortex WGCNA networks we designed two procedures aiming at evaluating their reproducibility at two levels.
On the one hand we wanted to assess whether the WGCNA topological overlap measure (TOM) matrix from the frontal cortex tissue was significantly reproduced in the WGCNA TOM matrix constructed for the Colauntoni dataset. For such purpose we applied a Mantel test [55] with the ape R package, version 3.4: this is a permutation test that, using two squared matrices of the same range (i.e. the two TOMs in this case), calculates a Z-statistic defined as the sum of the pairwise product of the lower triangles of the permuted TOMs. The test then compares the permuted distribution with the Z-statistic observed for the actual data and generates a p-value.
On the other hand, for evaluating the quality of each frontal cortex network’s module we constructed a signed WGCNA network on the Colauntoni dataset by using the same method employed for the construction of the signed network from our frontal cortex microarray gene expression profiles with beta value of 11, guaranteeing scale free topology property. The generated WGCNA network had 23 modules with sizes between 174 and 2293 genes. As we wanted to assess the level of replication of our frontal cortex co-expression network on the Colauntoni et al. network we carried out a preservation analysis by using the preservation R function implemented by WGCNA software. For that we focused on the Z-summary preservation estimate which is obtained by the aggregation of various estimates focused on two main aspects. On the one hand, estimates on how interaction patterns of genes within the modules, as they are seen in the reference tissue (i.e. in our case our frontal cortex samples), are preserved in the other tissue (i.e. the Colauntoni network). On the other hand, it includes as well clustering estimates which focus on how the genes which cluster together in the reference tissue maintain that clustering in the other dataset. The Z preservation summary is a mean of both estimates. And the estimates are obtained by a permutation analysis [53].
Gene set enrichment and pathway analysis with g:Profiler
We performed functional annotation and pathways analysis for the highly co-expressed genes within the FTD-genes containing modules to characterise their biological relevance assessing GO terms for biological processes (BPs), cellular components (CCs) and molecular functions (MFs) by means of the bioinformatics tool gProfiler (accessed in March and April 2015) [56]. We used the whole set of 19,152 transcripts as a tissue specific background and considered significant those GO terms with p < 0.05 based on gProfiler’s custom threshold g:SCS [56].
VisANT visualization
Interconnections between transcripts, defined by topological overlap measures (TOMs), of relevant modules containing FTD-genes were visualized using VisANT [57]. We only showed the strongest interconnected genes by visualizing TOMs greater than specific thresholds indicated for each module (explained in Figs. 2, 3, 4 and 5). We limited our analysis to relevant modules (= modules containing one or more FTD-genes with hub status [1-q < 0.1] and/or module membership [MM] values > 0.5) identified in frontal cortex.
Protein-protein interaction (PPI) analysis
We searched for currently known protein-protein interactors (PPIs) of the pure and spectrum FTD-genes and compared them with the lists of genes included in the modules containing FTD-genes identified in the frontal and temporal cortices. Briefly, a list of protein interactors associated with each FTD-gene was downloaded from Biogrid [58], IntAct and MINT [59] databases; such databases provide a constantly updated survey of PPIs based on manual curation of peer reviewed literature. We then filtered PPIs manually to remove interactions whose taxid was non-human. OnIy PPIs whose respective genes were expressed in the same modules containing the FTD-genes were considered of relevance based on the convergent evidence that they are: i) expressed in brain; ii) co-expressed with FTD-genes, and; iii) proven protein interactors of FTD-genes.
Acknowledgements
This work was supported by the MRC through the JPND RiMOD FTD programme and by the Wellcome Trust (grant no: 089698/Z/09/Z and 091673/Z/10/Z). The funding sources had no role in study design, collection, analysis and interpretation of data, neither in the writing of the report and in the decision to submit the article for publication. The United Kingdom Brain Expression Consortium (UKBEC) investigators are: Sebastian Guelfi, BSc, Department of Molecular Neuroscience, UCL Institute of Neurology, London, England; John Hardy, PhD, Department of Molecular Neuroscience, UCL Institute of Neurology, London, England; Adaikalavan Ramasamy, PhD, Department of Medical and Molecular Genetics, King’s College London, London, England; Mina Ryten, MD, Department of Molecular Neuroscience, UCL Institute of Neurology, London, England; Colin Smith, MD, Medical Research Council Sudden Death Brain and Tissue Bank, University of Edinburgh, Edinburgh, Scotland; Daniah Trabzuni, PhD, Department of Molecular Neuroscience, UCL Institute of Neurology, London, England; and Michael E. Weale, PhD, Department of Medical and Molecular Genetics, King’s College London, London, England.
