

Abstract
This article describes the discovery and development of the first highly selective, small molecule antagonist of the muscarinic acetylcholine receptor subtype I (mAChR1 or M1). An M1 functional, cell-based calcium-mobilization assay identified three distinct chemical series with initial selectivity for M1 versus M4. An iterative parallel synthesis approach was employed to optimize all three series in parallel, which led to the development of novel microwave-assisted chemistry and provided important take home lessons for probe development projects. Ultimately, this effort produced VU0255035, a potent (IC50 = 130 nM) and selective (>75-fold vs. M2-M5 and > 10 μM vs. a panel of 75 GPCRs, ion channels and transporters) small molecule M1 antagonist. Further profiling demonstrated that VU0255035 was centrally penetrant (BrainAUC/PlasmaAUC of 0.48) and active in vivo, rendering it acceptable as both an in vitro and in vivo MLSCN/ MLPCN probe molecule for studying and dissecting M1 function.
Keywords: muscarinic, acetylcholine, mAChR, M1, antagonist
I. Introduction
The Vanderbilt Screening Center for GPCRs, Ion Channels and Transporters, and the companion Chemistry Center, were established as members of the Molecular Libraries Screening Center Network (MLSCN) initiated and supported by the NIH Molecular Libraries Roadmap. The MLSCN is a nationwide consortium of facilities that provide high-throughput small molecule screening and medicinal chemistry expertise for the development of chemical probes for use as tools to explore biological targets/pathways for which small molecule tools are unavailable. Once probes are developed, they are available to the scientific community at no cost upon request, in an attempt to advance the biomedical community’s knowledge of a particular biological target and advance science. The Vanderbilt Center, employed novel, functional high-throughput screening technologies and protocols, such as the triple add paradigm, to identify agonists, antagonists and positive allosteric modulators in a single screen. The Chemistry Center employed, and continues to employ as a Specialized Chemistry Center within the MLPCN, Technology Enabled Synthesis (TES) to develop small molecule probes, relying on iterative solution phase parallel synthesis, microwave-assisted organic synthesis and high-throughput, mass-directed preparative HPLC purification, coupled with automated post-purification sample handling. Combining all of the above mentioned technologies and paradigms for screening, synthesis and DMPK evaluation affords an aggressive, expedited process for chemical lead optimization. This approach allows one synthetic chemists to support a chemical lead optimization effort with accelerated timelines delivering high quality probes in as little as 1-2 months from initiation of a probe development project.
Independently, the technologies and strategies described herein provide improvements for chemical lead optimization; however, when they become closely aligned with screening and DMPK resources in a ‘closed loop’ paradigm, the impact on probe discovery is exponential (Figure 1). Starting from an HTS hit, considerable attention is first devoted to library design, without question the most important component of a successful lead optimization effort. Library design changes over the course of a lead optimization campaign. The initial design strategy is to explode SAR around a screening hit and to be as diverse as possible with respect to monomer input and analog synthesis to rapidly identify productive changes for further optimization. In addition, this component of lead optimization is often conducted in parallel, wherein a single chemist will simultaneously synthesize diversity libraries around four to six hits to expediently identify the best leads for further optimization. After this initial diversity-oriented explosion, library design must become more focused in order to impact drug discovery goals: random libraries do not accelerate programs or lead to high quality probes. It is important to approach directed library design from a medicinal chemistry perspective and assemble the library as a collection of single compounds designed to address a particular issue. For example, the design of a 24-member library should involve careful thought regarding what would the first four single compounds to synthesize be to test a hypothesis, increase potency, improve PK, etc… Then, for each of the first four analogs synthesized, consider what the next four analogs should be if the first changes were productive or non-productive. This exercise in library design generates quality data that drives a lead optimization program towards probe compounds very quickly.
Figure 1.

Schematic overview of the TES approach to probe development. The iterative ‘closed loop’ process, coupled with close interaction with biologists, allows for a one-week turn-around for library design/synthesis and generation/dissemination of biological data.
