

Abstract
The post-genomic era has completed its first decade. During this time, we have seen an attempt to understand life not just through the study of individual isolated processes, but through the appreciation of the amalgam of complex networks, within which each process can influence others. Greatly benefiting this view has been the study of the epigenome, the set of DNA and histone protein modifications that regulate gene expression and the function of regulatory non-coding RNAs without altering the DNA sequence itself. Indeed, the availability of reference genome assemblies of many species has led to the development of methodologies such as ChIP-Seq and RNA-Seq that have allowed us to define with high resolution the genomic distribution of several epigenetic elements and to better comprehend how they are interconnected for the regulation of gene expression. In the last few years, the use of these methodologies in the cardiovascular field has contributed to our understanding of the importance of epigenetics in heart diseases, giving new input to this area of research. Here, we review recently acquired knowledge on the role of the epigenome in heart failure, and discuss the need of an epigenomics roadmap for cardiovascular disease.
Keywords: Epigenetics, Heart failure, Non-coding RNA, NGS
1. INTRODUCTION
Heart failure (HF) is a leading cause of morbidity and mortality in modern society. It is a common and final outcome of a variety of pathological conditions that affect myocardial function, such as cardiomyopathy, hypertension, aortic stenosis, myocardial infarction, diabetes mellitus, and intrinsic myocardial defects defined as primary cardiomyopathies [1]. HF is frequently accompanied by pathological remodeling of the heart, a phenomenon caused by cardiac hypertrophy, fibrosis, and inflammation. Underlying these pathological processes is a reprogramming of transcription within the cells that make up the heart. In cardiomyocytes, this reprogramming is characterized by the re-expression of “fetal” cardiac genes and by the repression of adult ones [2-4]. In particular, the re-expression of a set of fetal genes, including fetal myosin heavy chain (known in mouse as β-Mhc or Myh7, and in humans as α-MHC or MYH6), atrial natriuretic peptide (ANP, also known as Nppa) and brain natriuretic factor (BNF, also known Nppb), correlates with the clinical severity and prognosis of the pathology [5, 6].
Chromatin structure is a key element of gene expression control, regulating the accessibility of gene promoters to proteins of the transcription machinery (e.g., transcription factors, RNA polymerase) [7]. Regarding this, the degree of chromatin condensation and nucleosome mobility are two important structural parameters: active transcription is associated withpoorly condensed chromatin and with dynamic nucleosomes, whereas transcriptional inactivity is associated with a rigid chromatin structure due to high compaction and the presence of static nucleosomes [8]. Epigenetics is one important regulatory mechanism of chromatin structure, and thus of gene expression. This term describes a complex network of molecular modifications that act directly by altering the physical properties of histones, or indirectly through the recruitment of proteins or the synthesis of non-coding RNAs (ncRNAs) that influence chromatin organization [9, 10].
The last decade has seen the development of several methodologies that harness the power of next-generation high-throughput DNA sequencing (NGS) technology, such as chromatin immunoprecipitation combined with massively parallel DNA sequencing (ChIP-Seq), methyl-ChIP-Seq, and bisulfite sequencing [11]. These have allowed us to generate precise genome maps of the distribution of DNA and histone modifications. This information is indispensable for our understanding of how epigenetic mechanisms act to regulate gene expression and, hence, their role in physiology and disease [12]. Moreover, NGS technologies applied to RNA (RNA-Seq) have recently provided a more powerful tool for the analysis of the transcriptome, bringing to light the role of ncRNAs in regulating gene expression [13].
The use of these methodologies in the cardiovascular field is now helping us to understand the importance of epigenetics for HF, providing not only new input to the study of molecular mechanisms responsible for this pathology, but also opening up new therapeutic strategies based on the manipulation of the epigenome. Here, we will give an overview of recent discoveries and discuss the potential of targeting epigenetics for the treatment of HF.
2. THE ROLE OF DNA AND HISTONE MODIFICATIONS IN CARDIAC PATHOPHYSIOLOGY
2.1. Histone Acetylation: A Promising Therapeutic Target for HF
In eukaryotes, acetylation is one of the most common covalent post-translational modifications. This epigenetic mark occurs on the ε-amino group of lysine residues of histone and non-histone proteins. Histone acetylation plays a key role in gene expression by regulating the accessibility of gene promoters to transcription factors and complexes. The addition of an acetyl group to lysine residues of histone tails neutralizes their positive charge, weakening the binding of histones to DNA [14]. In addition, acetylation of histone H4 on lysine 16 inhibits the condensation of chromatin into a 30nm chromatin fiber [15]. As a result, chromatin acquires an open conformation that makes the DNA accessible to proteins of the transcriptional machinery. Moreover, acetylated histones can also act as docking sites for proteins involved in gene activation, such as the BET (bromodomain and extra-terminal) proteins, a family of four conserved proteins (Brd2, Brd3, Brd4, and the testis-specific BrdT) that bind acetylated histones through their bromodomain [16]. These proteins function as scaffolds for the assembly of macromolecular complexes involved in the recruitment and activation of the transcriptional machinery. For example, Brd4 positively regulates transcription by recruiting active cyclinT1 and Cdk9 to promoters. These proteins constitute two components of positive transcription elongation factor b (P-TEFb), a complex that promotes RNA polymerase (Pol) II processivity [17, 18].
Acetylation also occurs on non-histone proteins regulating the activity of histones. Important cellular processes regulated by acetylation of these non-histone proteins include mRNA splicing, mRNA transport, mRNA integrity, protein translation, and protein activity. Therefore, it is not surprising that non-histone protein acetylation is implicated in tumorigenesis and immune functions [19].
Acetylation level depends on the activity of two classes of enzymes: histone acetyl transferases (HATs) catalyze the transfer of the acetyl group of acetyl-CoA to the (-amino group of lysine; histone deacetylases (HDACs) remove the acetyl group. HDACs form a large family with at least 18 members classified into four classes according to their phylogenetic conservation: HDAC classes I, II, and IV are the classic HDACs and require electrophilic Zn2+ for their activity; in contrast, the seven sirtuins (SIRT) that make up class III use NAD+ as their cofactor for the deacetylation reaction [20, 21].