Abbreviations
- AD
Alzheimer’s disease
- AKT1
V-akt murine thymoma viral oncogene homolog 1
- ALS
amyotrophic lateral sclerosis
- ANXA7
annexin A7
- APP
amyloid beta (A4) precursor protein
- ARFGEF2
ADP ribosylation factor guanine nucleotide exchange factor 2
- ATN1
atrophin 1
- BPs
biological processes
- BTNL2
butyrophilin-Like 2
- BTRC
beta-transducin repeat containing E3 ubiquitin protein ligase
- bvFTD
behavioural variant FTD
- C9orf72
chromosome 9 open reading frame 72
- CAND1
Cullin-associated and neddylation-dissociated 1
- CCs
cellular components
- CD74
CD74 molecule, major histocompatibility complex, class II invariant chain
- CHMP2B
charged multivesicular body protein 2B
- CLTA
clathrin, light chain A
- COPB2
coatomer protein complex, subunit beta 2
- COPS3
COP9 signalosome subunit 3
- CREBBP
CREB binding protein
- CRKL
V-crk avian sarcoma virus CT10 oncogene homolog-like
- CTSC
catepsin C
- CUL2
Cullin 2
- CYBB
cytochrome b-245, beta polypeptide
- DOCK8
dedicator of cytokinesis 8
- EIF2B2
eukaryotic translation initiation factor 2B subunit beta
- ELAVL1
ELAV like RNA binding protein 1
- EP300
E1A binding protein p300
- ER
endoplasmic reticulum
- FCTX
frontal cortex
- FTD
frontotemporal dementia
- FTD-17
frontotemporal dementia linked to chromosome 17
- FTD-3
frontotemporal dementia linked to chromosome 3
- FTLD-FUS
frontotemporal lobar degeneration with FUS pathology
- FTLD-tau
frontotemporal lobar degeneration with tau pathology
- FTLD-TDP
frontotemporal lobar degeneration with TDP-43 pathology
- FTLD-UPS
frontotemporal lobar degeneration with p-62 (ubiquitin-proteasome system) pathology
- FUS
fused in sarcoma
- GO
gene ontology
- GRN
progranulin
- GWAS
genome wide association studies
- HCFC1
host cell factor C1
- HLA
human leukocyte antigen
- HLA-DMA
major histocompatibility complex, class II, DM alpha
- HLA-DMB
major histocompatibility complex, class II, DM beta
- HLA-DRA
major histocompatibility complex, class II, DR alpha
- HLA-DRB5
major histocompatibility complex, class II, DR beta 5
- HSPA13
heat shock protein family A (Hsp70) member 13
- IBMPFD
inclusion body myopathy with early-onset Paget disease and frontotemporal dementia
- KHSRP
KH-type splicing regulatory protein
- MAPT
microtubule associated protein tau
- MARK2
MAP/microtubule affinity-regulating kinase 2
- MARK4
MAP/microtubule affinity-regulating kinase 4
- MFs
molecular functions
- MHC
major histocompatibility complex
- MLL2
lysine (K)-specific methyltransferase
- MM
module membership
- MS
multiple sclerosis
- MYCBP2
MYC binding protein 2, E3 ubiquitin protein ligase
- NF1
neurofibromin 1
- NIPSNAP1
nipsnap homolog 1
- NRD1
nardilysin N-arginine dibasic convertase
- OPTN
optinuerin
- PBD
Paget’s disease of bone
- PLAA
phospholipase A2-activating protein
- PNFA
progressive non-fluent aphasia
- PPIs
protein-protein interactors
- PSMD12
proteasome 26S subunit, non-ATPase 12
- RAB11A
RAB11A, member RAS oncogene family
- RAB38
RAB38, member RAS oncogene family
- RTN3
reticulon 3
- SD
