

Abstract
Structure-based design, synthesis, and biological evaluation of a series of very potent HIV-1 protease inhibitors are described. In an effort to improve backbone ligand-binding site interactions, we have incorporated basic-amines at the C4 position of the bis-tetrahydrofuran (bis-THF) ring. We speculated that these substituents would make hydrogen bonding interactions in the flap region of HIV-1 protease. Synthesis of these inhibitors was performed diastereoselectively. A number of inhibitors displayed very potent enzyme inhibitory and antiviral activity. Inhibitors 25f, 25i, and 25j were evaluated against a number of highly-PI-resistant HIV-1 strains and they exhibited improved antiviral activity over darunavir. Two high resolution X-ray structures of 25f and 25g-bound HIV-1 protease revealed unique hydrogen bonding interactions with the backbone carbonyl group of Gly48 as well as with the backbone NH of Gly48 in the flap region of the enzyme active site. These ligand-binding site interactions are possibly responsible for their potent activity.
Keywords: HIV-1 protease inhibitors, Darunavir, amino-bis-THF, multidrug-resistant, design, synthesis, X-ray crystal structure, backbone binding
TOC Image
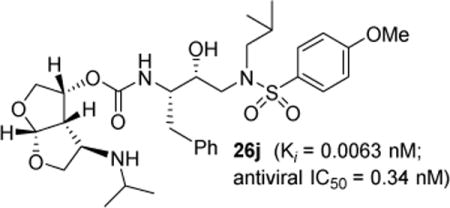
Introduction
Human immunodeficiency virus type 1 (HIV-1) protease is an essential enzyme critical to viral maturation and infectivity.1,2 Therefore, HIV-1 protease is an important drug-design target. The development of protease inhibitors and their introduction into the active antiretroviral therapy led to significant improvement of quality of life, enhanced HIV management, and renewed the mortality and morbidity of HIV/AIDS patients.3,4 Despite these improvements, the emergence of drug resistance, high pill burden, and drug side effects are raising serious concerns about the long-term prospects of HIV/AIDS management.5,6 The majority of currently approved HIV-1 protease inhibitors contain peptidic-like features which contributed to the poor solubility, oral bioavailability, high serum protein binding, and metabolism by liver microsomes.7,8
The development of new and improved anti-HIV-1 therapeutics, however, is faced with a variety of challenges different from the first and second line therapeutics. These include issues of selection pressure, emergence of multidrug-resistant HIV-1 variants, and cross – resistance.9,10,11 Over the years, our research efforts continued to focus on designing nonpeptide protease inhibitors that incorporate stereochemically defined, cyclic ether-derived, novel ligands.12,13,14 We reported the design and discovery of many HIV-1 protease inhibitors with intriguing structural features including the development of darunavir (1, Figure 1). In darunavir, we incorporated a 3(R), 3a(S), 6a(R)-bis-tetrahydrofuranyl urethane (bis-THF) and a hydroxyethylsulfonamide isostere.15,16 Darunavir exhibited excellent activity against a wide range of multidrug-resistant HIV-1 variants.17,18 Our X-ray structural studies of darunavir-bound HIV-1 protease revealed extensive hydrogen bonding interactions with the active site backbone atoms of HIV-1 protease.19 One of our major inhibitor design objectives is to promote maximum interactions, particularly hydrogen bonding interactions with backbone atoms. This ‘backbone binding concept’ emerged from the observation that there is minimal distortion of the protease backbone around the enzyme’s active site.20,21 The X-ray structure of the darunavir-HIV-1 protease complex showed that both oxygens of the bis-THF ligand formed strong hydrogen bonds with the backbone NH’s of Asp29 and Asp30 in the enzyme S2 subsite. Furthermore, the P2 carbonyl formed a strong hydrogen bond with the Gly27 backbone and P2′ amine functionality formed a strong hydrogen bond with the Asp30 NH in S2′-subsite. The backbone hydrogen bonds from the S2 to S2′-subsites are conceivably responsible for darunavir’s effectiveness against multidrug-resistant HIV-1 variants.15, 16
Figure 1.

Structure of HIV-1 protease inhibitors 1–4 and 25j.
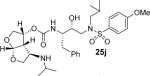
In an effort to further enhance ligand-binding site interactions, we subsequently incorporated an alkoxy substituent as well as fluorine at the C4 position of bis-THF ligand.22,23 Incorporation of a methoxy substituent at C4 led to inhibitor 3 which showed a water-mediated hydrogen bond with the amide NH of Gly-48 located at the flap of the HIV-1 protease. Also, introduction of gem-difluorines at the C4 position resulted in inhibitor 4 which exhibited strong non-bonded interactions with the carbonyl group of Gly48 through the gem-difluorides in the flap region of HIV-1 protease. These interactions appeared to be responsible for improved inhibitor affinity, antiviral activity against multidrug-resistant HIV-1 variants, and ability to cross the blood-brain-barrier.23,24 In the current studies, we plan to maintain all the key backbone hydrogen bonding interactions through the bis-THF oxygens. In addition, based upon the X-ray structure of 3-bound HIV-1 protease, we speculated that introduction of secondary amine functionality at the C4 position, as in inhibitor 25j, may lead to strong hydrogen bonding interactions with the Gly48 backbone atoms. Also, the alkyl side chain may fill in the hydrophobic pocket effectively in the S2 subsite. Overall, this improvement in backbone interactions and van der Waals interactions may not only result in improvement of antiviral activity against multidrug-resistant HIV-1 variants, but the basic amine may also improve the inhibitor’s pharmacological properties. Herein we report our enantioselective synthesis of C4-amine-derived bis-THF ligands using a 2,3-sigmatropic rearrangement as the key reaction. We incorporated these ligands into HIV-1 protease inhibitors and evaluated their biological properties. We also carried out X-ray structural studies of inhibitor-HIV-1 protease complexes to gain molecular insight into the ligand-binding site interactions.
Synthesis
Synthesis of the optically active C-4 azide derivative is shown in Scheme 1. The α,β-unsaturated isopropylidene derivative 5 was synthesized from commercially available D-mannitol as described by us previously.22 Dibal-H reduction of ester 5 provided an allylic alcohol which was reacted with tert-butylbromoacetate in the presence of cesium hydroxide in CH3CN at 23° C for 6 h to provide O-alkylation product 6 in 88% yield over two steps.25,26 Reaction of 6 with LiHMDS in THF at −40 °C to −30 °C for 1 h resulted in a sigmatropic rearrangement, providing α-hydroxy ester 7 as the major product in 62% yield.27,28
Scheme 1.
