

Abstract
Rapid adoption of next generation sequencing (NGS) in genomic medicine has been driven by low cost, high throughput sequencing and rapid advances in our understanding of the genetic bases of human diseases. Today, the NGS method has dominated sequencing space in genomic research, and quickly entered clinical practice. Because unique features of NGS perfectly meet the clinical reality (need to do more with less), the NGS technology is becoming a driving force to realize the dream of precision medicine. This article describes the strengths of NGS, NGS panels used in precision medicine, current applications of NGS in cytology, and its challenges and future directions for routine clinical use.
Keywords: Next generation sequencing, Gene panel, Precision medicine, Whole exome sequencing, Cancer.
THERE IS A CLINICAL NEED FOR NGS
Precision medicine is an emerging approach for disease prevention and treatment that takes individual variability into account [1]. To achieve “individual variability” requires analyzing multiple genes with little amounts of specimen inexpensively, quickly and sensitively. In late 20th century, Sanger sequencing was the most widely used sequencing method for approximately 25 years. More recently, Sanger sequencing has been supplanted by the next generation sequencing (NGS) technology. Compared with Sanger sequencing, NGS has many advantages, including: i) Speed – NGS is massively parallel, producing 500GB data in a single run on a single flow cell of HiSeq2500; ii) Cost- The massively parallel nature of NGS reduces sequencing time, manpower and reagents that translate into significant savings. For example, sequencing 1Mb DNA only costs $0.04 by using HiSeq2500, and $0.007 using HiSeq X Ten; iii) Sensitivity - NGS can reliably detect >1% mutations. This is critically important for detecting somatic mutations in the heterogeneous tumor samples; iv) Amount of sample - With the advances of library construction technology, NGS can perform well with the nanogram range of DNA. Both MiSeq and Ion PGM can sequence around 50 targeted genes with 10-50ng of FFPE DNA. This is particularly useful for the most accessible cytology specimens. In many clinical situations, the only available specimen is a fine needle core or aspiration biopsy or FFPE tissue slides, which do not provide enough DNA for classical Sanger sequencing; v) The number of targets - NGS technology can sequence multiple genes at a higher coverage. Since genomic research has facilitated the pace of target discovery for disease management, the numbers of genes that are associated with a disease phenotype and need to be assessed are increasing rapidly. Accordingly, there has been a marked increase in the number of targeted therapies approved for the treatment of patients with specific types of malignancies harboring specific types of sequence alterations [2].
In summary, NGS can accurately and sensitively sequence more target genes with less DNA, with reduced cost, time and labor as compared with Sanger sequencing. These tasks are sometimes either technically or practically not feasible for Sanger sequencing. A typical example is advanced or metastatic colorectal cancer that has wild-type KRAS, and therefore treated by anti-epidermal growth factor receptor (EGFR) antibodies such as cetuximab and panitumumab [3]. These patients frequently become resistant to the anti-EGFR therapy when activating mutations occur in either KRAS, NRAS, PIK3CA or BRAF. With a highly heterogeneous needle biopsy or a FFPE tissue slide, NGS can simultaneously test all possible mutations at >1% in these 4 genes as well as others to guide therapeutic decision-making. Such an analysis is difficult to achieve by Sanger sequencing, both practically (takes too long and costs too much) and technically (not enough tissue and low sensitivity).
NGS technology is still rapidly evolving. In the last decade, cost has rapidly decreased while throughput is continuously increasing with the release of new chemistry and new models (Fig. 1). Recently, the throughput of HiSeq2500 increased from 600GB to 1TB by the combination of newer V4 chemistry and a newer camera model which supports the higher cluster densities. On January 12th, 2015, Illumina announced the HiSeq3000/4000, which further increased throughput to 429GB/day, reduced cost to $20/GB compared with 167GB/day and $30/GB for V4 HiSeq2500, respectively. The continuous evolution of NGS technology in throughput and cost will undoubtedly further strengthen the position of NGS in precision medicine.
Fig. (1).
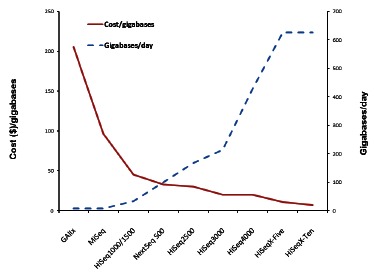
The Evolution of Sequencing Cost and Throughput with Different Sequencers. In the last decade, with the release of new sequencers, models and chemistries, the cost of sequencing has been rapidly decreasing, from over $200/gigabase (GAIIx) to under $10/gigabase (Hiseq X-Ten); while the throughput has been continuously increasing, from under 10 gigabases/day (GAIIx) to over 600 gigabases/day (Hiseq X-Ten).
CURRENTLY USED NGS PANELS IN PRECISION MEDICINE
NGS technology is rapidly making its way into clinical laboratories. Till now, most clinical applications have been in diagnostic testing for hereditary disorders and, more recently, for risk screening for hereditary cancers and therapeutic decision-making for somatic cancers. The testing contents have evolved from hotspot panels, actionable gene panels, and disease-focused panels to more comprehensive panels. Although exome and whole genome sequencing approaches are beginning to emerge, given the incomplete clinical annotation of the human genome, panel-based testing is more practical at the present time, and already holds a firm place in clinical applications [4]. (Fig. 2) shows the evolution of NGS based clinical testing from hotspot panels to whole genome sequencing. A brief summary of the current status of those panels is described below.
Fig. (2).

The Evolution of the NGS-Based Clinical Testing. NGS technologies are rapidly integrated into clinical applications. The hotspot panels, actionable gene panels, and disease-focused panels are commonly used in clinical testing. With further understanding and clinical annotation of the human genome, the exome and whole genome sequencing approaches will play more important role in clinical applications.
Hotspot Panels
The hotspot panel is a collection of frequently mutated hotspots that are either clinically actionable or with diagnostic/prognostic significance. Over the past several years, there has been a major shift in cancer diagnostics from physical and histological findings to additional assessment of targetable genomic mutations. In solid tumors, a good example where a hotspot panel used is lung cancer. Since the first approval of targeted drugs, like Tarceva (erlotinib) and Iressa (gefitinib), for non-small cell lung cancer (NSCLC) with activated EGFR mutations a decade ago, and recent approval of Xalkori (crizotinib) for patients with ALK gene fusions, routine genetic testing for somatic mutations from lung cancer biopsies has become the standard for providing optimal patient care [5-8]. In fact, the College of American Pathologists (CAP), the International Association for the Study of Lung Cancer (IASLC), and the Association for Molecular Pathology (AMP) issued a joint, evidence-based guideline establishing recommendations for molecular diagnostic testing in lung cancer [9]. Their recommendations were to test all patients with lung adenocarcinoma for EGFR and ALK abnormalities, regardless of clinical variables such as smoking history, gender, or ethnicity, to determine if tyrosine kinase or ALK inhibitor therapy may be beneficial.