semantic dementia
- SEC23A
Sec23 homolog A, COPII coat complex component
- SERINC1
serine incorporator 1
- SF3A1
splicing factor 3a, subunit 1
- SGTA
small glutamine-rich tetratricopeptide repeat (TPR)-containing, alpha
- SQSTM1
sequestosome 1
- SRCAP
Snf2-related CREBBP activator protein
- STAM
signal transducing adaptor molecule
- TARDBP
TAR-DNA binding protein 43
- TCTX
temporal cortex
- TLE3
transducin-like enhancer of split 3
- TMEM106B
transmembrane protein 106B
- TOM
topological overlap measure
- TTC37
tetratricopeptide repeat domain 37
- UBQLN1
ubiquilin 1
- UBQLN2
ubiquilin 2
- UKBEC
UK human brain expression consortium
- UPS
ubiquitin proteasome system
- USP9X
ubiquitin specific peptidase 9, X-linked
- VCP
valosin containing protein
- WGCNA
weighted gene co-expression network analysis
Additional files
This file includes additional and complementary text as well as supplementary figures. (PDF 1444 kb)
This file includes additional tables to complement and support the main text. (PDF 18446 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
RF designed the study, analysed and interpreted data, and drafted the manuscript. PF analysed and interpreted data, and helped in drafting the manuscript. JV helped in interpreting data, critically reviewed and commented on the manuscript and helped to revise the manuscript. JAB performed the Mantel test and constructed the replication network in different datasets and helped to revise the manuscript. SG extracted eQTL data from the Braineac repository and helped to revise the manuscript. JDW helped in the design of the study and critically reviewed the manuscript. PM helped in the design of the study, in interpreting data and critically reviewed the manuscript. MEW performed statistical analyses on the transcripts data used for network analysis and critically reviewed the manuscript. MR helped in designing the study, in data interpretation and helped during the revision process. JH designed the study, interpreted data and critically reviewed the manuscript. All authors read and approved the final manuscript.
Contributor Information
Raffaele Ferrari, Email: r.ferrari@ucl.ac.uk.
Paola Forabosco, Email: paola.forabosco@cnr.it.
Jana Vandrovcova, Email: jana.vandrovcova@csc.mrc.ac.uk.
Juan A. Botía, Email: j.botia@ucl.ac.uk
Sebastian Guelfi, Email: m.guelfi@ucl.ac.uk.
Jason D. Warren, Email: jason.warren@ucl.ac.uk
Parastoo Momeni, Email: parastoo@omixy.com.
Michael E. Weale, Email: michael.weale@kcl.ac.uk
Mina Ryten, Email: mina.ryten@ucl.ac.uk.
John Hardy, Email: j.hardy@ucl.ac.uk.
References
- 1.Ratnavalli E, Brayne C, Dawson K, Hodges JR. The prevalence of frontotemporal dementia. Neurology. 2002;58:1615–21. doi: 10.1212/WNL.58.11.1615. [DOI] [PubMed] [Google Scholar]
- 2.Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–54. doi: 10.1212/WNL.51.6.1546. [DOI] [PubMed] [Google Scholar]
- 3.Ferrari R, T.A., Momeni P., Molecular Genetics of Frontotemporal Dementia. In: eLS. John Wiley & Sons, Ltd: Chichester. 2013.