Another key feature of the ‘closed loop’ approach to lead optimization involves division of labor and the transfer of samples from medicinal chemists to the analytical chemists. In this paradigm, the medicinal chemists design and synthesize the compound libraries (24 to 96 compounds) and obtain analytical LCMS reports for each member of the library. At this point, the medicinal chemists transfers the crude samples to the analytical chemists who purify the libraries by mass-directed preparative HPLC to >98% using analytical-to-preparative software, perform all post-purification sample handling and coordinate submission of samples, in a 96-well plate format, to the biologists and DMPK personnel for screening (vide supra). If resources allow, this division of labor affords opportunities for the medicinal chemists to focus on library design, develop and optimize new chemistries and pursue multiple lead series in parallel.
The success of this paradigm hinges on rapid screening and dissemination of data to the medicinal chemists so that the next iteration of library synthesis can be initiated. To facilitate this, the delivery of compounds is coordinated with the biologists and assays are run within 24 hours after the compound libraries are delivered. Biological data is then returned within 48 hours of receipt of the libraries. This allows lead optimization to operate on a one week turn-around between the initiation of chemical synthesis and the generation of primary assay data. Secondary and or selectivity data typically trail primary data by 1-2 days. As these data trigger the need for DMPK information, DMPK data typically follows one week after the initial assay data is obtained. Overall, this expedited process parallels traditional singleton medicinal chemistry work flows, but generates data on hundreds of compounds in the time it used to take to evaluate just a few compounds. Moreover, this protocol allows a single synthetic chemist to support a chemical lead optimization effort with accelerated timelines delivering high quality probes in as little as 1-2 months from initiation of a probe development project.
During the first three years of the MLSCN, termed the pilot phase, Vanderbilt’s screening center focused exclusively on GPCRs, ion channels and transporter targets which lacked selective small molecule tools to study target function and to assess therapeutic potential. One such target which lacks the appropriate small molecule tools are the muscarinic acetylcholine receptors (mAChRs), and in this case, selective agonists and antagonists are required to advance the field. The muscarinic acetylcholine receptors (mACHRs) are members of the G Protein-Coupled Receptor (GPCR) family A that mediate the metabotropic actions of the neurotransmitter acetylcholine. To date, five distinct subtypes of mAChRs (M1-M5) have been cloned and sequenced. M1, M3 and M5 activate phospholipase C and calcium through Gq whereas M2 and M4 block the action of adenylyl cyclase through Gi/o. The cholinergic system, mediated by mAChRs, plays a critical role in a wide variety of CNS and peripheral functions including memory and attention mechanisms, motor control, nociception, regulation of sleep wake cycles, cardiovascular function, renal and gastrointestinal function and many others. As a result, agents that can selectively modulate the activity of mAChRs have the potential for therapeutic use in multiple pathological states. However, due to high sequence conservation within the orthosteric binding site of the five mAChR subtypes, it has been historically difficult to develop mAChR subtype selective ligands.
To date, the majority of reported muscarinic antagonists are unselective, such as a scopolamine 1 and atropine 2. Recently, pirenzapine, 3 has emerged as a relatively selective M1 receptor antagonist (20- to 50-fold versus M2-M5) and there are numerous reports of moderately selective M3 antagonists (20- to 50-fold versus M2) such as 4. Interestingly, the most selective M1 antagonist, MT7, 5, the 65 amino acid peptide, (>1,000-fold versus M2-M5) was derived from venom extracts of the green mamba snake (Figure 2). Based on brain expression and cellular localization, data from mAChR knock-out mice and clinical trials with muscarinic agents, the M1 mAChR subtype is an attractive molecular target for the treatment of Alzheimer’s disease (AD), Parkinson’s disease (PD) and dystonia due to its role in cognition and motor control. Indeed, pan-muscarinic agonists, such as the M1/M4 preferring xanomeline, showed efficacy in Phase III clinical trials in AD patients; however, activation of peripheral M2 and M3 receptors led to intolerable adverse side effects. Moreover, anti-cholinergic agents have also demonstrated efficacy in both PD and dystonia patients, and this benefit is believed to be derived from antagonism of the M1 mAChR subtype; however, the relative contributions from M4 are unclear. In order to probe the role of M1 antagonism as a potential therapeutic approach for Parkinson’s disease, dystonia and other movement disorders, potent small molecule mAChR antagonists are required with a high degree of M1 versus M4 selectivity for study both in vitro and in preclinical animal models.