There are two types of hotspot cancer panels currently available commercially to guide for treatment: one for the choice of therapy and the other for the amount of medication.
The AmpliSeq cancer panel V1, developed by the Life Technologies, represents one example of the former. This was the first commercially released hotspot cancer panel, and covers 739 clinically relevant hotspot mutations from 46 cancer genes, including well-established tumor suppressor genes and oncogenes. The product was designed for potential clinical application by including all EGFR, BRAF, KRAS and other clinically actionable hotspot mutations. Given its popularity, Illumina subsequently released a similar product -Truseq Amplicon cancer panel which targets 48 genes with 212 amplicons. In a recent large, prospective, multi-institution study of 1094 newly diagnosed cancer patients from Australia, tumor samples from 854 patients were successfully sequenced using the Illumina Truseq Amplicon cancer panel [10]. This study showed the reliability of the NGS technology to examine multiple gene loci across different tumor types in a single workflow. Clinically significant mutations were identified in 63% of patients in this study, and 26% patients had mutations with therapeutic implications.
Currently, the Ion AmpliSeq Cancer Panel V2 dominates the Hotspot panel market in the United States. This 50-gene hotspot panel maintains compatibility with FFPE samples while expanding the number of hotspot mutations to 2855. As expected, this panel has now been clinically validated and offered as a clinical test by several academic institutes and private laboratories, including the UCLA Clinical Microarray Lab and the Baylor Cancer Genetics Lab.
PGxOne™ pharmacogenomics test, developed by Admera Health, represents the second type of hotspot panel (http://www.admerahealth.com/pgxone/). This assay screens for 152 frequently mutated sites from 13 well-established pharmacogenomics genes that affect drug absorption, metabolism, or activity. These medically actionable and clinically relevant mutational data provide information for physicians to prescribe appropriate dose for effective treatment.
Actionable Gene Panels
The actionable gene panels evolved from hotspot panels by including all exons of targeted genes (or all clinical relevant regions) so that other pathogenic mutations outside frequently mutated sites can be interrogated. The common feature of these panels is to focus on actionable genes, such as EGFR, BRAF, KRAS, PIK3CA, NRAS, KIT and ALK, which are often targets of FDA-approved drugs in different tumor types. These testing results complement traditional cancer treatment tools, and expand treatment options by matching each patient with targeted therapies and clinical trials. These panels are currently offered by commercial vendors, academic institutes as well as private laboratories.
The first commercially released, small actionable gene panel is the TruSight Tumor panel, which enables clinical researchers to identify low-frequency mutations across 26 genes that are involved in targeted therapy for lung, colon, gastric, ovarian cancer and melanoma. This panel has been clinically validated and offered as a clinical test by several institutes. The V2 Comprehensive Cancer Gene Set offered by Washington University in St. Louis is a medium-sized, clinically actionable, customized cancer panel. This panel includes 42 clinically actionable cancer genes (20 for solid tumors, 16 for liquid tumors, and 6 for both) designed for assisting oncologists with stratification of disease subtypes and tailoring of effective personalized therapies. Foundation One, developed by Foundation Medicine, represents a comprehensive actionable gene panel. It interrogates the entire coding sequence of 236 cancer-related genes plus 47 introns from 19 genes which are often rearranged or altered in solid tumor cancers. These genes are known to be somatically altered in solid cancers based on recent scientific and clinical literature. This test provides more potential treatment options from not only FDA-approved targeted therapies, but also clinical trials.
The actionable gene panel has also been used for hereditary cancer to help risk assessment and patient management. Recently released from Myriad is a 25-gene panel named MyRisk, designed for the identification of clinically significant mutations impacting inherited risks for eight important cancers: breast, colorectal, ovarian, endometrial, gastric, melanoma, pancreatic, and prostate cancer. The test interpretation combines test results with personal/family cancer history for clinically actionable risk assessment, and provides specific medical management recommendations based on the guidelines of leading professional medical societies.
Disease-Focused Panels
The actionable gene panels are collections of well-studied actionable genes that are commonly involved in several diseases. Most of such panels interrogate somatic mutations to aid in therapeutic decision-making. The disease-focused panels are comprised of the genes for a particular disease. The latter panels are largely used for the germ line mutations to screen for the risk of inherited diseases, or to diagnose suspected genetic diseases. At present, the hereditary cancer panels are popular tests. Approximately 5-10% of all cancers are hereditary. More than 100 cancer susceptibility syndromes have been reported, including hereditary breast and ovarian cancer syndrome (HBOC), Lynch Syndrome, Cowden syndrome (CS) and Li-Fraumeni Syndrome (LFS). Many of these risk genes share molecular pathways and play a role in the repair of DNA damage, such as high risk gene BRCA1 and BRCA2, and modest risk gene BRIP1 and PALB2, which are all part of the Fanconi Anemia (FA)-BRCA Molecular Pathway and associated with increased risk of breast and ovarian cancer [11]. NGS-based screening for all of those genes for a particular cancer provides critical risk information for preventive management. These panels generally have a limited set of genes allowing multiplex and greater depth of coverage for increased analytical sensitivity and specificity, and decreased cost.
As of February 5th, 2015, ~7653 NGS-based clinical tests were available across the world (estimated using NGS as a term in the NIH Genetic Testing Registry, website www.ncbi.nlm.nih.gov), with about one-third in the commercial sector and two-thirds in academically affiliated clinical laboratories. There were 332 cancer panels from 115 laboratories. Of those, 227 tests from 91 laboratories were for hereditary cancer screening. (Table 1) summarizes a few popular, currently available hereditary cancer panels. Although fewer clinical laboratories have launched disease-focused NGS tests for somatic cancers, many laboratories are actively developing such tests. In the next few years, this is the area expected to expand quickly.
Table 1.
Examples of representative hereditary cancer panels.
