

Abstract
Osteoarthritis (OA) is a prevalent disease of articular joints and primarily characterized by degradation and calcification of articular cartilage. Presently, no effective treatment other than pain relief exists and patients ultimately need to undergo replacement surgery of the affected joint. During disease progression articular chondrocytes, the single cell type present in articular cartilage, show altered transcriptional profiles and undergo phenotypic changes that resemble the terminal differentiation route apparent in growth plate chondrocytes. Hence, given its prominent function in both regulating gene expression and maintaining cellular phenotypes, DNA methylation of CpG dinucleotides is intensively studied in the context of OA. An increasing number of studies have been published that employed a targeted approach on genes known to play a role in OA pathophysiology. As of such, it has become clear that OA responsive DNA methylation changes seem to mediate disease associated aberrant gene expression. Furthermore, established OA susceptibility alleles such as GDF5 and DIO2 appear to confer OA risk via DNA methylation and respective pathophysiological expression changes. In more recent years, genome wide profiling of DNA methylation in OA affected articular cartilage has emerged as a powerful tool to address the epigenetic changes in their entirety, which has resulted in the identification of putative patient subgroups as well as generic OA associated pathways.
Keywords: Articular cartilage, Data integration, DNA methylation, Osteoarthritis, Transcriptomics.
INTRODUCTION
Osteoarthritis (OA) is currently the most prevalent arthritic disease among the elderly population [1]. Patients are subject to impaired mobility, joint stiffness, pain and a significant decrease in quality of life. Presently, no cure exists and patients with end-stage disease ultimately need to undergo a joint replacement surgery of the affected joint. Subsequently, as disease incidence is increasing with the ongoing ageing population, the societal burden both in terms of disabling patients and economic concerns will continue to rise [2]. Pathophysiologically, the disease is primarily characterized by progressive degradation and calcification of cartilage in the articular joints, although in recent years it has become apparent that other tissues such as subchondral bone [3-5] and synovium [6-8] play substantial roles in OA pathology as well. The articular cartilage contains a hyaline type extracellular matrix (ECM) made up of collagens, proteoglycans and other structural proteins [9]. Articular chondrocytes, the single cell type present in articular cartilage, reside here and maintain tissue homeostasis by remodelling the ECM upon stresses and microtraumas [10]. To ensure cartilage integrity and cope with the challenges throughout life, the maturational arrested articular chondrocytes need to continuously employ, possibly error prone, adaptions to changes in the environment [11]. In order to facilitate these adaptations, articular chondrocytes are required to dynamically adjust expression of the appropriate genes, while maintaining their specific cellular phenotype [11]. In this regard, articular chondrocytes present in OA affected cartilage however, show increased expression of catabolic enzymes [12-14] and have seemingly lost their maturational arrested state [15-17], as they proliferate and regain growth-plate morphology [17], while degrading and calcifying the ECM of the articular cartilage [18, 19].
A substantial number of mechanisms are known that regulate gene expression [20] and cell fate persistence [21-23], commonly referred to as epigenetics. While there exists a considerable number of epigenetic layers, such as histone modifications, microRNAs and long non-coding RNAs, the most studied in OA is decidedly DNA methylation. Partly due to its seemingly, relatively stable epigenetic mark on gene expression and partly due to the readily available techniques to measure it. DNA methylation is the phenomenon in which the cytosine nucleotide that is located in cytosine-guanine (CpG) residue pairs gets methylated. Whether CpGs get methylated depend on the local genetic sequence [24], the cellular requirements [21] and environmental factors such the putative pathophysiological state of the respective tissue [24, 25]. The presence of methyl groups on CpGs is believed to interfere with binding of proteins to the DNA and subsequently affects transcription [26], which is marked by the correlation between the fraction of CpG methylation and gene expression [27], commonly referred to as expression quantitative trait methylation (eQTM) [24, 27]. As of such, tissue specific methylation profiles are assumed to maintain the respective transcriptional character and identity of distinct cell types. Interestingly, disease associated single nucleotide polymorphisms (SNPs) that influence aberrant expression, so called expression quantitative trait loci (eQTL), frequently seem to modulate their transcriptomic properties via DNA methylation quantitative trait loci (mQTL) [28, 29].
DNA METHYLATION IN OSTEOARTHRITIS
The involvement of DNA methylation in OA pathophysiology is becoming increasingly evident, reflected by the growing body of literature on the subject [13, 28-40]. However, given the cross-sectional nature of the reports that studied DNA methylation in OA affected articular cartilage, it is currently unclear whether the observed epigenetic differences precede disease onset or are merely consequence of the environmental changes that articular chondrocytes are subject to in OA. As of such, we here discuss two hypotheses that have been proposed to attribute distinct roles of epigenetics in OA pathophysiology.
Firstly, it is hypothesized that individuals with unfavourable epigenetic profiles will be more prone to develop OA and/or progress faster [41, 42]. As the local genetic sequence significantly influences the DNA methylation state of CpGs, these putative profiles likely arise due to the presence of OA susceptibility SNPs [43]. This would be in concordance with the identification of susceptibility genes in complex diseases such as OA, which not uncommonly assert their susceptibility via altered DNA methylation, depending on the presence of associated risk alleles [28, 29]. For example, OA risk alleles of SNPs in DIO2 and GDF5 appear to generate OA predisposing epigenetic profiles and modulate disease associated, epigenetically associated gene expression. The OA risk allele C of the rs225014 T>C SNP, which has been identified in a combined genome wide linkage and association study by our own group [44], marked not only allele specific expression of DIO2 [45], it additionally mediated epigenetic regulation of the gene [29]. More specifically, carriers of the risk allele showed increased transcriptional activity upon hypermethylation at a CpG located in a distal regulatory element. The risk allele T of the rs143383 C>T SNP [46-48], located in the 5’ untranslated region of GDF5, disrupts a CpG dinucleotide, of which has been shown that it modulates absolute GDF5 expression in articular cartilage [28]. Furthermore, the reported allele specific expression of GDF5 in heterozygous carriers marked by the rs143383 alleles [49] is affected by the extent of methylation of the respective CpG. Although increasing additional evidence is reported about OA susceptibility alleles, the exact biological mechanisms that confer OA susceptibility is unclear. More specifically, it is unclear whether epigenetic regulation of aberrant gene expression brought about by genetic variation is involved in disease aetiology or disease progression. We can however, conclude that epigenetic regulation of gene expression modulates OA susceptibility, at least at the DIO2 and GDF5 loci.
Alternatively, it has been suggested that age-related loss of epigenetic control [50] mediates the loss of the articular chondrocyte’s phenotype with ongoing OA, as life-long stresses and adaptations are expected to leave their mark on the epigenome [11]. Furthermore, given the altered cellular phenotype chondrocytes acquire in OA and given the role DNA methylation fulfils in maintaining cellular phenotypes, we advocate here that loss of epigenetic control causes reactivation of developmental pathways among articular chondrocytes that are subject to OA and subsequently mediate the morphological changes that are associated with affected articular chondrocytes.
