

Abstract
Radial deficiencies (RDs), defined as under/abnormal development or absence of any of the structures of the forearm, radial carpal bones and thumb, occur with a live birth incidence ranging from 1 out of 30,000 to 1 out 6,000 newborns and represent about one third/one fourth of all the congenital upper limb anomalies. About half of radial disorders have a mendelian cause and pattern of inheritance, whereas the remaining half appears sporadic with no known gene involved. In sporadic forms certain anomalies, such as thumb or radial hypoplasia, may occur either alone or in association with systemic conditions, like vertebral abnormalities or renal defects. All the cases with a mendelian inheritance are syndromic forms, which include cardiac defects (in Holt-Oram syndrome), bone marrow failure (in Fanconi anemia), platelet deficiency (in thrombocytopenia-absent-radius syndrome), ocular motility impairment (in Okihiro syndrome). The genetics of radial deficiencies is complex, characterized by genetic heterogeneity and high inter- and intra-familial clinical variability: this review will analyze the etiopathogenesis and the genotype/phenotype correlations of the main radial deficiency disorders in humans.
Keywords: Radial deficiency, Congenital upper limb anomalies, Thumb hypoplasia
INTRODUCTION
Radial deficiencies (RDs) are defined as any degree of congenital hypo/aplasia of thumb, thenar muscles, first metacarpal bone, radial carpal bones (scaphoid and trapezium) and/or radius, occurring with a live birth incidence ranging from 1 out of 30,000 to 1 out 6,000 newborns [1-3] and representing about one third/one fourth of the total incidence of the congenital upper limb anomalies [4-6]. The RDs explain the vast majority of the malformations of the radial side of the arm, whereas the frequency of other defects, i.e. thumb duplication or triphalangism, represent only about one tenth of the total [6]. In spite of the high clinical variability, ranging from a minimal hypoplasia of the thenar muscles to a severe shortening of the forearm with a complete radial absence, RDs can be all included into the embryological class of formation defects, for which the primary event is a localized developmental failure due to genetic or non genetic factors [5].
The ascertainment of the cause has become a main diagnostic step in the work up of the RDs, leading to the anticipation of many diagnosis in which the hand malformation is the first clue of a syndrome [7] and allowing the possibility of a prenatal diagnosis especially in familial cases when the genetic defect is already known [8]. For all those factors, it is clear that establishing an etiological diagnosis is highlybeneficial to the parents not only for the assignment of the disease prognosis, which includes also the associated malformations, the resulting disability and the lifelong risks, but also for the obvious reflection on the attributable recurrence risks. Genetic testing gives now an important contribution to the diagnosis of the mendelian forms, which however represent only about half of the total cases of RDs [6], whereas for the other half, mostly sporadic, the diagnosis remains only clinical due to the absence of a known causative gene involved. Moreover, the common clinical criteria which are usually employed for the differential diagnosis between the mendelian and the apparently sporadic forms, like the bilaterality or the presence of associated features in other organs, do not strictly apply in RDs: for example, one of the most paradigmatic case, the association of Vertebral defects, Anal atresia, Tracheo-Esophageal fistula with esophageal atresia, Cardiac defects, Renal and Limb anomalies (VACTERL), affects by definition multiple organs, has a limb involvement which is bilateral in about one fourth of the cases [9], but it is mostly sporadic with no responsible gene or genomic region involved [10]. The narrow border separating hereditary and sporadic RDs is underlined also by the often identical limb malformation of known mendelian (i.e. Holt Oram syndrome) and sporadic forms (isolated thumb hypoplasia), suggesting that the clinical diagnosis cannot rely on the limb phenotype only, but should require the evaluation of several organ districts. Primary RDs, however, mostly remain confined to the radial side of the arm, extending to the ulna only in the extreme forms when the radii are severely affected: for this reason, the finding of atypical features, such as an associated ulnar defect in mild RDs, should initially orient towards an alternative group of disorders. Moreover, the limb anomaly in RDs is not disease-specific (in Holt Oram for example, the phenotype can vary from a thumb hypoplasia to a complete absence of the thumb, radius and hypoplasia of the humerus with features of phocomelia) [11] and the radial and thumb involvement shows a high inter- and intra-familial variability. In addition, a distal-to-proximal (i.e. thumb to radius) progression of the anomaly is frequent but exceptions do exist, and, unlike the central and ulnar defects in which the progression is a constant finding, a complete radial aplasia can be accompanied by a moderate thumb involvement, for example in Thrombocytopenia Absent Radius (TAR) syndrome [12] or in VACTERL association (Fig. 1) [9]. Finally, a general feature of the RDs is the specificity of the involvement of other organs, featured by a non-random association with other malformations (i.e. kidney, heart, vertebrae) and a very rare belonging to complex phenotypes, for example those associated with intellectual disability, which are instead more common in cases with ulnar deficiency, often subtended by genomic microdeletion/microduplica- tions [13]. A parallel observation is also that of a general higher frequency of radial versus ulnar deficiencies in human diseases (three- to fourfold) [2], consistent with studies suggesting that the critical period for RDs in animal models occurs in a later and less lethal embryological stage, thus explaining by viability the higher frequency of disorders with a radial defect [14]. The higher vulnerability of the radial side and in particular of the thumb is well epitomized in the presence of more than 4,000 entries in the Online Mendelian Inheritance in Man for radial deficiency, thus representing the first target in limb disruption [15], consistent with the terminal position in the developmental scale [16] and with the paradigm of “the last to form/the first to be affected”, well known in human syndromes, in which intellectual disability, disrupting the latest formed system and the highest evolutionary function, results the most frequent defect in human syndromes.
Fig. (1).
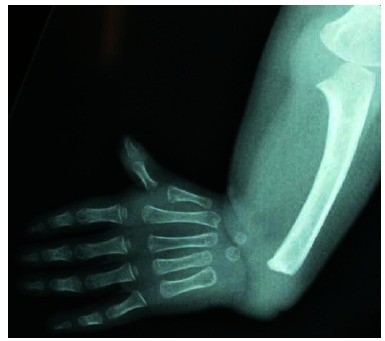
Anteroposterior X-ray showing the complete absence of the radius with the presence of the thumb.
Due to the importance of the embryology in the understanding the basis of human diseases with RDs, the next paragraph will present the main events in upper limb development.
Upper Limb Development
Beginning from the fourth week of development, HOX genes become up-regulated by retinoic acid in the fields where the limb buds will develop [17] and trigger the expression of T-box (TBX5), wingless-type MMTV (WNT), and fibroblast growth factor (FGF) proteins ensuring synchronized and progressive limb outgrowth [18]. The early limb bud is composed of the lateral migration of 2 layers of mesoderm (somitic and lateral plate) and outgrows into the overlying ectoderm [19]. Soon after the initial budding, the limb patterning proceeds along the three spatial axes: proximal-distal (PD) (shoulder-to-arm), anterior-posterior (AP) (radial-to-ulnar), and dorsal-ventral (DV) (back-to-palm). This specific pattern is under the tight control of spatio-temporally synchronized and simultaneously orchestrated signals of three important centers: (1) the Apical Ectodermal Ridge (AER), (2) the Zone of Polarizing Activity (ZPA) and (3) the non-AER ectoderm [20-22].
In the development of the proximal-distal axis the mesodermal FGF10, in conjunction with the ectodermal radical fringe (R-FNG) at the apical dorsal–ventral boundary induces ectodermal thickening to form the AER [23-25]. The AER, in turn, secretes WNT3 and several FGF proteins for maintaining the FGF10 expression in the underlying mesoderm, which in turn sustains the proliferation of a sub-AER cell population termed the progress zone [26]. The reciprocal loop of ectodermal and mesodermal FGF/WNT proteins maintains proximal–distal outgrowth [27].
The development in the dorsal-ventral axis is regulated by WNT7A secretion within the dorsal ectoderm. The WNT7A dorsalizes the underlying limb mesoderm through the induction of the Lim homeodomain transcription factor LMX1B [24, 28] and regulates the sonic hedgehog (shh) pathway, involved in the antero-posterior axis formation [29].
The development of the anterior-posterior (radio-ulnar) axis is controlled by the secretion of shh from the zone of polarizing activity (ZPA) along the posterior (ulnar) axis of the limb bud, inducing ulna formation in the forelimb and 4 ulnar-sided digits in the hand [30]. A progressive loss of SHH expression reduces limb outgrowth, volume, and width, mimicing the spectrum of ulnar longitudinal deficiencies [31]. When the loss of SHH reduces the FGF expression through the interruption of the SHH–FGF loop, the resulting phenotype involves a disruption of both ulnar and radial structures, particularly the thumb. This correlates with the common clinical presentation of RDs associated with ulnar deficiencies like severe symbrachydactylies or terminal transverse hemimelia. Finally, in cases in which the proliferation and ulnarization persists but there is a decrease in the FGF function, the resulting phenotype is similar to the RDs spectrum [32, 33]. All the complex interactions among the various morphogens in the development of the limb can be combined in several derangements and be reflected into a high phenotypic variety of malformations, which have been the object of several attempts of classification.
CLASSIFICATION OF RDs
The RDs belong to the “failure of formation” group of the classical classification of Swanson [34], which has been proposed in the sixties and adopted by the International Federation of Societies for Surgery of the Hand (IFSSH) with several modified versions [35, 36]. The main criteria inspiring this classification are essentially morphologic, with no reference on the etiology of the defect and no possibility to incorporate the progresses in the embryological understanding of the anomalies [36]. For example, in the last edition of the IFSSH classification (2009), including 1. Failure of formation; 2. Failure of differentiation; 3. Duplication; 4. Overgrowth; 5 Undergrowth; 6. Constriction band syndrome; 7. Generalized skeletal abnormalities, the separate groups of “Duplication,” or “Undergrowth”, when applied to the thumb (duplication or hypoplasia), are clearly redundant, since the two mechanisms are two faces of a “failure of formation” anomaly. Moreover, several other classification attempts have tried to focus on the aetiopathogenesis of upper limb anomalies, including a recent system grouping the defects on the basis of the embryologic stage [37] or according to the underlying molecular mechanism involved [38, 39]. All those attempts, however, had to face the problem of the wide spectrum of gravity and phenotypic variation of the single disorders (i.e. Holt-Oram syndrome) on one side, and of the presence of common molecular pathways disrupted by different insults, on the other.
For all this complexity, in the attempt to regulate the shaky ground of congenital upper limb anomalies, one of the last proposals unifies the failure of formation and differentiation into one category and puts the RDs into the malformations of the radial-ulnar (antero-posterior) axis divided into those involving the entire upper limb or only the hand plate [38]. The two forms obviously differ only in terms of gravity and not of pathogenesis, but the two classes of RDs are a proof of how ranking congenital anomalies according to their severity of expression is important because the extent of the involvement often determines the damaged function and provides a basis on which to guide treatment. An example of teratologic sequencing is the classification of thumb hypoplasia into five types of increasing severity [40]:
Type I Minimal shortening and narrowing of the bone and muscular structures;
Type II Mild underdevelopment of all structures; short bones; small diameters; mild thenar muscles hypoplasia; unstable thumb metacarplal-phalangeal (MCP) joint; narrowing of the first web space;
-
Type III; significant decrease in the thumb size; severe intrinsic and extrinsic muscle hypoplasia; unstable MCP joint; narrowing of the first web space
Subtype III A Stable carpal-metacarpal (CMC) joint
Subtype III B Unstable CMC joint;
Type IV Rudimentary thumb with no or very little metacarpal and no or very few muscles (pouce flottant)
Type V Complete aplasia of the thumb
This classification system, proposed by Manske and McCarroll in 1992 [40] on the original Blauth classification [41], aims at providing a common basis for prognostic information and for surgical decisions: types I, II, and IIIA deformities are treated with thumb reconstruction, while types IIIB, IV, and V deformities are treated with thumb ablation when present and index-finger pollicization.
Also the defects of the radius are classified according to a progressive skeletal deficiency, which are substantiated by a progressive radial angulation of the hand. The thumb is always affected, with a rough correlation in gravity with the radius, with several exceptions, as in TAR syndrome [42]. In parallel with the severity of the RD, also other radial structures, like muscles and tendons, can be affected and can contribute to the gravity of the functional impairment. A classification of the radial deficiency has evolved from the original presentation [43] to include the carpus and thumb [44] and finally to include proximal signs of insufficiency of the homerus and glenoid [45].
Type N. Normal radius and carpus with thumb deficiency
Type 0. Absent or hypoplastic carpal bones with normal length of the radius
Type I. A distal portion of the radius that is >2 mm shorter than the distal portion of the ulna.
Type II. The radius is hypoplastic in its entirety and often associated with bowing of the ulna.
Type III. The distal part of the radius is absent.
Type IV. The radius is completely absent.
Type V. Radial longitudinal deficiency with extremities with abnormal glenoid, absent proximal portion of the humerus, distal portion of the humerus articulates with ulna, radial-sided hand abnormalities.
The functional impairment in individuals with radial longitudinal deficiency can be influenced by several factors, including the bowing of the forearm, the deviation of the wrist, the limited or absent motility of the thumb, the impaired grip strength, which must all be considered and followed-up in the choice of a therapeutical (surgical or rehabilitative) strategy.
In the following paragraphs the main conditions associated with RDs will be reported. A tentative distinction has been made by grouping them according to their genetic basis into sporadic and mendelian forms. In the former group, all the present evidences supporting a genetic basis will be commented, whereas in the purely genetic forms an attempt to correlate the genotype with the phenotype will be made. For this reason, when dealing with entities like “thumb hypoplasia”, all the cases belonging to recognized syndromes are excluded.