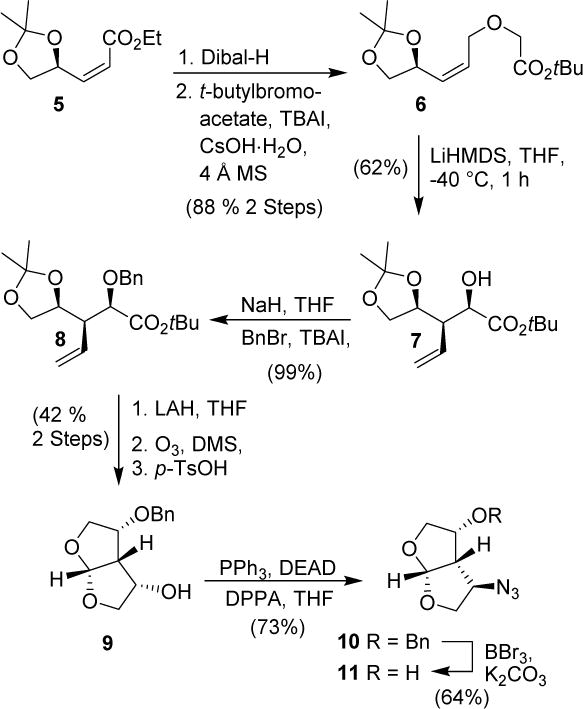
Synthesis of azido bis-THF derivatives
The α-hydroxy ester 7 was protected as the benzyl ether 8 in near quantitative yield. It was converted to bis-THF derivative in a three-step sequence involving, (1) reduction of 8 by LiAlH4 in THF at 0 °C to 23 °C to provide an alcohol; (2) ozonolytic cleavage of the olefin to form an aldehyde, and (3) reaction of the resulting crude aldehyde with p-TsOH in CH2Cl2 at 23 °C to provide bis-THF alcohol 9 in 42% yield over three steps. Alcohol 9 was converted to azide 10 under Mitsunobu conditions29 using diphenyl phosphorazidate (DPPA), triphenyl phosphine and diethyl azodicarboxylate (DEAD) at 0 °C to 23 °C for 18 h to provide azide 10 in 73% yield. Removal of the benzyl ether using BBr3 in the presence of K2CO3 at −78 °C to −20 °C for 2 h provided azidoalcohol 11 in 64% yield.30
Azide 10 was then converted to a number of amine derivatives as shown in Scheme 2. Catalytic hydrogenation of 10 over 10% Pd/C in the presence of di-tert-butyl dicarbonate (Boc2O) under a hydrogen filled balloon afforded Boc-derivative 12 in 98% yield. These reaction conditions did not remove benzyl ether. Reduction of azide 10 with Ph3P in aqueous THF at 23 ° C for 24 h provided the corresponding amine. Addition of methyl chloroformate in the presence of saturated aqueous NaHCO3 solution afforded methyl carbamate 13 in 71% yield. For the synthesis of various N-alkylated derivatives, Boc-derivative 12 was treated with NaH in THF at 0 °C for 30 min, and then MeI was added. The resulting mixture was warmed to 23 °C to provide N-methyl derivative 14 in 98% isolated yield. For the synthesis of N-ethyl derivative, Boc-derivative 12 was reacted with cesium hydroxide and ethyl iodide in DMF at 60 °C to provide the N-ethyl derivative 15 in 91% yield. Isopropyl derivative 16 was synthesized from azide 10 in a three step process involving reduction of the azide to the amine under hydrogenation conditions, reductive amination with acetone and sodium borohydride to afford the isopropyl amine, and finally Boc protection of the resulting amine with Boc anhydride.
Scheme 2.
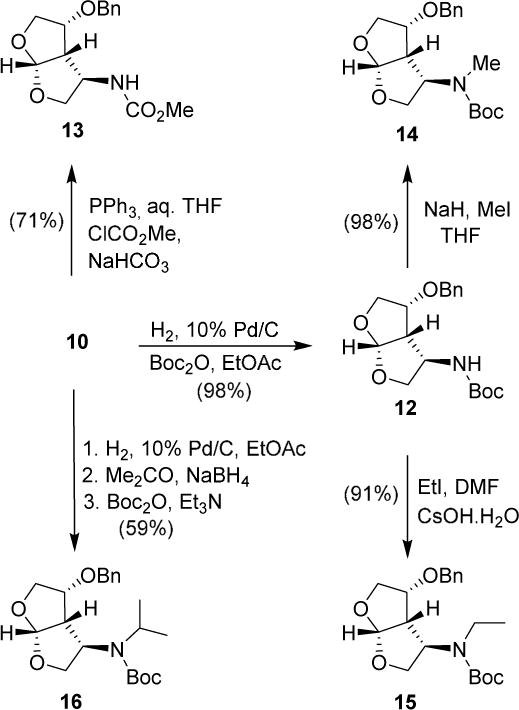
Synthesis of various C4-amino bis-THF derivatives
Various ligand alcohols were prepared by deprotection of benzyl ethers. As shown in Scheme 3, amine derivatives 12–16 were subjected to a catalytic amount of Pd(OH)2 in methanol under a hydrogen-filled balloon to provide alcohols 17–21 in good to excellent yields.
Scheme 3.

Synthesis of various aminoalcohol derivatives
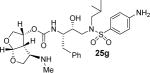
Various optically active ligand alcohols were converted to the corresponding activated mixed carbonates. As depicted in Scheme 4, treatment of ligand-alcohols (11, 17–21) with 4-nitrophenyl chloroformate and pyridine in CH2Cl2 at 0 °C to 23 °C for 12 h provided carbonates 22a–f in very good yields (71–91%). The syntheses of various inhibitors containing hydroxyethylaminesulfonamide isosteres with 4-methyoxysulfonamide (23) and 4-aminosulfonamide (24) as P2′-ligands are shown in Scheme 5. Reaction of amine 23 with activated carbonate 22a provided inhibitor 25a in good yield. Similarly, reaction of carbonates 22b–f and amine 23 provided inhibitors 25c–e, 25h. Inhibitor 25g was prepared by reaction of amine 24 and carbonate 22b followed by removal of the Boc group by treatment with TFA in CH2Cl2. Catalytic hydrogenation of the azide functionality in 25a over 10% Pd/C in ethyl acetate provided inhibitor 25b in excellent yield.
Scheme 4.
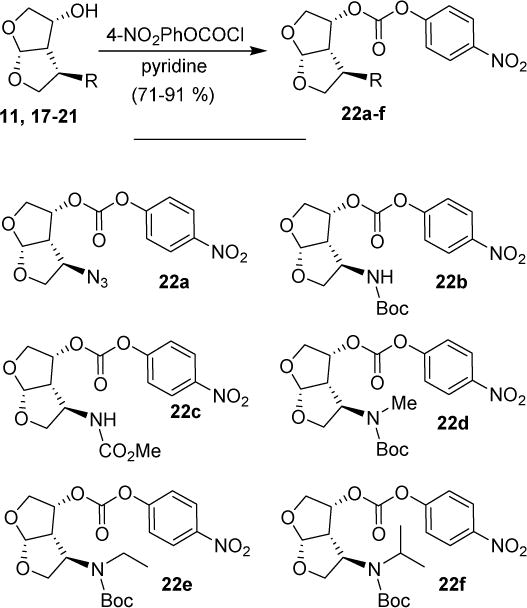
Synthesis of active carbonates of bis-THF ligands
Scheme 5.