EPIGENETIC CHANGES ACCOMPANY OSTEOARTHRITIS
A growing body of literature reports on altered DNA methylation at specific genes involved in the OA disease process, commonly at precise CpG sites located in regulatory elements near the respective genes (Table 1). Frequently, a cross-sectional study design is utilized to study OA associated methylation differences, either between healthy and affected or pairwise between preserved and macroscopically lesioned articular cartilage. Among others, catabolic and developmentally associated genes like MMP13, GDF5, SOX9, DIO2 and ADAMTS4 were shown to be differentially expressed between control and affected tissue, presumably mediated by differences in DNA methylation [13, 28-34]. Although it is not quite clear to what extent DNA methylation changes in OA development contribute to disease onset or progression, a growing number of studies investigates the mechanism by which such changes may affect chondrocyte function.
Table 1.
Overview of gene targeted DNA methylation studies in osteoarthritic articular cartilage.
| Gene | Methylation in OA | Expression in OA | Sample Size (OA, Control) | Joint | AZA | CpG Vector | Reference |
|---|---|---|---|---|---|---|---|
| COL9 | Increased | Down | 12, 10 | Hip | yes | Yes | [31] |
| GDF5 | Decreased | Up | 24, 19 | Knee and hip | yes | Yes | [28] |
| DIO2 | Increased | Up | 52, 52 | Knee and hip | yes | No | [29] |
| IL1β | Decreased | Up | 18, 12 | Hip | yes | Yes | [30] |
| MMP13 | Decreased | Up | 17, 12 | Hip | yes | Yes | [30] |
| iNOS | Decreased | Up | 13, 15 | Hip | no | Yes | [34] |
| SOX9 | Increased | Down | 9, 9 | Hip | yes | No | [32] |
| ADAMTS4 | Decreased | Up | 4, 1 | Hip | no | No | [33] |
| ADAMTS4 | Decreased | Up | 16, 10 | Hip | no | No | [13] |
| MMP13 | Decreased | Up | 16, 10 | Hip | no | No | [13] |
| MMP3 | Decreased | Up | 16, 10 | Hip | no | No | [13] |
| MMP9 | Decreased | Up | 16, 10 | Hip | no | No | [13] |
Consequently, various experimental set ups have been applied to confirm the mechanistic relation between DNA methylation and gene expression in the chondrocyte (Table 1). A commonly applied experiment that aims to validate observed eQTM loci, as has our own group, is the addition of 5-aza-2’-deoxycytidine (AZA) to the medium of cultured chondrocytes, either using immortalized cell lines or primary chondrocyte cultures [28-30, 32, 35]. AZA is a chemical agent that interferes with the addition of methyl groups on a genome wide scale to newly formed DNA strands during replication. Although valuable information can be gained from such experiments in a global sense, negative outcomes should not per se be considered as experimental falsifications of the earlier observed correlation between DNA methylation and expression, which is likely cell type and locus specific. As of such, not being able to validate earlier observed eQTM loci can very well arise from the fact that the entire genome gets demethylated upon prolonged AZA treatment, of which the transcriptomic consequences potentially overshadow the locus specific relation. Additionally, culturing cells in-vitro forces the cells to adapt to an artificial environment, which possibly only resembles the original tissues in a broad sense and subsequently disrupts the regulatory properties of cell type specific eQTM loci.
Another type of validation experiment being applied is cloning the genomic sequence, in which differential methylation was observed, into a CpG-free vector [51] containing a luciferase gene downstream of the multiple cloning site [30, 31, 34, 52]. Next, the vector is methylated in-vitro only at the cloned region and transfected into chondrocyte like cell-lines. Luciferase activity now depends on the transcriptional activity of the cloned region, which in turn depends on the methylated state of that respective region. Although using a CpG-free vector has great advantages over AZA treatment, as it does not induce genome-wide altered methylation, the technique will potentially only work for proximal promotors, as long-distance three dimensional genomic structures, which are often seen in gene expression regulation [53-55], depend on the complex interplay of the distant and proximal regulatory elements of genes. Again, as cell culturing is required to apply the technique, measuring promoter activity using a CpG free vector might be influenced by cellular adaptations to the artificial environment.
STRATIFICATION OF OSTEOARTHRITIS PATIENTS BY GENOME WIDE DNA METHYLATION PROFILING
In more recent years, multiple studies have reported on genome wide DNA methylation profiles of articular cartilage in the context of OA (Table 2), not least due to development of affordable genome wide DNA methylation arrays, such as the Illumina Infinium HumanMethylation450k BeadChip array. Genome wide profiling of DNA methylation in OA affected cartilage has revealed the presence of multiple OA patient strata, which are characterized by their respective methylation profiles. Firstly, Rushton et al. [37] and our group [38] have reported on the distinct genome wide DNA methylation profiles of knee and hip articular cartilage. Secondly, Rushton et al. [37] and Fernandez-Tajes et al. [36] have reported on a subgroup of patients defined by altered DNA methylation at inflammatory related genes.
Table 2.
Overview of current genome wide DNA methylation experiments in osteoarthritic articular cartilage.
| Sample Size (OA, Control) | Joint | Platform | OA Associated Pathways | Reference |
|---|---|---|---|---|
| 20, 25 | Knee | Illumina 27K | Inflammation, transcriptional regulation, ECM homeostasis | [36] |
| 21, 96 | Knee and hip | Illumina 450k | ECM homeostasis, ossification, inflammation, angiogenesis | [37] |
| 24, 24 | Hip | Illumina 450k | Development, angiogenesis, inflammation | [39] |
| 16, 16 | Knee | Agilent 244k | Development, catalytic activity | [40] |
| 31, 31 | Hip and knee | Illumina 450k | [38] | |
| 31, 31 | Hip and knee | Illumina 450k | Development, ECM homeostasis | [56] |
With respect to the first bifurcation of OA patients, the two types of joint specific articular cartilage are distinguished by large differentially methylated regions (DMRs), primarily at genes involved in development and cellular differentiation. Notably, over 10% of DMRs were observed in the four canonical homeobox (HOX) clusters while the spatiotemporal pattern present at the HOX clusters during development was absent, indicating that joint specific DMRs likely bear distinct functionality in adult articular cartilage [38]. Both studies that compared knee and hip articular cartilage reported on joint specific methylation patterns at the four HOX clusters and several homeotic cofactors, such as IRX3, SIX1, MEIS2, and DLX5. By analysis of transcriptomic data our group subsequently revealed that joint specific DMRs potentially mediate distinctive regenerative capacities of articular chondrocytes residing in different joints, given the developmental and regenerative nature of joint specific expression of genes located in the developmentally associated DMRs [38]. Although reports on joint specific methylation profiles might not reveal loci that are directly relevant for OA onset or progression, they undeniably expose the heterogeneity of the disease. These joint specific DNA methylation and associated transcriptomic profiles are concurrent with the reports of joint specific genetic OA susceptibility loci [57]. Henceforth, we can conclude that although a substantial amount of common features are present between knee and hip OA, the disease process and subsequent mode of action of putative therapeutic interventions might be different between the two joints.