Figure 2.

Structures of representative mAChR antagonists.
Based on this unmet need in the scientific community, our MLSCN Center initiated an effort to identify potent small molecule mAChR antagonists with high specificity for M1 for use as a chemical probe and lead for further optimization towards a novel therapeutic. Towards this goal, we optimized a functional, real-time cell-based calcium-mobilization assay employing an M1/CHO cell line (Z’ averaged 0.7), screened a 63,656 member MLSCN compound library and identified 2,179 primary M1 antagonist hits. Of these primary hits, 1,665 were available from Biofocus-DPI for re-test, and duplicate testing afforded 723 confirmed hits (43%). These compounds were then counter-screened against an mGluR4/CHO cell line which eliminated 9 hits. The remaining compounds were tested in triplicate in 10-point concentration response curves against both M1/CHO and M4/CHO cells to identify compounds with ~ 10-fold selectivity for M1 versus M4, our initial cutoff for a lead. While the vast majority of compounds displayed little or no selectivity for M1 versus M4, we identified three scaffolds that displayed ~10-fold selectivity for M1 versus M4 (Figure 3). The first scaffold 6 was a singleton hit based on a 3,6-disubstituted-[1,2,4]-triazolo[4,3-b]pyridazine scaffold with weak M1 antagonism, but selectivity versus M4 (M1 IC50 = 21 μM, M4 IC50 >150 μM). The second scaffold comprised two related structures based on a N-(4-(4-ethylpierazin-1-yl)phenyl amide scaffold, 7 (M1 IC50 = 0.49 μM, M4 IC50 = 7.9 μM) and 8 (M1 IC50 = 0.58 μM, M4 IC50 = 5.1 μM), which displayed ~16- to ~9-fold selectivity, respectively, for M1 versus M4. The third scaffold 9 was another singleton hit, based on an N-(3-piperazin-1-yl)-3-oxopropyl)benzo[c][1,2,5] thiadiazole-4-sulfonamide scaffold, that demonstrated a surprising >58-fold selectivity for M1 (IC50 = 2.55 μM) over M4 (IC50 >150 μM).
Figure 3.

M1 antagonist HTS leads 6-9 with selectivity versus M4 (~10- to >58-fold).
While this level of selectivity for the three series, coupled with diverse chemotypes, represented an attractive starting point, the HTS hits themselves did not meet the criteria for an MLSCN small molecule probe; therefore, we launched a chemical lead optimization campaign, based on an iterative parallel synthesis approach advancing all three series in parallel, to improve potency for M1 (M1 IC50 < 500 nM) while maintaining high selectivity versus M2-M5 (>50-fold) and physical properties that would provide utility as an in vitro and in vivo probe.