| Gene | BreastPanel | ColonPanel | OvaPanel | PancPanel | RenalPanel | EndometrialPanel |
|---|---|---|---|---|---|---|
| APC | ∆ | ● √ ∆ | ∆ | ● √ | ||
| ATM | ● √ | √ | ● √ | ● √ | ||
| AXIN2 | √ | |||||
| BARD1 | ● √ | ● √ | ||||
| BLM | √ | √ | √ | |||
| BMPR1A | ● | ● √ ∆ | ● | |||
| BRCA1 | ● √ ∆ | ∆ | ● √ ∆ | ● √ | √ | |
| BRCA2 | ● √ ∆ | ∆ | ● √ ∆ | ● † √ | √ | |
| BRIP1 | ● √ ∆ | ● √ ∆ | ||||
| CDC73 | ∆ | |||||
| CDH1 | ● √ ∆ | ● √ ∆ | ● √ ∆ | |||
| CDK4 | √ | |||||
| CDKN1C | ∆ | |||||
| CDKN2A | ∆ | ● √ | ||||
| CHEK2 | ● √ ∆ | ● √ ∆ | ● √ ∆ | √ | ||
| EPCAM | √ ∆ | ● √ ∆ | ● √ ∆ | ● √ | ● | ● √ |
| FAM175A | √ | √ | ||||
| FANCC | √ | √ | √ | |||
| FH | ● ∆ | |||||
| FLCN | ● ∆ | |||||
| GPC3 | ∆ | |||||
| HOXB13 | √ | √ | ||||
| MAX | ∆ | |||||
| MEN1 | ∆ | |||||
| MET | ● ∆ | |||||
| MIFT | ● | |||||
| MLH1 | √ ∆ | ● √ ∆ | ● √ ∆ | ● √ | ● | ● √ |
| MRE11A | ● √ | √ | ||||
| MSH2 | √ ∆ | ● √ ∆ | ● √ ∆ | ● √ | ● | ● √ |
| MSH6 | √ ∆ | ● √ ∆ | ● √ ∆ | ● √ | ● | ● √ |
| MUTYH | ● ∆ | ● √ ∆ | ● ∆ | √ | ||
| NBN | ● √ ∆ | ● √ ∆ | ||||
| NF1 | ● | ∆ | ||||
| PALB2 | ● √ ∆ | ● √ ∆ | ● √ | ∆ | ||
| PALLD | √ | |||||
| PMS1 | ∆ | ∆ | ∆ | |||
| Gene | BreastPanel | ColonPanel | OvaPanel | PancPanel | RenalPanel | EndometrialPanel |
| PMS2 | √ ∆ | ● √ ∆ | ● √ ∆ | ● √ | ● | ● √ |
| PRKAR1A | ∆ | |||||
| PTEN | ● √ ∆ | ● √ ∆ | ● √ ∆ | ● ∆ | ● √ ∆ | |
| RAD50 | ● √ | ● √ | ||||
| RAD51C | ● √ ∆ | ● √ ∆ | ||||
| RAD51D | ● √ ∆ | ● √ ∆ | ||||
| RET | ∆ | |||||
| SDHA | ● | |||||
| SDHAF2 | ∆ | |||||
| SDHB | ● ∆ | ∆ | ||||
| SDHC | ● | ∆ | ||||
| SDHD | ● ∆ | ∆ | ||||
| SMAD4 | ● √ ∆ | |||||
| STK11 | ● √ ∆ | ● √ ∆ | ● √ ∆ | ● √ | ||
| TMEM127 | ∆ | |||||
| TSC1 | ● | |||||
| TSC2 | ● | |||||
| TP53 | ● √ ∆ | ● √ ∆ | ● √ ∆ | ● √ | ● | √ ∆ |
| VHL | √ | ● ∆ | ∆ | |||
| WT1 | ∆ | |||||
| XRCC2 | √ | √ | √ | √ |
● Ambry Genetics; √ GeneDx; ∆ Baylor.
Comprehensive Panels
Although disease-focused panels have gained popularity, clinical laboratories are facing serious financial and practical challenges associated with 1) the development and validation of different disease-focused panels according to the American College of Medical Genetics and Genomics (ACMG) guidelines; 2) the limited number of clinical specimens required for clinical testing for any given disease at any given time; 3) the requirement to constantly update the content of existing panels. These challenges have led clinicians to wonder whether they should move directly to exome or whole genome sequencing. The question seems to be a relevant one, but laboratories hesitate to make the move when they have to face the hundreds of variants with unknown clinical significance from whole genome or whole exome sequencing approaches.
One compromise is to consider a more comprehensive panel that includes all genes associated with all diseases.
By taking full advantage of the high throughput nature of NGS technology, this approach would satisfy the simplicity of disease-targeted testing and also avoid interrogation of the majority of variants of unknown clinical significance. This “one for all” approach could minimize test development and validation efforts for multiple disease focused panels, maximize the multiplex capability by combining samples with different diseases in one assay, and reduce the frequency of required updates as new genes are identified. In practice, physicians could request testing using a specific disease focused sub-panel that is relevant to the patient’s phenotype. If that sub-panel does not yield expected results, additional analysis could be requested using the full panel, if clinically indicated.
Illumina’s TruSight One is an example of such a comprehensive panel,. This panel includes more than 60 well-established sub-panels and covers 4813 genes having known association with clinical phenotypes. This panel was designed to encompass the most commonly requested targeted gene panel assays, enabling laboratories to evaluate the gene targets in all of these panels with one physical assay. The Trusight One includes all exonic regions harboring disease-causing mutations identified based on information in the Human Gene Mutation Database (HGMD Professional), the Online Mendelian Inheritance in Man (OMIM) catalog, GeneTests.org, and other commercially available sequencing panels. Thus, this comprehensive panel analyzes all genes currently reviewed in clinical research settings, and could be used for any disease focused sub-panel testing after being completely validated in the clinical laboratory.
Whole Exome Sequencing
The whole exome represents the complete coding region of the genome. It is estimated to encompass only approximately 1-2% of the genome, yet contain approximately 85% of disease-causing pathogenic variants. While there is ongoing discussion about the readiness of whole exome sequencing (WES) for clinical applications, the ultimate adoption of this approach appears to be inevitable. Numerous studies have illustrated the power of WES in making new discoveries, such as the identification of a germline mutation in PALB2, a gene previously implicated in breast cancer risk, in an individual with familial pancreatic cancer [12], and a germline mutation in MAX in individuals with familial pheochromocytoma (PCC), which was not previously linked to familial PCC [13]. This unique power has made WES an ideal tool for testing the patients with undiagnosed diseases of suspected hereditary origin for possible elucidation of a cause of the disease.
A preliminary study of 250 patients with undiagnosed diseases demonstrated the utility of WES in mendelian disorders [14]. The results led to a genetic diagnosis for 62 of the 250 patients, 20 of whom had autosomal recessive diseases. The diagnosis yield was as high as 25% in resolving these hereditary disease diagnostic dilemmas. A few academic institutes have already been offering clinical WES, including the Baylor College of Medicine, Washington University of St. Louis, and UCLA.
Emory Genetics Laboratory (EGL) has developed a new generation of clinical whole exome sequencing test, named Medical EmExome, to provide enhanced coverage of medically relevant genes. This WES assay has >97% coverage of 22,000 genes with a mean read depth of 100X. Of the ~4600 disease-associated genes analyzed, 3000 have 100% coverage (≥20X) of all exons, which is significantly higher than other commercial whole exome sequencing tests. The Medical EmExome also provides the EmExome Boost Option, which allows clinicians to choose an EGL gene sub-panel relevant to the patient’s phenotype.
Although the WES approach is uniquely suitable for some patients with undiagnosed diseases or some of those patients with a negative result using a disease-focused panel, due to the limitations of WES, we do not anticipate the full range of clinical use of WES at the present time.