With respect to the second, another dichotomy is observed by cluster analysis of genome wide DNA methylation profiles of osteoarthritic articular cartilage [36, 37]. More specifically, putative patient subgroups have been defined by DNA methylation profiles of the affected cartilage that are enriched for differentially methylated CpGs located in or near genes involved in inflammation. The study of Fernandez-Tajes et al. used a sparse methylation array and a relatively small sample size consisting of only knee OA patients, which possibly explains why Rushton et al. observed substantially more differentially methylated CpGs (1,357 vs. 5,769) that separated the inflammatory cluster of patients in both knee and hip articular cartilage. While enrichment analysis of both studies revealed broadly similar enriched GO terms, being the inflammatory response and cytokine production, in the study of Fernandez-Tajes et al. this was only apparent among the hypomethylated CpGs and not among hypermethylated CpGs. Among the consistent inflammatory signals identified by the two studies among knee OA patients were multiple interleukin genes such as IL10, IL16 and IL19, but also developmental and ECM related genes such as RUNX2, FGFR1, COL6A3 and COL18A1. Noteworthy, in the study of Rushton et al. stratification by the inflammatory profile, using both hypo- and hypermethylated CpGs, of both knee and hip OA patients is observed, presumably mediated by overlapping pathways, albeit that only 3,496 out of 15,239 (23%) differentially methylated CpGs that separated OA hip patients overlapped with differentially methylated CpGs that separated OA knee patients [37]. Very recently, the group of Rushton et al. has further investigated the hypomethylated genes that are apparent among the inflammatory profiles in their hip OA patient cohort and report on specific zinc reporter genes that potentially mediate the patient stratification [58].
Intriguingly, when we performed GO term analysis on the genes that separated hip and knee OA patients consistently in the initial study of Rushton et al. and Fernandez-Tajes et al., we observed significant enrichment for ECM maintenance pathways (data not shown). Thus, while one of the reported clusters of OA patients is presumably characterized by an epigenetic inflammatory profile, a common underlying mechanisms appears defined by epigenetic regulation of ECM related genes, such as COL6A3, RUNX2, MMP13 and ADAMTS5. This proposition is additionally reflected by the fact that all studies report on the enrichment of ECM related pathways (Table 2) and by the analysis Fernandez-Tajes et al. performed on solely the hypermethylated CpGs, in which they also observe enrichment among ECM maintenance pathways. Finally, the study of Fernandez-Tajes et al. grossly compared methylation and additionally expression profiles between the OA subgroups. However, it is evident that the transcriptional consequences of these profiles need to be precisely elucidated, as modulating unfavourable epigenetic but subsequent transcriptomic profiles specifically can potentially attenuate disease onset or progression and might therefor serve as valuable therapeutic targets for the putative subtypes of OA. It should, however, be noted that of the five published genome wide DNA methylation studies in OA, only Fernandez-Tajes et al. and Rushton et al. have observed separate clustering of OA patients, while Jeffries et al. [39], Moazedi-Fuerst et al. [40] and our group [56] have not, warranting further research hereinto. Furthermore, not only discovery driven epigenomic profiling of articular cartilage is required to understand possible OA patient heterogeneity. In light of proposed OA subtypes in the literature, epigenetic interrogation of specific pathways by means of burden analyses in pathways such as those in estrogenic sensitivity [59] or apoptosis [60], might be a more powerful approach in the context of the large number of differentially methylated loci in OA reported by increasingly larger studies. In parallel, genome wide DNA methylation profiles of other joint tissues might additionally confer the proposed different OA subtypes.
CONSISTENT DNA METHYLATION DIFFERENCES BETWEEN OSTEOARTHRITIS AFFECTED AND CONTROL ARTICULAR CARTILAGE
Despite the putative segregation of distinct methylation profiles among OA affected joints [37, 38] and OA patients [36, 37], major communalities have been observed across the different genome wide studies (Table 2). Specifically, consistent enrichment of differential methylation among genes involved in development, as well as in collagen synthesis and other ECM maintenance pathways is reported by all studies, particularly among genes from the RUNX, COL and MMP families. Developmental processes thus appear entangled with OA associated degradation and calcification of articular cartilage during disease progression. Although our group has shown that the majority of differentially methylated CpGs do not associate in-cis with gene expression, enrichment analysis of OA responsive CpGs that did correlate with gene expression (87 CpGs, 70 genes) also revealed enrichment for ECM maintenance and developmental pathways, consisting of genes such as ROR2, WLS, VIT and SPP1 [56]. Nonetheless, genome wide DNA methylation profiles and the inherent differences between OA and control tissue need thus be interpreted with cause and preferably coupled with additional molecular measurements, such as gene expression data, to clarify the biological consequence. The 76% of genes of which expression did not correlate with in-cis DNA methylation could be regulated additionally by other epigenetic mechanisms, such as histon modifications or miRNA mediated silencing or alternatively, might possibly reflect remnants of early developmental or past wound healing processes.
In parallel, as reflected by OA risk alleles that affect DNA methylation mediated gene expression [28, 29] and the influential role of the genomic sequence on DNA methylation, partly reflected by the 40% of OA associated CpGs that are affected by the alleles of proximal SNPs [56], it is not unlikely that the total combination of minor genetic variants among OA patient contributes to putative unfavourable epigenomic and subsequent transcriptomic profiles. In this respect, the putative OA inflammatory subtype and the tissue specific mQTLs reported by our own group, could be the consequence of an inherent unfavourable epigenomic profile preceding disease onset. In line with the observed lack of heritability explained by traditional GWA approaches, these putative minor genetic variants might make up for the larger part of heritability in OA.
CONCLUSION
Genome wide DNA methylation profiling of OA affected articular cartilage has revealed widespread differences between OA and control tissues. Although the majority of CpGs do not associate with gene expression, CpGs of which we can consequently not conclude whether they play an active role in disease progression, reactivation of developmental pathways due to changes in epigenetic landscape is apparent in OA affected joints. Thus, we hypothesize here that to maintain healthy articular cartilage homeostasis throughout life, the respective chondrocytes utilize epigenetic mechanisms to transcriptomically adapt to the changing environment. Furthermore, the fact that gene expression changes occur via both hyper- and hypomethylation in OA affected articular cartilage, indicates that disease associated differential methylation is unlikely to be the product of a solely passive process. Lifelong adjustment of regulatory mechanisms, such as DNA methylation, is likely subject to stochastic error and subsequent accumulation of epigenetic modifications at developmental pathways, either via inaccurate restoration of the chondrocytes’ steady state or via an increasing number of epigenetic adaptations, seem to force chondrocytes towards terminal differentiation (Fig. 1). This proposition is additionally reflected by the morphological changes that occur in articular cartilage with ongoing OA, as reflected by degradation and calcification, mechanisms also observed among growth plate chondrocytes during development.