2. Optimization of an M1 selective antagonist screening hit based on a 3,6-disubstituted-[1,2,4]-triazolo[4,3-b]pyridazine scaffold
Classical conditions for the synthesis of 3,6-disubstituted-[1,2,4]triazolo[4,3-b]pyridazines such as 6 involve refluxing 3,6-dicholropyridazine 10 with an acylhydrazide 11 in toluene for 16 hours, or more typically 60 hours, to provide the 3-aryl-6-chloro-[1,2,4]triazolo[4,3-b]pyridazine 12 in yields less than 50% (Scheme 1). Introduction of the amino moiety in the 6-position was accomplished through an SNAr reaction employing either neat or steel bomb conditions at 100°-140°C for 8-30 hours to deliver analogs 13 in yields ranging from 40-70%. Moreover, previous efforts were focused on traditional medicinal chemistry approaches and the development of structure-activity-relationships (SAR), with little concern for achieving high chemical yields or reaction generality for either the heterocycle synthesis or the SNAr reaction. Interestingly, microwave-assisted organic synthesis has never before been applied to this heterocyclic system, and even more surprising when one considers a 1-6 day reaction time to deliver a single derivative of 6. Therefore, we quickly explored multiple parameters and developed general, high yielding microwave-assisted protocols to accelerate the synthesis of analogs of HTS lead 6. As shown in Scheme 1, catalytic HCl in EtOH with microwave irradiation reduced the reaction time to form 12 to only 10 minutes, from the classical 16-60 hours. Similarly, microwave irradiation accelerated the SNAr reaction, while also providing reaction generality, and resulted in another 10 minute reaction to afford the final 3,6-disubstituted-[1,2,4]triazolo[4,3-b]pyridazine analogs 13 of 6 in only 20 minute total reaction time in high yield.
Scheme 1.

Classical conditions and new, microwave-assisted conditions for the expedient and high-yielding synthesis of 3,6-disubstituted-[1,2,4]triazolo[4,3-b]pyridazines.
With a new, robust reaction sequence, HTS lead 6 was re-synthesized to confirm the activity and selectivity observed with the HTS stock solution. Evaluation of 6 against M1-M5 indicated that 6 was indeed a selective M1 antagonist (M1 IC50 = 23 μM, M2-M5 IC50 >>50 μM). Encouraged by this result, we employed an iterative parallel synthesis approach, employing our new MAOS protocols, to rapidly develop Structure-Activity-Relationships in an attempt to improve the M1 antagonist potency while maintaining selectivity for M2-M5. As shown in Figure 3, we simultaneously varied the substituents at the C-3 and C-6 positions, synthesizing small 12- to 24-members libraries employing the synthetic routes depicted in Scheme 1. Analogs of 6 were triaged in a single point 10 μM screen for the compound’s ability to decrease an EC80 concentration of acetylcholine. SAR for this series was rather ‘flat’, with subtle changes leading to a complete loss of M1 inhibitory activity. Out of ~60 analogs, only four demonstrated significant M1 antagonism; however, we managed to improve upon HTS hit 2. As shown in Figure 4, exploration of the C3 position identified both the 3-OMe phenyl derivative 14 and the 4-Me phenyl congener 15 as engendering more potency (M1 IC50 = 3.59 μM and 4.09 μM, respectively), while maintaining selectivity (M2-M5 IC50 >> 50 μM). When holding the 3-OMe phenyl moiety constant at C3 and exploring alternatively functionalized piperazines for the bromofuranoic amide at C6, we identified two piperazinyl piperazine analogs, 16 and 17 which maintained M1 antagonism (M1 IC50 = 3.99 μM and 6.64 μM, respectively) and selectivity (M2-M5 IC50 >> 50 μM). Moreover, these latter analogs, with basic amines, afforded improved solubility and physiochemical characteristics. Despite this improvement in M1 potency, this series did not meet MLSCN/MLPCN potency criteria (M1 IC50 < 500 nM) for a molecular probe.
Figure 4.

SAR plan and analogs of M1 antagonist HTS lead 6 with improved M1 potency and selectivity versus M2-M5.