SPORADIC FORMS
Thumb Hypoplasia
The hypoplasia is a rare thumb anomaly, characterized by varying degrees of incompletely developed first digital ray, contracted first web space, unstable metacarpo-phalangeal joint and thenar weakness [45], representing about 3.5%-4.6% of the congenital upper-limb malformations with an equal male to female gender distribution [44, 47, 48]. Isolated thumb hypoplasia, as part of the spectrum of RDs, represents its mildest form in comparison to the thumb hypoplasia and carpal anomalies (intermediate form) and to the absence or abnormality of all the radial structures (thumb, radial carpus, and radius), which can be placed at the extreme end of the spectrum [44, 49]. In a review by James et al. (2004) of a series of patients with thumb and radial deficiencies, the authors found that the thumb was always affected and concluded that the severity of the thumb deficiency was in general directly proportional to the severity of the radial deficiency, although in several cases exceptions existed [42].
However, the exact incidence of non-syndromic thumb hypoplasia is difficult to determine, since the isolated malformation does not differ from syndromic cases that in older studies might have gone undetected. In a recent report from a multidisciplinary group on upper limb anomalies, isolated thumb hypoplasia was found in 24/487 cases (4,9%); moreover, in 9 of those the anomaly was bilateral (1,8% of the total and 37.5% of the thumb anomalies) [6]. In other studies, the bilateral involvement is reported in about 60% of the affected patients, but when unilateral, the right hand is more commonly affected [50, 51]. It is possible that earlier studies overestimate the frequency of thumb hypoplasia, by including in this entity also syndromic cases, which in the past years might have escaped the diagnosis. Moreover, the issue of associated malformations is unresolved, since it appears to be a higher presence of vertebral and renal anomalies: epidemiological data from registries need extensive reports to be significant, and need specific details on the protocol used to reach the diagnosis, which is often difficult to achieve. It is however plausible to hypothesize that starting from thumb hypoplasia exists a continuum of signs and symptoms involving the vertebrae, the kidneys and other organs, which are diagnosed as VACTERL association only in cases in which the symptoms are very straightforward, like in the presence of a trachea-esophageal fistula, but, when individually present, might go undetected or cannot be diagnosed because they do not satisfy the molecular or clinical criteria for known syndromes. A recent study analyzing by array Comparative Genomic Hybridization (a-CGH) 54 patients with isolated and syndromic thumb and radial hypoplasia reported the presence in about 15% of the cases of different genomic rearrangements; however, the demonstration of their role in the pathogenesis of the disease was not conclusive and the authors could not rule out a coincidental finding [52].
VACTERL Association
The acronym VACTERL has replaced the previously used VATER, which was coined in the 70’s to describe the association specificity of a group of congenital malformations which included vertebral defects, anal atresia, tracheo-esophageal fistula with esophageal atresia (TEF/EA), radial and renal dysplasia [53]. Furthermore, the VATER association spectrum was expanded to include cardiac malformations, mainly ventricular septal defects (VSD), and any type of limb abnormality and the acronym has thus been extended to VACTERL [54, 55]. The name “association” rather than “syndrome” reflects the absence of a known common etiopathogenesis, but acknowledges the tendency of the above-mentioned malformations to occur together more frequently than expected. Population-based epidemiological studies in Europe and the United States have reported an incidence of VACTERL between 1 in 10,000–1 in 40,000 live births [56, 57]. Given the numerous overlapping conditions and the absence of confirmatory laboratory analysis, VACTERL association is typically considered a diagnosis of exclusion, for which the exact criteria for diagnosis in terms of the number and nature of anomalies included are controversial, although most clinicians and researchers require the presence of at least three component features for diagnosis [58].
Limb malformations are reported in approximately 40-50% of patients [58, 59] and are typically described as radial anomalies, including thumb and radial aplasia/hypoplasia with a wide range of severity (Fig. 2). In a recent study by Carli et al. (2014) on a cohort of 25 VACTERL patients selected for their upper limb involvement, the thumb was always affected: its function was severely impaired in 79% of all the cases and in about one third it was associated with aplasia of the radius. Approximately 75% of patients had a unilateral anomaly and when bilateral, the RD was always non symmetrical [9]. Of note, the authors describe that in two cases the radial anomaly was represented by a thumb duplication (in both cases the contralateral thumb was hypoplastic or absent), consistent with the above-reported hypothesis of a common embryological basis for thumb hypoplasia and duplication [38].
Fig. (2).
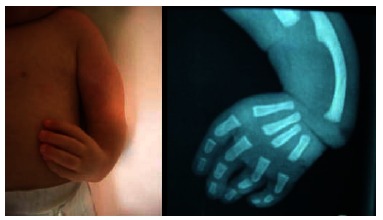
Clinical photograph (A) and anteroposterior X-ray (B) of a patient affected by VACTERL association showing a complete aplasia of both radius and thumb.
Among the other VACTERL malformations, the vertebral involvement is reported in approximately 60-80% of patients in a wide range of severity [56, 58-60], typically including segmentation defects, such as hemi- vertebrae, butterfly, wedge, fused, supernumerary or absent vertebrae, concurrent hypoplasia of both the odontoid process and coccyx [9]; in addition to frank vertebral anomalies, occult spinal dysraphisms like tethered spinal cord and philum lipoma may occur [9, 61, 62]. Imperforate anus/anal atresia occurs in approximately 55-90% of patients [56, 58-60], whereas cardiac malformations have been found in a recent study in about two third of VACTERL cases, with the most common anomaly represented by the ventricular septal defect, which was present in 18 of 31 patients (58%) [63]. Tracheo-esophageal fistula (TEF), with or without esophageal atresia, is reported in approximately 50-80% of patients with VACTERL and in about the same frequency of the renal anomalies [56, 58-60], which include unilateral renal agenesis (or bilateral in severe cases), horseshoe kidney, and cystic and/or dysplastic kidneys, sometimes accompanied by ureteral and GU anomalies [59, 64].
VACTERL association is a sporadic occurrence, which has lead to suggestions of environmental causative factors, even though several evidences have demonstrated the importance of the genetic contribution in the non-random anomalies’ association. Among those, the demonstration of an increased prevalence of core features in first-degree relatives of patients with VACTERL [59, 65], or numerical or structural chromosome aberrations, single gene mutations and mitochondrial dysfunction in individuals with the VATER/ VACTERL phenotype. The chromosomal anomalies which have been reported in the literature and reviewed by Reutter et al. (2013) [66] include: (I) deletions of 6q [67], distal 13q [68], 7q35qter [69], 5q11.2 [70], and 20q13.33 [71]; (II) duplication of 9q [72] and 22q11.21 [73]; (III) supernumerary der(22) syndrome [74]; (IV) mosaicism for supernumerary ring chromosome 12 [75] or 18 [76], and (V) partial monosomy 16p13.3pter/partial trisomy 16q22qter. [77] Single gene mutations have been described in few individuals: heterozygous de novo deletion in HOXD13 gene [78], haploinsufficiency of the LPP gene [79] and recessive mutations in the gene TNF receptor associated protein 1 (TRAP1), which were identified in three families with VACTERL association by whole exome sequencing [80]. Finally, there are six case reports published of individuals with VACTERL association in conjunction with mitochondrial dysfunction, as summarized recently by Siebel and Solomon in 2013 [81]. All those reports, however, refer to isolated individuals and account for only a small percentage of all the patients with VACTERL association, leaving the association still as a purely clinical diagnosis.
MENDELIAN FORMS
Fanconi Anemia
Fanconi Anemia (FA), firstly described in 1927 by Guido Fanconi [82] is a clinically and genetically heterogeneous disorder, which has been described in all races and ethnic groups and has been found with a higher frequency in Ashkenazi Jews and Afrikaners in South Africa. The disorder, inherited in an autosomal- (mostly) or X-linked recessive mode, has a worldwide prevalence of 1-5 per million and a heterozygous mutation carrier frequency estimated in about 1 out of 180 [83]. The life expectancy was initially calculated in about 20 years, but it has nowadays been extended to the third and fourth decade [84]. The majority of patients are diagnosed by the first decade of life, but individuals with atypical presentations (e.g. myelodysplasia) can be diagnosed in the adulthood.
FA is caused by a deficiency in the DNA repair featured by a genomic instability resulting in congenital abnormalities that may range from none (in one third of the cases) [85] to a full-blown phenotype involving any of the major organ systems [86], together with a cancer predisposition. To take note of the variability of the disease, overlapping with a wide range of disorders, including VACTERL and Holt Oram, an International Fanconi Anemia Registry (IFAR) was established at The Rockefeller University (www.rockefeller.edu/ fanconi/) in 1982 (now displayed by the Leiden Open Source Variation Database, LOVD v.3.0) (http://databases.lovd.nl/ shared/genes/), in which the recorded cases are confirmed with the cytogenetic and/or genetic testing, thus allowing a careful recording of the phenotypic spectrum of “true” cases of FA.
RDs are generally the first sign of the presence of the syndrome: they are most frequently bilateral, with a wide range of abnormalities ranging from bilateral absent thumbs and radii to a unilateral hypoplastic or bifid thumb [87]. The clinical suspicion must be raised when in addition to the radial involvement is present any of the following signs, like pre- and postnatal growth retardation resulting in short stature, microcephaly, microphtalmia, malformations of the kidneys (e.g. renal aplasia or hypoplasia, horseshoe kidney), heart, gastrointestinal (e.g. duodenal atresia), endocrinopathies (glucose/insulin abnormalities, growth hormone insufficiency and hypothyroidism), abnormalities of ears and hearing loss, abnormalities of the skin (hyper- or hypopigmentation and cafe-au-lait spots), developmental disabilities and neurological abnormalities. Due to the high risk of cancer (about 9% of FA patients develop leukemia, 7% a myelodysplastic syndrome, 5% have solid tumors, most commonly of the aerodigestive tract or gynecological [88]), a RD, especially when bilateral, even if isolated, has to raise the suspicion of FA and prompt the clinician to perform specific tests for the disease (see next paragraph). In a review of all the reported cases from 1928 to 2009, Shimamura and Alter describe a thumb involvement in 35% of the cases: they can be hypo/aplasic, duplicated, triphalangeal, low set. Upper limbs are involved in unilateral or bilateral fashion in 35% of the cases. Absence or hypoplasia of the thumbs is reported in 7% of the cases, a generic “hand” involvement is reported in a further 5% of the cases (flat thenar eminence, absent first metacarpal, clinodactyly, polydactyly) and in 1% of the cases a dysplastic and short ulna is described. As stated by the authors, those features are clearly an underestimate of the real frequency, since, especially in the early cases, a thorough phenotypic description was frequently missing [89].
FA is caused by defects of a complex of nuclear proteins (a multi-subunit E3 ubiquitin ligase) involved in the FA-BReast CAncer (BRCA) pathway to repair the damaged DNA. A key event in the activation of the FA-BRCA pathway is the monoubiquitination of the FANCD2 protein in the S phase after the DNA damage: in FA the deficiency of the DNA repair system in the removal of the DNA interstrand crosslinks (mostly pyrimidine dimers) results in spontaneous chromosome breakages, hypersensitivity to DNA damage, hypermutability due to allele-loss mutations, intensive breakage at telomeric sequences and increase in chromosome end fusions [84]. Chromosome breakages are diagnosed by the use of cytogenetic tests performed on blood samples where T- lymphocytes are cultured 72 hours in the presence of crosslinking agents, i.e. diepoxybutane, mitomycin C or cisplatinum. These agents cause growth inhibition, cell cycle arrest and chromosomal breakages, which are quantified in Giemsa-stained (not banded) metaphase spreads by analyzing at least two slides and 25 cells per slide for a total of 50 metaphases. The analysis should record chromatid gaps, breaks, tri/quadriradial chromosomes and interchange-type aberrations, whereas dicentric, acentric and ring chromosomes should not be included in the final analysis. Cells from FA patient should show an increased proportion of chromosomal breakages when stimulated at 50nM, 150nM and 300nM MMC. At the highest concentration all cells generally manifest aberrations; on the contrary, healthy subjects’ cells show chromosomal damage only at 300nM, with an average of 30% of cells affected with 1 to ≤5 breakage event/cell [90].
From the genetic point of view, FA shows extremely high genetic heterogeneity and the list of genes involved in the disease increases over time. Up to now, the complementation groups involved in FA and having a role in FA-BRCA pathway to repair damaged DNA are the following: FANCA (maps in 16q24.3), FANCB (Xp22.2), FANCC (9q22.32), FANCD1 (BRCA2, 13q13.1), FANCD2 (3p25.3), FANCE (6p21.31), FANCF (11p14.3), FANCG (XRCC9, 9p13.3), FANCI (15q26.1), FANCJ (BRIP1, 17q23.2), FANCL (PHF9, 2p16.1), FANCM (14q21.2), FANCN (PALB2, 16p12.2), FANCO (RAD51C, 17q22), FANCP (SLX4, 16p13.3) and FANCQ (XPF, 16p13.12) [84, 91-95]. Moreover, the complexity of such genetic heterogeneity is not aided by the presence of genotype–phenotype correlations pinpointing a specific gene involved: in terms of effects on the clinical phenotype, it is now thought that the type of the underlying mutation (e.g. frameshift vs. missense) is more predictive than the affected gene [96]. FANCA mutations account for the majority of FA cases, with the exception of black South African patients for whom the disease is due to a founder mutation represented by a homozygous deletion (637_643delTACCGCC) in the FANCG gene in 80% of the cases [97]. Approximately 85% of patients worldwide are classified into subtypes A, C, or G, about 8% of patients are FANCE and FANCF and the remaining have one of the other subtypes. [84] Cases of spontaneous genetic reversion have been described, leading to hematopoietic mosaicism: in revertant cells, the presence of a functional FA gene confers growth advantage, favoring the progressive replacement of defective cells in the bone marrow [98].