Fig. (1).
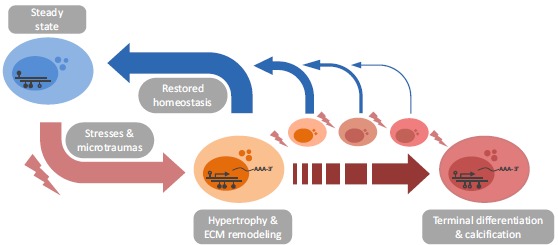
Schematic overview of the proposed loss of epigenetic control among articular chondrocytes. Articular cartilage is subject to lifelong challenges which requires the respective chondrocytes to dynamically adapt gene expression in order to return to and maintain homeostasis and subsequent tissue integrity. This is seemingly accomplished by using epigenetic modifications, reflected by OA associated DNA methylation differences, which are likely not reversed flawlessly upon returning to a steady state. We propose that accumulation of these epigenetic adaptations will eventually lead to altered cellular phenotypes which are unable to return to a healthy, steady state chondrocyte. In parallel, articular chondrocytes of OA patients might already bear an unfavorable epigenomic profile preceding disease onset, which implies that these chondrocytes are more prone to enter the active ECM remodeling state and/or might be less able to return to a steady state. Finally, independent of OA associated epigenetic changes in articular chondrocytes, joint and patient specific epigenomic profiles seem to modulate our proposed mechanism.
In conclusion, studying genome wide DNA methylation in OA has proven to serve as an excellent proxy to probe the underlying pathological cellular processes in OA. In light of the inherent genetically and consequent epigenetically complex nature of OA pathology, the genome wide efforts of recent years that have reported on the DNA methylation landscape of OA affected articular cartilage have delivered valuable insight, insight that would not have become apparent by mere gene targeted approaches.
FUTURE PROSPECTS OF EPIGENETICS IN OSTEOARTHRITIS
Given that differences in the methylome of OA affected compared to healthy or preserved articular cartilage do not imply downstream effects, reflected by the relatively small number of in-cis eQTM loci reported by our group, stresses the need for integration of DNA methylation data with other types of molecular profiling. It is of great interest to the field that the disease relevant, tissue specific epigenomic and transcriptomic QTL profiles are characterised, as they might reveal the predisposing, complex genetic architecture that underlies OA susceptibility. Our own group has undertaken the first step herein and has reported on the effects of SNPs on OA associated DNA methylation and transcriptional differences, as well as on the direct relation between methylation and expression on a genome wide scale [56]. Moreover, the reported genetic variants will potentiate the power of GWA studies, as the multiple testing penalty is substantially reduced when only the functional SNPs, in terms of regulation of transcription, are addressed as opposed to the entire genome. Likely a larger number of SNPs are of relevance, as we have not addressed long distance effects and larger sample sizes are possibly required to gain additional, robust understanding of the reported differences and associations, a point also raised by others [61]. In order to test whether indeed loss of epigenetic control confers the pathophysiological changes are apparent with OA progression, longitudinal studies that address the contribution of ageing in altered DNA methylation are required, preferably specifically at genes of which transcriptional changes potentially modulate the disease process. Confirmation of this hypothesis might potentially reveal the genetic drivers of OA.
In light of the systemic, but not unidirectional differences in DNA methylation on a genome wide scale that are apparent with OA progression in affected cartilage, it seems that systemically targeting DNA methylation for clinical purposes in OA is farfetched. Moreover, the widespread epigenetic differences that accompany OA suggestively affect expression of genes primarily involved in developmental processes, such as endochondral ossification. Aside from the substantial scientific challenge to locus specifically modulate DNA methylation, it seems more pragmatic to directly address the mRNA or protein molecules of the respective genes and pathways. While this is a difficult task on its own, specifically and locus specifically targeting DNA methylation in our view will unlikely serve a clinical purpose for OA in the near future.
However, aside from the putative limited role of epigenetics in curing OA, it may serve as an important biomarker when measured in clinically available tissues. As has also been shown for a number of molecular markers in blood or serum [62-64], it is apparent that transcriptomic data can purposely be used to identify symptomatic OA patients using mRNA extracted from peripheral blood mononuclear cells [60]. For example, the blood transcriptomic profiles of OA patients are enriched for genes involved in apoptosis, which were subsequently shown to reflect the pathophysiological state of the articular cartilage [60]. Hence, in light of the responsiveness of the epigenome to environmental changes and its relation with gene expression it is expected epigenetics can fulfil a similar purpose. Moreover, hereby not only clinical associations can be constructed, but also important insight is given into the complex disease process. Preliminary results from our own group indicate that indeed DNA methylation might serve as a powerful biomarker for OA progression. More specifically, as little as four CpGs were needed to distinguish fast progressing from non-progressing OA patients with 76% accuracy [65].
It should be noted that we have here primarily discussed DNA methylation in articular cartilage in the context of OA, not in the last place due to the fact that degradation of articular cartilage is the primary feature of OA. However, it is clear that other joint tissues are also involved in the disease process as a whole. Other disease relevant tissues, such as the synovium [66-68] and to some extent the subchondral bone [69], have been addressed in light of other muscoskeletal or rheumatic pathologies and indicate compelling prospects for OA research. In this regard, preliminary data from a small study revealed DNA methylation differences between subchondral bone adjacent to varying degrees of damaged articular cartilage of knee OA patients, indicating that epigenetic regulation is likely involved in the pathophysiological interplay between subchondral bone and articular cartilage [69].
Considering the proposed subgroups of OA patients, the initial genome wide DNA methylation studies discussed here have reported on putative stratification of OA patients based on inflammatory profiles present in articular cartilage only. However, given the increasingly important role of secondary tissues in OA, it is possible that subgroups of patients can be defined by genome wide DNA methylation profiling of those respective tissues. Consequently, the OA research field might elucidate other proposed OA patient subgroups, such as distinct differential estrogenic responses advocated by Herrero-Beaumont et al. [59].
ACKNOWLEDGEMENTS
Declared none.
LIST OF ABBREVIATIONS
- OA
= Osteoarthritis
- ECM
= Extracellular matrix
- CpG
= Cytosine-guanine dinucleotide
- SNP
= Single nucleotide polymorphism
- eQTM
= Expression quantitative trait methylation
- eQTL
= Expression quantitative trait locus
- mQTL
= Methylation quantitative trait locus
- AZA
= 5-aza-2’-deoxycytidine
- DMR
= Differentially methylated region
- HOX
= Homeobox
CONFLICT OF INTEREST
The author(s) confirm that this article content has no conflict of interest.