Much knowledge on the role of histone acetylation in the heart comes from studies on HDACs and HATs that showed how these enzymes are key players in the gene expression reprogramming underlying HF. The HAT E1A-associated nuclear protein p300 (p300) is a master transcription factor for cardiac hypertrophy. p300 expression is increased in hypertrophied and failing human hearts. In mice models, modulation of its expression interferes with cardiac function: low overexpression of p300 causes cardiac hypertrophy, whereas its downregulation interferes with pressure overload-induced hypertrophy [22]. The picture is more complex for HDACs. HDAC9 and HDAC5 (two class II HDACs) act as negative regulators of cardiac hypertrophy, repressing the activity of MEF2c, a transcription factor that regulates the expression of pro-hypertrophic genes [23, 24]. SIRT6 also has an anti-hypertrophic activity due to its ability to inhibit the pro-hypertrophic insulin-like growth factor (IGF)-Akt signaling pathway. SIRT6 represses the transcription of key IGF signaling-related genes, deacetylating lysine 9 of histone H3 assembled on promoters of those genes. SIRT6 is recruited to these regulatory elements through the interaction with c-Jun [25]. On the other hand, the role of class I HDACs is not completely clear. Trivedi et al. reported that HDAC2 had a pro-hypertrophic function, blocking the expression of anti- hypertrophic gene Inpp5f [26], whereas Montogomery et al. found that HDAC1 and HDAC2 were essential for normal growth and morphogenesis of the heart, but not for the hypertrophic response to infused isoproterenol—a β-adrenergic agonist that causes cardiac hypertrophy—or to transverse aortic constriction (TAC)—a surgical procedure that induces cardiac hypertrophy through pressure overload [27]. These discrepancies in phenotype could be due to the different strategies used for generating the studied knockout mice and/or their genetic background [20, 27].
Interestingly, it was also found that the acetylation of non-histone proteins regulated the development of hypertrophy: SIRT3 protected the heart from oxidative stress during a pro-hypertrophic stimulus, through deacetylation of forkhead box O3a (Foxo3a). This promoted the nuclear localization of Foxo3a, which in turn enhanced the transcription of antioxidant genes [28] (Table 1). Overall, these reports show that acetylation is a mechanism that regulates pathological pathways of HF, acting at multiple levels to modulate chromatin structure and the activity of non-histone proteins.
Table 1.
Cardiac phenotype of genetically modified HDAC mice.
| HDAC Class | Role in Heart | Phenotype of Mice Models |
|---|---|---|
| I | ||
| HDAC 1 and HDAC2 | unclear | Trivedi et al. described that cardiac-specific double knock-out Hdac1/2 mice have dilated cardiomyopathy, arrhythmias, and neonatal lethality, accompanied by up-regulation of genes encoding skeletal muscle-specific myofibrillar proteins and calcium channels. Montogomery et al. did not detect any alteration of cardiac function of knock-out mice (see text). |
| II | ||
| HDAC 5 | anti-hypertrophic | Major sensibility to the development of cardiac hypertrophy and a failure in responding to pro-hypertrophy stimuli, such as TAC and calcineurin activation. |
| HDAC 9 | anti-hypertrophic | Major sensibility to the development of cardiac hypertrophy and a failure in responding to pro-hypertrophy stimuli, such as TAC and calcineurin activation. |
| III | ||
| SIRT6 | anti-hypertrophic | Cardiac-specific double knock-out Sirt6 mice developed cardiac hypertrophy and heart failure; Sirt6 transgenic mice were protected from hypertrophic stimuli, such as TAC and isoproterenol infusion. |
| SIRT3 | anti-hypertrophic | Sirt3 knock-out mice had an increased heart weight/body weight (HW/BW) ratio and interstitial fibrosis at 8 weeks of age; cardiac-specific Sirt3 transgenic mice were protected from hypertrophic stimuli, such angiotensin II infusion and isoproterenol infusion. |
Further steps have been taken more recently toward better understanding of how histone acetylation regulates gene expression changes in HF, by studying BET proteins. Brd2, Brd3, and Brd4 are expressed in adult heart; JQ1—a selective inhibitor of BET proteins—had a cardioprotective effect in mice subjected to TAC or injected with phenylephrine (PE). Moreover, Brd4 knockdown in neonatal rat ventricular cardiomyocytes interfered with PE-induced hypertrophy. Gene-expression profiling and ChIP-Seq studies revealed that BET proteins promote Pol II pause–release and transcription elongation during pathological cardiac hypertrophy [29, 30] (Fig. 1). These findings support an important role for Brd4 in HF and indicate a clinical potential for bromodomain-inhibiting drugs, strengthening previous reports on the therapeutic potential of histone acetylation in HF [31, 32]. However, the development of epigenetics-based HF therapy targeting the histone acetylation pathway is limited by the pleiotropic effect of these drugs due to the ubiquitous expression of their targets. Further studies are needed to identify molecular targets of histone acetylation that are specific for the heart. To this end, definition of the genes and proteins regulated by acetylation in HF might result invaluable.
Fig. (1).
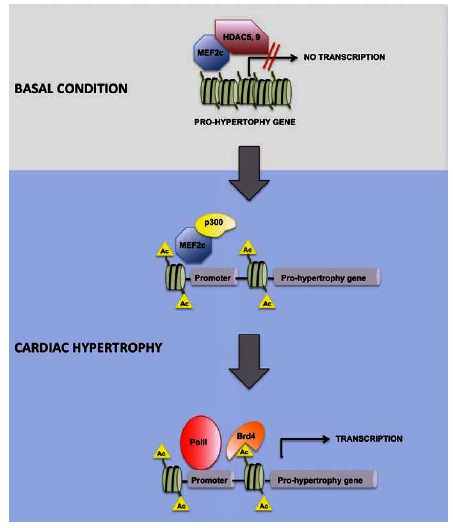
Model of the potential collaboration between Mef2c and BRD4 in promoting the expression of pro-hypertrophic genes. In the basal condition, pro-hypertrophic genes are silenced by the interaction of Mef2c with HDAC5 or HDAC9. In response to a hypertrophic stimulus, HDACs detach from Mef2c, which acetylates histones assembled at promoter regions of pro-hypertrophic genes, through the binding of histone acetyltransferase p300. In turn, BRD4 binds the acetylated promoter, promoting transcription elongation.