Next Generation Sequencing panels for FA have been developed and are offered by different companies and hospitals across the world (for example the Molecular Genetics Laboratory of the Cincinnati Children's Hospital Medical Center (http://www.cincinnatichildrens.org) has a panel including 16 genes: FANCA, FANCB, FANCC, BRCA2, FANCD2, FANCE, FANCF, FANCG, FANCI, BRIP1, FANCL, FANCM, PALB2, SLX4, RAD51C, ERCC4). Whole exome sequencing has also been applied for the diagnosis of FA, and by the use of this technique a novel (c.989_995del, p.H330LfsX2) mutation in FANCA gene has been identified [99].
Holt-Oram Syndrome
The Holt-Oram syndrome (HOS), firstly described by Holt and Oram in 1960, is a clinically heterogeneous disorder essentially characterized by upper limb abnormalities and heart defects [100], with a prevalence of 1:135,615 births [11].
Upper limb deficiency is variable, but most patients belong to the spectrum of RDs: the abnormalities are often but not always bilateral and asymmetrical, with the left side being more severely affected [101]. The thumb, which is the most commonly affected structure, may be triphalangeal and have a finger-like appearance (5-finger hand), hypoplastic or completely absent. Carpal abnormalities may be the only evidence of the syndrome, displaying a higher specificity for HOS than the changes in the thumb itself [102]. Radial hypoplasia is more frequent than agenesis, whereas ulnar aplasia/hypoplasia and humeral hypoplasia/phocomelia occur in 24.6% and 42.6% of HOS patients, respectively (Fig. 3) [11, 103]. Posteriorly and laterally protuberant medial epicondyles of the humerus were seen in several patients. Furthermore, most patients present narrow and sloping shoulders due to the short clavicles and the hypoplasia of the head of the humerus. Other features include prominent acromioclavicular joint, pectus excavatum and pectoralis major hypoplasia [103]. Several other skeletal anomalies have been anedoctally described, like cervical vertebral fusion [104] or hypoplasia of the toe phalanges [105] (in this latter case the feet anomaly was not segregating with the disease in other affected family members).
Fig. (3).
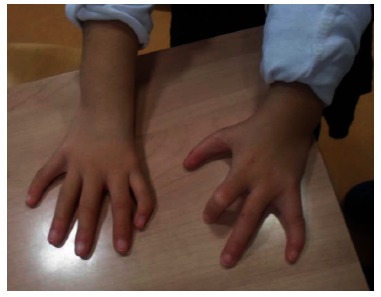
Clinical photograph of a patient with Holt-Oram syndrome with right non opposable triphalangeal thumb and left thumb aplasia treated by index finger pollicization.
Congenital heart malformations are reported in 75% of individuals with HOS: the most commonly observed are ostium secundum atrial septal defect (ASD) and ventricular septal defect (VSD), although almost every type of cardiac malformation has been reported, either alone or as part of multiple defects [11, 106, 107]. Individuals with HOS, with or without a congenital heart malformation, are at risk for disturbances of the cardiac conduction that may present even at birth with sinus bradycardia and atrioventricular blocks.
The HOS is inherited as an autosomal dominant disorder with very high penetrance. The gene for HOS, which was linked to the chromosome 12q24 regione, was identified as TBX5, a member of T-box family playing a key role in limb and cardiac development [108]. Recently, TBX5 was shown to have a role in the embryogenesis of the sternum and as a result, sternum defects can be observed in HOS patients [109].
Patients with a clinical diagnosis of HOS show a mutation in TBX5 in 74% of cases [110]; in a range between 30% to 85% of those cases the mutation arises de novo [110, 111]. The mutation spectrum in the TBX5 is wide [112], with more than 90 germline mutations described so far (The Human Gene Mutation Database, http://www.hgmd.cf.ac.uk/ ac/index.php), including 37 nonsense and missense mutations, 22 small deletions, 9 small insertions, 1 small indel, 8 gross deletion, 3 gross insertions, 2 complex, 1 regulatory, and 12 splice site mutations (up-to-dated to February 2015). The dominant phenotype of HOS appears to result from a null allele and haploinsufficiency of TBX5 caused by truncating mutations, but occasionally sequence variations can lead to the extension of the TBX5 protein. TBX5 function is considered to be gene dosage sensitive since under- and overexpression cause the same phenotype. Some mutations have been shown to impair the nuclear localization of the TBX5 protein or disrupt the TBX5 interaction with co-factors and downstream target genes [113, 114].
Truncating TBX5 mutations cause substantial abnormalities in both limb and heart [115], but there is no genotype/phenotype correlation between the different truncating mutations. In contrast, missense mutations in the T-box domain of the TBX5 gene produce distinct phenotypes ranging from significant cardiac malformations but only minor skeletal abnormalities (c.238G>A, p.G80R; c.505G>A p.G169R), to extensive upper limb malformations but less significant cardiac abnormalities (missense mutations in codon 237, c.709C>T, p.R237W; c.710G>A, p.R237Q). Also mutations located outside the T-box domain show a similar genotype-phenotype correlation, determining predominantly heart or limb defects according to the mutation. Germinal mosaicism has also been reported in one case report in which an unaffected man fathered 4 offspring with HOS by 3 different women [116]. Although no other cases have been described in the literature, we have found a family with 2 affected offspring from unaffected parents. Obviously, this issue is of particular relevance for genetic counseling for the attribution of a correct recurrence risk.
Duane-Radial Ray Syndrome or Okihiro Syndrome
Duane-Radial ray syndrome (DRRS) is a rare disorder whose prevalence is unknown, and with a few families described so far. The disease is also called “Okihiro syndrome” according to the clinician who firstly described it [117], although other authors had already reported the syndrome using the term DR syndrome, where D stands for Duane anomaly and deafness and R identifies the radial and renal manifestations of the condition [118].
The syndrome is featured by upper limb and ocular anomalies, these latter characterized by a congenital eye movement disorder (called Duane anomaly or Duane retraction syndrome, DRS), due to a developmental failure of cranial nerve VI (the abducens nerve which controls eye movement), resulting in absent or decreased abduction (Type 1 DRS), adduction (Type 2 DRS) or both (Type 3 DRS), and of narrowing of the palpebral fissure and retraction of the globe when adduction is attempted. The disorder is more frequent in females (about 60% of cases) and in more than half of the cases it is bilateral. When the disorder is monolateral, usually the left side is the affected one [119, 120].
The upper limb abnormalities include a RD which can range from a mild hypoplasia of the thenar eminence to the absence of the radius and thumb, whereas the thumb anomalies can vary from aplasia to hypoplasia, triphalageal thumb or duplication (Fig. 4).
Fig. (4).
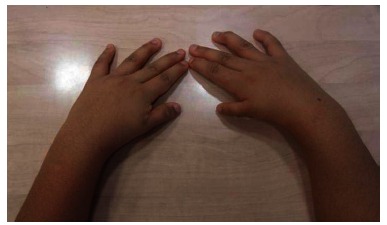
Clinical photograph of bilateral thumb hypoplasia in a patient affected by Okihiro syndrome.
Autopsy and MRI studies of patients have revealed hypoplasia or absence of the sixth nerve nucleus on the affected side, the ipsilateral lateral rectus being innervated by branches of the oculomotor nerve. Sensorineural hearing loss and spinal and other skeletal abnormalities also occur, and polydactyly, hemifacial microsomia with skin tags, cardiac defects, and Hirschsprung disease have also been reported [117, 121]. Other minor features have been observed: hypertelorism, epicanthic folds, slit-like openings of the external auditory meatus, flat feet and large sandal gaps, that may help discriminate DRRS from HOS if no Duane anomaly is present [122]. Kidney defects, including mild malrotation, ectopia, horseshoe kidney, renal hypoplasia, vesico-ureteral reflux and bladder diverticula do not belong to the typical spectrum of the DRRS but have been described in combination with upper limb and ocular anomalies. This association was earlier thought to be a distinct entity and referred to as 'acro-renal-ocular syndrome' [123], but it is now considered as a continuum of the DRRS spectrum of anomalies, due to the finding in patients with acro-renal-ocular syndrome of heterozygous mutations in SALL4 (Spalt-like transcription factor 4), the causative gene of the DRRS [124]. SALL4, localized on chromosome 20q13, belongs to the SALL family of zinc finger transcription factors and consists of 4 exons containing three highly conserved C2H2 double zinc finger domains [125]. SALL4 mutations are located in exon 2 and, to a lesser extent, in exon 3 and consist of highly penetrant (95%), heterozygous small and gross deletions, small insertions, small indels, short duplications, nonsense mutations (Gln652*, Ser763*, Arg831*, Arg865*, Arg905*) [126, 127]. The phenotype of larger deletions (not extending into other genes) is not significantly different from that caused by almost all truncating point mutations [128], which are all expected to result in nonsense-mediated mRNA decay. One missense mutation, His888Arg, has been reported in a mild form of the syndrome [129]. Mutations in SALL4 have been also reported in patients with an initial diagnosis of VACTERL (Gln166*) and HOS (Lys175*, Arg617*, Val754Met, Glu889*), which was later revised to DDRS [126].
A mouse model shows that Sall4 is regulated by Tbx5 transcription factor and that the two genes contribute to patterning and morphogenesis of the anterior forelimb and heart, therefore explaining the shared phenotypes between DDRD and HOS, which is associated with mutations in the TBX5 gene. In situ hybridization analysis of Sall4 expression during mouse embryogenesis displays a prominent localization in the midbrain and branchial arches and suggests that a dosage reduction of Sall4 might disrupt the abducens nerve development [130, 131].
Thrombocytopenia-absent Radius Syndrome
The thrombocytopenia-absent radius syndrome (TAR) was firstly defined as a syndrome in a study by Hall et al. (1969), who presented the clinical findings of a cohort of 40 patients [132]. It is a rare disease, with an incidence of approximately 1 in 240 000 birth [133], characterized by the absence of the radius in each forearm with the presence of the thumbs (this feature distinguishes TAR from other syndromes such as Fanconi anemia) (Fig. 5) and by reduction in the number of platelets (<50 platelets/nL) due to a decreased bone marrow production. Affected individuals frequently present with bleeding episodes in the first year of life that become less severe over time; in some cases the platelet levels become normal [132, 134]. TAR syndrome is also featured by short stature and additional skeletal anomalies, whose severity varies from absence of radii to virtual absence of upper limbs, with or without lower limb defects such as hip dysplasia, genua vara, and bowing of the long bones [12, 135, 136]. In a series of 34 TAR patients, renal and cardiac anomalies have been described in 23% and 15% of cases, respectively [137].
Fig. (5).
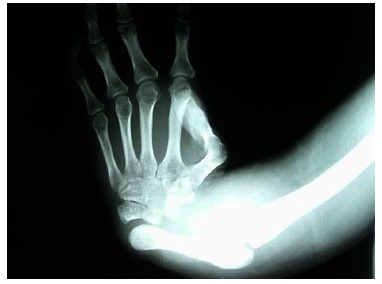
Anteroposterior X-ray of a patient affected by TAR syndrome showing complete aplasia of the radius and a triphalangeal thumb.
The presence of families with several affected individuals born to unaffected parents was initially considered an hallmark of an autosomal recessive mode of inheritance [138], but a more complex pattern was proposed in 2007 when a common interstitial microdeletion of 200 kb on chromosome 1q21.1, encompassing 11 genes, was detected in all 30 patients with the diagnosis of TAR syndrome, and it was excluded in the controls. In 25% of the investigated patients, the deletion occurred de novo, but in the other 75% of the cases the deletion was inherited from one unaffected parent, suggesting that TAR syndrome is associated with a microdeletion on 1q21.1, which is considered necessary but not sufficient to cause the phenotype and requires the effect of an additional modifier (mTAR) for its full-blown phenotypic expression in the syndrome [12]. In order to identify the causative allele, Albers et al. [135] performed an exome sequencing in five unrelated TAR cases with a 1q21.1 deletion but were not able to identify any rare deleterious protein-coding variant. The authors then analyzed all the rare (Minor Allele Frequency, MAF <.05) variants mapping in the critical region, regardless of their predicted consequences, as potentially causative. This allowed them to identify a low-frequency (MAF= .03) Single Nucleotide Polymorphsim (SNP) in the 5’UTR region of the gene RBM8A in four of the TAR syndrome cases under analysis and another low-frequency SNP (MAF= .004) in the first intron of the same gene in the last case. The findings were confirmed in further 48 individuals with TAR syndrome and a 1q21.1 deletion, in which the presence of the 5’UTR and intronic SNPs were found in 35 and 11 cases, respectively. The two TAR cases with no deletion were shown to carry the 5’UTR SNP in compound heterozygosity with a RBM8A 4bp frameshift insertion and a nonsense mutation, respectively, thus defining the nature of TAR syndrome as bi-allelic featured by a null allele (caused by either a deletion or a nonsense/frameshift mutation) and a SNP in regulatory regions (5’UTR or first intron) [135].
The causative gene is the RNA binding motif protein 8A -RBM8A-, which encodes the Y14, a protein with a conserved RNA-binding motif. Y14 is found predominantly in the nucleus and it is involved in several important cellular functions including nuclear export and subcellular localization of specific transcripts, translational enhancement and nonsense-mediated RNA decay [139-141]. No biallelic RBM8A gene mutations have been reported, suggesting that the complete loss of the Y14 protein is lethal, probably due to its involvement in basic cellular functions [142].
Lacrimo-auriculo-dento-digital Syndrome
The lacrimo-auriculo-dento-digital (LADD) syndrome, also known as Levy-Hollister syndrome, is a very a rare condition with about 60 cases described so far. The syndrome can be associated with multiple congenital anomalies, characterized by abnormalities of the lacrimal and/or salivary system and anomalies of the ears (cup-shaped, with mixed hearing loss), teeth and limbs [143, 144]. Limb defects mostly involve the thumbs, ranging from total aplasia to hypoplastic, digitalized triphalangeal or duplicated thumbs. In addition to those cardinal features, facial dysmorphisms, malformations of the kidney and respiratory system and abnormal genitalia have been reported [145].