REFERENCES
- 1.Bijlsma J.W., Berenbaum F., Lafeber F.P. Osteoarthritis: an update with relevance for clinical practice. Lancet. 2011;377(9783):2115–2126. doi: 10.1016/S0140-6736(11)60243-2. [DOI] [PubMed] [Google Scholar]
- 2.Woolf A.D., Erwin J., March L. The need to address the burden of musculoskeletal conditions. Best Pract. Res. Clin. Rheumatol. 2012;26(2):183–224. doi: 10.1016/j.berh.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 3.Thysen S., Luyten F.P., Lories R.J. Loss of Frzb and Sfrp1 differentially affects joint homeostasis in instability-induced osteoarthritis. Osteoarthritis Cartilage. 2015;23(2):275–279. doi: 10.1016/j.joca.2014.10.010. [DOI] [PubMed] [Google Scholar]
- 4.Cui Z., Xu C., Li X., Song J., Yu B. Treatment with recombinant lubricin attenuates osteoarthritis by positive feedback loop between articular cartilage and subchondral bone in ovariectomized rats. Bone. 2015;74:37–47. doi: 10.1016/j.bone.2014.12.065. [DOI] [PubMed] [Google Scholar]
- 5.Binks D.A., Gravallese E.M., Bergin D., Hodgson R.J., Tan A.L., Matzelle M.M., McGonagle D., Radjenovic A. Role of vascular channels as a novel mechanism for subchondral bone damage at cruciate ligament entheses in osteoarthritis and inflammatory arthritis. Ann. Rheum. Dis. 2015;74(1):196–203. doi: 10.1136/annrheumdis-2013-203972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Lent P.L., Blom A.B., Schelbergen R.F., Slöetjes A., Lafeber F.P., Lems W.F., Cats H., Vogl T., Roth J., van den Berg W.B. Active involvement of alarmins S100A8 and S100A9 in the regulation of synovial activation and joint destruction during mouse and human osteoarthritis. Arthritis Rheum. 2012;64(5):1466–1476. doi: 10.1002/art.34315. [DOI] [PubMed] [Google Scholar]
- 7.Lories R.J., Corr M., Lane N.E. To Wnt or not to Wnt: the bone and joint health dilemma. Nat. Rev. Rheumatol. 2013;9(6):328–339. doi: 10.1038/nrrheum.2013.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ford C.E., Qian Ma S.S., Quadir A., Ward R.L. The dual role of the novel Wnt receptor tyrosine kinase, ROR2, in human carcinogenesis. Int. J. Cancer. 2013;133(4):779–787. doi: 10.1002/ijc.27984. [DOI] [PubMed] [Google Scholar]
- 9.Hosseini S.M., Wu Y., Ito K., van Donkelaar C.C. The importance of superficial collagen fibrils for the function of articular cartilage. Biomech. Model. Mechanobiol. 2014;13(1):41–51. doi: 10.1007/s10237-013-0485-0. [DOI] [PubMed] [Google Scholar]
- 10.Lotz M., Loeser R.F. Effects of aging on articular cartilage homeostasis. Bone. 2012;51(2):241–248. doi: 10.1016/j.bone.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goldring M.B., Marcu K.B. Epigenomic and microRNA-mediated regulation in cartilage development, homeostasis, and osteoarthritis. Trends Mol. Med. 2012;18(2):109–118. doi: 10.1016/j.molmed.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Struglics A., Larsson S., Pratta M.A., Kumar S., Lark M.W., Lohmander L.S. Human osteoarthritis synovial fluid and joint cartilage contain both aggrecanase- and matrix metalloproteinase-generated aggrecan fragments. Osteoarthritis Cartilage. 2006;14(2):101–113. doi: 10.1016/j.joca.2005.07.018. [DOI] [PubMed] [Google Scholar]
- 13.Roach H.I., Yamada N., Cheung K.S., Tilley S., Clarke N.M., Oreffo R.O., Kokubun S., Bronner F. Association between the abnormal expression of matrix-degrading enzymes by human osteoarthritic chondrocytes and demethylation of specific CpG sites in the promoter regions. Arthritis Rheum. 2005;52(10):3110–3124. doi: 10.1002/art.21300. [DOI] [PubMed] [Google Scholar]
- 14.Hashimoto K., Otero M., Imagawa K., de Andrés M.C., Coico J.M., Roach H.I., Oreffo R.O., Marcu K.B., Goldring M.B. Regulated transcription of human matrix metalloproteinase 13 (MMP13) and interleukin-1β (IL1B) genes in chondrocytes depends on methylation of specific proximal promoter CpG sites. J. Biol. Chem. 2013;288(14):10061–10072. doi: 10.1074/jbc.M112.421156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aigner T., Dudhia J. Phenotypic modulation of chondrocytes as a potential therapeutic target in osteoarthritis: a hypothesis. Ann. Rheum. Dis. 1997;56(5):287–291. doi: 10.1136/ard.56.5.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Facchini A., Borzì R.M., Olivotto E., Platano D., Pagani S., Cetrullo S., Flamigni F. Role of polyamines in hypertrophy and terminal differentiation of osteoarthritic chondrocytes. Amino Acids. 2012;42(2-3):667–678. doi: 10.1007/s00726-011-1041-9. [DOI] [PubMed] [Google Scholar]
- 17.Sun M.M., Beier F. Chondrocyte hypertrophy in skeletal development, growth, and disease. Birth Defects Res. C Embryo Today. 2014;102(1):74–82. doi: 10.1002/bdrc.21062. [DOI] [PubMed] [Google Scholar]
- 18.Bantsimba-Malanda C., Cottet J., Netter P., Dumas D., Mainard D., Magdalou J., Vincourt J.B. Chondrocalcin is internalized by chondrocytes and triggers cartilage destruction via an interleukin-1β-dependent pathway. Matrix Biol. 2013;32(7-8):443–451. doi: 10.1016/j.matbio.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 19.van C.A., Blaney D.E., Garcia D.V, van G.E., van den Berg, W., Goumans M.J., Ten D.P., van der Kraan, P. The high affinity ALK1-ligand BMP9 induces a hypertrophy-like state in chondrocytes that is antagonized by TGFbeta1. Osteoarthritis Cartilage. 2015;23(6):985–995. doi: 10.1016/j.joca.2015.02.007. [DOI] [PubMed] [Google Scholar]