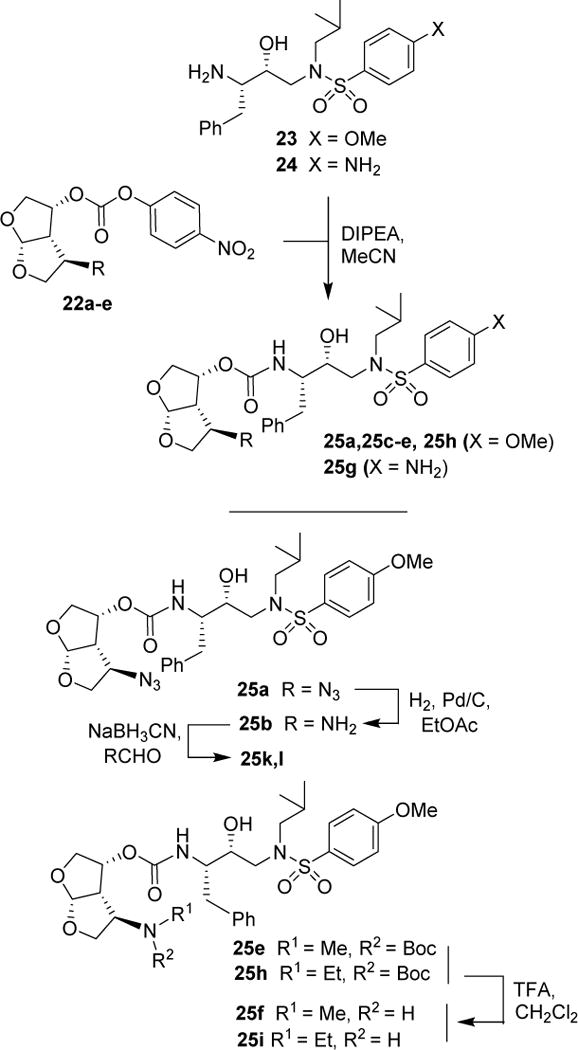
Synthesis of HIV-1 protease inhibitors 25a–l
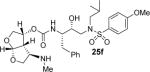
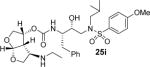
Inhibitor 25f was prepared by removal of the Boc-group from inhibitor 25d by treatment with TFA in CH2Cl2 at 23 °C for 2 h. Similarly, inhibitor 25i and 25j were prepared by removal of the Boc group by treatment with TFA in CH2Cl2 at 23 °C for 2–4 h. Inhibitors 25k and 25l containing dimethyl and diethyl amine functionalities were prepared by reductive amination of amine 25b with paraformaldehyde and acetaldehyde respectively to provide 25k and 25l (50–53% yield).
Results and Discussions
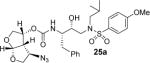
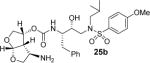
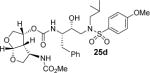
All inhibitors were evaluated in an HIV-1 protease inhibitory assay according to the protocol described by Toth and Marshall.31 Compounds that exhibited potent Ki values, were then selected for further evaluation in an antiviral assay in MT-2 human T-lymphocytes exposed to HIV-1LAI.17 Inhibitor structures and biological results are shown in Table 1. As can be seen, C-4 substituted amino-bis-THF ligand-derived inhibitors exhibited very potent enzyme inhibitory and antiviral activity. The azide containing inhibitor 25a displayed very good enzymatic and antiviral activity (entry 1). The corresponding amine-derived inhibitor 25b showed significant reduction in potency. The Boc-amine derivative 25c, however, gained substantial potency over the amine derivative 25b (entries 2 and 3). The corresponding methyl carbamate 25d has shown comparable activity to Boc-derivative 25c. N-methylation of the Boc-derivative provided inhibitor 25e with nearly 10-fold loss in antiviral activity, indicating that the NH group may be important for potency (entries 3 and 5). Inhibitor 25f with a methylamine functionality at C4 turned out to be a potent inhibitor (Ki = 1.5 pM and antiviral IC50 35 nM). The corresponding 4-amino sulfonamide derivative 25g, however, displayed reduction of enzyme inhibitory and antiviral activity (entry 7). We have examined the effect of steric bulk on the amine functionality. The Boc-ethyl derivative 25h showed comparable enzyme inhibitory activity to the methyl derivative 25e (entries 5 and 8). The ethylamine derivative 25i showed slightly improved antiviral IC50 value (22 nM) over the methyl derivative 25f (IC50 value 35 nM, entries 6 and 9). A sterically demanding isopropyl amine derivative 25j showed over 60-fold improvement in antiviral activity over the methyl and ethyl derivatives (entries 6 and 9). The dimethyl amine derivative 25k and diethylamine derivative 25l were significantly more potent than the corresponding methylamine and ethylamine derivatives 25f and 25i respectively. This result indicates that the dialkylamine is most likely involved in enhanced van der Waals interactions in the S2 subsite of HIV-1 protease. It appears that the C4-position of the bis-THF can accommodate a basic amine functionality with further improvement in antiviral activity compared to unsubstituted bis-THF derivatives.
Table 1.
Enzymatic inhibitory and antiviral activity of inhibitors
| Entry | Inhibitor | Ki(nM) | IC50(nM)a,b |
|---|---|---|---|
| 1. |
|
0.0039 | 4.4 ±0.5 |
| 2. |
|
0.53 | 48 ± 6.5 |
| 3. |

|
0.014 | 4.4 ± 0.9 |
| 4. |
|
0.024 | 3.7±0.5 |
| 5. |

|
0.012 | 37 ±7.7 |
| 6. |
|
0.0015 | 35 ±2.8 |
| 7. |
|
0.0099 | 480 ± 31 |
| 8. |

|
0.048 | nt |
| 9. |
|
0.027 | 22 ± 1.1 |
| 10. |
|
0.0063 | 0.34± 0.2 |
| 11. |
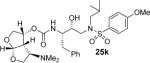
|
0.014 | 4.4±0.7 |
| 12. |
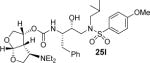
|
0.014 | 7.2 ±0.4 |
Human T-lymphoid (MT-2) cells were exposed to 100 TCID50 values of HIV-1LAI, and cultured in the presence of each PI, and IC50 values were determined using the MTT assay. Darunavir exhibited Ki,= 16 pM, IC50 = 3.0 nM.
nt = not tested.
Inhibitors 25f, 25i, and 25j with basic amine functionalities on the bis-THF ring showed potent antiviral activity. We speculated that the improvement of antiviral IC50 is possibly due to additional hydrogen bonding interactions as well as increased lipophilicity of the inhibitors due to improved van der Waals interactions in active site. We selected these inhibitors for further evaluation against highly darunavir-resistant and multi-PI-resistant HIV-1 variants, HIV-1DRVRP10, HIV-1DRVRP20 and HIV-1DRVRP51. Antiviral assays using human MT-4 cells exposed to these PI-resistant variants showed that all three inhibitors displayed comparable EC50 values ranging from 21 nM to 39 nM against HIV-1DRVRP10 and HIV-1DRVRP20. Fold changes for inhibitors 25f and 25i were very low (1 to 2.6) against HIV-1DRVRP10 and HIV-1DRVRP20. While inhibitor 25j showed potent antiviral EC50 against HIV-1NL4-3, its fold-changes against these strains were significantly higher (21 to 75-fold). Interestingly, sterically less demanding methyl amine derivative 25f was equipotent against HIVWT (EC50 = 0.029 μM) and HIV-1DEVRP20 (EC50 = 0.032 μM), however, inhibitor 25j with isopropylamine displayed nearly 23-fold poorer in antiviral potency against HIV-1DRVRP20 (EC50 = 0.024 μM) compared to that against the wild-type HIVNL4-3 (EC50 = 0.001 μM). Darunavir in the same assay exhibited EC50 value of 0.097 μM against HIV-1DRVRP20. Both inhibitors 25i and 25j showed comparable EC50 value against highly PI-resistant strain HIV-1DRVRP51 and fold changes were 15-fold and 260-fold respectively, indicating higher fold changes with bulkier amine substituent (isopropyl vs. methyl). The fold changes for darunavir was significantly greater. Inhibitors 25i and 25j showed 10-fold improvement in EC50 values over darunavir. It turned out that inhibitor 25i with a smaller ethylamine at C4 position of bis-THF has a greater genetic barrier than inhibitor 25j with an isopropyl amine at C4. Furthermore, it appears that incorporation of basic amine at C4 leads to inhibitors with greater genetic barrier than unsubstituted inhibitor, such as darunavir. The overall improvement of antiviral activity of amine-derived inhibitors is possibly due to their enhanced backbone interactions as well as van der Waals interactions in the S2 subsite.