2.2. Is Histone Methylation a New Layer of Epigenetic Regulation of HF?
Histone methylation occurs on all three basic residues (i.e., arginine, lysine, and histidine). Most studies on methylation have focused on the first two. Lysine (K) can be mono- (me1), di- (me2), and tri- (me3) methylated on the (-amino group, whereas arginine can be mono- (me1) and di- (me2) methylated on the guanidine group, with addition of two methyl groups occurring symmetrically or asymmetrically. The role of methylated lysine and arginine for gene expression depends on the position of the residue on the histone and on the degree of methylation [33, 34]. For example, H3K36me3 is present at the gene body of transcriptionally active genes and has a key role in transcription elongation, whereas di- and tri-methylation of H3K9 (H3K9me2 and H3K9me3, respectively) promote gene silencing through the recruitment of heterochromatin protein 1 (HP1), a family of three highly conserved proteins (HP1α, HP1β, and HP1γ) involved in chromatin condensation [35]. H3K4me1, outside of the promoter and on the first exon, marks enhancers whose activity depends on the presence of other histone modifications: the epigenetic signature H3K4me1 plus H3K27ac marks active enhancers, whereas H3K4me1 plus H3K27me3 are found at “poised” enhancers [36, 37].
Histone methylation at lysines is a dynamic epigenetic alteration regulated by lysine-specific methyltransferases (KMTs) and demethylases (KDMs). Over 50 of these enzymes have been identified in humans. The higher number of enzymes involved in histone methylation compared with histone acetylation and DNA methylation reflects the greater complexity of the epigenetic language [38]. Although this muddles the understanding of their function, it opens up the possibility of designing more-specific epigenetic therapies than those based on the use of HDAC/DNMT inhibitors [39].
Histone methylation is an important player in cellular processes such as cell cycle, cell differentiation, and DNA repair. Alteration of histone methylation has been linked with several human diseases, including cancer and neuronal disorders [40]. Despite this, only in recent years have we begun to study the role of methylation in HF. Genome-wide histone methylation profiling of cardiomyocytes isolated from TAC mice revealed that histone methylation contributes to the definition of an epigenetic signature that distinguishes genes modulated in cardiac hypertrophy; that also study identified a large set of putative enhancers whose activity changed upon induction of HF, and revealed a role for myocyte enhancer factor MEF2C and MEF2A in regulating these enhancers [41]. A number of studies on histone-methylating enzymes have shown the key role of some of these in regulating the cardiac gene expression program in physiological and pathological conditions. JMJD2A/ KMD4—a histone demethylase that catalyzes the demethylation of H3K9me3 and H3K36me3—is required for cardiac hypertrophy by cooperating with SRF and myocardin, triggering the expression of FHL1 (four and a half LIM domains protein 1), which is involved in mediating the hypertrophic response in TAC mice [42]. PAX-interacting protein 1 (PTIP)—an essential co-factor for methylation of histone H3 at lysine 4—is required to maintain heart homeostasis. Its activity is necessary for the expression of Kv channel-interacting protein 2 (Kcnip2) [43]. It has also been proposed that the loss of H3K9me2 and H3K9me3 at the promoters of ANF and BNP in human failing myocardium is responsible for the re-expression of these genes, and that this process is under the control of HDAC4. The model proposed is that HDAC4 recruits SUV39H1—a histone methyltransferase responsible for di- and tri-methylation of lysine 9 of histone H3—to the promoters of ANF and BNP under normal conditions, whereas in failing myocardium, these protein interactions are disrupted by nuclear export of HDAC4, upon phosphorylation by Ca²⁺/calmodulin-dependent protein kinase II (CaMKII)δB [44].
1.3. Is DNA Methylation a New Epigenetic Marker of Gene Expression Reprogramming in HF?
In higher eukaryotes, DNA methylation occurs preferentially at the fifth carbon of cytosine in CpG dinucleotides. These CpG sites map to intragenic regions, such as promoters and gene bodies, as well as to intergenic regions, including enhancers and repetitive elements [45]. Recent genome-wide methylation profiling has revealed that the effect of DNA methylation on gene expression depends on which genetic element is affected by this epigenetic mark: methylation at promoters is generally associated with transcription repression, whereas DNA methylation within the gene body is positively correlated with transcription activation [46, 47]. Furthermore, there is evidence to suggest that gene body methylation might be involved in pre-mRNA splicing [48]. Finally, CpG methylation at enhancers causes their inactivation [49-51].
The molecular mechanisms through which DNA methylation regulates gene expression are not completely understood. Although there is much evidence that methylation of DNA blocks gene expression by interfering with the binding of transcription factors or the recruitment of methyl-CpG (mCpG)-binding proteins (MBPs) that trigger the inactivation of chromatin [52], it is not clear how this epigenetic mark can promote transcription. In recent years, a dogma of DNA methylation that has fallen is its irreversibility. DNA can be actively demethylated through direct deamination of 5-methyl cytosine (5-mC)—mediated by activation-induced deaminase (AID)/APOBEC-family cytosine deaminases—or by oxidation of 5-mC to 5-hydroxymethylcytosine (5-hmC)—catalyzed by ten-eleven translocation (TET) enzymes—followed by deamination by base excision repair enzymes [53]. Several studies are bringing to light how 5-hmC is not just the intermediate of the demethylation reaction, but can also function as a sixth nucleobase. High-resolution genome-wide mapping of 5-hmC in differentiated cells has linked this DNA modification with transcription activation [54-56]. However, there are other studies in pluripotent cells that have found this DNA modification to be associated with transcription repression [55, 57]. An explanation for this contradiction could be that the function of 5-hmC is chromatin-context dependent.