- 20.Jaenisch R., Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet. 2003;33(Suppl.):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 21.Slieker R.C., Bos S.D., Goeman J.J., Bovée J.V., Talens R.P., van der Breggen R., Suchiman H.E., Lameijer E.W., Putter H., van den Akker E.B., Zhang Y., Jukema J.W., Slagboom P.E., Meulenbelt I., Heijmans B.T. Identification and systematic annotation of tissue-specific differentially methylated regions using the Illumina 450k array. Epigenetics Chromatin. 2013;6(1):26. doi: 10.1186/1756-8935-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laurent L., Wong E., Li G., Huynh T., Tsirigos A., Ong C.T., Low H.M., Kin Sung K.W., Rigoutsos I., Loring J., Wei C.L. Dynamic changes in the human methylome during differentiation. Genome Res. 2010;20(3):320–331. doi: 10.1101/gr.101907.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lister R., Pelizzola M., Dowen R.H., Hawkins R.D., Hon G., Tonti-Filippini J., Nery J.R., Lee L., Ye Z., Ngo Q.M., Edsall L., Antosiewicz-Bourget J., Stewart R., Ruotti V., Millar A.H., Thomson J.A., Ren B., Ecker J.R. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462(7271):315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gutierrez-Arcelus M., Lappalainen T., Montgomery S.B., Buil A., Ongen H., Yurovsky A., Bryois J., Giger T., Romano L., Planchon A., Falconnet E., Bielser D., Gagnebin M., Padioleau I., Borel C., Letourneau A., Makrythanasis P., Guipponi M., Gehrig C., Antonarakis S.E., Dermitzakis E.T. Passive and active DNA methylation and the interplay with genetic variation in gene regulation. eLife. 2013;2:e00523. doi: 10.7554/eLife.00523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramos Y.F., den Hollander W., Bovée J.V., Bomer N., van der Breggen R., Lakenberg N., Keurentjes J.C., Goeman J.J., Slagboom P.E., Nelissen R.G., Bos S.D., Meulenbelt I. Genes involved in the osteoarthritis process identified through genome wide expression analysis in articular cartilage; the RAAK study. PLoS One. 2014;9(7):e103056. doi: 10.1371/journal.pone.0103056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang H., Maurano M.T., Qu H., Varley K.E., Gertz J., Pauli F., Lee K., Canfield T., Weaver M., Sandstrom R., Thurman R.E., Kaul R., Myers R.M., Stamatoyannopoulos J.A. Widespread plasticity in CTCF occupancy linked to DNA methylation. Genome Res. 2012;22(9):1680–1688. doi: 10.1101/gr.136101.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wagner J.R., Busche S., Ge B., Kwan T., Pastinen T., Blanchette M. The relationship between DNA methylation, genetic and expression inter-individual variation in untransformed human fibroblasts. Genome Biol. 2014;15(2):R37. doi: 10.1186/gb-2014-15-2-r37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reynard L.N., Bui C., Canty-Laird E.G., Young D.A., Loughlin J. Expression of the osteoarthritis-associated gene GDF5 is modulated epigenetically by DNA methylation. Hum. Mol. Genet. 2011;20(17):3450–3460. doi: 10.1093/hmg/ddr253. [DOI] [PubMed] [Google Scholar]
- 29.Bomer N., den Hollander W., Ramos Y.F., Bos S.D., van der Breggen R., Lakenberg N., Pepers B.A., van Eeden A.E., Darvishan A., Tobi E.W., Duijnisveld B.J., van den Akker E.B., Heijmans B.T., van Roon-Mom W.M., Verbeek F.J., van Osch G.J., Nelissen R.G., Slagboom P.E., Meulenbelt I. Underlying molecular mechanisms of DIO2 susceptibility in symptomatic osteoarthritis. Ann. Rheum. Dis. 2015;74(8):1571–1579. doi: 10.1136/annrheumdis-2013-204739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hashimoto K., Otero M., Imagawa K., de Andrés M.C., Coico J.M., Roach H.I., Oreffo R.O., Marcu K.B., Goldring M.B. Regulated transcription of human matrix metalloproteinase 13 (MMP13) and interleukin-1β (IL1B) genes in chondrocytes depends on methylation of specific proximal promoter CpG sites. J. Biol. Chem. 2013;288(14):10061–10072. doi: 10.1074/jbc.M112.421156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Imagawa K., de Andrés M.C., Hashimoto K., Itoi E., Otero M., Roach H.I., Goldring M.B., Oreffo R.O. Association of reduced type IX collagen gene expression in human osteoarthritic chondrocytes with epigenetic silencing by DNA hypermethylation. Arthritis Rheumatol. 2014;66(11):3040–3051. doi: 10.1002/art.38774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim K.I., Park Y.S., Im G.I. Changes in the epigenetic status of the SOX-9 promoter in human osteoarthritic cartilage. J. Bone Miner. Res. 2013;28(5):1050–1060. doi: 10.1002/jbmr.1843. [DOI] [PubMed] [Google Scholar]
- 33.Cheung K.S., Hashimoto K., Yamada N., Roach H.I. Expression of ADAMTS-4 by chondrocytes in the surface zone of human osteoarthritic cartilage is regulated by epigenetic DNA de-methylation. Rheumatol. Int. 2009;29(5):525–534. doi: 10.1007/s00296-008-0744-z. [DOI] [PubMed] [Google Scholar]
- 34.de Andrés M.C., Imagawa K., Hashimoto K., Gonzalez A., Roach H.I., Goldring M.B., Oreffo R.O. Loss of methylation in CpG sites in the NF-κB enhancer elements of inducible nitric oxide synthase is responsible for gene induction in human articular chondrocytes. Arthritis Rheum. 2013;65(3):732–742. doi: 10.1002/art.37806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Loeser R.F., Im H.J., Richardson B., Lu Q., Chubinskaya S. Methylation of the OP-1 promoter: potential role in the age-related decline in OP-1 expression in cartilage. Osteoarthritis Cartilage. 2009;17(4):513–517. doi: 10.1016/j.joca.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fernández-Tajes J., Soto-Hermida A., Vázquez-Mosquera M.E., Cortés-Pereira E., Mosquera A., Fernández-Moreno M., Oreiro N., Fernández-López C., Fernández J.L., Rego-Pérez I., Blanco F.J. Genome-wide DNA methylation analysis of articular chondrocytes reveals a cluster of osteoarthritic patients. Ann. Rheum. Dis. 2014;73(4):668–677. doi: 10.1136/annrheumdis-2012-202783. [DOI] [PubMed] [Google Scholar]