To obtain molecular insight into the inhibitor-HIV-1 protease interactions, we have determined the X-ray structures of the methylamine derivative 25f and HIV-1 protease complex as well as inhibitor 25g and HIV-1 complex. The structures were refined at 1.34 Å and 1.29 Å resolution, respectively. The crystal structures contain the protease dimer and the inhibitors are seen in two orientations related by a 180° rotation with 60/40% relative occupancies. The overall structures are comparable to the structure of HIV-1 protease with darunavir,32 showing root mean square differences of 0.16 Å and 0.13 Å for Cα atoms, respectively, for the two structures. Differences between equivalent Cα atoms are less than 0.5 Å. The 2Fo-Fc and omit Fo-Fc maps are shown for the inhibitors in both X-ray structures (please see supporting information). The 2Fo-Fc map is contoured at a level of 1.0 σ and the omit map is contoured at 2.0 σ. The two inhibitors differ in the substituents of the phenyl ring at P2′, where inhibitor 25f has a methoxy group and inhibitor 25g has an amino group. Both inhibitors maintained all the key interactions observed in darunavir bound HIV-1. In addition, the C4-NH was shown to form a unique hydrogen bond with the carbonyl of Gly48 shown in Figure 2 for inhibitor 25f. This interaction was absent for the oxymethyl oxygen of the related inhibitor, 3 (pdb code: 3QAA).22 The C4-NH also formed a water-mediated hydrogen bonding interactions to amide NH of Gly48. Similar inhibitor and enzyme backbone interactions along with increased van der Waals interactions due to isopropyl group may have significantly contributed towards the increased antiviral activity of inhibitor 25j. Of particular note, inhibitor 25j incorporates a basic amine functionality which may improve pharmacological properties of this class of inhibitors.
Figure 2.
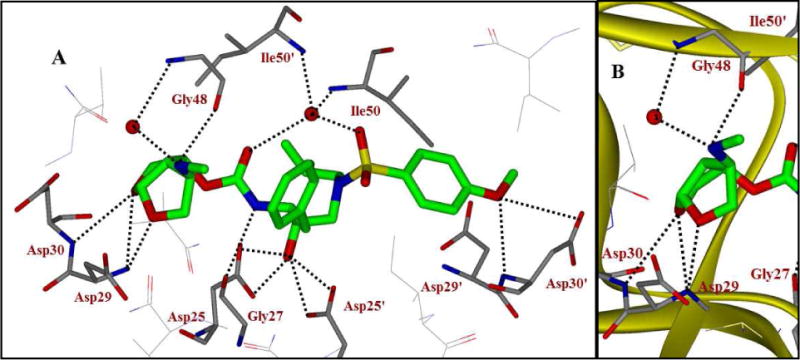
A. X-ray structure of inhibitor 25f-bound HIV-1 protease (PDB code: 5BRY). Hydrogen bonding interactions are shown as dotted lines. B. The main backbone interactions with Gly48 in the flap region and Asp29 and Asp30 in the S2 subsite are shown.
There are several major types of antiretroviral drugs for treating HIV/AIDS. These drugs interfere with different steps in HIV replication. While the use of antiretroviral therapy (ART) treatment and guidelines are updated constantly with the availability of new drugs, PIs continue to be an important element of current ART. Integrase and reverse transcriptase inhibitors are also widely used in ART regimens. The ART regimens are usually well tolerated, however each drug can have serious side effects. Unlike PIs, one major advantage of integrase inhibitors is that they do not require boosting. However, serious side effects including hepatotoxicity, GI disorders, skin disorders, thrombocytopenia, and renal failure are known to occur.33 Depending upon situation, in such a scenario, HIV/AIDS patients have to switch to other key drugs, such as PIs. Also, certain patients receiving long-term ART regimens containing integrase inhibitors tend to harbor HIV-1 variants resistant to integrase inhibitors. Furthermore, patients with heavily-ART regimens-experience tend to have drug-failure with various PIs even with DRV. Thus, more potent PIs with a “high genetic barrier” are critical to effective long-term treatment of HIV/AIDS.
It has been documented that darunavir potently inhibits the replication of not only wild-type HIV-1 but also multidrug-resistant HIV-1 variants. However, the emergence of DRV-resistant HIV-1 variants has been reported in vitro and in vivo and patients harboring such DRV-resistant HIV-1 variants have had treatment failure.34,35 Regarding the emergence of DRV-resistant HIV-1 variants, all three C-4 amino derivatives appeared to be more potent against the DRV-resistant HIV-1 variants. For example, the fold difference in the IC50 value of 25f against HIV-1DRVRP20 compared to the IC50 value of 25f against the wild-type HIV-1NL4-3 was only 1.1, while the fold difference in the case of DRV was as much as 37.3. Moreover, the fold-differences in the case of 25i and 25j with HIV-1DRVRP51 were 15 and 260, respectively, while that in the case of DRV with HIV-1DRVRP51 was as much as 1,346. These data signify that the three C4-modified compounds remain highly active against the DRV-resistant HIV-1 variants. The ligand-binding site interactions of the substituted derivatives are different in the S2-subsite. As shown in Figure 2, the C4-NH of inhibitor 25f forms a unique hydrogen bond with the carbonyl of Gly48. The C4-NH also forms water-mediated hydrogen bonding interactions to amide NH of Gly48. These interactions, which were absent in the case of DRV,17 may explain why these inhibitors 25f, 25i, and 25j remain active against the DRV-resistant HIV-1 variants (e.g., HIV-1DRVRP20 and HIV-1DRVRP51; Table 2).
Table 2.