LADD is transmitted in an autosomal dominant fashion and it caused by the heterozygous mutations in the fibroblast growth factor (FGF) and fibroblast growth factor receptors (FGFR), molecules activating different signaling cascades which have critical roles in development, repair, metabolism and neuronal activities both during the embryological development and in the adult organism. More specifically, 3 FGF-related genes have been demonstrated to be associated to the syndrome, two of which are FGFR (FGFR2 and FGFR3), whereas one is a FGFR2b ligand (FGF10) [146].
Animal models have shown that FGF10 and FGFR2b are involved in the control of branching during the development of lung, pancreas, mammary gland, thyroid, lacrimal and salivary glands. Moreover, Fgf10 knockout mice are characterized by a complete truncation of the fore- and hindlimbs. Detailed roles of Fgf10 are reported in the review by Itoh and Ohta [147]. In contrast, FGFR3 is involved in skeletal formation since mice in which the FGFR3 is selectively inactivated show different phenotypes according to the FGFR3 isoform which is targeted (i.e. skeletal overgrowth, decreased bone mineral density [148].
Acrofacial Dysostosis 1, Nager Type
Nager acrofacial dysostosis (NAFD) has been recognized as a specific entity by Nager and de Reynier (1948), but had been probably reported before by Slingenberg (1908) [149, 150]. It is a clinically heterogeneous group of disorders characterized by craniofacial malformations and radial deficiency. The incidence of Nager syndrome appears to be low, with an estimate of 3/1,000,000 in Finland [151].
The pre-axial limb anomalies are very variable and often asymmetric. The thumb anomalies include hypoplasia or aplasia, duplication, limited movement and symphalangism. The radial hypoplasia or aplasia is often associated with a proximal radioulnar synostosis. Lower limb involvement is usually mild and rare: talipes, hypoplastic hallux and other toes and absence of creases at the toes [152].
The craniofacial involvement is mainly characterized by severe micrognathia, frequently requiring the placement of a tracheostomy in the early childhood, malar hypoplasia and cleft palate. Due to these anomalies, feeding and breathing problems are observed in infants with Nager syndrome. The major facial features of Nager syndrome include downslanting palpebral fissures, absent eyelashes, eyelid coloboma, midface retrusion and malformed ears. Conductive hearing loss caused by defects in the middle ear is present in about 60% of patients, and may cause speech delay. Less commonly, affected individuals have abnormalities of the heart, kidneys, genitalia and urinary tract [153].
By an exome sequencing approach, the haploinsufficiency of the SF3B4 (Splicing Factor 3B, subunit 4) gene has been identified as a cause of Nager syndrome [154]. The SF3B4 gene encodes a highly conserved protein implicated in mRNA splicing and bone morphogenic protein (BMP) signaling, a pathway involved in early embryogenesis and skeletal development. In the study conducted be Bernier et al. (2012), SF3B4 mutations were found in 20 of the 35 (57%) families, including five of the seven families with more than one individual affected by Nager syndrome, with frameshift mutations being the most frequent genetic alterations [154]. Three nonsense mutations and one T > C transition destroying the translation start signal was subsequently described in 2013 [144]. Other 6 novel mutations were described by Petit [155], and a new mutation affecting a splice site (c.35-2A>G) was detected in a fetus with multiple malformations including congenital diaphragmatic hernia (first report) and a marked shortening of the upper limbs with predominant involvement of the radial ray [156].
Townes-Brocks Syndrome
Townes-Brocks syndrome (TBS), firstly described in 1972 by Townes and Brocks [157], is a genetic condition with a variable pattern of malformation including imperforate anus, abnormally shaped ears and thumb anomalies, which can be hypoplastic, duplicated or triphalangeal (Fig. 6). Other possible signs and symptoms include kidney and heart abnormalities, mild to profound hearing loss, eye anomalies and genital malformations [158]. These features vary among affected individuals, even within the same family. Intellectual disability or learning problems have also been reported in about 20 percent of affected patients and occasionally endocrine abnormalities, i.e. growth hormone deficiency and hypothyroidism [159]. The prevalence is unknown, partly because the clinical diagnosis of Townes-Brocks syndrome is often complicated by an overlap with VACTERL association, which may lead to an over-ascertainment of TBS prevalence. Martinez-Frias estimated the prevalence at 1:250,000 but he did not use stringent diagnostic criteria for TBS [160].
Fig. (6).
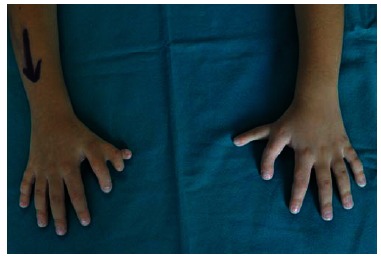
Clinical photograph of bilateral thumb duplication in a patient affected by Townes-Brocks Syndrome.
Townes-Brocks is caused by heterozygous mutations in SALL1 located on chromosome 16q12.1 and encoding a C2H2 zinc finger transcription factor implied in organogenesis and cell differentiation [161]. More than 70 mutations, including nonsense, frameshift, small insertions and deletions have been described so far (HMGD database): the majority occur within a mutational “hot spot region” of 802 bp (from nt 764 to 1565) located between the glutamine-rich interaction domain and the most amino-terminal double zinc finger domain in exon 2, resulting in the truncation of the protein upstream of the DNA binding domain [162]. The haploinsufficiency was the initially proposed disease-causing mechanism [163], but experimental and animal data suggested a dominant negative mechanism [164]. Moreover, the rarity of the condition is paralleled by the lack of unequivocal data to support the previous genotype-phenotype correlations. For example, developmental delay, already mentioned as an occasional feature of the syndrome, is mostly accounted for by genomic deletions encompassing SALL1 and involving also other genes in 16q12.1 [158]. Finally, the homozygous c.3160C>T, p.R1054* SALL1 mutation in 2 female siblings has been associated with a severe phenotype featured by multiple congenital anomalies, central nervous system defects, cortical blindness, and profound developmental delay [165].
Baller-Gerold Syndrome
Baller-Gerold syndrome (BGS) is a very rare condition, originally described by Baller (1950) and Gerold (1959) [166, 167]. It is characterized by the association of coronal craniosynostosis with radial ray anomalies (oligodactyly, hypo/aplasia of the thumb and/or hypo/aplasia of the radius), associated with facial dysmorphisms (brachycephaly, ocular exophthalmia, frontal bossing, nasal hypoplasia, small mouth, ogival palate). An inconstant sign is poikiloderma (areas of hypo/hyperpigmentation and telangectasias), which can appear during the first months of life. A hallmark is the failure to thrive, with the final height stabilized around -4 SD. Patellar aplasia or hypoplasia can be observed during childhood and results in genu recurvatum and knee instability. Intelligence is usually normal [168]. Patients have a predisposition to cancer, in particular osteosarcoma and lymphoma [169]. The prevalence of Baller-Gerold syndrome is unknown; it is probably less than 1:1,000,000.
Baller-Gerold syndrome is an autosomal recessive condition caused by mutations in the RECQL4 gene that maps to chromosome 8q24.3 [170]. RECQL4 is a member of the RecQ helicase gene family whose function is to maintain the genomic stability [171].
Baller–Gerold syndrome is allelic to two other conditions, for which mutations of the RECQL4 gene have been demostrated: Rothmund-Thomson syndrome, characterized by radial ray defects, poikiloderma, short stature, sparse scalp hair, sparse or absent eyelashes and/or eyebrows, juvenile cataract, skeletal abnormalities, premature aging and a predisposition to cancer, and RAPADILINO syndrome, characterized by pre- and post-natal growth retardation and radial defects, such as hypoplasia and aplasia of thumbs and radius. Although these disorders historically represent distinct genetic entities, clinical and genetic analysis have delineated a common genetic basis (RECQL4-related syndromes) and a highly variable expressivity with significant overlaps in the associated phenotypic manifestations, although precise genotype-phenotype correlations are still missing [172]. The only suggested association is the increased risk of cancer, especially osteosarcoma, in RTS patients harboring at least one truncating mutation in the RECQL4 gene [173].
FINAL REMARKS
Identifying a fetus or a newborn with a RD means having already significantly restricted the field of diagnosis: as evident in the previous paragraphs, the selective involvement of the radial side of the upper limb is subtended by just a few disorders, which contrasts with the frequent involvement of the radial side (especially the thumb) in several malformation syndromes. Those latter, however, variably involve also the other fingers and/or the ulnar side of the forearm and consist of more generalized and non-specific clinical pictures, which need to be carefully differentiated by the true RDs: for example, the finding of hypoplasia or syndactylies involving other fingers together with the thumb should cast doubts on the diagnosis of a “true” RD syndrome. On the other hand, the severity of the RD, which measures the involvement of the radius and/or the thumb, although essential to establish a general prognosis in terms of quality of life, has no relation with the etiology of the malformation: most of the RDs show all the spectrum of gravity of thumb and radial involvement, including also thumb duplication, and can not be differentiated by the limb phenotype only. Also the pattern of associated malformations and disorders is often overlapping, requiring a standard clinical approach to the RDs, which includes cardiac and renal ultrasound, costo-vertebral radiographs and blood cell count as a first screen. Finally, the recourse to the genetic testing for the confirmation of the clinical diagnosis of a syndrome with RD not always leads to a definitive assignment, being burdened by a wide shadow of negative test results, especially when the phenotypes are not typical. Those cases include a grey zone of tests performed for the exclusion of genetic diseases with a severe prognosis (i.e. DEB test for Fanconi anemia), wrong diagnosis, atypical phenotypes, non-mendelian disorders, but probably also cases for which the gene involved is not yet known. The incresing diagnostic yield of the new techniques for genetic testing will help in the future in refining the molecular diagnosis of a larger number of patients with RDs and differentiate more and more the true mendelian from the sporadic/multifactorial etiologies.
ACKNOWLEDGEMENTS
The authors thank the collegues of the Multidisciplinary Group on Congenital Hand Malformations at the University Hospital of Modena, Italy for helpful discussions. I.S. is granted by Programma di ricerca Regione-Universita` 2010–2012, Strategic Programme ‘‘Next-generation sequencing and gene therapy to diagnose and cure rare diseases in Regione Emilia Romagna (RER)’’.
CONFLICT OF INTEREST
The author(s) confirm that this article content has no conflict of interest.
FINANCIAL DISCLOSURE
No external funding was secured for this study and the authors have no financial relationships relevant to this article to disclose.