Despite the fact that DNA methylation is implicated in many cellular processes, such as cell development, genomic stability, and gene imprinting, and that aberrant DNA methylation has been associated with several human diseases, such as cancer and neuronal disorders [58], only in the last few years has the role of this mark been investigated in the heart. DNA methylation profiling of patients with idiopathic dilated cardiomyopathy (DCM) showed that the profile of this epigenetic mark was altered in DCM and that it was linked to repression of two genes—lymphocyte antigen 75 (LY75) and adenosine receptor A2A (ADORA2A)—implicated in adaptive or maladaptive pathways in HF [59]. Another genome-wide study carried out on end-stage cardiomyopathy patients showed an alteration of CpG methylation at promoters and gene bodies. Interestingly, that study revealed changes in DNA methylation level on repetitive elements, suggesting a role in HF [60]. However, it remains to be elucidated how these sequences are involved in this pathology and how changes in the level of DNA methylation regulates gene activity.
Findings in humans have been supported by a recent study showing that the cardiomyocytes of mice subjected to chronic pressure overload stress have a methylation profile similar to that observed at the neonatal stage, and that DNA methylation changes occur on the regulatory sequences of genes involved in processes altered in HF, such as cardiac development and energy metabolism [61]. Moreover, treatment of adult male rats with 5-aza-2′deoxycytidine—an inhibitor of DNA methylation—ameliorated cardiac function after induction of cardiac hypertrophy with norepinephrine [62]. Overall, these studies showed that post-mitotic cardiomyocytes have a dynamic DNA methylation profile, and that the pattern of methylation may define the pathological gene expression program of HF. Further studies are needed to delineate the causal effect of DNA methylation changes in disease and the molecular mechanisms that govern this process. Moreover, although there are few studies aimed at elucidating the role of DNA methylation in HF, the part played by 5-hmC remains unknown. Therefore, studies are required to investigate also this DNA modification in the heart.
3. LONG NON-CODING RNA: A NEW PLAYER IN HEART FAILURE
The advent of tools for high-throughput RNA sequencing (RNA-Seq) has led to the identification of long-non coding RNA (lncRNA) as an additional level of epigenetic regulation. LncRNAs are transcripts more than 200 nucleotides long that do not encode proteins or that have a coding potential of less than 100 amino acids. The mechanisms of action of lncRNAs are not completely clear. However, studies conducted to date have revealed how they might function as regulators of chromatin organization or at transcriptional and post-transcriptional levels, such as mRNA processing, stability, and translation [10]. For example, the lncRNA Kcnq1ot1 represses the transcription of the gene kcnq1, recruiting to its promoter G9a and two members of the polycomb repressive complex 2 (PRC2)—Ezh2 and Suz12—responsible for tri-methylation of H3K9 and H3K27 [63]. The lncRNA NRON inhibits the activity of NFAT, preventing its transport to the nucleus by interfering with the binding of importins, a family of nuclear transport proteins [64]. Finally, the lncRNAs obtained from the transcription of short interspersed elements (SINEs)—a class of repetitive DNA elements—block the transcription of heat shock genes, preventing the formation of pre-initiation complex through the binding with Pol II [65].
LncRNAs have a key role in several biological processes, such as cell cycle, cell differentiation, and imprinting. Thus, it is not surprising that more and more evidence is accumulating on the involvement of lncRNAs in many pathologies, such as cancer, viral infection, and neurodegenerative disorders (e.g., spinocerebellar ataxia, amyotrophic lateral sclerosis, and fragile X syndrome) [66]. Recent reports have also suggested an important role for these transcripts in several aspect of cardiac biology, ranging from development to pathologies such as myocardial infarction and HF. The involvement of lncRNAs in heart development is supported by studies on the lncRNAs Fendrr (fetal-lethal noncoding developmental regulatory RNA) and Bvh (Braveheart). Fendrr is expressed in posterior mesoderm and is required for correct heart development. Knockout mice had an abnormal heart function due to thinning of the ventricular wall caused by impaired cardiomyocyte proliferation. At the molecular level, Fendrr was involved in defining the gene expression program of heart development, promoting either gene activation or silencing through the interaction with PRC2 and TrxG/MLL, which tri-methylate lysine 27 and lysine 4 of histone H3, respectively [67]. Bvh was found essential for cardiomyocyte differentiation and involved in the upstream regulation of Mesp1 (Mesoderm posterior protein 1) [68].
Regarding heart disease, genetic studies have linked the lncRNA MIAT (myocardial associated transcript) with myocardial infarction [69]. Studies on the locus of fetal MHC identified a cluster of lncRNA transcripts in this locus: lncRNAs named myosin-chain-associated RNA transcripts (Myheart or Mhrt) had an anti-hypertrophic action, silencing the guest gene. Mhrts were downregulated in TAC mice, and their overexpression improved cardiac function after pressure overload. Transcriptional repression promoted by Mhrts was driven by the Brg1-HDAC-Parp chromatin complex (Fig. 2) [70].
Fig. (2).
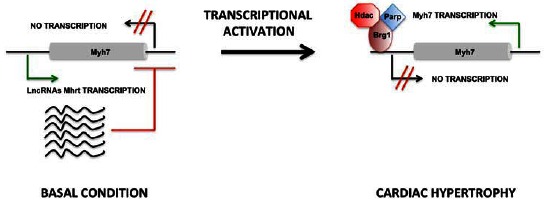
Schematic on the role of Mhrts long noncoding RNAs in cardiac hypertrophy. In the basal condition, the locus of Myh7 encodes a cluster of lncRNAs known as Mhrts (myosin heavy-chain-associated RNA transcripts), which repress the transcription of Myh7. In response to a hypertrophic stimulus, the Brg1-HDAC-Parp chromatin repressor complex inhibits the transcription of Mhrts, causing transcription activation of Myh7.