Antiviral Activity of 25f, 25i and 25j against Multidrug Resistant HIV-1 Variants.
| Virus | DRV | EC50 ± SD, (μM) (fold change)a,b
|
Inhibitor 25j | |
|---|---|---|---|---|
| Inhibitor 25f | Inhibitor 25i | |||
| HIV-1NL4-3 | 0.0026 ±0.0006 | 0.029 ±0.003 | 0.015 ±0.003 | 0.001 ±0.0033 |
| HIV-1DRV RP10 | 0.031 ±0.005 (12) | nd | 0.039 ±0.009 (2.6) | 0.021 ±0.004 (21) |
| HIV-1DRVRP20 | 0.097 ±0.051 (37.3) | 0.032 ±0.003 (1.1) | nd | 0.024 ±0.013 (23) |
| HIV-1DRV Rp51 | 3.5 ±1.4 (1346) | nd | 0.23 ±0.08 (15) | 0.26 ±0.12 (260) |
MT-4 cells (1 × 104) were exposed to 50 TC1D50S of wild-type HIV-lNL4-3, HIV-1DRVRP10, HIV-lDRVRP20, or HIV-lDRVRP51 and cultured in the presence of various concentrations of each PI, and the IC50 values were determined using the XTT assay. The amino acid substitutions identified in protease of HIV-lDRVRP10, HIV-lDRVRP20 and HIV-lDRVRP51 compared to HIV-1NL4-3 include L101/I15V/K20R/L241/V321/M361/M46L/L63P/V82A/L89M; L101/115V/K20R/L241/V321/M361/M46L/L63P/A71T/V82A/L89M and L101/115V/K20R/L241/V321/L33F/M36I/M46L/I54M/L63P/K70Q/V82I/I84V/L89M, respectively.
All assays were conducted in triplicate, and the data shown respresent mean values (±1 standard deviation) derived from the results of two independent experiments. nd, not determined.
Of particular note, cleavage site mutations within the retroviral polyprotein, the substrate for HIV-1 protease, are known to play critical roles in HIV-1’s acquisition of resistance to various protease inhibitors.36 In this regard, when we successfully selected DRV-resistant HIV-1 variants (e.g., HIV-1DRVRP20 and HIV-1DRVRP51) using the mixture of 8 multi-drug resistant HIV-1 variants as a starting viral population,35 no additional cleavage site mutations within the polyprotein were identified.35 Instead, H219Q and I223V substitutions, located in the CypA binding loop of the Gag protein emerged in early selection stages. Plus, a unique combination of V32I, L33F, I54M, and I84V substitutions, which appeared to be responsible for the loss of DRV’s protease dimerization inhibition activity,37,38 was identified within. Although, how these amino acid substitutions contribute to HIV-1’s acquisition of resistance to DRV remains to be further elucidated, the C4-modification in the bis-THF moiety of DRV may shed light in the better understanding of the mechanism of DRV resistance and further design of more potent and resistance-repellant protease inhibitors.
Conclusion
We have designed and synthesized a series of HIV-1 protease inhibitors containing various C4-amine derivatives. One of objectives of these studies was to incorporate basic amine functionalities that can form hydrogen bonding interactions with the backbone atoms in the flap region of the HIV-1 protease. We have investigated a range of amine functionalities. The synthesis of the corresponding amine-bis-THF ligands was carried out stereoselectively and in an optically active manner. The synthesis involved isopropylidene-D-glyceraldehyde as the optically active starting material and a 2,3-sigmatropic rearrangement as the key reaction which installed two chiral centers with high diasteroselectivity. In general, mono- or disubstituted amine-derivatives on the bis-THF ligand showed excellent enzyme inhibitory and antiviral activity. Inhibitor 25j with a C4-isopropylamine functionality exhibited the best results. It showed marked antiviral activity with an IC50 value of 0.34 nM, showing over 10-fold improvement compared to darunavir. Inhibitors 25f, 25i, and 25j were evaluated against a panel of multidrug-resistant HIV-1 variants and these inhibtors showed improved antiviral activity over darunavir. We have obtained high resolution X-ray crystal structures of related inhibitors 25f and 25g bound to HIV-1 protease. These structures revealed that the C4-NH formed a direct backbone hydrogen bonding interaction with Gly48 carbonyl in the S2-subsite. Furthermore, the C4-NH formed water-mediated hydrogen bonds with Gly48 backbone NH. The improved antiviral activity of 25j against multidrug-resistant HIV-1 variants is possibly due to similar backbone hydrogen bonding interactions as well as hydrophobic interaction due to the isopropyl group. The basic amine functionality on the inhibitor may improve pharmacological properties. Further studies aimed at optimization of ligand-binding site interactions and improving drug-like properties of these inhibitors are currently in progress.
Experimental Section
General
All anhydrous solvents were obtained according to the following procedures: diethyl ether and tetrahydrofuran (THF) were distilled from sodium/benzophenone under argon; dichloromethane from calcium hydride. All other solvents were reagent grade. All moisture sensitive reactions were carried out in a flame-dried flask under argon atmosphere. Column chromatography was performed with Whatman 240–400 mesh silica gel under low pressure of 3–5 psi. Thin layer chromatography was carried out with E. Merck silica gel 60-F-254 plates. Yields refer to chromatographically and spectroscopically pure compounds. 1H NMR and 13C NMR spectra were recorded on a Varian Inova-300 (300 MHz and 75 MHz, respectively), Bruker Avance ARX- 400 (400 MHz and 100 MHz), and Bruker Avance DRX-500 (500 MHz and 125 MHz). High and low resolution mass spectra were carried out by the Mass Spectroscopy Center at Purdue University. The purity of all test compounds was determined by HRMS and HPLC analysis. All test compounds showed ≥95% purity.
(2Z)-t-Butyl 2-((3-((4S)-2,2-dimethyl-1,3-dioxolan-4-yl))allyloxy)acetate (6)
Diisobutyl aluminum hydride (1 M in CH2Cl2, 27.5 mL, 27.5 mmol,) was slowly added to a cold solution (−78 °C) of ethyl (2Z)-3-[(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]-2-propenoate (5), (5.5 g, 27.5 mmol) in dichloromethane (100.0 mL). The solution was allowed to stir for 15 min at −78 °C (a color change from colorless to yellow and back to colorless indicates that the reaction is complete). A saturated solution of sodium potassium tartrate (20 mL) was added and the reaction mixture was warmed to room temperature. The reaction was stirred until both layers were transparent. The organic layer was separated and the aqueous layer was extracted with dichloromethane (3 × 15 mL). The organic layers were combined, washed with brine and dried over MgSO4. The solid was filtered out and the organic layer was concentrated under vacuum to obtain the desired allyl alcohol. The crude mixture was taken to the next step without purification. Rf = 0.27 (40% ethyl acetate/hexanes). 1H NMR (400 MHz, CDCl3) δ 5.84−5.81 (m, 1H), 5.55 (t, J = 8.5 Hz, 1H), 4.85 (q, J = 9.6 Hz, 1H), 4.28 (dd, J = 6.8, 7.3 Hz, 1H), 4.18 (d, J = 4.8 Hz, 1H), 4.08 (t, J = 6.5 Hz, 1H), 3.56 (t, J = 9.0 Hz, 1H), 2.12 (bs, 1H), 1.41 (s, 3H), 1.38 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 133.1, 129.4, 109.4, 71.8, 69.4, 58.5, 26.6, 25.8.