- 37.Rushton M.D., Reynard L.N., Barter M.J., Refaie R., Rankin K.S., Young D.A., Loughlin J. Characterization of the cartilage DNA methylome in knee and hip osteoarthritis. Arthritis Rheumatol. 2014;66(9):2450–2460. doi: 10.1002/art.38713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.den Hollander W., Ramos Y.F., Bos S.D., Bomer N., van der Breggen R., Lakenberg N., de Dijcker W.J., Duijnisveld B.J., Slagboom P.E., Nelissen R.G., Meulenbelt I. Knee and hip articular cartilage have distinct epigenomic landscapes: implications for future cartilage regeneration approaches. Ann. Rheum. Dis. 2014;73(12):2208–2212. doi: 10.1136/annrheumdis-2014-205980. [DOI] [PubMed] [Google Scholar]
- 39.Jeffries M.A., Donica M., Baker L.W., Stevenson M.E., Annan A.C., Humphrey M.B., James J.A., Sawalha A.H. Genome-wide DNA methylation study identifies significant epigenomic changes in osteoarthritic cartilage. Arthritis Rheumatol. 2014;66(10):2804–2815. doi: 10.1002/art.38762. [DOI] [PubMed] [Google Scholar]
- 40.Moazedi-Fuerst F.C., Hofner M., Gruber G., Weinhaeusel A., Stradner M.H., Angerer H., Peischler D., Lohberger B., Glehr M., Leithner A., Sonntagbauer M., Graninger W.B. Epigenetic differences in human cartilage between mild and severe OA. J. Orthop. Res. 2014;32(12):1636–1645. doi: 10.1002/jor.22722. [DOI] [PubMed] [Google Scholar]
- 41.Reynard L.N., Loughlin J. The genetics and functional analysis of primary osteoarthritis susceptibility. Expert Rev. Mol. Med. 2013;15:e2. doi: 10.1017/erm.2013.4. [null.]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reynard L.N., Loughlin J. Genetics and epigenetics of osteoarthritis. Maturitas. 2012;71(3):200–204. doi: 10.1016/j.maturitas.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 43.Rakyan V.K., Down T.A., Balding D.J., Beck S. Epigenome-wide association studies for common human diseases. Nat. Rev. Genet. 2011;12(8):529–541. doi: 10.1038/nrg3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meulenbelt I., Min J.L., Bos S., Riyazi N., Houwing-Duistermaat J.J., van der Wijk H-J., Kroon H.M., Nakajima M., Ikegawa S., Uitterlinden A.G., van Meurs J.B., van der Deure W.M., Visser T.J., Seymour A.B., Lakenberg N., van der Breggen R., Kremer D., van Duijn C.M., Kloppenburg M., Loughlin J., Slagboom P.E. Identification of DIO2 as a new susceptibility locus for symptomatic osteoarthritis. Hum. Mol. Genet. 2008;17(12):1867–1875. doi: 10.1093/hmg/ddn082. [DOI] [PubMed] [Google Scholar]
- 45.Bos S.D., Bovée J.V., Duijnisveld B.J., Raine E.V., van Dalen W.J., Ramos Y.F., van der Breggen R., Nelissen R.G., Slagboom P.E., Loughlin J., Meulenbelt I. Increased type II deiodinase protein in OA-affected cartilage and allelic imbalance of OA risk polymorphism rs225014 at DIO2 in human OA joint tissues. Ann. Rheum. Dis. 2012;71(7):1254–1258. doi: 10.1136/annrheumdis-2011-200981. [DOI] [PubMed] [Google Scholar]
- 46.Southam L., Rodriguez-Lopez J., Wilkins J.M., Pombo-Suarez M., Snelling S., Gomez-Reino J.J., Chapman K., Gonzalez A., Loughlin J. An SNP in the 5′-UTR of GDF5 is associated with osteoarthritis susceptibility in Europeans and with in vivo differences in allelic expression in articular cartilage. Hum. Mol. Genet. 2007;16(18):2226–2232. doi: 10.1093/hmg/ddm174. [DOI] [PubMed] [Google Scholar]
- 47.Chapman K., Takahashi A., Meulenbelt I., Watson C., Rodriguez-Lopez J., Egli R., Tsezou A., Malizos K.N., Kloppenburg M., Shi D., Southam L., van der Breggen R., Donn R., Qin J., Doherty M., Slagboom P.E., Wallis G., Kamatani N., Jiang Q., Gonzalez A., Loughlin J., Ikegawa S. A meta-analysis of European and Asian cohorts reveals a global role of a functional SNP in the 5′ UTR of GDF5 with osteoarthritis susceptibility. Hum. Mol. Genet. 2008;17(10):1497–1504. doi: 10.1093/hmg/ddn038. [DOI] [PubMed] [Google Scholar]
- 48.Valdes A.M., Evangelou E., Kerkhof H.J., Tamm A., Doherty S.A., Kisand K., Tamm A., Kerna I., Uitterlinden A., Hofman A., Rivadeneira F., Cooper C., Dennison E.M., Zhang W., Muir K.R., Ioannidis J.P., Wheeler M., Maciewicz R.A., van Meurs J.B., Arden N.K., Spector T.D., Doherty M. The GDF5 rs143383 polymorphism is associated with osteoarthritis of the knee with genome-wide statistical significance. Ann. Rheum. Dis. 2011;70(5):873–875. doi: 10.1136/ard.2010.134155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Egli R.J., Southam L., Wilkins J.M., Lorenzen I., Pombo-Suarez M., Gonzalez A., Carr A., Chapman K., Loughlin J. Functional analysis of the osteoarthritis susceptibility-associated GDF5 regulatory polymorphism. Arthritis Rheum. 2009;60(7):2055–2064. doi: 10.1002/art.24616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Talens R.P., Christensen K., Putter H., Willemsen G., Christiansen L., Kremer D., Suchiman H.E., Slagboom P.E., Boomsma D.I., Heijmans B.T. Epigenetic variation during the adult lifespan: cross-sectional and longitudinal data on monozygotic twin pairs. Aging Cell. 2012;11(4):694–703. doi: 10.1111/j.1474-9726.2012.00835.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Klug M., Rehli M. Functional analysis of promoter CpG methylation using a CpG-free luciferase reporter vector. Epigenetics. 2006;1(3):127–130. doi: 10.4161/epi.1.3.3327. [DOI] [PubMed] [Google Scholar]
- 52.Reynard L.N., Bui C., Syddall C.M., Loughlin J. CpG methylation regulates allelic expression of GDF5 by modulating binding of SP1 and SP3 repressor proteins to the osteoarthritis susceptibility SNP rs143383. Hum. Genet. 2014;133(8):1059–1073. doi: 10.1007/s00439-014-1447-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zuin J., Dixon J.R., van der Reijden M.I., Ye Z., Kolovos P., Brouwer R.W., van de Corput M.P., van de Werken H.J., Knoch T.A., van IJcken W.F., Grosveld F.G., Ren B., Wendt K.S. Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc. Natl. Acad. Sci. USA. 2014;111(3):996–1001. doi: 10.1073/pnas.1317788111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smyk M., Szafranski P., Startek M., Gambin A., Stankiewicz P. Chromosome conformation capture-on-chip analysis of long-range cis-interactions of the SOX9 promoter. Chromosome Res. 2013;21(8):781–788. doi: 10.1007/s10577-013-9386-4. [DOI] [PubMed] [Google Scholar]