Finally, recent studies integrating transcriptomic and epigenomic data identified a set of lncRNAs transcribed from enhancers during cardiomyocyte differentiation [71]. Interestingly, a large set of these embryonic-stage genetic elements was found re-expressed in stressed heart. Thus, the recapitulation of the fetal gene expression program in HF concerns not only coding genes but also non-coding enhancer transcripts (known as eRNA). Gene expression profiling of mouse myocardium at different stages of development revealed a number of lncRNAs modulated in hypertrophic myocardium of adult mice, and indicated that the recapitulation of embryonic gene expression concerns only eRNA [72].
CONCLUSION
During the last two decades, much progress has been made in the understanding of the molecular and cellular processes that contribute to HF. Despite this, a cure for this pathology is not available yet.
The above-described studies support an important role for epigenetics in causing the gene expression changes responsible for HF. The findings open up the possibility of developing novel therapeutic strategies for HF based on interference with the epigenetic changes occurring in the pathology. Indeed, many papers on histone deacetylase inhibitors have shown how they prevent cardiac hypertrophy in vitro and in vivo. However, we are far from using HDAC inhibitors clinically for the treatment of HF because of their toxicity. This problem could be overcome through the design of molecules capable of selectively blocking the epigenetic changes occurring in the specific disease. To do this, we will need to better define the “HF epigenome”. Thus, there is the need for an epigenomics roadmap, the objectives of which should be focused on defining how epigenetic elements are modified in the pathology, elucidating the molecular pathways that drive these changes, and determining if the environmental factors impacting heart function, such as diet, lifestyle, and metabolic diseases act on the heart through the modulation of epigenetic mechanisms.
Finally, because disease-causing mutations affecting genes belonging to an epigenetic pathway can be passed on to future generations, genome-wide association studies represent a powerful tool for the investigation of HF etiology and may lead to the development of improved clinical management of HF patients, opening up the possibility of novel diagnostic procedures and personalized treatment strategies designed on individual epigenetic profiles.
ACKNOWLEDGEMENTS
Declared none.
LIST OF ABBREVIATIONS
- β-MHC/MYH7
= Fetal myosin heavy chain
- ANP
= Atrial natriuretic peptide
- BNF
= Brain natriuretic factor
- MEF2c
= Myocyte-specific enhancer factor 2C, also known as MADS box transcription enhancer factor 2, polypeptide C
- SRF
= Serum response factor
- NFAT
= Nuclear factor of activated T-cells
- MIAT
= Myocardial associated transcript
- p300
= E1A-associated nuclear protein p300
- HDAC
= Histone deacetylase
- SIRT
= Sirtuin
- BET proteins
= Bromodomain (BRD) and extra-C terminal domain (BET) proteins
CONFLICT OF INTEREST
The author(s) confirm that this article content has no conflict of interest.
REFERENCES
- 1.McMurray J.J. Clinical practice. Systolic heart failure. N. Engl. J. Med. 2010;362(3):228–238. doi: 10.1056/NEJMcp0909392. [DOI] [PubMed] [Google Scholar]
- 2.Booz G.W., Day J.N., Baker K.M. Interplay between the cardiac renin angiotensin system and JAK-STAT signaling: role in cardiac hypertrophy, ischemia/reperfusion dysfunction, and heart failure. J. Mol. Cell. Cardiol. 2002;34(11):1443–1453. doi: 10.1006/jmcc.2002.2076. [DOI] [PubMed] [Google Scholar]
- 3.Muth J.N., Bodi I., Lewis W., Varadi G., Schwartz A. A Ca(2+)-dependent transgenic model of cardiac hypertrophy: A role for protein kinase Calpha. Circulation. 2001;103(1):140–147. doi: 10.1161/01.CIR.103.1.140. [DOI] [PubMed] [Google Scholar]
- 4.Foshay K., Rodriguez G., Hoel B., Narayan J., Gallicano G.I. JAK2/STAT3 directs cardiomyogenesis within murine embryonic stem cells in vitro. Stem Cells. 2005;23(4):530–543. doi: 10.1634/stemcells.2004-0293. [DOI] [PubMed] [Google Scholar]
- 5.Battistoni A., Rubattu S., Volpe M. Circulating biomarkers with preventive, diagnostic and prognostic implications in cardiovascular diseases. Int. J. Cardiol. 2012;157(2):160–168. doi: 10.1016/j.ijcard.2011.06.066. [DOI] [PubMed] [Google Scholar]
- 6.Feldman A.M., Weinberg E.O., Ray P.E., Lorell B.H. Selective changes in cardiac gene expression during compensated hypertrophy and the transition to cardiac decompensation in rats with chronic aortic banding. Circ. Res. 1993;73(1):184–192. doi: 10.1161/01.RES.73.1.184. [DOI] [PubMed] [Google Scholar]
- 7.Wu J.I., Lessard J., Crabtree G.R. Understanding the words of chromatin regulation. Cell. 2009;136(2):200–206. doi: 10.1016/j.cell.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lanctôt C., Cheutin T., Cremer M., Cavalli G., Cremer T. Dynamic genome architecture in the nuclear space: regulation of gene expression in three dimensions. Nat. Rev. Genet. 2007;8(2):104–115. doi: 10.1038/nrg2041. [DOI] [PubMed] [Google Scholar]
- 9.Bernstein B.E., Meissner A., Lander E.S. The mammalian epigenome. Cell. 2007;128(4):669–681. doi: 10.1016/j.cell.2007.01.033. [DOI] [PubMed] [Google Scholar]
- 10.Wang K.C., Chang H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell. 2011;43(6):904–914. doi: 10.1016/j.molcel.2011.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park P.J. ChIP-seq: advantages and challenges of a maturing technology. Nat. Rev. Genet. 2009;10(10):669–680. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feinberg A.P. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447(7143):433–440. doi: 10.1038/nature05919. [DOI] [PubMed] [Google Scholar]