To a round bottom flask contianing flame dried 4Å molecular sieves (6.0 g) and acetonitrile (200 mL) was added a solution of (2Z)-3-((4S)-2,2-Dimethyl-1,3-dioxolan-4-yl)prop-2-en-1-ol (27.5 mmol) in acetonitrile (20.0 mL), followed by t-butylbromoacetate (4.9 mL, 33.0 mmol), tetrabutylammonium iodide (1.0 g, 2.75 mmol) and cesium hydroxide monohydrate (6.90 g, 41.3 mmol) at room temperature. The reaction was allowed to stir for 6 h. The solid was filtered out and the solvent was concentrated under vacuum; the residue was purified by flash column chromatography (5% ethyl acetate/hexanes) to afford 6 (6.60 g, 88% yield, 2 steps) as a colorless oil. Rf = 0.57 (30% ethyl acetate/hexanes). 1H NMR (400 MHz, CDCl3) δ 5.78–5.74 (m, 1H), 5.61 – 5.66 (m, 1H), 4.83 (q, J = 7.4 Hz, 1H), 4.16–4.20 (m, 2H), 4.09 (dd, J = 6.2, 2.0 Hz, 1H), 3.94 (d, J = 3.1 Hz, 2H), 3.54 (t, J = 8.1 Hz, 1H), 1.47 (s, 9H), 1.34 (s, 3H), 1.31 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 169.3, 131.4, 129.5, 109.3, 81.6, 71.9, 69.4, 67.7, 66.5, 28.0, 26.6, 25.8. LRMS-ESI (m/z) 273 (M+H).
(2R,3R)-t-Butyl 3-(((4S)-2,2-dimethyl-1,3-dioxolan-4-yl))-2-hydroxypent-4-enoate (7)
A solution of LiHMDS (36.0 mL 1 M in THF, 36.0 mmol) was added to a cold solution (−45 °C) of 6 (6.60 g, 24.2 mmol) in THF (100 mL). LiHMDS was added at a rate that did not exceed −40 °C. The reaction mixture was allowed to warm slowly to −30 °C over 1 h. The reaction was quenched with saturated ammonium chloride (20 mL) and extracted with ethyl acetate (3×20 mL) after warming to room temperature. The organic layers were combined, washed with brine, dried over anhydrous MgSO4 and concentrated under vacuum. The residue was purified with a 5 – 10 percent gradient of ethyl acetate/hexanes. The desired rearranged product 7 (4.10 g, 62% yield) was obtained as a colorless oil. Rf = 0.49 (30% ethyl acetate/hexanes). (1H NMR (400 MHz, CDCl3) δ 5.88–5.79 (m, 1H), 5.20 (dd, J = 17.1 Hz, 10.3, 2H), 4.32 (q, J = 6.8 Hz, 1H), 4.14 (q, J = 4.6 Hz, 2.4 Hz, 1H), 4.09 (dd, J = 8.1 Hz, 6.1 Hz, 1H), 3.85 (t, J = 7.7 Hz, 1H), 3.04 (d, J = 4.6 Hz, 1H), 2.61–2.56 (m, 1H), 1.47 (s, 9H), 1.43 (s, 3H), 1.37 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 172.5, 132.4, 119.6, 109.1, 82.7, 76.0, 71.2, 67.3, 50.3, 27.9, 26.8, 25.4. LRMS-ESI (m/z) 295 (M+Na).
(2R,3S)-t-Butyl 2-(benzyloxy)-3-((S)-2,2-dimethyl-1,3-dioxolan-4-yl)pent-4-enoate (8)
To a cold (0 °C) solution of 7 (4.1 g, 14.9 mmol), benzyl bromide (2.30 mL, 19.37 mmol) and tetrabutyl ammonium iodide (0.50 g, 1.50 mmol) in THF (50.0 mL) was added sodium hydride (0.78 g, 19.37 mmol). The reaction was warmed to 23 °C and allowed to stir for 3 h. Upon completion the reaction was quenched with saturated ammonium chloride (10.0 mL). The reaction was extracted with ethyl acetate (3×10 mL). The organic layers were combined, washed with brine and dried over anhydrous sodium sulfate. The solvent was reduced under vacuum and the residue was purified on silica gel to obtain 8 (5.3 g, 99% yield) as a colorless oil. Rf = 0.61 (20% ethyl acetate/hexanes). 1H NMR (400 MHz, CDCl3) δ 7.35–7.29 (m, 5H), 5.95–5.85 (m, 1H), 5.18 (dd, J = 17.2, 10.3 Hz, 2H) 4.75 (d, J = 11.5 Hz, 1H), 4.33 (d. J = 11.5 Hz, 1 H), 4.20 (q, J = 7.3, 1H), 3.85 (d, J = 3.5 Hz, 1H), 3.77 (dd, J = 6.1, 2.0 Hz, 1H), 3.66 (t, J = 7.7 Hz, 1H), 2.64–2.58 (m, 1H), 1.47 (s, 9H), 1.39 (s, 3H), 1.33 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 169.9, 137.3, 133.5, 128.3, 128.2, 127.9, 119.1, 108.9, 81.7, 78.2, 75.6, 72.3, 67.2, 50.3, 28.1, 26.6, 25.4. LRMS-ESI (m/z) 385 (M+Na).
(3S,3aS,4R,6aR)-4-(Benzyloxy)hexahydrofuro[2,3-b]furan-3-ol (9)
To a cold (0 °C) solution of 8 (5.3 g, 14.8 mmol) in THF (50.0 mL) was added lithium aluminum hydride (1.3 g, 31.3 mmol). The reaction was allowed to stir for 1 h at 23 °C after which the reaction was cooled to 0 °C and quenched by adding excess ethyl acetate, 1 N NaOH (0.5 mL), H2O (0.5 mL). After a white precipitate formed magnesium sulfate was added and stirred for 15 min. The reaction mixture was filtered and concentrated under vacuum.
The crude mixture was taken up in CH2Cl2/MeOH (20.0 ml, 4:1), cooled to −78 °C and a stream of O3 was bubbled through the solution until a blue color persisted. Upon consumption of the starting material, argon was bubbled through the blue solution until the solution became clear. Dimethyl sulfide (0.13 mL, 1.75 mmol) was added to the reaction and the mixture was warmed to room temperature and stirred an additional 3 h. The reaction mixture was carefully concentrated at (0 °C), then 5 mL of CH2Cl2, p-TsOH (30.0 mg) and MeOH (0.5 μL) were added to the residue and the mixture was stirred for 6 h at room temperature. The reaction was again carefully concentrated and the residue was purified on silica gel (20% ether/hexanes to 50% ether/hexanes) to afford compound 9, (1.5 g, 42 % yield 2 steps) as a colorless oil. Rf = 0.52 (60% ethyl acetate/hexanes). 1H NMR (400 MHz, CDCl3) δ 7.38–7.29 (m, 5H), 5.82 (d, J = 5.3 Hz, 1H), 4.56–4.46 (m, 4H), 4.12 (d, J = 10.2 Hz, 1H), 4.03 – 3.96 (m, 2H) 3.61 – 3.57 (dd, J = 6.9, 6.8 Hz, 1H), 2.91 (dd, J = 5.3, 5.4 Hz, 1H), 2.16 (bs, 1H). 13C NMR (100 MHz, CDCl3) δ 137.7, 128.4, 127.7, 127.7, 109.2, 78.6, 74.2, 73.2, 71.1, 68.9, 55.4. LRMS-ESI (m/z) 237 (M+H).