REFERENCES
- 1.Goldfarb C.A., Wall L., Manske P.R. Radial longitudinal deficiency: the incidence of associated medical and musculoskeletal conditions. J. Hand Surg. Am. 2006;31(7):1176–1182. doi: 10.1016/j.jhsa.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 2.Koskimies E., Lindfors N., Gissler M., Peltonen J., Nietosvaara Y. Congenital upper limb deficiencies and associated malformations in Finland: a population-based study. J. Hand Surg. Am. 2011;36(6):1058–1065. doi: 10.1016/j.jhsa.2011.03.015. [DOI] [PubMed] [Google Scholar]
- 3.Pakkasjärvi N., Koskimies E., Ritvanen A., Nietosvaara Y., Mäkitie O. Characteristics and associated anomalies in radial ray deficiencies in Finland--a population-based study. Am. J. Med. Genet. A. 2013;161A(2):261–267. doi: 10.1002/ajmg.a.35707. [DOI] [PubMed] [Google Scholar]
- 4.Giele H., Giele C., Bower C., Allison M. The incidence and epidemiology of congenital upper limb anomalies: a total population study. J. Hand Surg. Am. 2001;26(4):628–634. doi: 10.1053/jhsu.2001.26121. [DOI] [PubMed] [Google Scholar]
- 5.Kozin S.H. Upper-extremity congenital anomalies. J. Bone Joint Surg. Am. 2003;85-A(8):1564–1576. doi: 10.2106/00004623-200308000-00021. [DOI] [PubMed] [Google Scholar]
- 6.Carli D., Fairplay T., Ferrari P., Sartini S., Lando M., Garagnani L., Di Gennaro G.L., Di Pancrazio L., Bianconi G., Elmakky A., Bernasconi S., Landi A., Percesepe A. Genetic Basis of Congenital Upper Limb Anomalies: Analysis of 487 Cases of a Specialized Clinic. Birth Defects Res. Part A. 2013;97:798–805. doi: 10.1002/bdra.23212. [DOI] [PubMed] [Google Scholar]
- 7.Naderi N., McCurdy M.T., Reed R.M. Holt-Oram: when the key to a broken heart is in the hand. BMJ Case Rep. 2014;2014:bcr2014203851. doi: 10.1136/bcr-2014-203851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kennelly M.M., Moran P. A clinical algorithm of prenatal diagnosis of Radial Ray Defects with two and three dimensional ultrasound. Prenat. Diagn. 2007;27(8):730–737. doi: 10.1002/pd.1770. [DOI] [PubMed] [Google Scholar]
- 9.Carli D., Garagnani L., Lando M., Fairplay T., Bernasconi S., Landi A., Percesepe A. VACTERL (vertebral defects, anal atresia, tracheoesophageal fistula with esophageal atresia, cardiac defects, renal and limb anomalies) association: disease spectrum in 25 patients ascertained for their upper limb involvement. J. Pediatr. 2014;164(3):458–62.e1, 2. doi: 10.1016/j.jpeds.2013.09.033. [DOI] [PubMed] [Google Scholar]
- 10.Solomon B.D., Pineda-Alvarez D.E., Hadley D.W., Hansen N.F., Kamat A., Donovan F.X., Chandrasekharappa S.C., Hong S.K., Roessler E., Mullikin J.C., NISC Comparative Sequencing Program Exome Sequencing and High-Density Microarray Testing in Monozygotic Twin Pairs Discordant for Features of VACTERL Association. Mol. Syndromol. 2013;4(1-2):27–31. doi: 10.1159/000345406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barisic I., Boban L., Greenlees R., Garne E., Wellesley D., Calzolari E., Addor M.C., Arriola L., Bergman J.E., Braz P., Budd J.L., Gatt M., Haeusler M., Khoshnood B., Klungsoyr K., McDonnell B., Nelen V., Pierini A., Queisser-Wahrendorf A., Rankin J., Rissmann A., Rounding C., Tucker D., Verellen-Dumoulin C., Dolk H. Holt Oram syndrome: a registry-based study in Europe. Orphanet J. Rare Dis. 2014;9(1):156. doi: 10.1186/s13023-014-0156-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klopocki E., Schulze H., Strauss G., Ott C.E., Hall J., Trotier F., Fleischhauer S., Greenhalgh L., Newbury-Ecob R.A., Neumann L.M., Habenicht R., König R., Seemanova E., Megarbane A., Ropers H.H., Ullmann R., Horn D., Mundlos S. Complex inheritance pattern resembling autosomal recessive inheritance involving a microdeletion in thrombocytopenia-absent radius syndrome. Am. J. Hum. Genet. 2007;80(2):232–240. doi: 10.1086/510919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keeling S.L., Lee-Jones L., Thompson P. Interstitial deletion 4q32-34 with ulnar deficiency: 4q33 may be the critical region in 4q terminal deletion syndrome. Am. J. Med. Genet. 2001;99(2):94–98. doi: 10.1002/1096-8628(2000)9999:999<00::AID-AJMG1134>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 14.Kato H., Ogino T., Minami A., Ohshio I. Experimental study of radial ray deficiency. J. Hand Surg. [Br.] 1990;15(4):470–476. doi: 10.1016/0266-7681(90)90094-K. [DOI] [PubMed] [Google Scholar]
- 15.Raynaud A. Developmental mechanism involved in the embryonic reduction of limbs in reptiles. Int. J. Dev. Biol. 1990;34(1):233–243. [PubMed] [Google Scholar]
- 16.Oberg K.C. Review of the molecular development of the thumb: digit primera. Clin. Orthop. Relat. Res. 2014;472(4):1101–1105. doi: 10.1007/s11999-013-3008-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burke A.C., Nelson C.E., Morgan B.A., Tabin C. Hox genes and the evolution of vertebrate axial morphology. Development. 1995;121(2):333–346. doi: 10.1242/dev.121.2.333. [DOI] [PubMed] [Google Scholar]
- 18.Ng J.K., Kawakami Y., Büscher D., Raya A., Itoh T., Koth C.M., Rodríguez Esteban C., Rodríguez-León J., Garrity D.M., Fishman M.C., Izpisúa Belmonte J.C. The limb identity gene Tbx5 promotes limb initiation by interacting with Wnt2b and Fgf10. Development. 2002;129(22):5161–5170. doi: 10.1242/dev.129.22.5161. [DOI] [PubMed] [Google Scholar]
- 19.Daluiski A., Yi S.E., Lyons K.M. The molecular control of upper extremity development: implications for congenital hand anomalies. J. Hand Surg. Am. 2001;26(1):8–22. doi: 10.1053/jhsu.2001.9419. [DOI] [PubMed] [Google Scholar]
- 20.Mariani F.V., Martin G.R. Deciphering skeletal patterning: clues from the limb. Nature. 2003;423(6937):319–325. doi: 10.1038/nature01655. [DOI] [PubMed] [Google Scholar]
- 21.Niswander L. Pattern formation: old models out on a limb. Nat. Rev. Genet. 2003;4(2):133–143. doi: 10.1038/nrg1001. [DOI] [PubMed] [Google Scholar]
- 22.Tickle C. Making digit patterns in the vertebrate limb. Nat. Rev. Mol. Cell Biol. 2006;7(1):45–53. doi: 10.1038/nrm1830. [DOI] [PubMed] [Google Scholar]
- 23.Laufer E., Dahn R., Orozco O.E., Yeo C.Y., Pisenti J., Henrique D., Abbott U.K., Fallon J.F., Tabin C. Expression of Radical fringe in limb-bud ectoderm regulates apical ectodermal ridge formation. Nature. 1997;386(6623):366–373. doi: 10.1038/386366a0. [DOI] [PubMed] [Google Scholar]
- 24.Rodriguez-Esteban C., Schwabe J.W., De La Peña J., Foys B., Eshelman B., Izpisúa Belmonte J.C. Radical fringe positions the apical ectodermal ridge at the dorsoventral boundary of the vertebrate limb. Nature. 1997;386(6623):360–366. doi: 10.1038/386360a0. [DOI] [PubMed] [Google Scholar]
- 25.Zakany J., Zacchetti G., Duboule D. Interactions between HOXD and Gli3 genes control the limb apical ectodermal ridge via Fgf10. Dev. Biol. 2007;306(2):883–893. doi: 10.1016/j.ydbio.2007.03.517. [DOI] [PubMed] [Google Scholar]
- 26.Boehm B., Westerberg H., Lesnicar-Pucko G., Raja S., Rautschka M., Cotterell J., Swoger J., Sharpe J. The role of spatially controlled cell proliferation in limb bud morphogenesis. PLoS Biol. 2010;8(7):e1000420. doi: 10.1371/journal.pbio.1000420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barrow J.R., Thomas K.R., Boussadia-Zahui O., Moore R., Kemler R., Capecchi M.R., McMahon A.P. Ectodermal Wnt3/beta-catenin signaling is required for the establishment and maintenance of the apical ectodermal ridge. Genes Dev. 2003;17(3):394–409. doi: 10.1101/gad.1044903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Riddle R.D., Ensini M., Nelson C., Tsuchida T., Jessell T.M., Tabin C. Induction of the LIM homeobox gene Lmx1 by WNT7a establishes dorsoventral pattern in the vertebrate limb. Cell. 1995;83(4):631–640. doi: 10.1016/0092-8674(95)90103-5. [DOI] [PubMed] [Google Scholar]
- 29.Yang Y., Niswander L. Interaction between the signaling molecules WNT7a and SHH during vertebrate limb development: dorsal signals regulate anteroposterior patterning. Cell. 1995;80(6):939–947. doi: 10.1016/0092-8674(95)90297-X. [DOI] [PubMed] [Google Scholar]
- 30.Riddle R.D., Johnson R.L., Laufer E., Tabin C. Sonic hedgehog mediates the polarizing activity of the ZPA. Cell. 1993;75(7):1401–1416. doi: 10.1016/0092-8674(93)90626-2. [DOI] [PubMed] [Google Scholar]
- 31.Zhu J., Nakamura E., Nguyen M.T., Bao X., Akiyama H., Mackem S. Uncoupling Sonic hedgehog control of pattern and expansion of the developing limb bud. Dev. Cell. 2008;14(4):624–632. doi: 10.1016/j.devcel.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun X., Mariani F.V., Martin G.R. Functions of FGF signalling from the apical ectodermal ridge in limb development. Nature. 2002;418(6897):501–508. doi: 10.1038/nature00902. [DOI] [PubMed] [Google Scholar]
- 33.Tabin C., Wolpert L. Rethinking the proximodistal axis of the vertebrate limb in the molecular era. Genes Dev. 2007;21(12):1433–1442. doi: 10.1101/gad.1547407. [DOI] [PubMed] [Google Scholar]
- 34.Swanson A.B. A classification for congenital malformations of the hand. Acad. Med. Bull New Jersey. 1964;10:166–169. [Google Scholar]
- 35.De Smet L., IFSSH. International Federation for Societies for Surgery of the Hand JSSH. Japanese Society for Surgery of the Hand Classification for congenital anomalies of the hand: the IFSSH classification and the JSSH modification. Genet. Couns. 2002;13(3):331–338. [PubMed] [Google Scholar]
- 36.Manske P.R., Oberg K.C. Classification and developmental biology of congenital anomalies of the hand and upper extremity. J. Bone Joint Surg. Am. 2009;91(Suppl. 4):3–18. doi: 10.2106/JBJS.I.00008. [DOI] [PubMed] [Google Scholar]
- 37.Chung M.S. Congenital differences of the upper extremity: classification and treatment principles. Clin. Orthop. Surg. 2011;3(3):172–177. doi: 10.4055/cios.2011.3.3.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oberg K.C., Feenstra J.M., Manske P.R., Tonkin M.A. Developmental biology and classification of congenital anomalies of the hand and upper extremity. J. Hand Surg. Am. 2010;35(12):2066–2076. doi: 10.1016/j.jhsa.2010.09.031. [DOI] [PubMed] [Google Scholar]
- 39.Zuniga A., Zeller R., Probst S. The molecular basis of human congenital limb malformations. Wiley Interdiscip. Rev. Dev. Biol. 2012;1(6):803–822. doi: 10.1002/wdev.59. [DOI] [PubMed] [Google Scholar]
- 40.Manske P.R., McCarroll H.R., Jr Reconstruction of the congenitally deficient thumb. Hand Clin. 1992;8(1):177–196. [PubMed] [Google Scholar]
- 41.Blauth W. [The hypoplastic thumb]. Arch. Orthop. Unfallchir. 1967;62(3):225–246. doi: 10.1007/BF00416751. [German]. [DOI] [PubMed] [Google Scholar]
- 42.James M.A., Green H.D., McCarroll H.R., Jr, Manske P.R. The association of radial deficiency with thumb hypoplasia. J. Bone Joint Surg. Am. 2004;86-A(10):2196–2205. doi: 10.2106/00004623-200410000-00010. [DOI] [PubMed] [Google Scholar]
- 43.Bayne L.G., Klug M.S. Long-term review of the surgical treatment of radial deficiencies. J. Hand Surg. Am. 1987;12(2):169–179. doi: 10.1016/S0363-5023(87)80267-8. [DOI] [PubMed] [Google Scholar]
- 44.James M.A., McCarroll H.R., Jr, Manske P.R. The spectrum of radial longitudinal deficiency: a modified classification. J. Hand Surg. Am. 1999;24(6):1145–1155. doi: 10.1053/jhsu.1999.1145. [DOI] [PubMed] [Google Scholar]
- 45.Goldfarb C.A., Manske P.R., Busa R., Mills J., Carter P., Ezaki M. Upper-extremity phocomelia reexamined: a longitudinal dysplasia. J. Bone Joint Surg. Am. 2005;87(12):2639–2648. doi: 10.2106/JBJS.D.02011. [DOI] [PubMed] [Google Scholar]
- 46.Castriota-Scanderbeg A. Dallapiccola, B. Abnormal Skeletal Phenotypes: From Simple Signs to Complex Diagnoses. Berlin, Heidelberg: Springer-Verlag; 2005. pp. 437–441. [Google Scholar]
- 47.Flatt A.E. The care of congenital hand anomalies. 1994. [Google Scholar]
- 48.Abdel-Ghani H., Amro S. Characteristics of patients with hypoplastic thumb: a prospective study of 51 patients with the results of surgical treatment. J. Pediatr. Orthop. B. 2004;13(2):127–138. doi: 10.1097/00009957-200403000-00013. [DOI] [PubMed] [Google Scholar]