- 13.Wang Z., Gerstein M., Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009;10(1):57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang X., Hayes J.J. Acetylation mimics within individual core histone tail domains indicate distinct roles in regulating the stability of higher-order chromatin structure. Mol. Cell. Biol. 2008;28(1):227–236. doi: 10.1128/MCB.01245-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shogren-Knaak M., Ishii H., Sun J.M., Pazin M.J., Davie J.R., Peterson C.L. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311(5762):844–847. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- 16.Prinjha R.K., Witherington J., Lee K. Place your BETs: the therapeutic potential of bromodomains. Trends Pharmacol. Sci. 2012;33(3):146–153. doi: 10.1016/j.tips.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 17.Jang M.K., Mochizuki K., Zhou M., Jeong H.S., Brady J.N., Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell. 2005;19(4):523–534. doi: 10.1016/j.molcel.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 18.Yang Z., Yik J.H., Chen R., He N., Jang M.K., Ozato K., Zhou Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell. 2005;19(4):535–545. doi: 10.1016/j.molcel.2005.06.029. [DOI] [PubMed] [Google Scholar]
- 19.Spange S., Wagner T., Heinzel T., Krämer O.H. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int. J. Biochem. Cell Biol. 2009;41(1):185–198. doi: 10.1016/j.biocel.2008.08.027. [DOI] [PubMed] [Google Scholar]
- 20.Haberland M., Montgomery R.L., Olson E.N. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat. Rev. Genet. 2009;10(1):32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vaquero A., Sternglanz R., Reinberg D. NAD+-dependent deacetylation of H4 lysine 16 by class III HDACs. Oncogene. 2007;26(37):5505–5520. doi: 10.1038/sj.onc.1210617. [DOI] [PubMed] [Google Scholar]
- 22.Wei J.Q., Shehadeh L.A., Mitrani J.M., Pessanha M., Slepak T.I., Webster K.A., Bishopric N.H. Quantitative control of adaptive cardiac hypertrophy by acetyltransferase p300. Circulation. 2008;118(9):934–946. doi: 10.1161/CIRCULATIONAHA.107.760488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang C.L., McKinsey T.A., Chang S., Antos C.L., Hill J.A., Olson E.N. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell. 2002;110(4):479–488. doi: 10.1016/S0092-8674(02)00861-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang S., McKinsey T.A., Zhang C.L., Richardson J.A., Hill J.A., Olson E.N. Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol. Cell. Biol. 2004;24(19):8467–8476. doi: 10.1128/MCB.24.19.8467-8476.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sundaresan N.R., Vasudevan P., Zhong L., Kim G., Samant S., Parekh V., Pillai V.B., Ravindra P.V., Gupta M., Jeevanandam V., Cunningham J.M., Deng C.X., Lombard D.B., Mostoslavsky R., Gupta M.P. The sirtuin SIRT6 blocks IGF-Akt signaling and development of cardiac hypertrophy by targeting c-Jun. Nat. Med. 2012;18(11):1643–1650. doi: 10.1038/nm.2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trivedi C.M., Luo Y., Yin Z., Zhang M., Zhu W., Wang T., Floss T., Goettlicher M., Noppinger P.R., Wurst W., Ferrari V.A., Abrams C.S., Gruber P.J., Epstein J.A. Hdac2 regulates the cardiac hypertrophic response by modulating Gsk3 beta activity. Nat. Med. 2007;13(3):324–331. doi: 10.1038/nm1552. [DOI] [PubMed] [Google Scholar]
- 27.Montgomery R.L., Davis C.A., Potthoff M.J., Haberland M., Fielitz J., Qi X., Hill J.A., Richardson J.A., Olson E.N. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007;21(14):1790–1802. doi: 10.1101/gad.1563807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sundaresan N.R., Gupta M., Kim G., Rajamohan S.B., Isbatan A., Gupta M.P. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Invest. 2009;119(9):2758–2771. doi: 10.1172/JCI39162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anand P., Brown J.D., Lin C.Y., Qi J., Zhang R., Artero P.C., Alaiti M.A., Bullard J., Alazem K., Margulies K.B., Cappola T.P., Lemieux M., Plutzky J., Bradner J.E., Haldar S.M. BET bromodomains mediate transcriptional pause release in heart failure. Cell. 2013;154(3):569–582. doi: 10.1016/j.cell.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spiltoir J.I., Stratton M.S., Cavasin M.A., Demos-Davies K., Reid B.G., Qi J., Bradner J.E. BET acetyl-lysine binding proteins control pathological cardiac hypertrophy. . J Mol Cell Cardiol. . 2013;63 :175 –179.. doi: 10.1016/j.yjmcc.2013.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Antos C.L., McKinsey T.A., Dreitz M., Hollingsworth L.M., Zhang C.L., Schreiber K., Rindt H., Gorczynski R.J., Olson E.N. Dose-dependent blockade to cardiomyocyte hypertrophy by histone deacetylase inhibitors. J. Biol. Chem. 2003;278(31):28930–28937. doi: 10.1074/jbc.M303113200. [DOI] [PubMed] [Google Scholar]
- 32.Gallo P., Latronico M.V., Gallo P., Grimaldi S., Borgia F., Todaro M., Jones P., Gallinari P., De Francesco R., Ciliberto G., Steinkühler C., Esposito G., Condorelli G. Inhibition of class I histone deacetylase with an apicidin derivative prevents cardiac hypertrophy and failure. Cardiovasc. Res. 2008;80(3):416–424. doi: 10.1093/cvr/cvn215. [DOI] [PubMed] [Google Scholar]
- 33.Kouzarides T. Histone methylation in transcriptional control. Curr. Opin. Genet. Dev. 2002;12(2):198–209. doi: 10.1016/S0959-437X(02)00287-3. [DOI] [PubMed] [Google Scholar]