3. Optimization of a M1 selective antagonist screening hit based on an N-(4-(4-ethylpierazin-1-yl)phenyl amide scaffold
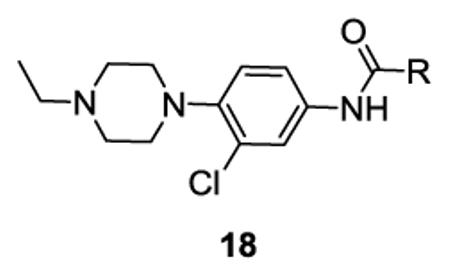
Analogues of 7 and 8 were synthesized in a library format, and both requisite anilines, 3-chloro-(4-(4-ethylpierazin-1yl)aniline and (4-(4-ethylpierazin-1yl)aniline, were commercially available and acylated under standard conditions employing polymer-supported reagents and scavengers to afford 24-member libraries of analogues based on either 7 (M1 IC50 = 0.49 μM, M4 IC50 = 7.9 μM) or 8 (M1 IC50 = 0.58 μM, M4 IC50 = 5.1 μM). In the initial lead optimization phase, we prepare a 24-member library employing a diversity set of acid chlorides containing aromatic, alphatic, polar, basic and acidic moieties in order to rapidly probe the breadth and scope of the SAR; subsequent libraries will be more focused. As the chemistry was straightforward, we elected to re-synthesize the parent compounds 7 and 8 within the library. All analogues were purified by mass-guided HPLC to analytical purity. Surprisingly, all analogues synthesized within the first 24-member library, as well as the re-synthesized parent 8, were found to be inactive on M1. Moreover, upon re-synthesis within a library, 7 lost considerable efficacy as an M1 antagonist (M1 IC50 = 13 μM), but still displayed ~10-fold selectivity versus M4 (IC50 >150 μM). Not surprisingly, analysis of the original screening samples 7 and 8 indicated that there were several impurities in the wells, and we elected not to pursue a complex deconvolution exercise. Despite these findings, the strategy of employing library synthesis and exploding SAR around a primary HTS hit proved advantageous for 7, as analogues 18 proved to possess intriguing mAChR selectivity profiles (Table 1).
Table 1.
Potency and selectivity of analogs 18 of the HTS lead 7.
| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| Cmpd | R | M1 IC50 (μM)a |
M2 IC50 (μM)a |
M3 IC50 (μM)a |
M4 IC50 (μM)a |
M5 IC50 (μM)a |
| 7 |
|
13.2 | >150 | >150 | >150 | >150 |
| 18a |
|
>150 | >150 | >150 | >150 | >150 |
| 18b |
|
4.6 | >150 | >150 | >150 | >150 |
| 18c |
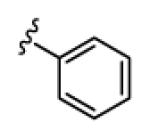
|
5.0 | >150 | >150 | 66 | >150 |
| 18d |
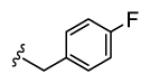
|
5.6 | >150 | >150 | >150 | >150 |
| 18e |
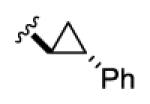
|
1.15 | 29 | 24 | 20 | 13 |
| 18f |
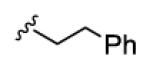
|
1.1 | 52 | 70 | 18 | 7.6 |
| 18g |
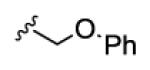
|
3.3 | >150 | >150 | >150 | >150 |
| 18h |
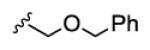
|
18.8 | >150 | >150 | >150 | >150 |
| 18i |
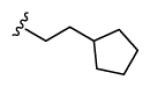
|
0.44 | 3.5 | 3.1 | >150 | 1.1 |
IC50s are an average of three independent experiments using rat mAChR (CHO) cell lines.