Whole Genome Sequencing
Whole-genome sequencing (WGS) represents the next step in the progression to complete elucidation of the genomic determinants of a patient’s heritable make-up, and thus is the most comprehensive tool for future clinical application. It is expected to provide full coverage of all protein coding regions like WES as well as intronic and other noncoding regions associated with inherited diseases. With the recent release of Illumina HiSeq X Ten, a human genome can be sequenced at 30x coverage under $1000. Thus, the cost of sequencing is not a barrier for clinical WGS anymore.
Nevertheless, although the decreasing financial cost of sequencing has made the WGS technology more accessible, some technical and clinical interpretation issues remain unresolved. Researchers at the Stanford University School of Medicine have found that significant challenges must be overcome before WGS can be routinely used clinically [15]. In particular, they found that the use of WGS was associated with incomplete coverage of inherited disease genes, low reproducibility of detection of genetic variation with the highest potential clinical effects, and uncertainty about clinically reportable findings. It appears that there are still technical challenges and "considerable" human resource needs in order to interpret and validate the data returned by WGS.
However, the WGS will become an ultimate tool in routine clinical practice in the future. As an early exercise, Illumina has already offered Trugenome clinical WGS services in its CLIA-certified, CAP-accredited Clinical Services Laboratory to help identify the underlying genetic cause of an undiagnosed rare genetic disease, or determine a patient’s carrier status and genetic predisposition towards adult-onset diseases. Clinicians can also use Illumina’s laboratory to generate whole-genome sequencing data and use their own expertise for the clinical interpretation of the WGS findings.
CLINICAL APPLICATIONS OF NGS IN CYTOLOGY
In clinical practice, a cytology specimen, particularly from minimally invasive fine needle aspiration (FNA), is typically the first and easiest specimen available for clinical testing. In some instances, cytology specimens are the only material available when tumor size, location or comorbid conditions preclude concurrent core needle or excisional biopsy [16]. In fact, FNA procedures have been included in the recommended guidelines for the diagnosis of thyroid carcinomas, lung carcinomas and sarcomas [17]. It is clear that FNA is emerging as one of the most important tools in pathological diagnosis and molecular analysis for personalized medicine. However, a well-known common challenge for using cytology specimens for Sanger sequencing is the limited amount of, sometimes degraded, DNA. Whether we can effectively use the limited amount of cytology specimens for NGS applications has significant implications in patient care.
Recent improvement of FNA procedures and technological advancement in constructing a DNA library with the small amount of DNA have made NGS technology applicable to cytology specimens in the clinical setting. Several studies with FNA and other cytology specimens have established its feasibility in different cancers as briefed below.
Lung Cancer
Lung cancer is the leading cause of cancer-related death worldwide. About 85% lung cancers are NSCLC. The majority of NSCLC are diagnosed at an advanced stage, after missing the best time for surgical resection. Therefore, the diagnosis and therapeutic decision for lung cancer heavily rely on minimally invasive procedures, either small biopsies or cytology samples.
Lung cancer has the most available targeted therapies. The targeted genes include EGFR, BRAF, KRAS, ALK, and ROS1 [18, 19]. More potential targets, such as PIK3CA, FGFR1 and DDR2, are in the clinical trials. Therefore, the number of predictive biomarkers for novel targeted drugs entering into clinical practice is expected to rapidly increase. Thus, the NGS technology is superior to current standard methodologies in lung cancer treatment.
The Ion AmpliSeq Colon and Lung Cancer Panel has been applied to the detection of targeted gene mutations using 38 lung adenocarcinomas cytology specimens [20]. That study simultaneously assessed 504 mutational hotspots from 22 selected lung cancer-associated genes. Of the 38 cases, 36 were successfully sequenced (95%). Twenty-four out of 36 cases identified at least one mutation. Many of the mutated genes were well known driver genes such as EGFR, KRAS, PIK3CA, BRAF, TP53, and PTEN. Of those, EGFR and KRAS mutations were found in 6/36 and 10/36 cases, respectively, and were independently confirmed by Sanger sequencing or high resolution melting analysis. The data suggested that NGS can be reliably applied on cytology specimens with high sensitivity, specificity and reproducibility.
Thyroid Cancer
Thyroid cancer is the most common malignancy of endocrine organs. Its incidence is steadily increasing in the United States and worldwide [21]. Thyroid cancer typically occurs in thyroid nodules. FNA followed by cytological examination is an accurate and cost effective diagnostic method for evaluating thyroid nodules. This commonly used approach allows detecting cancer or establishing a diagnosis of a benign nodule in most cases. However, in approximately 25% of nodules, the diagnosis cannot be established and consequently classified as indeterminate by FNA cytology, hampering clinical management of these patients [22]. Because thyroid cancer evolves and progresses through multiple genetic and epigenetic aberrations, the detection of somatic mutations in thyroid cancer by NGS offers the potential to improve the accuracy of cancer diagnosis and prognosis in thyroid nodules, in addition to being helpful for therapeutic purposes [23, 24].
In a study of 1056 FNA samples, Nikiforov et al. demonstrated the clinical utility of panel-based molecular testing in the diagnosis and management of thyroid FNA samples with indeterminate cytology [25]. Subsequently, Nikiforova et al. validated a large series of neoplastic and non-neoplastic thyroid samples using the Ion Personal Genome Machine sequencer and their custom-designed, targeted NGS ThyroSeq panel [26]. In this study, they sequenced 228 frozen, formalin-fixed, and fine-needle aspiration thyroid samples representing all major types of thyroid cancer, using the Ion Torrent amplicon-based sequencing approach. They chose the amplicon-based approach because 1) this approach allows the use of 10 ng of input DNA for efficient amplification of genomic regions of interest, and 2) it works well with partially degraded DNA due to the small size of amplicons. Thus, this approach is ideal for cytology specimens. Their results in their validation study showed that ThyroSeq delivered an overall success rate of 99.6% for analysis of multiple mutations by NGS. Only 1 out of 51 (2%) routine FNA samples failed the NGS sequencing, suggesting that the vast majority of FNA samples should be amenable to such analysis [26]. Recently, using the second version of their targeted Thyroseq NGS assay, they also showed that this NGS assay allows a highly accurate diagnosis of cancer in thyroid nodules given a FNA cytologic diagnosis of follicular neoplasm/suspicious for a follicular neoplasm [27].
Further, to test if NGS has added value for the diagnosis of thyroid FNA specimens with indeterminate cytology, Le Mercier et al. retrospectively analyzed 34 indeterminate FNA samples using the AmpliSeq cancer panel V2. Mutations in BRAF, NRAS, KRAS and PTEN that are known to be involved in thyroid cancer biology were detected in 5 of the 7 malignant cases, giving a 71% sensitivity of this molecular test for the diagnosis of malignancy [28]. This study demonstrated that the detection of mutations known to be involved in thyroid cancer can improve the sensitivity of thyroid FNA diagnosis.