- 4.Lam BY, Halliday GM, Irish M, Hodges JR, Piguet O. Longitudinal white matter changes in frontotemporal dementia subtypes. Hum Brain Mapp. 2014;35:3547–57. doi: 10.1002/hbm.22420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Möller C, Dieleman N, van der Flier WM, Versteeg A, Pijnenburg Y, Scheltens P, et al. More atrophy of deep gray matter structures in frontotemporal dementia compared to Alzheimer’s disease. J Alzheimers Dis. 2014;44:635–47. doi: 10.3233/JAD-141230. [DOI] [PubMed] [Google Scholar]
- 6.Mahoney CJ, Malone IB, Ridgway GR, Buckley AH, Downey LE, Golden HL, et al. White matter tract signatures of the progressive aphasias. Neurobiol Aging. 2013;34:1687–99. doi: 10.1016/j.neurobiolaging.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mahoney CJ, Ridgway GR, Malone IB, Downey LE, Beck J, Kinnunen KM, et al. Profiles of white matter tract pathology in frontotemporal dementia. Hum Brain Mapp. 2014;35:4163–79. doi: 10.1002/hbm.22468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mahoney CJ, Beck J, Rohrer JD, Lashley T, Mok K, Shakespeare T, et al. Frontotemporal dementia with the C9ORF72 hexanucleotide repeat expansion: clinical, neuroanatomical and neuropathological features. Brain. 2012;135:736–50. doi: 10.1093/brain/awr361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perry DC, Sturm VE, Seeley WW, Miller BL, Kramer JH, Rosen HJ. Anatomical correlates of reward-seeking behaviours in behavioural variant frontotemporal dementia. Brain. 2014;137:1621–6. doi: 10.1093/brain/awu075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Halliday G, Bigio EH, Cairns NJ, Neumann M, Mackenzie IR, Mann DM. Mechanisms of disease in frontotemporal lobar degeneration: gain of function versus loss of function effects. Acta Neuropathol. 2012;124:373–82. doi: 10.1007/s00401-012-1030-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rohrer JD, Warren JD. Phenotypic signatures of genetic frontotemporal dementia. Curr Opin Neurol. 2011;24:542–9. doi: 10.1097/WCO.0b013e32834cd442. [DOI] [PubMed] [Google Scholar]
- 12.van der Zee J, Gijselinck I, Dillen L, Van Langenhove T, Theuns J, Engelborghs S, et al. A pan-European study of the C9orf72 repeat associated with FTLD: geographic prevalence, genomic instability, and intermediate repeats. Hum Mutat. 2013;34:363–73. doi: 10.1002/humu.22244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hardy J, Rogaeva E. Motor neuron disease and frontotemporal dementia: sometimes related, sometimes not. Exp Neurol. 2013;262:75–83. doi: 10.1016/j.expneurol.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 14.Deming Y, Cruchaga C. TMEM106B: a strong FTLD disease modifier. Acta Neuropathol. 2014;127:419–22. doi: 10.1007/s00401-014-1249-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Deerlin VM, Sleiman PM, Martinez-Lage M, Chen-Plotkin A, Wang LS, Graff-Radford NR, et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet. 2010;42:234–9. doi: 10.1038/ng.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ferrari R, Hernandez DG, Nalls MA, Rohrer JD, Ramasamy A, Kwok JB, et al. Frontotemporal dementia and its subtypes: a genome-wide association study. Lancet Neurol. 2014;13:686–99. doi: 10.1016/S1474-4422(14)70065-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Forabosco P, Ramasamy A, Trabzuni D, Walker R, Smith C, Bras J, et al. Insights into TREM2 biology by network analysis of human brain gene expression data. Neurobiol Aging. 2013;34:2699–714. doi: 10.1016/j.neurobiolaging.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Colantuoni C, Lipska BK, Ye T, Hyde TM, Tao R, Leek JT, et al. Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature. 2011;478:519–23. doi: 10.1038/nature10524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oldham MC, Konopka G, Iwamoto K, Langfelder P, Kato T, Horvath S, et al. Functional organization of the transcriptome in human brain. Nat Neurosci. 2008;11:1271–82. doi: 10.1038/nn.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller JA, Horvath S, Geschwind DH. Divergence of human and mouse brain transcriptome highlights Alzheimer disease pathways. Proc Natl Acad Sci U S A. 2010;107:12698–703. doi: 10.1073/pnas.0914257107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–78. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sugino K, Hempel CM, Miller MN, Hattox AM, Shapiro P, Wu C, et al. Molecular taxonomy of major neuronal classes in the adult mouse forebrain. Nat Neurosci. 2006;9:99–107. doi: 10.1038/nn1618. [DOI] [PubMed] [Google Scholar]