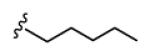
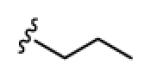
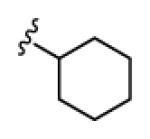
Table 1 highlights SAR and mAChR selectivity for analogs 18 of HTS hit 7. In general, SAR was rather flat for this series. Truncation of the pentyl side chain of 7 to simpler aliphatic chains, such as n-propyl 18a, led to a total loss of M1 antagonist activity. Cyclization to form a cyclohexyl ring, as in 18b, afforded a selective M1 antagonist (M1 IC50 = 4.6 μM, >32-fold selective versus M2-M5), and a 3-fold increase in potency relative to HTS lead 7. The phenyl analog 18c maintained M1 activity relative to 18b, but mAChR selectivity at M4 began to erode. However, conversion to a benzyl moiety 9d once again maintained M1 activity (M1 IC50 = 5.6 μM) and also displayed >26-fold selectivity for M2-M5 (IC50s >150 μM). Further chain homologation to the phenethyl congener 18f afforded a low micromolar potency M1 antagonist (M1 EC50 = 1.1 μM) with high mAChR subtype selectivity (47-fold versus M2, 63-fold versus M3, 16- fold versus M4 and 6.9-fold versus M5). Introduction of a cyclic constraint in the form of a cyclopropyl moiety in the phenethyl chain as in 18e provided a compound with an in vitro profile roughly equivalent to 18f. Incorporation of an oxygen atom in the phenylether as in 18g provided an M1 antagonist of modest potency (M1 IC50 = 3.3 μM), but with >45-fold selectivity versus M2-M5. Replacement of the phenyl moiety with a cyclopentyl group afforded compound 18i, with an M1 IC50 of 441 nM and with >340-fold selectivity versus M4, but modest selectivity versus M2, M3 and M5 (7.9-fold, 7-fold, and 2.4-fold, respectively). Compound 18i possessed the potentcy requirements for an MLSCN/MLPCN M1 antagonist probe molecule (affinity/activity >500 nM) as well as the required selectivity (>10-fold selectivity) versus M4 (>340-fold selectivity). When evaluated against other receptors and enzymes, 18i displayed no significant ancillary pharmacology.
Attention now focused on examining mAChR subtype selectivity in binding assays to determine if the functional selectivity was mirrored in competition radioligand binding experiments and to determine whether 18i was binding at the orthosteric versus an allosteric binding site. For these experiments, we evaluated the ability of 18i to displace [3H]-N-methylscopolamine ([3H]-NMS), an orthosteric radioligand, versus all five mAChR subtypes with atropine as a positive control. In the event, 18i was shown to possess an M1 Ki of 12.7 nM with selectivity versus M2-M5 (6- to 35-fold) and atropine controls demonstrated pan-mAChR antagonism as anticipated (Kis for M1-M5 of 0.56 nM to 2.8 nM). We found that the functional M1 versus M4 selectivity was mirrored in the radioligand competition binding experiment, but the fold-selectivity had diminished ~10-fold.
In order to confirm 18i’s activity in an alternate signaling pathway modulated by M1 and to further elucidate its binding mode, phosphoinostitide (PI) hydrolysis studies and Schild analysis were performed on 18i. 18i causes a dose-dependent rightward shift of the ACh concentration-resposnse curve in a PI hydrolysis experiment which translates in a Schild analysis to a Kd of 10 nM and a slope of 0.98+0.10. These data strongly support the [3H]-NMS binding data and indicate that 18i is an orthosteric M1 antagonist; however, they do not rule out a binding mode wherein 18i partially overlaps with the orthosteric binding site which could account for the observed competitive binding with [3H]-NMS and high M1 versus M4 subtype selectivity. Nor do these data rule out the possibility that 18i is in fact binding to a non-overlapping allosteric site which causes a conformational exclusion of the orthosteric ligand binding site. Mutagenesis and off-rate experiments are planned to address these possibilities.
In summary of this series, an MLSCN/MLPCN M1 antagonist chemical probe development project afforded 18i (VU0359517), a selective M1 versus M4 (>340-fold functional selectivity) orthosteric antagonist which meets the criteria for a small molecule MLSCN/MLPCN chemical probe. Our hit-to-lead strategy of iterative library synthesis to explode SAR and to re-synthesize HTS hits within the first generation libraries proved highly beneficial, as the initial HTS hits 7 and 8 lost considerable activity upon re-synthesis and evaluation. Had we employed a more traditional approach wherein HTS ‘hits’ were first re-synthesized and evaluated prior to generating analogues, these series would not have been pursued further, and 18i (VU0359517) would not have been identified. Clearly, serendipity played a major role in the success of this lead optimization strategy, but this is a high risk approach that must be judiciously employed based on the chemistries involved, the assay capacity and the overall cost. While 18i (VU0359517) is an important tool to dissect the individual contribution of M1 versus M4 antagonism, the muscarinic field still required a potent M1 antagonist probe that was highly selective versus M2-M5.