(3R,3aS,4R,6aR)-3-Azido-4-(benzyloxy)hexahydrofuro[2,3-b]furan (10)
To a cold (0 °C) solution of 9 (0.18 g, 0.76 mmol) and triphenylphosphine (0.40 g, 1.52 mmol) in THF (5.0 mL) was added diethyl azodicarboxylate (40 % in toluene) (0.66 mL, 1.52 mmol) followed by diphenylphosphoryl azide (0.33 mL, 1.52 mmol). The reaction was allowed to stir 18 h before diluting with ethyl acetate (10 mL) and quenched with a saturated solution of sodium bicarbonate (10 mL). The reaction was extracted with ethyl acetate (3×15 ml). The organic layers were combined, washed with brine and dried over anhydrous sodium sulfate. The solvent was concentrated under vacuum and the crude mixture was purified on silica gel using 5% ethyl acetate/hexanes to obtain 10 (0.15 g, 73% yield) as colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.45 – 7.23 (m, 5H), 5.79 (d, J = 5.1 Hz, 1H), 4.55 (d, J = 5.1 Hz, 2H), 4.41 – 4.35 (d, J = 4.3 Hz, 1H), 4.32 – 4.21 (m, 1H), 4.10 (dd, J = 10.1 Hz, 4.4 Hz, 1H), 4.04 – 3.94 (m, 2H), 3.65 (dd, J = 9.2, 7.9, 1H), 2.97 – 2.93 (m, 1H). 13C NMR (100 MHz, CDCl3) δ 137.0, 128.6, 128.1, 127.7, 108.6, 76.6, 73.9, 72.7, 70.8, 61.2, 51.8.
(3R,3aS,4R,6aR)-4-Azidohexahydrofuro[2,3-b]furan-3-ol (11)
Benzyl ether 10 (124 mg, 0.47 mmol) was dissolved in dry DCM under argon. K2CO3 (164 mg, 1.19 mmol, 2.5 eq) was added and stirred for 30 minutes at room temperature. The reaction was cooled to −78°C and BBr3 (1 M in DCM, 0.52 mL, 0.52 mmol, 1.1 eq) was added dropwise. The reaction was allowed to warm slowly from −78°C to −20°C and then maintained at −20°C for a total of 2 hours. Upon consumption of starting material identified via TLC analysis, the crude reaction mixture was loaded onto a column and was purified via flash column chromatography on silica gel (30% ethyl acetate/hexanes to 50% ethyl acetate/hexanes) to afford compound 11 (51 mg, 64%) as a yellow residue. Rf = 0.13 (30% ethyl acetate/hexanes) 1H NMR (400 MHz, CDCl3) δ 5.78 (d, J= 5.2 Hz, 1H), 4.60–4.52 (m, 1H), 4.41 (dt J=4.7, 6.1 Hz, 1H), 4.09 (dd, J= 10.1, 4.4 Hz, 1H), 4.03–3.96 (m, 2H), 3.63 (dd, J= 9.6, 6.6 Hz, 1H) 2.91–2.85 (m,1H) 13C NMR (100 MHz, CDCl3) δ 108.8, 74.0, 73.2, 69.9, 60.9, 53.2. LRMS-ESI (m/z) 237 (M+H).
t-Butyl (3R,3aS,4R,6aR)-4-(benzyloxy)hexahydrofuro[2,3-b]furan-3-ylcarbamate (12)
To a round bottom flask charged with 10 (0.09 g, 0.36 mmol) and Boc2O (0.09 g, 0.43 mmol) in ethyl acetate (5.0 mL) (under argon) was added 10% Pd/C (10% by wt.). The mixture was stirred under an atmosphere of hydrogen for 18 h at 1 atm. Upon completion the reaction mixture was filtered through a plug of silica. The solvent was concentrated under vacuum and purified on silica gel using 20% ethyl acetate/hexanes to obtain 12 (0.12 g, 98% yield) as white solid. Rf = 0.61 (40% ethyl acetate/hexanes). 1H NMR (300 MHz, CDCl3) δ 7.48 – 7.26 (m, 5H), 5.76 (d, J = 5.2 Hz, 1H), 4.90 – 4.67 (m, 2H), 4.55 (d, J = 11.6, 2H), 4.24 (dd, J = 15.8, 7.2, 1H), 4.06 (dd, J = 9.7, 4.4, 1H), 3.96 (dd, J = 9.4, 6.7, 1H), 3.84 (d, J = 9.7, 1H), 3.69 (dd, J = 9.3, 7.5, 1H), 2.85 – 2.81 (m, 1H), 1.45 (s, 9H). 13C NMR (75 MHz, CDCl3) δ 155.0, 137.5, 128.5, 127.9, 108.7, 79.7, 76.8, 75.3, 72.5, 71.3, 52.8, 52.4, 28.4. LRMS-ESI (m/z) 358 (M+Na).
Methyl (3R,3aS,4R,6aR)-4-(benzyloxy)hexahydrofuro[2,3-b]furan-3-ylcarbamate (13)
(3R,3aS,4R,6aR)-3-azido-4-(benzyloxy)hexahydrofuro[2,3-b]furan (10) (0.05 g, 0.19 mmol) and triphenyl phosphine (0.06 g, 0.230 mmol) was dissolved in THF/H2O (4.0 mL, 3:1) and stirred at 23 °C for 24 h. After the consumption of the starting material, saturated sodium bicarbonate (1.0 mL) was added followed by methyl chloroformate (0.05 mL, 0.65 mmol) and the reaction was stirred for 6 h. The reaction was diluted and extracted with ethyl acetate (3×5 mL). The organic layers were combined, washed with brine and concentrated under vacuum. The crude mixture was purified on silica gel using 20 % ethyl acetate/hexanes to obtain 13 (40.0 mg, 71% yield). Rf = 0.24 (20 % ethyl acetate/hexanes). 1H NMR (300 MHz, CDCl3) δ 7.46 – 7.23 (m, 5H), 5.77 (d, J = 5.2 Hz, 1H), 5.15 (d, J = 7.4 Hz, 1H), 4.79 (d, J = 11.5 Hz, 1H), 4.59–4.53 (m, 2H), 4.25 (dd, J = 15.8 Hz, 7.6 Hz, 1H), 4.06 (dd, J = 9.9 Hz, 4.4 Hz, 1H), 3.98 (dd, J = 9.3 Hz, 6.8 Hz, 1H), 3.86 (d, J = 9.9 Hz, 1H), 3.68 (s, 4H), 2.93 – 2.79 (m, 1H). 13C NMR (75 MHz, CDCl3) δ 156.3, 137.4, 128.5, 127.9, 108.6, 76.8, 75.0, 72.5, 71.3, 52.8, 52.1
t-Butyl (3R,3aS,4R,6aR)-4-(benzyloxy)hexahydrofuro[2,3-b]furan-3-yl(methyl)carbamate (14)
To a cold (0 °C) solution of 12 (0.03 g, 0.10 mmol) in THF (5.0 mL) was added NaH (0.01 g, 0.19 mmol) and methyl iodide (12.0 μL, 0.19 mmol). The mixture was warmed to room temperature and stirred for 2 h. Upon completion, the solution was cooled to 0 °C and saturated ammonium chloride (1.0 mL) was added. The reaction was diluted and extracted with ethyl acetate (3×15 ml). The organic layers were combined, washed with brine and dried over anhydrous sodium sulfate. The solvent was concentrated under vacuum and the crude mixture was purified on silica gel using 20% ethyl acetate/hexanes to obtain 14 (34.0 mg, 98% yield). Rf = 0.48 (40% ethyl acetate/hexanes). 1H NMR (300 MHz, CDCl3) δ 7.64 – 7.23 (m, 5H), 5.81 (d, J = 5.3 Hz, 1H), 5.44 – 5.01 (m, 1H), 4.70 (bs, 1H), 4.49 (d, J = 11.7 Hz, 1H), 4.23 (dd, J = 15.0 Hz, 8.3 Hz, 1H), 4.11 (dd, J = 10.1 Hz, 6.3 Hz, 1H), 4.01 – 3.89 (m, 2H), 3.63 (t, J = 8.6 Hz, 1H), 2.86 (bs, 1H), 2.81 (s, 3H), 1.45 (s, 9H). 13C NMR (75 MHz, CDCl3) δ 155.1, 137.5, 128.4, 127.8, 109.2, 80.0, 76.5, 74.0, 72.4, 70.43, 52.9, 52. 4, 50.6, 30.0, 28.4.