- 34.Bernstein B.E., Kamal M., Lindblad-Toh K., Bekiranov S., Bailey D.K., Huebert D.J., McMahon S., Karlsson E.K., Kulbokas E.J., III, Gingeras T.R., Schreiber S.L., Lander E.S. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120(2):169–181. doi: 10.1016/j.cell.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 35.Lachner M., O’Carroll D., Rea S., Mechtler K., Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001;410(6824):116–120. doi: 10.1038/35065132. [DOI] [PubMed] [Google Scholar]
- 36.Zentner G.E., Tesar P.J., Scacheri P.C. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res. 2011;21(8):1273–1283. doi: 10.1101/gr.122382.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Creyghton M.P., Cheng A.W., Welstead G.G., Kooistra T., Carey B.W., Steine E.J., Hanna J., Lodato M.A., Frampton G.M., Sharp P.A., Boyer L.A., Young R.A., Jaenisch R. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA. 2010;107(50):21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cloos P.A., Christensen J., Agger K., Helin K. Erasing the methyl mark: histone demethylases at the center of cellular differentiation and disease. Genes Dev. 2008;22(9):1115–1140. doi: 10.1101/gad.1652908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arrowsmith C.H., Bountra C., Fish P.V., Lee K., Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat. Rev. Drug Discov. 2012;11(5):384–400. doi: 10.1038/nrd3674. [DOI] [PubMed] [Google Scholar]
- 40.Greer E.L., Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012;13(5):343–357. doi: 10.1038/nrg3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Papait R., Cattaneo P., Kunderfranco P., Greco C., Carullo P., Guffanti A., Viganò V., Stirparo G.G., Latronico M.V., Hasenfuss G., Chen J., Condorelli G. Genome-wide analysis of histone marks identifying an epigenetic signature of promoters and enhancers underlying cardiac hypertrophy. Proc. Natl. Acad. Sci. USA. 2013;110(50):20164–20169. doi: 10.1073/pnas.1315155110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang T., Kohlhaas M., Backs J., Mishra S., Phillips W., Dybkova N., Chang S., Ling H., Bers D.M., Maier L.S., Olson E.N., Brown J.H. CaMKIIdelta isoforms differentially affect calcium handling but similarly regulate HDAC/MEF2 transcriptional responses. J. Biol. Chem. 2007;282(48):35078–35087. doi: 10.1074/jbc.M707083200. [DOI] [PubMed] [Google Scholar]
- 43.Stein A.B., Jones T.A., Herron T.J., Patel S.R., Day S.M., Noujaim S.F., Milstein M.L., Klos M., Furspan P.B., Jalife J., Dressler G.R. Loss of H3K4 methylation destabilizes gene expression patterns and physiological functions in adult murine cardiomyocytes. J. Clin. Invest. 2011;121(7):2641–2650. doi: 10.1172/JCI44641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hohl M., Wagner M., Reil J.C., Müller S.A., Tauchnitz M., Zimmer A.M., Lehmann L.H., Thiel G., Böhm M., Backs J., Maack C. HDAC4 controls histone methylation in response to elevated cardiac load. J. Clin. Invest. 2013;123(3):1359–1370. doi: 10.1172/JCI61084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Suzuki M.M., Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat. Rev. Genet. 2008;9(6):465–476. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- 46.Deaton A.M., Webb S., Kerr A.R., Illingworth R.S., Guy J., Andrews R., Bird A. Cell type-specific DNA methylation at intragenic CpG islands in the immune system. Genome Res. 2011;21(7):1074–1086. doi: 10.1101/gr.118703.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maunakea A.K., Nagarajan R.P., Bilenky M., Ballinger T.J., D’Souza C., Fouse S.D., Johnson B.E., Hong C., Nielsen C., Zhao Y., Turecki G., Delaney A., Varhol R., Thiessen N., Shchors K., Heine V.M., Rowitch D.H., Xing X., Fiore C., Schillebeeckx M., Jones S.J., Haussler D., Marra M.A., Hirst M., Wang T., Costello J.F. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466(7303):253–257. doi: 10.1038/nature09165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maunakea A.K., Chepelev I., Cui K., Zhao K. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 2013;23(11):1256–1269. doi: 10.1038/cr.2013.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hon G.C., Rajagopal N., Shen Y., McCleary D.F., Yue F., Dang M.D., Ren B. Epigenetic memory at embryonic enhancers identified in DNA methylation maps from adult mouse tissues. Nat. Genet. 2013;45(10):1198–1206. doi: 10.1038/ng.2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lister R., Pelizzola M., Dowen R.H., Hawkins R.D., Hon G., Tonti-Filippini J., Nery J.R., Lee L., Ye Z., Ngo Q.M., Edsall L., Antosiewicz-Bourget J., Stewart R., Ruotti V., Millar A.H., Thomson J.A., Ren B., Ecker J.R. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462(7271):315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stadler M.B., Murr R., Burger L., Ivanek R., Lienert F., Schöler A., van Nimwegen E., Wirbelauer C., Oakeley E.J., Gaidatzis D., Tiwari V.K., Schübeler D. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011;480(7378):490–495. doi: 10.1038/nature10716. [DOI] [PubMed] [Google Scholar]
- 52.Hendrich B., Tweedie S. The methyl-CpG binding domain and the evolving role of DNA methylation in animals. Trends Genet. 2003;19(5):269–277. doi: 10.1016/S0168-9525(03)00080-5. [DOI] [PubMed] [Google Scholar]
- 53.Tahiliani M., Koh K.P., Shen Y., Pastor W.A., Bandukwala H., Brudno Y., Agarwal S., Iyer L.M., Liu D.R., Aravind L., Rao A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324(5929):930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ficz G., Branco M.R., Seisenberger S., Santos F., Krueger F., Hore T.A., Marques C.J., Andrews S., Reik W. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature. 2011;473(7347):398–402. doi: 10.1038/nature10008. [DOI] [PubMed] [Google Scholar]