4. Optimization of a M1 selective antagonist screening hit based on an N-(3-piperazin-1-yl)-3-oxopropyl)benzo[c][1,2,5]thiadiazole-4-sulfonamide scaffold
While the vast majority of our HTS hits displayed no selectivity for M1 versus M4, compound 9, based on a N-(3-piperazin-1-yl)-3-oxopropyl)benzo[c][1,2,5] thiadiazole-4-sulfonamide scaffold, demonstrated a surprising >58-fold selectivity for M1 (IC50 = 2.55 μM) over M4 (IC50 >150 μM). Encouraged by this result, we explored the selectivity of 9 versus M2-M5 and discovered that unlike the other two lead series 6 and 7, 9 was >58-fold selective for M1 versus M2- M5 as well; however, 9 did not meet the potency criteria for an MLSCN/MLPCN probe (IC50 < 500 nM). Our plan for optimization of 9 involved a multi-dimensional library approach (Figure 5); however, like series 7, SAR could be described as ‘flat’ or ‘shallow’, with no tolerance for changes to the linker, regioisomeric attachment of the linker to the 1,2,5-thiadiazole, divergent amines or alternatively aryl and/or heteroaryl piperazine amides. After synthesizing and evaluating ~90 analogs, only two analogs besides 9, both regiosiomeric pyridylpiperazine amides 19 and 20, afforded any significant M1 antagonist activity.
Figure 5.

SAR plan and analogs of M1 antagonist HTS lead 9 with improved M1 potency and selectivity versus M2-M5.
Importantly, 9 (VU0255035) exceeded the potency criteria (IC50 <500 nM) for an MLSCN/MLPCN M1 anatgonist probe with an M1 IC50 of 130 nM. When evaluated against M2-M5, we were pleased to see that VU0255035 was highly selective (M2-M5 IC50 >10 μM) providing >75-fold functional selectivty for M1. Compound concentrations were solubility limited, but little inhibition of M2-M5 was observed with VU0255035 up to 150 μM in some experiments. Compound 9 (VU0255035) represents the most functionally potent and selective small molecule M1 antagonist (MW =432) ever reported, and rivals the mAChR selectivity profile of the macropeptide MT7. The functional mAChR selectivity was mirrored in radioligand binding assay with [3H]-NMS affording 45- to 159-fold selectivity versus M2-M5.
Based on the ‘flat’ SAR and the unprecedented selectivity for a small molecule mAChR antagonist, we assumed, based on our past experience with allosteric ligands, that VU0255035 was in fact an allosteric antagonist. As we had done with 18i, we performed phosphoinostitide (PI) hydrolysis studies and Schild analysis on VU0255035 to confirm its activity in an alternate signaling pathway modulated by M1 and to further elucidate its binding mode. VU0255035 caused a dose-dependent rightward shift of the ACh concentration-resposnse curve in a PI hydrolysis experiment which translates in a Schild analysis to a Kd of 16 nM and a slope of 0.90+0.08. These data support the [3H]-NMS binding data and indicate that VU0255035 is an orthosteric M1 antagonist; however, they do not rule out a binding mode wherein VU0255035 partially overlaps with the orthosteric binding site which could account for the observed competitive binding with [3H]-NMS and high M1 versus M2-M5 subtype selectivity, especially considering the slope was 0.90 instead of 1.0. Nor do these data rule out the possibility that VU0255035 is in fact binding to a non-overlapping allosteric site which causes a conformational exclusion of the orthosteric ligand binding site. However, subsequent mutagenesis experiments confirmed that VU0255035 is an orthosteric antagonist. In the Y381A mutant, wherein ACh loses affinity, VU0255035 antagonism of M1 mAChR responses are right shifted, suggesting VU0255035 is an orthosteric antagonist – despite the high mAChR subtype-selectivity.