- 55.Sanyal A., Lajoie B.R., Jain G., Dekker J. The long-range interaction landscape of gene promoters. Nature. 2012;489(7414):109–113. doi: 10.1038/nature11279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.den Hollander W., Ramos Y.F., Bomer N., Elzinga S., van der Breggen R., Lakenberg N., de Dijcker W.J., Suchiman H.E., Duijnisveld B.J., Houwing-Duistermaat J.J., Slagboom P.E., Bos S.D., Nelissen R.G., Meulenbelt I. Transcriptional associations of osteoarthritis-mediated loss of epigenetic control in articular cartilage. Arthritis Rheumatol. 2015;67(8):2108–2116. doi: 10.1002/art.39162. [DOI] [PubMed] [Google Scholar]
- 57.Zeggini E., Panoutsopoulou K., Southam L., Rayner N.W., Day-Williams A.G., Lopes M.C., Boraska V., Esko T., Evangelou E., Hoffman A., Houwing-Duistermaat J.J., Ingvarsson T., Jonsdottir I., Jonnson H., Kerkhof H.J., Kloppenburg M., Bos S.D., Mangino M., Metrustry S., Slagboom P.E., Thorleifsson G., Raine E.V., Ratnayake M., Ricketts M., Beazley C., Blackburn H., Bumpstead S., Elliott K.S., Hunt S.E., Potter S.C., Shin S.Y., Yadav V.K., Zhai G., Sherburn K., Dixon K., Arden E., Aslam N., Battley P.K., Carluke I., Doherty S., Gordon A., Joseph J., Keen R., Koller N.C., Mitchell S., O’Neill F., Paling E., Reed M.R., Rivadeneira F., Swift D., Walker K., Watkins B., Wheeler M., Birrell F., Ioannidis J.P., Meulenbelt I., Metspalu A., Rai A., Salter D., Stefansson K., Stykarsdottir U., Uitterlinden A.G., van Meurs J.B., Chapman K., Deloukas P., Ollier W.E., Wallis G.A., Arden N., Carr A., Doherty M., McCaskie A., Willkinson J.M., Ralston S.H., Valdes A.M., Spector T.D., Loughlin J., arcOGEN Consortium. arcOGEN Collaborators Identification of new susceptibility loci for osteoarthritis (arcOGEN): a genome-wide association study. Lancet. 2012;380(9844):815–823. doi: 10.1016/S0140-6736(12)60681-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rushton M.D., Young D.A., Loughlin J., Reynard L.N. Differential DNA methylation and expression of inflammatory and zinc transporter genes defines subgroups of osteoarthritic hip patients. Ann. Rheum. Dis. 2015;74(9):1778–1782. doi: 10.1136/annrheumdis-2014-206752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Herrero-Beaumont G., Roman-Blas J.A., Castañeda S., Jimenez S.A. Primary osteoarthritis no longer primary: three subsets with distinct etiological, clinical, and therapeutic characteristics. Semin. Arthritis Rheum. 2009;39(2):71–80. doi: 10.1016/j.semarthrit.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 60.Ramos Y.F., Bos S.D., Lakenberg N., Böhringer S., den Hollander W.J., Kloppenburg M., Slagboom P.E., Meulenbelt I. Genes expressed in blood link osteoarthritis with apoptotic pathways. Ann. Rheum. Dis. 2014;73(10):1844–1853. doi: 10.1136/annrheumdis-2013-203405. [DOI] [PubMed] [Google Scholar]
- 61.Loughlin J., Reynard L.N. Osteoarthritis: Epigenetics of articular cartilage in knee and hip OA. Nat. Rev. Rheumatol. 2015;11(1):6–7. doi: 10.1038/nrrheum.2014.189. [DOI] [PubMed] [Google Scholar]
- 62.Meulenbelt I., Kloppenburg M., Kroon H.M., Houwing-Duistermaat J.J., Garnero P., Hellio Le Graverand M.P., Degroot J., Slagboom P.E. Urinary CTX-II levels are associated with radiographic subtypes of osteoarthritis in hip, knee, hand, and facet joints in subject with familial osteoarthritis at multiple sites: the GARP study. Ann. Rheum. Dis. 2006;65(3):360–365. doi: 10.1136/ard.2005.040642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Meulenbelt I., Kloppenburg M., Kroon H.M., Houwing-Duistermaat J.J., Garnero P., Hellio-Le Graverand M.P., DeGroot J., Slagboom P.E. Clusters of biochemical markers are associated with radiographic subtypes of osteoarthritis (OA) in subject with familial OA at multiple sites. The GARP study. Osteoarthritis Cartilage. 2007;15(4):379–385. doi: 10.1016/j.joca.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 64.van Spil W.E., Jansen N.W., Bijlsma J.W., Reijman M., DeGroot J., Welsing P.M., Lafeber F.P. Clusters within a wide spectrum of biochemical markers for osteoarthritis: data from CHECK, a large cohort of individuals with very early symptomatic osteoarthritis. Osteoarthritis Cartilage. 2012;20(7):745–754. doi: 10.1016/j.joca.2012.04.004. [DOI] [PubMed] [Google Scholar]
- 65.Ramos Y.F., Hollander W.D., Lakenberg N., van der Breggen R., Bomer N., Kroon H., Kloppenburg M., Slagboom P., Meulenbelt I. Risk prediction using epigenetic profiles in blood of osteoarthritis patients. Osteoarthritis Cartilage. 2015;23(20):A73–A74. doi: 10.1016/j.joca.2015.02.150. [DOI] [Google Scholar]
- 66.Karouzakis E., Trenkmann M., Gay R.E., Michel B.A., Gay S., Neidhart M. Epigenome analysis reveals TBX5 as a novel transcription factor involved in the activation of rheumatoid arthritis synovial fibroblasts. J. Immunol. 2014;193(10):4945–4951. doi: 10.4049/jimmunol.1400066. [DOI] [PubMed] [Google Scholar]
- 67.Nakano K., Boyle D.L., Firestein G.S. Regulation of DNA methylation in rheumatoid arthritis synoviocytes. J. Immunol. 2013;190(3):1297–1303. doi: 10.4049/jimmunol.1202572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Park S.H., Kim S.K., Choe J.Y., Moon Y., An S., Park M.J., Kim D.S. Hypermethylation of EBF3 and IRX1 genes in synovial fibroblasts of patients with rheumatoid arthritis. Mol. Cells. 2013;35(4):298–304. doi: 10.1007/s10059-013-2302-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang Y., Fukui N., Yahata M., Lee M. Genome-wide DNA methylation profiling of osteoarthritic subchondral bone. OsteoarthritisCartilage. 2015;23(20):A195. doi: 10.1016/j.joca.2015.02.984. [DOI] [Google Scholar]