Pancreatic Cancer
Pancreatic cancer represents the fourth-highest cause of cancer death in the United States with the lowest survival rate among the most common cancers (~6%). Many genetic alterations have been associated with the development of pancreatic cancer. The four most frequently mutated genes are oncogene KRAS and tumor suppressor genes CDKN2A/p16, SMAD4 and TP53 [29]. These signature genes have been used as tumor markers for the diagnosis of pancreatic adenocarcinoma. The combination of cytological evaluation and tumor marker mutational analysis, especially for inconclusive cases, can potentially enhance the diagnostic power.
To explore the performance of NGS in the diagnosis of pancreatic ductal adenocarcinoma (PDAC) using FNA specimens, Dario de Biase et al. analyzed KRAS mutations, which have been reported in >95% PDAC, by using Sanger sequencing (considered as a gold standard technique for DNA sequence analysis), allele specific locked nucleic acid PCR (ASLNAqPCR) and 454 Next Generation Sequencing (454 GS-Junior platform, Roche) [30]. Sixty specimens from endoscopic ultrasonography FNA were analyzed for KRAS exon 2 and exon 3 mutations. Sanger sequencing delivered a clinical sensitivity for the detection of the KRAS mutation of 42.1%, ASLNAqPCR of 52.8% and 454 GS-Junior of 73.7%. The study not only demonstrated the feasibility of FNA for NGS, but also showed a better accuracy compared to other classical techniques.
The feasibility of NGS testing with cytology specimens has now been established. We anticipate that successful application of cytology specimens will further facilitate the clinical utility of NGS in cancer.
CHALLENGES, SOLUTIONS AND FUTURE DIRECTIONS
NGS technology has had a dramatic impact on precision medicine from risk assessment to early diagnosis, prognosis and treatment. Successful application of NGS technology to cytology specimens can further enhance its power in the disease management. However, there are several key challenges that impede the wide adoption of NGS in clinical laboratories. Addressing the following challenges can pave the way for gene panels, WES, and ultimately WGS testing in the daily practice of precision medicine.
Lack of Evidence Base for NGS Tests
Although there are many examples of the beneficial impact of NGS tests, overall, we have insufficient evidence-based framework to convince the FDA to approve, insurance companies to cover, and physicians to use those tests. This is perhaps the biggest challenge for NGS tests to fully penetrate the many facets of clinical care in a timely fashion [31].
In this regard, the NGS community may benefit from partnering with public health agencies and private sectors to collectively address this fundamental question. This effort requires data curation from the primary scientific literature, carrying out expensive and time-consuming clinical trials. By forming partnerships, we can enrich our knowledge and resources, increase in efficiency and reduce financial burden for a given institute.
The Office of Public Health Genomics at the U.S. Centers for Disease Control and Prevention has developed a framework for evaluating emerging genetic tests. Their evaluation includes four key areas: analytic validity (how accurately and reliably the test measures the genotype of interest), clinical validity (how consistently and accurately the test detects or predicts the intermediate or final outcomes of interest), clinical utility (how likely the test is to significantly improve patient outcomes), and ethical, legal, and social implications that may arise in the context of using the genomic tests. Through partnership and networking, the NGS community can use this framework as a basis to further develop a specific and comprehensive plan to provide evidence base for NGS tests.
Lack of Understanding of NGS Tests
There is a growing sentiment that uptake of genomic medicine is slow because health care providers and community in general lack understanding of NGS tests. This will directly reduce the number of orders, and affect insurance coverage and FDA clearance for these tests. Therefore, enhanced NGS education is the key not only for health care providers, but also for other related professions including policy-makers, regulators, lawyers, investors and insurance underwriters. In addition to classic educational means, such as conferences, publications and media, the NGS education effort should start from the schools, i.e., the Universities should train more qualified genomic teachers, have more genomic medicine major and offer more genomic courses. For pathologists in training, understanding of at least the basics of genomic tests and interpretation of the results of these tests in medicine is currently being encouraged [32-34].
A combination of better education in NGS tests as well as better tools for clinical decision support will speed up the NGS adoption. Several efforts are under way to track and make the latest information on genomic tests. The NIH Genetic Testing Registry is a National Institutes of Health’s (NIH)-funded, new centralized database for comprehensive genetic test information, including its purpose, target populations, methods, analytical validity, clinical validity, clinical utility, ordering information, laboratory location, contact information, certifications and licenses. The Pharmacogenomics Knowledge Base (PharmGKB) is an online resource that includes information on potentially clinically actionable gene-drug associations and genotype-phenotype relationships. Much of the information is manually curated from the published literature and is used to write evidence summaries and pharmacogenomic-based drug dosing guidelines. (Table 2) lists a few key websites that curate available genomic tests and the evidence to support their use. These tools will help physicians to understand and order such tests.
Table 2.
Genomic resources for genomic tests and data interpretation.
| Website Name | URL Address | Brief Description |
|---|---|---|
| PharmKGB | http://www.pharmgkb.org | A pharmacogenomics knowledge resource that encompasses clinical information including dosing guidelines and drug labels, potentially clinically actionable gene-drug associations and genotype-phenotype relationships. |
| Genetic testing registry | http://www.ncbi.nlm.nih.gov/gtr/ | Central location for voluntary submission of genetic test information by providers; includes information on test methodology, validity, evidence of the test’s usefulness, and laboratory contacts and credentials; currently includes 19,000 tests for 4,500 conditions and 3000 genes as of June 26, 2014 |
| GAPP Finder | http://64.29.163.162:8080/GAPPKB/topicStartPage.do | A searchable database of genetic tests and genomic applications in transition from research to clinical and public health practice. The search query can include disease, genes, drug, test, etc. Includes 547 tests as of June 26,2014 |
| FDA Pharmacogenomic Biomarkers | http://www.fda.gov/drugs/scienceresearch/researchareas/pharmacogenetics/ucm083378.htm | A list of FDA-approved drugs with pharmacogenomic information in their labeling. includes 161 biomarker-drug pairs as of June 26, 2014 |
| EGAPP | http://www.egappreviews.org/about.htm | A resource for evidence regarding the validity and utility of genetic tests for clinical practice and recommendations on implementation of genetic tests from professional organizations and advisory committees |
| Evidence aggregator | http://64.29.163.162:8080/GAPPKB/evidencerStartPage.do | An application that facilitates searching for evidence reports, systematic reviews, guidelines related to the use of genetic tests and other genomic applications |
Lack of Clinically Annotated Genetic Variants for Accurate Data Interpretation
Today, the bottleneck of genomic diagnostics has moved from data acquisition to data interpretation. An important challenge of efficiently translating NGS data into actionable information for clinicians is the lack of understanding of the impact of most genetic variants on human health and disease. Understanding these variants require massive sources of genomic and phenotypic data and shared efforts in studying variants [4]. This will take many years and requires a lot of collective effort. The International Collaboration for Clinical Genomics is working closely with NCBI to develop standards, to assist clinical laboratories in sharing their data and to develop approaches to curate the shared data.