Prior to conducting further in vitro and in vivo experiments, the selectivity of VU0255035 was evaluated against larger panels of molecular targets. In both the UNC Psychoactive Drug Screen and against a large panel of GPCRs, ion channels, transporters and kinases, VU0255035 was devoid of significant ancillary pharmacology (no Kis or IC50s <10 μM). At this point, we elected to evaluate the ability of VU0255035 to block the potentiation of carbachol (CCh)-induced NMDAR currents in hippocampal CA1 pyramidal cells. Application of 10 μM CCh elicited a strong NMDA-evoked current, which was completely blocked by 5 μM VU0255035. Alone, VU0255035 had no effect. This study demonstrated that VU0255035 engages the M1 receptor in a native tissue preparation.
Based on these data, we performed a plasma:brain study to determine if VU0255035 would afford brain exposure when dosed systemically. Male Sprauge Dawley rates were dosed with 10 mg/kg (i.p.) VU0255035, and good brains levels of VU0255035 were achieved providing a BrainAUC/PlasmaAUC of 0.48. Studies have shown that the M1 mAChR subtype is responsible for pilocarpine-induced seizures. Thus, we performed studies to determine if our M1-selective antagonist VU0255035 could block pilocarpine-induced seizures in vivo and improve survival. In the event, pilocarpine was administered (280 mg/kg), followed after 40 minutes by VU0255035 at 10 mg/kg (i.p.) or vehicle. After 4 hours, 5/8 (67.5%) of the mice receiving pilocarpine/vehicle died as compared to only 2/8 (25%) pilocarpine/ VU0255035 treated animals at 24 hours. Moreover, VU0255035 had a statistically significant effect reducing seizure count; thus, VU0255035 antagonizes the M1 receptor in vivo. Thus, VU0255035 is a highly selective and brain penetrant M1 antagonist in vitro and in vivo probe.
5. Summary and Outlook
In summary, we have reviewed the probe development process at the Vanderbilt Screening Center for GPCRs, Ion Channels and Transporters, and the companion Chemistry Center, which led to the discovery of VU0255035 is a highly selective and brain penetrant M1 antagonist in vitro and in vivo probe. In short order, three distinct chemical series were evaluated and optimized affording weak, but selective M1 antagonists based on a 3,6-disubstituted-[1,2,4]-triazolo[4,3-b]pyridazine scaffold, or a potent M1 versus M4 selective antagonists based on a N-(4-(4-ethylpierazin-1-yl)phenyl amide scaffold , or finally, the most highly selective small molecule M1 antagonist ever described, based on a N-(3-piperazin-1-yl)-3-oxopropyl)benzo[c][1,2,5] thiadiazole-4-sulfonamide scaffold from which VU0255035 originated. With a selective M1 antagonist in vitro and in vivo probe, the role of M1 can now be dissected in a number of disease states where M1 is thought to play a critical role such as Parkinson’s disease, dystonia and fragile X syndrome to name only a few. As an MLPCN probe compound, VU0255035 is freely available to any investigator upon request, so we are hoping this will lead to an explosion of innovative basic science concerning the M1 receptor.
5. References
- [1].Kumar C, Madison V. AKT crystal structure and AKT-specific inhibitors. Oncogene. 2005;24(50):7493–501. doi: 10.1038/sj.onc.1209087. [DOI] [PubMed] [Google Scholar]
- [2].Cheng Jin Q, Lindsley Craig W., Cheng George Z, Yang Hua, Nicosia Santo V. The Akt/PKB pathway: molecular target for cancer drug discovery. Oncogene. 2005;24(50):7482–92. doi: 10.1038/sj.onc.1209088. [DOI] [PubMed] [Google Scholar]