- 49.Blauth W., Schneider-Sickert F. Congenital deformities of the hand: an atlas on their surgical treatment. 1981. [DOI] [Google Scholar]
- 50.James M.A., McCarroll H.R., Jr, Manske P.R. Characteristics of patients with hypoplastic thumbs. J. Hand Surg. Am. 1996;21(1):104–113. doi: 10.1016/S0363-5023(96)80162-6. [DOI] [PubMed] [Google Scholar]
- 51.Riley S.A. An overview of radial longitudinal deficiency. Curr. Orthop. Pract. 2008;9:655–659. doi: 10.1097/BCO.0b013e3283154d60. [DOI] [Google Scholar]
- 52.Vergult S., Hoogeboom A.J., Bijlsma E.K., Sante T., Klopocki E., De Wilde B., Jongmans M., Thiel C., Verheij J.B., Perez-Aytes A., Van Esch H., Kuechler A., Barge-Schaapveld D.Q., Sznajer Y., Mortier G., Menten B. Complex genetics of radial ray deficiencies: screening of a cohort of 54 patients. Genet. Med. 2013;15(3):195–202. doi: 10.1038/gim.2012.120. [DOI] [PubMed] [Google Scholar]
- 53.Quan L., Smith D.W. The VATER association. Vertebral defects, Anal atresia, T-E fistula with esophageal atresia, Radial and Renal dysplasia: a spectrum of associated defects. J. Pediatr. 1973;82(1):104–107. doi: 10.1016/S0022-3476(73)80024-1. [DOI] [PubMed] [Google Scholar]
- 54.Temtamy S.A., Miller J.D. Extending the scope of the VATER association: definition of the VATER syndrome. J. Pediatr. 1974;85(3):345–349. doi: 10.1016/S0022-3476(74)80113-7. [DOI] [PubMed] [Google Scholar]
- 55.Nora A.H., Nora J.J. A syndrome of multiple congenital anomalies associated with teratogenic exposure. Arch. Environ. Health. 1975;30(1):17–21. doi: 10.1080/00039896.1975.10666626. [DOI] [PubMed] [Google Scholar]
- 56.Botto L.D., Khoury M.J., Mastroiacovo P., Castilla E.E., Moore C.A., Skjaerven R., Mutchinick O.M., Borman B., Cocchi G., Czeizel A.E., Goujard J., Irgens L.M., Lancaster P.A., Martínez-Frías M.L., Merlob P., Ruusinen A., Stoll C., Sumiyoshi Y. The spectrum of congenital anomalies of the VATER association: an international study. Am. J. Med. Genet. 1997;71(1):8–15. doi: 10.1002/(SICI)1096-8628(19970711)71:1<8::AID-AJMG2>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 57.Khoury M.J., Cordero J.F., Greenberg F., James L.M., Erickson J.D. A population study of the VACTERL association: evidence for its etiologic heterogeneity. Pediatrics. 1983;71(5):815–820. [PubMed] [Google Scholar]
- 58.Källén K., Mastroiacovo P., Castilla E.E., Robert E., Källén B. VATER non-random association of congenital malformations: study based on data from four malformation registers. Am. J. Med. Genet. 2001;101(1):26–32. doi: 10.1002/ajmg.1201. [DOI] [PubMed] [Google Scholar]
- 59.Solomon B.D., Pineda-Alvarez D.E., Raam M.S., Bous S.M., Keaton A.A., Vélez J.I., Cummings D.A. Analysis of component findings in 79 patients diagnosed with VACTERL association. Am. J. Med. Genet. A. 2010;152A(9):2236–2244. doi: 10.1002/ajmg.a.33572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weaver D.D., Mapstone C.L., Yu P.L. The VATER association. Analysis of 46 patients. Am. J. Dis. Child. 1986;140(3):225–229. doi: 10.1001/archpedi.1986.02140170051027. [DOI] [PubMed] [Google Scholar]
- 61.Kuo M.F., Tsai Y., Hsu W.M., Chen R.S., Tu Y.K., Wang H.S. Tethered spinal cord and VACTERL association. J. Neurosurg. 2007;106(3) Suppl.:201–204. doi: 10.3171/ped.2007.106.3.201. [DOI] [PubMed] [Google Scholar]
- 62.O’Neill B.R., Yu A.K., Tyler-Kabara E.C. Prevalence of tethered spinal cord in infants with VACTERL. J. Neurosurg. Pediatr. 2010;6(2):177–182. doi: 10.3171/2010.5.PEDS09428. [DOI] [PubMed] [Google Scholar]
- 63.Cunningham B.K., Hadley D.W., Hannoush H., Meltzer A.C., Niforatos N., Pineda-Alvarez D., Sachdev V., Warren-Mora N., Solomon B.D. Analysis of cardiac anomalies in VACTERL association. Birth Defects Res. A Clin. Mol. Teratol. 2013;97(12):792–797. doi: 10.1002/bdra.23211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Solomon B.D., Raam M.S., Pineda-Alvarez D.E. Analysis of genitourinary anomalies in patients with VACTERL (Vertebral anomalies, Anal atresia, Cardiac malformations, Tracheo-Esophageal fistula, Renal anomalies, Limb abnormalities) association. Congenit. Anom. (Kyoto) 2011;51(2):87–91. doi: 10.1111/j.1741-4520.2010.00303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bartels E., Jenetzky E., Solomon B.D., Ludwig M., Schmiedeke E., Grasshoff-Derr S., Schmidt D., Märzheuser S., Hosie S., Weih S., Holland-Cunz S., Palta M., Leonhardt J., Schäfer M., Kujath C., Rissmann A., Nöthen M.M., Reutter H., Zwink N. Inheritance of the VATER/VACTERL association. Pediatr. Surg. Int. 2012;28(7):681–685. doi: 10.1007/s00383-012-3100-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Reutter H., Ludwig M. VATER/VACTERL Association: Evidence for the Role of Genetic Factors. Mol. Syndromol. 2013;4(1-2):16–19. doi: 10.1159/000345300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McNeal R.M., Skoglund R.R., Francke U. Congenital anomalies including the VATER association in a patient with del(6)q deletion. J. Pediatr. 1977;91(6):957–960. doi: 10.1016/S0022-3476(77)80903-7. [DOI] [PubMed] [Google Scholar]
- 68.Walsh L.E., Vance G.H., Weaver D.D. Distal 13q Deletion Syndrome and the VACTERL association: case report, literature review, and possible implications. Am. J. Med. Genet. 2001;98(2):137–144. doi: 10.1002/1096-8628(20010115)98:2<137::AID-AJMG1022>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 69.Zen P.R., Riegel M., Rosa R.F., Pinto L.L., Graziadio C., Schwartz I.V., Paskulin G.A. Esophageal stenosis in a child presenting a de novo 7q terminal deletion. Eur. J. Med. Genet. 2010;53(5):333–336. doi: 10.1016/j.ejmg.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 70.de Jong E.M., Douben H., Eussen B.H., Felix J.F., Wessels M.W., Poddighe P.J., Nikkels P.G., de Krijger R.R., Tibboel D., de Klein A. 5q11.2 deletion in a patient with tracheal agenesis. Eur. J. Hum. Genet. 2010;18(11):1265–1268. doi: 10.1038/ejhg.2010.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Solomon B.D., Pineda-Alvarez D.E., Hadley D.W., Keaton A.A., Agochukwu N.B., Raam M.S., Carlson-Donohoe H.E., Kamat A., Chandrasekharappa S.C. De novo deletion of chromosome 20q13.33 in a patient with tracheo-esophageal fistula, cardiac defects and genitourinary anomalies implicates GTPBP5 as a candidate gene. Birth Defects Res. A Clin. Mol. Teratol. 2011;91(9):862–865. doi: 10.1002/bdra.20821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Aynaci F.M., Celep F., Karagüzel A., Baki A., Yildiran A. A case of VATER association associated with 9qh+. Genet. Couns. 1996;7(4):321–322. [PubMed] [Google Scholar]
- 73.Schramm C., Draaken M., Bartels E., Boemers T.M., Aretz S., Brockschmidt F.F., Nöthen M.M., Ludwig M., Reutter H. De novo microduplication at 22q11.21 in a patient with VACTERL association. Eur. J. Med. Genet. 2011;54(1):9–13. doi: 10.1016/j.ejmg.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 74.Prieto J.C., Garcia N.M., Elder F.F., Zinn A.R., Baker L.A. Phenotypic expansion of the supernumerary derivative (22) chromosome syndrome: VACTERL and Hirschsprung’s disease. J. Pediatr. Surg. 2007;42(11):1928–1932. doi: 10.1016/j.jpedsurg.2007.07.030. [DOI] [PubMed] [Google Scholar]
- 75.Cinti R., Priolo M., Lerone M., Gimelli G., Seri M., Silengo M., Ravazzolo R. Molecular characterisation of a supernumerary ring chromosome in a patient with VATER association. J. Med. Genet. 2001;38(2):E6. doi: 10.1136/jmg.38.2.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.van der Veken L.T., Dieleman M.M., Douben H., van de Brug J.C., van de Graaf R., Hoogeboom A.J., Poddighe P.J., de Klein A. Low grade mosaic for a complex supernumerary ring chromosome 18 in an adult patient with multiple congenital anomalies. Mol. Cytogenet. 2010;3:13. doi: 10.1186/1755-8166-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yamada K., Uchiyama A., Arai M., Kubodera K., Yamamoto Y., Orii K.O., Nagasawa H., Masuno M., Kohno Y. Severe upper airway stenosis in a boy with partial monosomy 16p13.3pter and partial trisomy 16q22qter. Congenit. Anom. (Kyoto) 2009;49(2):85–88. doi: 10.1111/j.1741-4520.2009.00228.x. [DOI] [PubMed] [Google Scholar]
- 78.Garcia-Barceló M.M., Wong K.K., Lui V.C., Yuan Z.W., So M.T., Ngan E.S., Miao X.P., Chung P.H., Khong P.L., Tam P.K. Identification of a HOXD13 mutation in a VACTERL patient. Am. J. Med. Genet. A. 2008;146A(24):3181–3185. doi: 10.1002/ajmg.a.32426. [DOI] [PubMed] [Google Scholar]
- 79.Arrington C.B., Patel A., Bacino C.A., Bowles N.E. Haploinsufficiency of the LIM domain containing preferred translocation partner in lipoma (LPP) gene in patients with tetralogy of Fallot and VACTERL association. Am. J. Med. Genet. A. 2010;152A(11):2919–2923. doi: 10.1002/ajmg.a.33718. [DOI] [PubMed] [Google Scholar]
- 80.Saisawat P., Kohl S., Hilger A.C., Hwang D.Y., Yung Gee H., Dworschak G.C., Tasic V., Pennimpede T., Natarajan S., Sperry E., Matassa D.S., Stajić N., Bogdanovic R., de Blaauw I., Marcelis C.L., Wijers C.H., Bartels E., Schmiedeke E., Schmidt D., Märzheuser S., Grasshoff-Derr S., Holland-Cunz S., Ludwig M., Nöthen M.M., Draaken M., Brosens E., Heij H., Tibboel D., Herrmann B.G., Solomon B.D., de Klein A., van Rooij I.A., Esposito F., Reutter H.M., Hildebrandt F. Whole-exome resequencing reveals recessive mutations in TRAP1 in individuals with CAKUT and VACTERL association. Kidney Int. 2014;85(6):1310–1317. doi: 10.1038/ki.2013.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Siebel S., Solomon B.D. Mitochondrial Factors and VACTERL Association-Related Congenital Malformations. Mol. Syndromol. 2013;4(1-2):63–73. doi: 10.1159/000346301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fanconi G. Familiaere infantile perniziosaartige Anaemie (pernizioeses Blutbild und Konstitution). Jahrb. Kinderheilk. 1927;117:257–280. [Google Scholar]
- 83.Rosenberg P.S., Tamary H., Alter B.P. How high are carrier frequencies of rare recessive syndromes? Contemporary estimates for Fanconi Anemia in the United States and Israel. Am. J. Med. Genet. A. 2011;155A(8):1877–1883. doi: 10.1002/ajmg.a.34087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schneider M., Chandler K., Tischkowitz M., Meyer S. Fanconi anaemia: genetics, molecular biology, and cancer - implications for clinical management in children and adults. Clin. Genet. 2014 doi: 10.1111/cge.12517. [DOI] [PubMed] [Google Scholar]
- 85.Giampietro P.F., Verlander P.C., Davis J.G., Auerbach A.D. Diagnosis of Fanconi anemia in patients without congenital malformations: an international Fanconi Anemia Registry Study. Am. J. Med. Genet. 1997;68(1):58–61. doi: 10.1002/(SICI)1096-8628(19970110)68:1<58::AID-AJMG11>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 86.Auerbach A.D. Fanconi anemia and its diagnosis. Mutat. Res. 2009;668(1-2):4–10. doi: 10.1016/j.mrfmmm.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wilks D.J., Kay S.P., Bourke G. Fanconi’s anaemia and unilateral thumb polydactyly--don’t miss it. J. Plast. Reconstr. Aesthet. Surg. 2012;65(8):1083–1086. doi: 10.1016/j.bjps.2012.02.015. [DOI] [PubMed] [Google Scholar]
- 88.Alter B.P. Cancer in Fanconi anemia, 1927-2001. Cancer. 2003;97(2):425–440. doi: 10.1002/cncr.11046. [DOI] [PubMed] [Google Scholar]
- 89.Shimamura A., Alter B.P. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev. 2010;24(3):101–122. doi: 10.1016/j.blre.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Oostra A.B., Nieuwint A.W., Joenje H., de Winter J.P. Diagnosis of Fanconi Anemia: Chromosomal Breakage Analysis. Anemia, 2012. [DOI] [PMC free article] [PubMed]