At present, building comprehensive, constantly updated genomic databases is an immediate solution to address current challenge. Progress has been made with the recent launch of several public and private initiatives. ClinGen is a NIH-funded clinical genome resource to document which genetic variants play a role in disease and those that are relevant to patient care (http://clinicalgenome.org/). Its primary goal is to share genomic and phenotypic data through a centralized database in a standardized clinical annotation and interpretation. ClinGen is directly linked to ClinVar, a public database specifically focused on relationships among human variations and phenotypes with supporting evidence to help interpretation of clinically relevant mutations [35]. The PharmacoGenomic Mutation Database (PGMD. http:// www.biobase-international.com/product/pgmd) is a commercial resource for identifying all published genetic variants that have been shown to affect drug response in patients, thus guiding physicians to select appropriate drug and dose for maximum benefit and minimum side effect. Another resource with a focus of somatic mutations is My Cancer Genome (http://www.mycancergenome.org). This personalized cancer knowledge resource gathers up-to-date, well-established cancer mutation information, related therapeutic implications, and available clinical trials, making a convenient one-stop-shopping tool for physicians. For germline mutations, the Human Gene Mutation Database Professional (HGMD Pro) is a mature commercial resource providing comprehensive data on human germline mutations, particularly useful for hereditary disease risk screening and diagnosis.
Given the current challenge on accurate annotation of genetic variants, some third-parties provide genome interpretive services to assist clinicians in understanding the genetic variants and its clinical relevance to treatment. This nascent field currently includes startup companies like Knome (http://www.knome.com), Silicon Valley Biosystems (http://www.svbio.com) and Omicia (http://www.omicia.com). These companies offer software, computer infrastructure, and services required to process, analyze, and produce tailored diagnostic reports.
Initiatives in Development of Guidelines for Clinical NGS
The accurate interpretation of genetic variants identified by NGS is one thing, how to report the findings is another. One of the issues facing laboratories who offer genetic testing is how to report the variants that are unrelated to the indication for testing, such as risk of developing cancer or the risk of developing other genetic diseases or conditions like neurologic or psychiatric illnesses. These findings may have an impact not only on the individual patient but also on immediate family members. This issue is particularly significant for WES and WGS testing.
It is necessary to develop an ethical and legal framework for clinical NGS testing, including procedures for informed consent, and reporting incidental findings and returning the data to patients and their families. The ACMG developed a statement as a guide to obtaining informed consent in clinical NGS [36], which is a critical prerequisite for clinical NGS testing to educate the patient about NGS-based testing, including expectations from the test prior to performing the test. The ACMG also proposed recommendations for reporting of incidental findings in clinical exome and genome sequencing, which were updated after discussion, including in the oncology community, since implications of incidental or secondary findings apply also to cancer genome sequencing. [37-40].
To provide a general guidance for the development and interpretation of NGS-based tests, several professional organizations have developed guidelines for the practice of clinical NGS, including the AMP [41], and the Centers for Disease Control, in the United States [42]. Similar quality assurance guidelines for NGS in diagnostic pathology are also being established in Europe [43]. The ACMG has also developed a position statement for the detection of germline mutations by whole exome and genome sequencing [44], and for the validation of NGS methods and platforms, monitoring NGS testing, data interpretation and reporting [45]. In March 2015, the ACMG issued a joint consensus statement with the AMP for the interpretation of sequence variants, recommending use of the categories “pathogenic”, “likely pathogenic”, “uncertain significance”, “likely benign” and “benign” [46]. All above guidelines can be applied to interpret germline as well as somatic variants to guide cancer risk assessments and treatments. However, before comprehensive and consensus guidelines are established, we need to balance privacy issues with the potential advantages and drawbacks of sharing genetic data with patients and their relatives.
ACKNOWLEDGEMENTS
Declared none.
CONFLICT OF INTEREST
The author(s) confirm that this article content has no conflict of interest.
REFERENCES
- 1.National Research Council Toward precision medicine: building a knowledge network for biomedical research and a new taxonomy of disease. [PubMed]
- 2.Garraway L.A. Genomics-driven oncology: framework for an emerging paradigm. J. Clin. Oncol. 2013;31(15):1806–1814. doi: 10.1200/JCO.2012.46.8934. [DOI] [PubMed] [Google Scholar]
- 3.Karapetis C.S., Khambata-Ford S., Jonker D.J., O’Callaghan C.J., Tu D., Tebbutt N.C., Simes R.J., Chalchal H., Shapiro J.D., Robitaille S., Price T.J., Shepherd L., Au H.J., Langer C., Moore M.J., Zalcberg J.R. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 2008;359(17):1757–1765. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- 4.Rehm H.L. Disease-targeted sequencing: a cornerstone in the clinic. Nat. Rev. Genet. 2013;14(4):295–300. doi: 10.1038/nrg3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lynch T.J., Bell D.W., Sordella R., Gurubhagavatula S., Okimoto R.A., Brannigan B.W., Harris P.L., Haserlat S.M., Supko J.G., Haluska F.G., Louis D.N., Christiani D.C., Settleman J., Haber D.A., Haber D.A. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004;350(21):2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 6.Soda M., Choi Y.L., Enomoto M., Takada S., Yamashita Y., Ishikawa S., Fujiwara S., Watanabe H., Kurashina K., Hatanaka H., Bando M., Ohno S., Ishikawa Y., Aburatani H., Niki T., Sohara Y., Sugiyama Y., Mano H. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 7.Kwak E.L., Bang Y.J., Camidge D.R., Shaw A.T., Solomon B., Maki R.G., Ou S.H., Dezube B.J., Jänne P.A., Costa D.B., Varella-Garcia M., Kim W.H., Lynch T.J., Fidias P., Stubbs H., Engelman J.A., Sequist L.V., Tan W., Gandhi L., Mino-Kenudson M., Wei G.C., Shreeve S.M., Ratain M.J., Settleman J., Christensen J.G., Haber D.A., Wilner K., Salgia R., Shapiro G.I., Clark J.W., Iafrate A.J. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N. Engl. J. Med. 2010;363(18):1693–1703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gambacorti-Passerini C., Messa C., Pogliani E.M. Crizotinib in anaplastic large-cell lymphoma. N. Engl. J. Med. 2011;364(8):775–776. doi: 10.1056/NEJMc1013224. [DOI] [PubMed] [Google Scholar]