- 23.Winden KD, Oldham MC, Mirnics K, Ebert PJ, Swan CH, Levitt P, et al. The organization of the transcriptional network in specific neuronal classes. Mol Syst Biol. 2009;5:291. doi: 10.1038/msb.2009.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang L, Tsai CC. Atrophin proteins: an overview of a new class of nuclear receptor corepressors. Nucl Recept Signal. 2008;6:e009. doi: 10.1621/nrs.06009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nisoli I, Chauvin JP, Napoletano F, Calamita P, Zanin V, Fanto M, et al. Neurodegeneration by polyglutamine Atrophin is not rescued by induction of autophagy. Cell Death Differ. 2010;17:1577–87. doi: 10.1038/cdd.2010.31. [DOI] [PubMed] [Google Scholar]
- 26.Chen M, Xia X, Zhu X, Cao J, Xu D, Ni Y, et al. Expression of SGTA correlates with neuronal apoptosis and reactive gliosis after spinal cord injury. Cell Tissue Res. 2014;358:277–88. doi: 10.1007/s00441-014-1946-1. [DOI] [PubMed] [Google Scholar]
- 27.Kiyokawa E, Mochizuki N, Kurata T, Matsuda M. Role of Crk oncogene product in physiologic signaling. Crit Rev Oncog. 1997;8:329–42. doi: 10.1615/CritRevOncog.v8.i4.30. [DOI] [PubMed] [Google Scholar]
- 28.Zwart W, Griekspoor A, Kuijl C, Marsman M, van Rheenen J, Janssen H, et al. Spatial separation of HLA-DM/HLA-DR interactions within MIIC and phagosome-induced immune escape. Immunity. 2005;22:221–33. doi: 10.1016/j.immuni.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 29.Min SW, Cho SH, Zhou Y, Schroeder S, Haroutunian V, Seeley WW, et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron. 2010;67:953–66. doi: 10.1016/j.neuron.2010.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sarnat HB, Flores-Sarnat L. Infantile tauopathies: Hemimegalencephaly; tuberous sclerosis complex; focal cortical dysplasia 2; ganglioglioma. Brain Dev. 2015;37:553–562. doi: 10.1016/j.braindev.2014.08.010. [DOI] [PubMed] [Google Scholar]
- 31.Schwalbe M, Biernat J, Bibow S, Ozenne V, Jensen MR, Kadavath H, et al. Phosphorylation of human Tau protein by microtubule affinity-regulating kinase 2. Biochemistry. 2013;52:9068–79. doi: 10.1021/bi401266n. [DOI] [PubMed] [Google Scholar]
- 32.Trinczek B, Brajenovic M, Ebneth A, Drewes G. MARK4 is a novel microtubule-associated proteins/microtubule affinity-regulating kinase that binds to the cellular microtubule network and to centrosomes. J Biol Chem. 2004;279:5915–23. doi: 10.1074/jbc.M304528200. [DOI] [PubMed] [Google Scholar]
- 33.Spillantini MG, Goedert M. Tau pathology and neurodegeneration. Lancet Neurol. 2013;12:609–22. doi: 10.1016/S1474-4422(13)70090-5. [DOI] [PubMed] [Google Scholar]
- 34.Brady RM, Zinkowski RP, Binder LI. Presence of tau in isolated nuclei from human brain. Neurobiol Aging. 1995;16:479–86. doi: 10.1016/0197-4580(94)00170-6. [DOI] [PubMed] [Google Scholar]
- 35.Sultan A, Nesslany F, Violet M, Bégard S, Loyens A, Talahari S, et al. Nuclear tau, a key player in neuronal DNA protection. J Biol Chem. 2011;286:4566–75. doi: 10.1074/jbc.M110.199976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Violet M, Delattre L, Tardivel M, Sultan A, Chauderlier A, Caillierez R, et al. A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front Cell Neurosci. 2014;8:84. doi: 10.3389/fncel.2014.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frazão JB, Thain A, Zhu Z, Luengo M, Condino-Neto A, Newburger PE. Regulation of CYBB gene expression in human phagocytes by a distant upstream NF-kappaB binding site. J Cell Biochem. 