- 91.Moldovan G.L., D’Andrea A.D. How the fanconi anemia pathway guards the genome. Annu. Rev. Genet. 2009;43:223–249. doi: 10.1146/annurev-genet-102108-134222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vaz F., Hanenberg H., Schuster B., Barker K., Wiek C., Erven V., Neveling K., Endt D., Kesterton I., Autore F., Fraternali F., Freund M., Hartmann L., Grimwade D., Roberts R.G., Schaal H., Mohammed S., Rahman N., Schindler D., Mathew C.G. Mutation of the RAD51C gene in a Fanconi anemia-like disorder. Nat. Genet. 2010;42(5):406–409. doi: 10.1038/ng.570. [DOI] [PubMed] [Google Scholar]
- 93.Stoepker C., Hain K., Schuster B., Hilhorst-Hofstee Y., Rooimans M.A., Steltenpool J., Oostra A.B., Eirich K., Korthof E.T., Nieuwint A.W., Jaspers N.G., Bettecken T., Joenje H., Schindler D., Rouse J., de Winter J.P. SLX4, a coordinator of structure-specific endonucleases, is mutated in a new Fanconi anemia subtype. Nat. Genet. 2011;43(2):138–141. doi: 10.1038/ng.751. [DOI] [PubMed] [Google Scholar]
- 94.Kim Y., Lach F.P., Desetty R., Hanenberg H., Auerbach A.D., Smogorzewska A. Mutations of the SLX4 gene in Fanconi anemia. Nat. Genet. 2011;43(2):142–146. doi: 10.1038/ng.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bogliolo M., Schuster B., Stoepker C., Derkunt B., Su Y., Raams A., Trujillo J.P., Minguillón J., Ramírez M.J., Pujol R., Casado J.A., Baños R., Rio P., Knies K., Zúñiga S., Benítez J., Bueren J.A., Jaspers N.G., Schärer O.D., de Winter J.P., Schindler D., Surrallés J. Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am. J. Hum. Genet. 2013;92(5):800–806. doi: 10.1016/j.ajhg.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Neveling K., Endt D., Hoehn H., Schindler D. Genotype-phenotype correlations in Fanconi anemia. Mutat. Res. 2009;668(1-2):73–91. doi: 10.1016/j.mrfmmm.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 97.Feben C., Kromberg J., Wainwright R., Stones D., Poole J., Haw T., Krause A. Hematological consequences of a FANCG founder mutation in Black South African patients with Fanconi anemia. Blood Cells Mol. Dis. 2015;54(3):270–274. doi: 10.1016/j.bcmd.2014.11.011. [DOI] [PubMed] [Google Scholar]
- 98.Soulier J., Leblanc T., Larghero J., Dastot H., Shimamura A., Guardiola P., Esperou H., Ferry C., Jubert C., Feugeas J.P., Henri A., Toubert A., Socié G., Baruchel A., Sigaux F., D’Andrea A.D., Gluckman E. Detection of somatic mosaicism and classification of Fanconi anemia patients by analysis of the FA/BRCA pathway. Blood. 2005;105(3):1329–1336. doi: 10.1182/blood-2004-05-1852. [DOI] [PubMed] [Google Scholar]
- 99.Zheng Z., Geng J., Yao R.E., Li C., Ying D., Shen Y., Ying L., Yu Y., Fu Q. Molecular defects identified by whole exome sequencing in a child with Fanconi anemia. Gene. 2013;530(2):295–300. doi: 10.1016/j.gene.2013.08.031. [DOI] [PubMed] [Google Scholar]
- 100.Goldfarb C.A., Wall L.B. Holt-Oram syndrome. J. Hand Surg. Am. 2014;39(8):1646–1648. doi: 10.1016/j.jhsa.2014.02.015. [DOI] [PubMed] [Google Scholar]
- 101.Smith A.T., Sack G.H., Jr, Taylor G.J. Holt-Oram syndrome. J. Pediatr. 1979;95(4):538–543. doi: 10.1016/S0022-3476(79)80758-1. [DOI] [PubMed] [Google Scholar]
- 102.Poznanski A.K., Gall J.C., Jr, Stern A.M. Skeletal manifestations of the Holt-Oram syndrome. Radiology. 1970;94(1):45–53. doi: 10.1148/10.1148/94.1.45. [DOI] [PubMed] [Google Scholar]
- 103.Newbury-Ecob R.A., Leanage R., Raeburn J.A., Young I.D. Holt-Oram syndrome: a clinical genetic study. J. Med. Genet. 1996;33(4):300–307. doi: 10.1136/jmg.33.4.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Brassington A.M., Sung S.S., Toydemir R.M., Le T., Roeder A.D., Rutherford A.E., Whitby F.G., Jorde L.B., Bamshad M.J. Expressivity of Holt-Oram syndrome is not predicted by TBX5 genotype. Am. J. Hum. Genet. 2003;73(1):74–85. doi: 10.1086/376436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Garavelli L., De Brasi D., Verri R., Guareschi E., Cariola F., Melis D., Calcagno G., Salvatore F., Unger S., Sebastio G., Albertini G., Rivieri F., Soli F., Superti-Furga A., Gentile M. Holt-Oram syndrome associated with anomalies of the feet. Am. J. Med. Genet. A. 2008;146A(9):1185–1189. doi: 10.1002/ajmg.a.32170. [DOI] [PubMed] [Google Scholar]
- 106.Baban A., Postma A.V., Marini M., Trocchio G., Santilli A., Pelegrini M., Sirleto P., Lerone M., Albanese S.B., Barnett P., Boogerd C.J., Dallapiccola B., Digilio M.C., Ravazzolo R., Pongiglione G. Identification of TBX5 mutations in a series of 94 patients with Tetralogy of Fallot. Am. J. Med. Genet. A. 2014;164A(12):3100–3107. doi: 10.1002/ajmg.a.36783. [DOI] [PubMed] [Google Scholar]
- 107.Baban A., Pitto L., Pulignani S., Cresci M., Mariani L., Gambacciani C., Digilio M.C., Pongiglione G., Albanese S. Holt-Oram syndrome with intermediate atrioventricular canal defect, and aortic coarctation: functional characterization of a de novo TBX5 mutation. Am. J. Med. Genet. A. 2014;164A(6):1419–1424. doi: 10.1002/ajmg.a.36459. [DOI] [PubMed] [Google Scholar]
- 108.Basson C.T., Bachinsky D.R., Lin R.C., Levi T., Elkins J.A., Soults J., Grayzel D., Kroumpouzou E., Traill T.A., Leblanc-Straceski J., Renault B., Kucherlapati R., Seidman J.G., Seidman C.E. Mutations in human TBX5 cause limb and cardiac malformation in Holt-Oram syndrome. Nature Genet., 1997, 15, 30-35. Note. Nat. Genet. 1997;15:411. doi: 10.1038/ng0197-30. [Erratum]. [DOI] [PubMed] [Google Scholar]
- 109.Bickley S.R., Logan M.P. Regulatory modulation of the T-box gene Tbx5 links development, evolution, and adaptation of the sternum. Proc. Natl. Acad. Sci. USA. 2014;111(50):17917–17922. doi: 10.1073/pnas.1409913111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.McDermott D.A., Bressan M.C., He J., Lee J.S., Aftimos S., Brueckner M., Gilbert F., Graham G.E., Hannibal M.C., Innis J.W., Pierpont M.E., Raas-Rothschild A., Shanske A.L., Smith W.E., Spencer R.H., St John-Sutton M.G., van Maldergem L., Waggoner D.J., Weber M., Basson C.T. TBX5 genetic testing validates strict clinical criteria for Holt-Oram syndrome. Pediatr. Res. 2005;58(5):981–986. doi: 10.1203/01.PDR.0000182593.95441.64. [DOI] [PubMed] [Google Scholar]
- 111.Huang T. Current advances in Holt-Oram syndrome. Curr. Opin. Pediatr. 2002;14(6):691–695. doi: 10.1097/00008480-200212000-00008. [DOI] [PubMed] [Google Scholar]
- 112.Heinritz W., Shou L., Moschik A., Froster U.G. The human TBX5 gene mutation database. Hum. Mutat. 2005;26(4):397. doi: 10.1002/humu.9375. [DOI] [PubMed] [Google Scholar]
- 113.Cross S.J., Ching Y.H., Li Q.Y., Armstrong-Buisseret L., Spranger S., Lyonnet S., Bonnet D., Penttinen M., Jonveaux P., Leheup B., Mortier G., Van Ravenswaaij C., Gardiner C.A. The mutation spectrum in Holt-Oram syndrome. J. Med. Genet. 2000;37(10):785–787. doi: 10.1136/jmg.37.10.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mori A.D., Bruneau B.G. TBX5 mutations and congenital heart disease: Holt-Oram syndrome revealed. Curr. Opin. Cardiol. 2004;19(3):211–215. doi: 10.1097/00001573-200405000-00004. [DOI] [PubMed] [Google Scholar]
- 115.Basson C.T., Huang T., Lin R.C., Bachinsky D.R., Weremowicz S., Vaglio A., Bruzzone R., Quadrelli R., Lerone M., Romeo G., Silengo M., Pereira A., Krieger J., Mesquita S.F., Kamisago M., Morton C.C., Pierpont M.E., Müller C.W., Seidman J.G., Seidman C.E. Different TBX5 interactions in heart and limb defined by Holt-Oram syndrome mutations. Proc. Natl. Acad. Sci. USA. 1999;96(6):2919–2924. doi: 10.1073/pnas.96.6.2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Braulke I., Herzog S., Thies U., Zoll B. Holt-Oram syndrome in four half-siblings with unaffected parents: brief clinical report. Clin. Genet. 1991;39(4):241–244. doi: 10.1111/j.1399-0004.1991.tb03021.x. [DOI] [PubMed] [Google Scholar]
- 117.Okihiro M.M., Tasaki T., Nakano K.K., Bennett B.K. Duane syndrome and congenital upper-limb anomalies. A familial occurrence. Arch. Neurol. 1977;34(3):174–179. doi: 10.1001/archneur.1977.00500150060012. [DOI] [PubMed] [Google Scholar]
- 118.Temtamy S.A., Shoukry A.S., Ghaly I., El-Meligy R., Boulos S.Y. The Duane radial dysplasia syndrome: an autosomal dominant disorder. Birth Defects Orig. Artic. Ser. 1975;XI(5):344–345. [Google Scholar]
- 119.Marshman W.E., Schalit G., Jones R.B., Lee J.P., Matthews T.D., McCabe S. Congenital anomalies in patients with Duane retraction syndrome and their relatives. J. AAPOS. 2000;4(2):106–109. doi: 10.1067/mpa.2000.103439. [DOI] [PubMed] [Google Scholar]
- 120.Ye X.C., Pegado V., Patel M.S., Wasserman W.W. Strabismus genetics across a spectrum of eye misalignment disorders. Clin. Genet. 2014;86(2):103–111. doi: 10.1111/cge.12367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hayes A., Costa T., Polomeno R.C. The Okihiro syndrome of Duane anomaly, radial ray abnormalities, and deafness. Am. J. Med. Genet. 1985;22(2):273–280. doi: 10.1002/ajmg.1320220208. [DOI] [PubMed] [Google Scholar]
- 122.Kohlhase J., Schubert L., Liebers M., Rauch A., Becker K., Mohammed S.N., Newbury-Ecob R., Reardon W. Mutations at the SALL4 locus on chromosome 20 result in a range of clinically overlapping phenotypes, including Okihiro syndrome, Holt-Oram syndrome, acro-renal-ocular syndrome, and patients previously reported to represent thalidomide embryopathy. J. Med. Genet. 2003;40(7):473–478. doi: 10.1136/jmg.40.7.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Halal F., Homsy M., Perreault G. Acro-renal-ocular syndrome: autosomal dominant thumb hypoplasia, renal ectopia, and eye defect. Am. J. Med. Genet. 1984;17(4):753–762. doi: 10.1002/ajmg.1320170406. [DOI] [PubMed] [Google Scholar]
- 124.Kohlhase J., Heinrich M., Schubert L., Liebers M., Kispert A., Laccone F., Turnpenny P., Winter R.M., Reardon W. Okihiro syndrome is caused by SALL4 mutations. Hum. Mol. Genet. 2002;11(23):2979–2987. doi: 10.1093/hmg/11.23.2979. [DOI] [PubMed] [Google Scholar]
- 125.Al-Baradie R., Yamada K., St Hilaire C., Chan W.M., Andrews C., McIntosh N., Nakano M., Martonyi E.J., Raymond W.R., Okumura S., Okihiro M.M., Engle E.C. Duane radial ray syndrome (Okihiro syndrome) maps to 20q13 and results from mutations in SALL4, a new member of the SAL family. Am. J. Hum. Genet. 2002;71(5):1195–1199. doi: 10.1086/343821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Borozdin W., Wright M.J., Hennekam R.C., Hannibal M.C., Crow Y.J., Neumann T.E., Kohlhase J. Novel mutations in the gene SALL4 provide further evidence for acro-renal-ocular and Okihiro syndromes being allelic entities, and extend the phenotypic spectrum. J. Med. Genet. 2004;41(8):e102. doi: 10.1136/jmg.2004.019505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Borozdin W., Graham J.M., Jr, Böhm D., Bamshad M.J., Spranger S., Burke L., Leipoldt M., Kohlhase J. Multigene deletions on chromosome 20q13.13-q13.2 including SALL4 result in an expanded phenotype of Okihiro syndrome plus developmental delay. Hum. Mutat. 2007;28(8):830. doi: 10.1002/humu.9502. [DOI] [PubMed] [Google Scholar]
- 128.Borozdin W., Boehm D., Leipoldt M., Wilhelm C., Reardon W., Clayton-Smith J., Becker K., Mühlendyck H., Winter R., Giray O., Silan F., Kohlhase J. SALL4 deletions are a common cause of Okihiro and acro-renal-ocular syndromes and confirm haploinsufficiency as the pathogenic mechanism. J. Med. Genet. 2004;41(9):e113. doi: 10.1136/jmg.2004.019901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Miertus J., Borozdin W., Frecer V., Tonini G., Bertok S., Amoroso A., Miertus S., Kohlhase J. A SALL4 zinc finger missense mutation predicted to result in increased DNA binding affinity is associated with cranial midline defects and mild features of Okihiro syndrome. Hum. Genet. 2006;119(1-2):154–161. doi: 10.1007/s00439-005-0124-7. [DOI] [PubMed] [Google Scholar]
- 130.Kohlhase J., Chitayat D., Kotzot D., Ceylaner S., Froster U.G., Fuchs S., Montgomery T., Rösler B. SALL4 mutations in Okihiro syndrome (Duane-radial ray syndrome), acro-renal-ocular syndrome, and related disorders. Hum. Mutat. 2005;26(3):176–183. doi: 10.1002/humu.20215. [DOI] [PubMed] [Google Scholar]