- 9.Lindeman N.I., Cagle P.T., Beasley M.B., Chitale D.A., Dacic S., Giaccone G., Jenkins R.B., Kwiatkowski D.J., Saldivar J.S., Squire J., Thunnissen E., Ladanyi M., College of American Pathologists International Association for the Study of Lung Cancer and Association for Molecular Pathology Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J. Mol. Diagn. 2013;15(4):415–453. doi: 10.1016/j.jmoldx.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 10.Wong S.Q., Fellowes A., Doig K., Ellul J., Bosma T.J., Irwin D., Vedururu R., Tan A.Y., Weiss J., Chan K.S., Lucas M., Thomas D.M., Dobrovic A., Parisot J.P., Fox S.B. Assessing the clinical value of targeted massively parallel sequencing in a longitudinal, prospective population-based study of cancer patients. Br. J. Cancer. 2015;112(8):1411–1420. doi: 10.1038/bjc.2015.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.King M.C., Marks J.H., Mandell J.B., New York Breast Cancer Study Group Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science. 2003;302(5645):643–646. doi: 10.1126/science.1088759. [DOI] [PubMed] [Google Scholar]
- 12.Jones S., Hruban R.H., Kamiyama M., Borges M., Zhang X., Parsons D.W., Lin J.C., Palmisano E., Brune K., Jaffee E.M., Iacobuzio-Donahue C.A., Maitra A., Parmigiani G., Kern S.E., Velculescu V.E., Kinzler K.W., Vogelstein B., Eshleman J.R., Goggins M., Klein A.P. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009;324(5924):217. doi: 10.1126/science.1171202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mannelli M., Castellano M., Schiavi F., Filetti S., Giacchè M., Mori L., Pignataro V., Bernini G., Giachè V., Bacca A., Biondi B., Corona G., Di Trapani G., Grossrubatscher E., Reimondo G., Arnaldi G., Giacchetti G., Veglio F., Loli P., Colao A., Ambrosio M.R., Terzolo M., Letizia C., Ercolino T., Opocher G., Italian Pheochromocytoma/Paraganglioma Network Clinically guided genetic screening in a large cohort of italian patients with pheochromocytomas and/or functional or nonfunctional paragangliomas. J. Clin. Endocrinol. Metab. 2009;94(5):1541–1547. doi: 10.1210/jc.2008-2419. [DOI] [PubMed] [Google Scholar]
- 14.Yang Y., Muzny D.M., Reid J.G., Bainbridge M.N., Willis A., Ward P.A., Braxton A., Beuten J., Xia F., Niu Z., Hardison M., Person R., Bekheirnia M.R., Leduc M.S., Kirby A., Pham P., Scull J., Wang M., Ding Y., Plon S.E., Lupski J.R., Beaudet A.L., Gibbs R.A., Eng C.M. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med. 2013;369(16):1502–1511. doi: 10.1056/NEJMoa1306555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dewey F.E., Grove M.E., Pan C., Goldstein B.A., Bernstein J.A., Chaib H., Merker J.D., Goldfeder R.L., Enns G.M., David S.P., Pakdaman N., Ormond K.E., Caleshu C., Kingham K., Klein T.E., Whirl-Carrillo M., Sakamoto K., Wheeler M.T., Butte A.J., Ford J.M., Boxer L., Ioannidis J.P., Yeung A.C., Altman R.B., Assimes T.L., Snyder M., Ashley E.A., Quertermous T. Clinical interpretation and implications of whole-genome sequencing. JAMA. 2014;311(10):1035–1045. doi: 10.1001/jama.2014.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yung R.C., Otell S., Illei P., Clark D.P., Feller-Kopman D., Yarmus L., Askin F., Gabrielson E., Li Q.K. Improvement of cellularity on cell block preparations using the so-called tissue coagulum clot method during endobronchial ultrasound-guided transbronchial fine-needle aspiration. Cancer Cytopathol. 2012;120(3):185–195. doi: 10.1002/cncy.20199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kanagal-Shamanna R., Portier B.P., Singh R.R., Routbort M.J., Aldape K.D., Handal B.A., Rahimi H., Reddy N.G., Barkoh B.A., Mishra B.M., Paladugu A.V., Manekia J.H., Kalhor N., Chowdhuri S.R., Staerkel G.A., Medeiros L.J., Luthra R., Patel K.P. Next-generation sequencing-based multi-gene mutation profiling of solid tumors using fine needle aspiration samples: promises and challenges for routine clinical diagnostics. Mod. Pathol. 2014;27(2):314–327. doi: 10.1038/modpathol.2013.122. [DOI] [PubMed] [Google Scholar]
- 18.Mok T.S. Personalized medicine in lung cancer: what we need to know. Nat. Rev. Clin. Oncol. 2011;8(11):661–668. doi: 10.1038/nrclinonc.2011.126. [DOI] [PubMed] [Google Scholar]
- 19.Pao W., Hutchinson K.E. Chipping away at the lung cancer genome. Nat. Med. 2012;18(3):349–351. doi: 10.1038/nm.2697. [DOI] [PubMed] [Google Scholar]
- 20.Scarpa A., Sikora K., Fassan M., Rachiglio A.M., Cappellesso R., Antonello D., Amato E., Mafficini A., Lambiase M., Esposito C., Bria E., Simonato F., Scardoni M., Turri G., Chilosi M., Tortora G., Fassina A., Normanno N. Molecular typing of lung adenocarcinoma on cytological samples using a multigene next generation sequencing panel. PLoS One. 2013;8(11):e80478. doi: 10.1371/journal.pone.0080478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vigneri R., Malandrino P., Vigneri P. The changing epidemiology of thyroid cancer: why is incidence increasing? Curr. Opin. Oncol. 2015;27(1):1–7. doi: 10.1097/CCO.0000000000000148. [DOI] [PubMed] [Google Scholar]
- 22.Gharib H. Changing trends in thyroid practice: understanding nodular thyroid disease. Endocr. Pract. 2004;10(1):31–39. doi: 10.4158/EP.10.1.31. [DOI] [PubMed] [Google Scholar]
- 23.Nikiforov Y.E., Nikiforova M.N. Molecular genetics and diagnosis of thyroid cancer. Nat. Rev. Endocrinol. 2011;7(10):569–580. doi: 10.1038/nrendo.2011.142. [DOI] [PubMed] [Google Scholar]