2015;116:2008–17. doi: 10.1002/jcb.25155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Griggs BL, Ladd S, Saul RA, DuPont BR, Srivastava AK. Dedicator of cytokinesis 8 is disrupted in two patients with mental retardation and developmental disabilities. Genomics. 2008;91:195–202. doi: 10.1016/j.ygeno.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tangye SG. T cells require DOCK8 for flexibility and function. J Exp Med. 2014;211:2482–3. doi: 10.1084/jem.21113insight2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Amor S, Woodroofe MN. Innate and adaptive immune responses in neurodegeneration and repair. Immunology. 2014;141:287–91. doi: 10.1111/imm.12134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frake RA, Ricketts T, Menzies FM, Rubinsztein DC. Autophagy and neurodegeneration. J Clin Invest. 2015;125:65–74. doi: 10.1172/JCI73944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Inuzuka M, Hayakawa M, Ingi T. Serinc, an activity-regulated protein family, incorporates serine into membrane lipid synthesis. J Biol Chem. 2005;280:35776–83. doi: 10.1074/jbc.M505712200. [DOI] [PubMed] [Google Scholar]
- 43.Bernstein HG, Stricker R, Dobrowolny H, Steiner J, Bogerts B, Trübner K, et al. Nardilysin in human brain diseases: both friend and foe. Amino Acids. 2013;45:269–78. doi: 10.1007/s00726-013-1499-8. [DOI] [PubMed] [Google Scholar]
- 44.Wu S, Zhu W, Nhan T, Toth JI, Petroski MD, Wolf DA. CAND1 controls in vivo dynamics of the cullin 1-RING ubiquitin ligase repertoire. Nat Commun. 2013;4:1642. doi: 10.1038/ncomms2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79:416–38. doi: 10.1016/j.neuron.2013.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.http://hbatlas.org/ (accessed May 2015).
- 47.http://www.braineac.org/ (accessed May 2015).
- 48.Trabzuni D, Ryten M, Walker R, Smith C, Imran S, Ramasamy A, et al. Quality control parameters on a large dataset of regionally dissected human control brains for whole genome expression studies. J Neurochem. 2011;119:275–82. doi: 10.1111/j.1471-4159.2011.07432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Millar T, Walker R, Arango JC, Ironside JW, Harrison DJ, MacIntyre DJ, et al. Tissue and organ donation for research in forensic pathology: the MRC Sudden Death Brain and Tissue Bank. J Pathol. 2007;213:369–75. doi: 10.1002/path.2247. [DOI] [PubMed] [Google Scholar]
- 50.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4:Article17. doi: 10.2202/1544-6115.1128. [DOI] [PubMed] [Google Scholar]
- 52.Bettencourt C, Ryten M, Forabosco P, Schorge S, Hersheson J, Hardy J, et al. Insights from cerebellar transcriptomic analysis into the pathogenesis of ataxia. JAMA Neurol. 2014;71:831–9. doi: 10.1001/jamaneurol.2014.756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Langfelder P, Luo R, Oldham MC, Horvath S. Is my network module preserved and reproducible? PLoS Comput Biol. 2011;7:e1001057. doi: 10.1371/journal.pcbi.1001057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miller JA, Cai C, Langfelder P, Geschwind DH, Kurian SM, Salomon DR, et al. Strategies for aggregating gene expression data: the collapseRows R function. BMC Bioinformatics. 2011;12:322. doi: 10.1186/1471-2105-12-322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mantel N. The detection of disease clustering and a generalized regression approach. Cancer Res. 1967;27:209–20. [PubMed] [Google Scholar]
- 56.Reimand J, Arak T, Vilo J. g:Profiler--a web server for functional interpretation of gene lists (2011 update) Nucleic Acids Res. 2011;39:W307–15. doi: 10.1093/nar/gkr378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hu Z. Using VisANT to analyze networks. Curr Protoc Bioinformatics. 2014;8:8.8.1–8.8.39. doi: 10.1002/0471250953.bi0808s45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.http://thebiogrid.org/ (accessed May 2015).
- 59.http://www.imexconsortium.org/ (accessed May 2015).