- 55.Wutz A., Gribnau J. X inactivation Xplained. Curr. Opin. Genet. Dev. 2007;17(5):387–393. doi: 10.1016/j.gde.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 56.Tsagaratou A., Äijö T., Lio C.W., Yue X., Huang Y., Jacobsen S.E., Lähdesmäki H., Rao A. Dissecting the dynamic changes of 5-hydroxymethylcytosine in T-cell development and differentiation. Proc. Natl. Acad. Sci. USA. 2014;111(32):E3306–E3315. doi: 10.1073/pnas.1412327111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu H., D’Alessio A.C., Ito S., Wang Z., Cui K., Zhao K., Sun Y.E., Zhang Y. Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes Dev. 2011;25(7):679–684. doi: 10.1101/gad.2036011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Robertson K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005;6(8):597–610. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- 59.Haas J., Frese K.S., Park Y.J., Keller A., Vogel B., Lindroth A.M., Weichenhan D., Franke J., Fischer S., Bauer A., Marquart S., Sedaghat-Hamedani F., Kayvanpour E., Köhler D., Wolf N.M., Hassel S., Nietsch R., Wieland T., Ehlermann P., Schultz J.H., Dösch A., Mereles D., Hardt S., Backs J., Hoheisel J.D., Plass C., Katus H.A., Meder B. Alterations in cardiac DNA methylation in human dilated cardiomyopathy. EMBO Mol. Med. 2013;5(3):413–429. doi: 10.1002/emmm.201201553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Movassagh M., Choy M.K., Knowles D.A., Cordeddu L., Haider S., Down T., Siggens L., Vujic A., Simeoni I., Penkett C., Goddard M., Lio P., Bennett M.R., Foo R.S. Distinct epigenomic features in end-stage failing human hearts. Circulation. 2011;124(22):2411–2422. doi: 10.1161/CIRCULATIONAHA.111.040071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gilsbach R., Preissl S., Grüning B.A., Schnick T., Burger L., Benes V., Würch A., Bönisch U., Günther S., Backofen R., Fleischmann B.K., Schübeler D., Hein L. Dynamic DNA methylation orchestrates cardiomyocyte development, maturation and disease. Nat. Commun. 2014;5:5288. doi: 10.1038/ncomms6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xiao D., Dasgupta C., Chen M., Zhang K., Buchholz J., Xu Z., Zhang L. Inhibition of DNA methylation reverses norepinephrine-induced cardiac hypertrophy in rats. Cardiovasc. Res. 2014;101(3):373–382. doi: 10.1093/cvr/cvt264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pandey R.R., Mondal T., Mohammad F., Enroth S., Redrup L., Komorowski J., Nagano T., Mancini-Dinardo D., Kanduri C. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol. Cell. 2008;32(2):232–246. doi: 10.1016/j.molcel.2008.08.022. [DOI] [PubMed] [Google Scholar]
- 64.Willingham A.T., Orth A.P., Batalov S., Peters E.C., Wen B.G., Aza-Blanc P., Hogenesch J.B., Schultz P.G. A strategy for probing the function of noncoding RNAs finds a repressor of NFAT. Science. 2005;309(5740):1570–1573. doi: 10.1126/science.1115901. [DOI] [PubMed] [Google Scholar]
- 65.Geisler S., Coller J. RNA in unexpected places: long non-coding RNA functions in diverse cellular contexts. Nat. Rev. Mol. Cell Biol. 2013;14(11):699–712. doi: 10.1038/nrm3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Qureshi I.A., Mehler M.F. Emerging roles of non-coding RNAs in brain evolution, development, plasticity and disease. Nat. Rev. Neurosci. 2012;13(8):528–541. doi: 10.1038/nrn3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Grote P., Wittler L., Hendrix D., Koch F., Währisch S., Beisaw A., Macura K., Bläss G., Kellis M., Werber M., Herrmann B.G. The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev. Cell. 2013;24(2):206–214. doi: 10.1016/j.devcel.2012.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Klattenhoff C.A., Scheuermann J.C., Surface L.E., Bradley R.K., Fields P.A., Steinhauser M.L., Ding H., Butty V.L., Torrey L., Haas S., Abo R., Tabebordbar M., Lee R.T., Burge C.B., Boyer L.A. Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell. 2013;152(3):570–583. doi: 10.1016/j.cell.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ishii N., Ozaki K., Sato H., Mizuno H., Saito S., Takahashi A., Miyamoto Y., Ikegawa S., Kamatani N., Hori M., Saito S., Nakamura Y., Tanaka T. Identification of a novel non-coding RNA, MIAT, that confers risk of myocardial infarction. J. Hum. Genet. 2006;51(12):1087–1099. doi: 10.1007/s10038-006-0070-9. [DOI] [PubMed] [Google Scholar]
- 70.Han P., Li W., Lin C.H., Yang J., Shang C., Nurnberg S.T., Jin K.K., Xu W., Lin C.Y., Lin C.J., Xiong Y., Chien H.C., Zhou B., Ashley E., Bernstein D., Chen P.S., Chen H.S., Quertermous T., Chang C.P. A long noncoding RNA protects the heart from pathological hypertrophy. Nature. 2014;514(7520):102–106. doi: 10.1038/nature13596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ounzain S., Pezzuto I., Micheletti R., Burdet F., Sheta R., Nemir M., Gonzales C., Sarre A., Alexanian M., Blow M.J., May D., Johnson R., Dauvillier J., Pennacchio L.A. Functional importance of cardiac enhancer-associated noncoding RNAs in heart development and disease. J. Mol. Cell Cardiol. 2014;76 :55–70.. doi: 10.1016/j.yjmcc.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Matkovich S.J., Edwards J.R., Grossenheider T.C., de Guzman Strong C., Dorn G.W., II Epigenetic coordination of embryonic heart transcription by dynamically regulated long noncoding RNAs. Proc. Natl. Acad. Sci. USA. 2014;111(33):12264–12269. doi: 10.1073/pnas.1410622111. [DOI] [PMC free article] [PubMed] [Google Scholar]