- 24.Xing M., Haugen B.R., Schlumberger M. Progress in molecular-based management of differentiated thyroid cancer. Lancet. 2013;381(9871):1058–1069. doi: 10.1016/S0140-6736(13)60109-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nikiforov Y.E., Ohori N.P., Hodak S.P., Carty S.E., LeBeau S.O., Ferris R.L., Yip L., Seethala R.R., Tublin M.E., Stang M.T., Coyne C., Johnson J.T., Stewart A.F., Nikiforova M.N. Impact of mutational testing on the diagnosis and management of patients with cytologically indeterminate thyroid nodules: a prospective analysis of 1056 FNA samples. J. Clin. Endocrinol. Metab. 2011;96(11):3390–3397. doi: 10.1210/jc.2011-1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nikiforova M.N., Wald A.I., Roy S., Durso M.B., Nikiforov Y.E. Targeted next-generation sequencing panel (ThyroSeq) for detection of mutations in thyroid cancer. J. Clin. Endocrinol. Metab. 2013;98(11):E1852–E1860. doi: 10.1210/jc.2013-2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nikiforov Y.E., Carty S.E., Chiosea S.I., Coyne C., Duvvuri U., Ferris R.L., Gooding W.E., Hodak S.P., LeBeau S.O., Ohori N.P., Seethala R.R., Tublin M.E., Yip L., Nikiforova M.N. Highly accurate diagnosis of cancer in thyroid nodules with follicular neoplasm/suspicious for a follicular neoplasm cytology by ThyroSeq v2 next-generation sequencing assay. Cancer. 2014;120(23):3627–3634. doi: 10.1002/cncr.29038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Le Mercier M., D’Haene N., De Nève N., Blanchard O., Degand C., Rorive S., Salmon I. Next-generation sequencing improves the diagnosis of thyroid FNA specimens with indeterminate cytology. Histopathology. 2015;66(2):215–224. doi: 10.1111/his.12461. [DOI] [PubMed] [Google Scholar]
- 29.Ryan D.P., Hong T.S., Bardeesy N. Pancreatic adenocarcinoma. N. Engl. J. Med. 2014;371(11):1039–1049. doi: 10.1056/NEJMra1404198. [DOI] [PubMed] [Google Scholar]
- 30.de Biase D., Visani M., Baccarini P., Polifemo A.M., Maimone A., Fornelli A., Giuliani A., Zanini N., Fabbri C., Pession A., Tallini G. Next generation sequencing improves the accuracy of KRAS mutation analysis in endoscopic ultrasound fine needle aspiration pancreatic lesions. PLoS One. 2014;9(2):e87651. doi: 10.1371/journal.pone.0087651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McCarthy J.J., McLeod H.L., Ginsburg G.S. Genomic medicine: a decade of successes, challenges, and opportunities. Sci. Transl. Med. 2013;5(189):189sr4. doi: 10.1126/scitranslmed.3005785. [DOI] [PubMed] [Google Scholar]
- 32.Haspel R.L., Arnaout R., Briere L., Kantarci S., Marchand K., Tonellato P., Connolly J., Boguski M.S., Saffitz J.E. A call to action: training pathology residents in genomics and personalized medicine. Am. J. Clin. Pathol. 2010;133(6):832–834. doi: 10.1309/AJCPN6Q1QKCLYKXM. [DOI] [PubMed] [Google Scholar]
- 33.ASCP ASCP. Training Residents in Genomics website. www.pathology learning.org/trig (Accessed 2010).
- 34.Schrijver I., Natkunam Y., Galli S., Boyd S.D. Integration of genomic medicine into pathology residency training: the stanford open curriculum. J. Mol. Diagn. 2013;15(2):141–148. doi: 10.1016/j.jmoldx.2012.11.003. [DOI] [PubMed] [Google Scholar]
- 35.Landrum M.J., Lee J.M., Riley G.R., Jang W., Rubinstein W.S., Church D.M., Maglott D.R. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42(Database issue):D980–D985. doi: 10.1093/nar/gkt1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.ACMG Board of Directors Points to consider for informed consent for genome/exome sequencing. Genet. Med. 2013;15(9):748–749. doi: 10.1038/gim.2013.94. [DOI] [PubMed] [Google Scholar]
- 37.Green R.C., Berg J.S., Grody W.W., Kalia S.S., Korf B.R., Martin C.L., McGuire A.L., Nussbaum R.L., O’Daniel J.M., Ormond K.E., Rehm H.L., Watson M.S., Williams M.S., Biesecker L.G., American College of Medical Genetics and Genomics ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet. Med. 2013;15(7):565–574. doi: 10.1038/gim.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.ACMG updates recommendations on “opt-out” for genome sequencing return of results. American College of Medical Genetics and Genomics. www.acmg.net/docs/Release_ACMGUpdates Recommendations_ final.pdf (Accessed 2010). 2010.
- 39.Parsons D.W., Roy A., Plon S.E., Roychowdhury S., Chinnaiyan A.M. Clinical tumor sequencing: an incidental casualty of the American College of Medical Genetics and Genomics recommendations for reporting of incidental findings. J. Clin. Oncol. 2014;32(21):2203–2205. doi: 10.1200/JCO.2013.54.8917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.ACMG Board of Directors ACMG policy statement: updated recommendations regarding analysis and reporting of secondary findings in clinical genome-scale sequencing. Genet. Med. 2015;17(1):68–69. doi: 10.1038/gim.2014.151. [DOI] [PubMed] [Google Scholar]
- 41.Schrijver I., Aziz N., Farkas D.H., Furtado M., Gonzalez A.F., Greiner T.C., Grody W.W., Hambuch T., Kalman L., Kant J.A., Klein R.D., Leonard D.G., Lubin I.M., Mao R., Nagan N., Pratt V.M., Sobel M.E., Voelkerding K.V., Gibson J.S. Opportunities and challenges associated with clinical diagnostic genome sequencing: a report of the Association for Molecular Pathology. J. Mol. Diagn. 2012;14(6):525–540. doi: 10.1016/j.jmoldx.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gargis A.S., Kalman L., Berry M.W., Bick D.P., Dimmock D.P., Hambuch T., Lu F., Lyon E., Voelkerding K.V., Zehnbauer B.A., Agarwala R., Bennett S.F., Chen B., Chin E.L., Compton J.G., Das S., Farkas D.H., Ferber M.J., Funke B.H., Furtado M.R., Ganova-Raeva L.M., Geigenmüller U., Gunselman S.J., Hegde M.R., Johnson P.L., Kasarskis A., Kulkarni S., Lenk T., Liu C.S., Manion M., Manolio T.A., Mardis E.R., Merker J.D., Rajeevan M.S., Reese M.G., Rehm H.L., Simen B.B., Yeakley J.M., Zook J.M., Lubin I.M. Assuring the quality of next-generation sequencing in clinical laboratory practice. Nat. Biotechnol. 2012;30(11):1033–1036. doi: 10.1038/nbt.2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dubbink H.J., Deans Z.C., Tops B.B., van Kemenade F.J., Koljenović S., van Krieken H.J., Blokx W.A., Dinjens W.N., Groenen P.J. Next generation diagnostic molecular pathology: critical appraisal of quality assurance in Europe. Mol. Oncol. 2014;8(4):830–839. doi: 10.1016/j.molonc.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.ACMG Board of Directors Points to consider in the clinical application of genomic sequencing. Genet. Med. 2012;14(8):759–761. doi: 10.1038/gim.2012.74. [DOI] [PubMed] [Google Scholar]
- 45.Rehm H.L., Bale S.J., Bayrak-Toydemir P., Berg J.S., Brown K.K., Deignan J.L., Friez M.J., Funke B.H., Hegde M.R., Lyon E., Working Group of the American College of Medical Genetics and Genomics Laboratory Quality Assurance Commitee ACMG clinical laboratory standards for next-generation sequencing. Genet. Med. 2013;15(9):733–747. doi: 10.1038/gim.2013.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W.W., Hegde M., Lyon E., Spector E., Voelkerding K., Rehm H.L., ACMG Laboratory Quality Assurance Committee Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]