- 131.Koshiba-Takeuchi K., Takeuchi J.K., Arruda E.P., Kathiriya I.S., Mo R., Hui C.C., Srivastava D., Bruneau B.G. Cooperative and antagonistic interactions between Sall4 and Tbx5 pattern the mouse limb and heart. Nat. Genet. 2006;38(2):175–183. doi: 10.1038/ng1707. [DOI] [PubMed] [Google Scholar]
- 132.Hall J.G., Levin J., Kuhn J.P., Ottenheimer E.J., van Berkum K.A., McKusick V.A. Thrombocytopenia with absent radius (TAR). Medicine (Baltimore) 1969;48(6):411–439. doi: 10.1097/00005792-196948060-00001. [DOI] [PubMed] [Google Scholar]
- 133.Martínez-Frías M.L., Bermejo Sánchez E., García García A., Pérez Fernández J.L., Cucalón Manzanos F., Calvo Aguilar M.J., Ripalda Crespo M.J. [An epidemiological study of the thrombocytopenia with radial aplasia syndrome (TAR) in Spain]. An. Esp. Pediatr. 1998;49(6):619–623. [PubMed] [Google Scholar]
- 134.Hedberg V.A., Lipton J.M. Thrombocytopenia with absent radii. A review of 100 cases. Am. J. Pediatr. Hematol. Oncol. 1988;10(1):51–64. doi: 10.1097/00043426-198821000-00010. [DOI] [PubMed] [Google Scholar]
- 135.Albers C.A., Paul D.S., Schulze H., Freson K., Stephens J.C., Smethurst P.A., Jolley J.D., Cvejic A., Kostadima M., Bertone P., Breuning M.H., Debili N., Deloukas P., Favier R., Fiedler J., Hobbs C.M., Huang N., Hurles M.E., Kiddle G., Krapels I., Nurden P., Ruivenkamp C.A., Sambrook J.G., Smith K., Stemple D.L., Strauss G., Thys C., van Geet C., Newbury-Ecob R., Ouwehand W.H., Ghevaert C. Compound inheritance of a low-frequency regulatory SNP and a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nat. Genet. 2012;44(4):435–439, S1-S2. doi: 10.1038/ng.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bottillo I., Castori M., De Bernardo C., Fabbri R., Grammatico B., Preziosi N., Scassellati G.S., Silvestri E., Spagnuolo A., Laino L., Grammatico P. Prenatal diagnosis and post-mortem examination in a fetus with thrombocytopenia-absent radius (TAR) syndrome due to compound heterozygosity for a 1q21.1 microdeletion and a RBM8A hypomorphic allele: a case report. BMC Res. Notes. 2013;22(6):376. doi: 10.1186/1756-0500-6-376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Greenhalgh K.L., Howell R.T., Bottani A., Ancliff P.J., Brunner H.G., Verschuuren-Bemelmans C.C., Vernon E., Brown K.W., Newbury-Ecob R.A. Thrombocytopenia-absent radius syndrome: a clinical genetic study. J. Med. Genet. 2002;39(12):876–881. doi: 10.1136/jmg.39.12.876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hall J.G. Thrombocytopenia and absent radius (TAR) syndrome. J. Med. Genet. 1987;24(2):79–83. doi: 10.1136/jmg.24.2.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kim V.N., Kataoka N., Dreyfuss G. Role of the nonsense-mediated decay factor hUpf3 in the splicing-dependent exon-exon junction complex. Science. 2001;293(5536):1832–1836. doi: 10.1126/science.1062829. [DOI] [PubMed] [Google Scholar]
- 140.Lykke-Andersen J., Shu M.D., Steitz J.A. Communication of the position of exon-exon junctions to the mRNA surveillance machinery by the protein RNPS1. Science. 2001;293(5536):1836–1839. doi: 10.1126/science.1062786. [DOI] [PubMed] [Google Scholar]
- 141.Palacios I.M., Gatfield D., St Johnston D., Izaurralde E. An eIF4AIII-containing complex required for mRNA localization and nonsense-mediated mRNA decay. Nature. 2004;427(6976):753–757. doi: 10.1038/nature02351. [DOI] [PubMed] [Google Scholar]
- 142.Albers C.A., Newbury-Ecob R., Ouwehand W.H., Ghevaert C. New insights into the genetic basis of TAR (thrombocytopenia-absent radii) syndrome. Curr. Opin. Genet. Dev. 2013;23(3):316–323. doi: 10.1016/j.gde.2013.02.015. [DOI] [PubMed] [Google Scholar]
- 143.Levy W.J. Mesoectodermal dysplasia. A new combination of anomalies. Am. J. Ophthalmol. 1967;63(5):978–982. doi: 10.1016/0002-9394(67)90043-8. [DOI] [PubMed] [Google Scholar]
- 144.Hollister D.W., Klein S.H., De Jager H.J., Lachman R.S., Rimoin D.L. The lacrimo-auriculo-dento-digital syndrome. J. Pediatr. 1973;83(3):438–444. doi: 10.1016/S0022-3476(73)80268-9. [DOI] [PubMed] [Google Scholar]
- 145.Bamforth J.S., Kaurah P. Lacrimo-auriculo-dento-digital syndrome: evidence for lower limb involvement and severe congenital renal anomalies. Am. J. Med. Genet. 1992;43(6):932–937. doi: 10.1002/ajmg.1320430605. [DOI] [PubMed] [Google Scholar]
- 146.Rohmann E., Brunner H.G., Kayserili H., Uyguner O., Nurnberg G., Lew E.D., Dobbie A., Eswarakumar V.P., Uzumcu A., Ulubil-Emeroglu M., Leroy J.G., Li Y. Mutations in different components of FGF signaling in LADD syndrome. 2006. [DOI] [PubMed]
- 147.Itoh N., Ohta H. Fgf10: a paracrine-signaling molecule in development, disease, and regenerative medicine. Curr. Mol. Med. 2014;14(4):504–509. doi: 10.2174/1566524014666140414204829. [DOI] [PubMed] [Google Scholar]
- 148.Eswarakumar V.P., Schlessinger J. Skeletal overgrowth is mediated by deficiency in a specific isoform of fibroblast growth factor receptor 3. Proc. Natl. Acad. Sci. USA. 2007;104(10):3937–3942. doi: 10.1073/pnas.0700012104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Nager F.R., de Reynier J.P. Das Gehoerorgan bei den angeborenen Kopfmissbildungen. Pract. Otorhinolaryng. 1948;10(Suppl. 2):1–128. [Google Scholar]
- 150.Slingenberg B. Misbildungen von Extremitaeten. Virchows Arch. Path. Anat. 1908;193:1–92. doi: 10.1007/BF01991532. [DOI] [Google Scholar]
- 151.Halonen K., Hukki J., Arte S., Hurmerinta K. Craniofacial structures and dental development in three patients with Nager syndrome. J. Craniofac. Surg. 2006;17(6):1180–1187. doi: 10.1097/01.scs.0000246494.08700.ab. [DOI] [PubMed] [Google Scholar]
- 152.Wieczorek D. Human facial dysostoses. Clin. Genet. 2013;83(6):499–510. doi: 10.1111/cge.12123. [DOI] [PubMed] [Google Scholar]
- 153.Czeschik J.C., Voigt C., Alanay Y., Albrecht B., Avci S., Fitzpatrick D., Goudie D.R., Hehr U., Hoogeboom A.J., Kayserili H., Simsek-Kiper P.O., Klein-Hitpass L., Kuechler A., López-González V., Martin M., Rahmann S., Schweiger B., Splitt M., Wollnik B., Lüdecke H.J., Zeschnigk M., Wieczorek D. Clinical and mutation data in 12 patients with the clinical diagnosis of Nager syndrome. Hum. Genet. 2013;132(8):885–898. doi: 10.1007/s00439-013-1295-2. [DOI] [PubMed] [Google Scholar]
- 154.Bernier F.P., Caluseriu O., Ng S., Schwartzentruber J., Buckingham K.J., Innes A.M., Jabs E.W., Innis J.W., Schuette J.L., Gorski J.L., Byers P.H., Andelfinger G., Siu V., Lauzon J., Fernandez B.A., McMillin M., Scott R.H., Racher H., Majewski J., Nickerson D.A., Shendure J., Bamshad M.J., Parboosingh J.S., FORGE Canada Consortium Haploinsufficiency of SF3B4, a component of the pre-mRNA spliceosomal complex, causes Nager syndrome. Am. J. Hum. Genet. 2012;90(5):925–933. doi: 10.1016/j.ajhg.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Petit F., Escande F., Jourdain A.S., Porchet N., Amiel J., Doray B., Delrue M.A., Flori E., Kim C.A., Marlin S., Robertson S.P., Manouvrier-Hanu S., Holder-Espinasse M. Nager syndrome: confirmation of SF3B4 haploinsufficiency as the major cause. Clin. Genet. 2014;86(3):246–251. doi: 10.1111/cge.12259. [DOI] [PubMed] [Google Scholar]
- 156.Castori M., Bottillo I., D’Angelantonio D., Morlino S., De Bernardo C., Scassellati Sforzolini G., Silvestri E., Grammatico P. A 22-Week-Old Fetus with Nager Syndrome and Congenital Diaphragmatic Hernia due to a Novel SF3B4 Mutation. Mol. Syndromol. 2014;5(5):241–244. doi: 10.1159/000365769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Townes P.L., Brocks E.R. Hereditary syndrome of imperforate anus with hand, foot, and ear anomalies. J. Pediatr. 1972;81(2):321–326. doi: 10.1016/S0022-3476(72)80302-0. [DOI] [PubMed] [Google Scholar]
- 158.Miller E.M., Hopkin R., Bao L., Ware S.M. Implications for genotype-phenotype predictions in Townes-Brocks syndrome: case report of a novel SALL1 deletion and review of the literature. Am. J. Med. Genet. A. 2012;158A(3):533–540. doi: 10.1002/ajmg.a.34426. [DOI] [PubMed] [Google Scholar]
- 159.Lawrence C., Hong-McAtee I., Hall B., Hartsfield J., Rutherford A., Bonilla T., Bay C. Endocrine abnormalities in Townes-Brocks syndrome. Am. J. Med. Genet. A. 2013;161A(9):2266–2273. doi: 10.1002/ajmg.a.36104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Martínez-Frías M.L., Bermejo Sánchez E., Arroyo Carrera I., Pérez Fernández J.L., Pardo Romero M., Burón Martínez E., Hernández Ramón F. [The Townes-Brocks syndrome in Spain: the epidemiological aspects in a consecutive series of cases]. An. Esp. Pediatr. 1999;50(1):57–60. [PubMed] [Google Scholar]
- 161.Kohlhase J., Wischermann A., Reichenbach H., Froster U., Engel W. Mutations in the SALL1 putative transcription factor gene cause Townes-Brocks syndrome. Nat. Genet. 1998;18(1):81–83. doi: 10.1038/ng0198-81. [DOI] [PubMed] [Google Scholar]
- 162.Botzenhart E.M., Bartalini G., Blair E., Brady A.F., Elmslie F., Chong K.L., Christy K., Torres-Martinez W., Danesino C., Deardorff M.A., Fryns J.P., Marlin S., Garcia-Minaur S., Hellenbroich Y., Hay B.N., Penttinen M., Shashi V., Terhal P., Van Maldergem L., Whiteford M.L., Zackai E., Kohlhase J. Townes-Brocks syndrome: twenty novel SALL1 mutations in sporadic and familial cases and refinement of the SALL1 hot spot region. Hum. Mutat. 2007;28(2):204–205. doi: 10.1002/humu.9476. [DOI] [PubMed] [Google Scholar]
- 163.Kohlhase J. SALL1 mutations in Townes-Brocks syndrome and related disorders. Hum. Mutat. 2000;16(6):460–466. doi: 10.1002/1098-1004(200012)16:6<460::AID-HUMU2>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 164.Kiefer S.M., Robbins L., Barina A., Zhang Z., Rauchman M. SALL1 truncated protein expression in Townes-Brocks syndrome leads to ectopic expression of downstream genes. Hum. Mutat. 2008;29(9):1133–1140. doi: 10.1002/humu.20759. [DOI] [PubMed] [Google Scholar]
- 165.Vodopiutz J., Zoller H., Fenwick A.L., Arnhold R., Schmid M., Prayer D., Müller T., Repa A., Pollak A., Aufricht C., Wilkie A.O., Janecke A.R. Homozygous SALL1 mutation causes a novel multiple congenital anomaly-mental retardation syndrome. J. Pediatr. 2013;162(3):612–617. doi: 10.1016/j.jpeds.2012.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Baller F. Radiusaplasie und Inzucht. Z. Menschl. Vererb. Konstitutionsl. 1950;29:782–790. [Google Scholar]
- 167.Gerold M. [Healing of a fracture in an unusual case of congenital anomaly of the upper extremities]. Zentralbl. Chir. 1959;84(21):831–834. [PubMed] [Google Scholar]
- 168.Galea P., Tolmie J.L. Normal growth and development in a child with Baller-Gerold syndrome (craniosynostosis and radial aplasia). J. Med. Genet. 1990;27(12):784–787. doi: 10.1136/jmg.27.12.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Debeljak M., Zver A., Jazbec J. A patient with Baller-Gerold syndrome and midline NK/T lymphoma. Am. J. Med. Genet. A. 2009;149A(4):755–759. doi: 10.1002/ajmg.a.32736. [DOI] [PubMed] [Google Scholar]
- 170.Van Maldergem L., Siitonen H.A., Jalkh N., Chouery E., De Roy M., Delague V., Muenke M., Jabs E.W., Cai J., Wang L.L., Plon S.E., Fourneau C., Kestilä M., Gillerot Y., Mégarbané A., Verloes A. Revisiting the craniosynostosis-radial ray hypoplasia association: Baller-Gerold syndrome caused by mutations in the RECQL4 gene. J. Med. Genet. 2006;43(2):148–152. doi: 10.1136/jmg.2005.031781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Bachrati C.Z., Hickson I.D. RecQ helicases: guardian angels of the DNA replication fork. Chromosoma. 2008;117(3):219–233. doi: 10.1007/s00412-007-0142-4. [DOI] [PubMed] [Google Scholar]
- 172.Kellermayer R. The versatile RECQL4. Genet. Med. 2006;8(4):213–216. doi: 10.1097/01.gim.0000214457.58378.1a. [DOI] [PubMed] [Google Scholar]
- 173.Wang L.L., Gannavarapu A., Kozinetz C.A., Levy M.L., Lewis R.A., Chintagumpala M.M., Ruiz-Maldanado R., Contreras-Ruiz J., Cunniff C., Erickson R.P., Lev D., Rogers M., Zackai E.H., Plon S.E. Association between osteosarcoma and deleterious mutations in the RECQL4 gene in Rothmund-Thomson syndrome. J. Natl. Cancer Inst. 2003;95(9):669–674. doi: 10.1093/jnci/95.9.669. [DOI] [PubMed] [Google Scholar]