t-Butyl (3R,3aS,4R,6aR)-4-(benzyloxy)hexahydrofuro[2,3-b]furan-3-yl(ethyl)carbamate (15)
To a round bottom flask charged with activated molecular sieves (0.5g) was added DMF (2.0 mL), 12 (0.29g, 0.86 mmol), ethyl iodide (0.15 mL, 1.82 mmol) and CsOH·H2O (0.31g, 1.82 mmol). The mixture was heated to 60 °C for 12 h. Upon completion, the reaction mixture was diluted with ethyl acetate and filtered through a plug of celite. The organic layer was washed with water and dried over anhydrous sodium sulfate. After purification by flash chromatography the product (15) was obtained as a white solid (284 mg, 91 % yield). Rf = 0.45 (20 % ethyl acetate/hexanes). 1H NMR (400 MHz, CDCl3) δ 7.42 – 7.28 (m, 5H), 5.81 (d, J = 5.2 Hz, 1H), 4.94 (bs, 1H), 4.69 (d, J = 11.0 Hz, 1H), 4.50 (d, J = 11.7 Hz, 1H), 4.24 – 4.18 (m, 1H), 4.13 (dd, J = 9.7, 6.9 Hz, 1H), 3.96 – 3.92 (m, 2H), 3.63 (t, J = 8.7 Hz, 1H), 3.33 – 3.15 (m, 2H), 3.00 – 2.98 (m, 1H) 1.46 (s, 9H), 1.14 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 154.8, 137.5, 128.4, 127.8, 127.6, 109.2, 79.7, 76.6, 74.0, 72.5, 70.1, 56.3, 51.1, 38.5, 28.4, 15.1
t-Butyl ((3R,3aR,4R,6aR)-4-(benzyloxy)hexahydrofuro[2,3-b]furan-3-yl)(isopropyl)carbamate (16)
To a solution of azide (56.0 mg, 0.21 mmol) in ethyl acetate (2 mL) was added 10% Pd–C (5 mg) at 23 °C under argon. The argon balloon was replaced with a H2 balloon, and the reaction mixture was stirred for 2 h under H2 atmosphere. The reaction mixture was filtered through Celite, washed with ethyl acetate, and concentrated under reduced pressure, and the resulting residue was directly used for the next step.
To the crude amine, reagent grade acetone (0.25 mL, 3.0 mmol) was added and stirred for 24 h at room temperature. Acetone was removed by rotary evaporation and reconstituted with 100% ethanol (0.25 mL). To the resulting mixture was added NaBH4 (16.0 mg, 0.42 mmol) and the reaction mixture stirred for an additional 2 h. The reaction was then quenched with sat. NaHCO3 and then extracted with CH2Cl2 (3 × 2 mL). The combined extracts were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure.
Above crude isopropyl derivative was dissolved in CH2Cl2, Et3N (73 μL, 0.44 mmol) and di-tert-butyl dicarbonate (57.0 mg, 0.26 mmol) were added and stirred at room temperature overnight. The mixture is concentrated in vacuo and the residue was purified by silica gel column chromatography (15% EtOAc/hexanes) to obtain the product 16 47.0 mg (yield 59%, over three steps). 1H NMR (400 MHz, CDCl3) δ 7.38–7.32 (m, 5H), 5.83 (d, J = 4.8 Hz, 1H), 4.77–4.58 (m, 1H), 4.63 (d, J = 12.1 Hz, 1H), 4.52 (d, J = 12.1 Hz, 1H), 4.22–4.12 (m, 2H), 3.98–3.84 (m, 3H), 3.67–3.59 (m, 1H), 3.09–3.02 (m, 1H), 1.48 (s, 9H), 1.19 (d, J = 6.9 Hz, 3H), 1.16 (d, J = 6.9 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 154.9, 137.6, 128.5, 128.0, 127.9, 109.7, 82.2, 80.1, 74.7, 72.7, 69.9, 51.3, 47.7, 29.7, 28.6; LRMS (ESI), m/z 378 (M + H)+.
General Procedure for O-benzyl deprotection
To a round bottom flask charged with the indicated starting material (12 – 16) in methanol (5.0 mL) (under argon) was added Pd(OH)2 (10 mol%). The mixture was stirred under a 1 atm (hydrogen balloon) for 18 h. Upon completion the reaction mixture was filtered through a plug of silica. The solvent was concentrated under vacuum and the crude mixture was purified on silica gel using 20% – 30% ethyl acetate/hexanes to obtain the desired alcohols (17–21).
t-Butyl (3R,3aS,4R,6aR)-4-hydroxyhexahydrofuro[2,3-b]furan-3-ylcarbamate (17)
Following the general procedure outlined above, alcohol 17 was prepared in 95% yield. Rf = 0.24 (60 % ethyl acetate/hexanes). 1H NMR (300 MHz, CDCl3) δ 5.74 (d, J = 5.2, 1H), 4.97 (d, J = 8.7 Hz, 1H), 4.60 – 4.41 (m, 2H), 4.15 (dd, J = 9.6 Hz, 5.8 Hz, 1H), 4.03 (dd, J = 9.0 Hz, 6.8 Hz, 1H), 3.73 (dd, J = 9.7 Hz, 2.9 Hz, 1H), 3.60 (t, J = 8.6 Hz, 1H), 2.74 (s, 1H), 2.1 (bs, 1H) 1.43 (s, 9H). 13C NMR (75 MHz, CDCl3) δ 155.9, 109.1, 80.5, 73.5, 72.7, 69.9, 55.3, 50.9, 28.3
Methyl (3R,3aS,4R,6aR)-4-hydroxyhexahydrofuro[2,3-b]furan-3-ylcarbamate (18)
Following the general procedure outlined above, alcohol 18 was prepared in 72% yield. Rf = 0.16 (60 % ethyl acetate/hexanes).1H NMR (300 MHz, CDCl3) δ 5.76 (d, J = 5.2 Hz, 1H), 5.21 (bs, 1H), 4.66 – 4.44 (m, 2H), 4.16 (dd, J = 9.7 Hz, 5.6 Hz, 1H), 4.04 (dd, J = 9.0 Hz, 6.8 Hz, 1H), 3.89 (d, J = 3.2 Hz, 1H), 3.78 (dd, J = 9.8 Hz, 2.4 Hz, 1H), 3.72 – 3.53 (m, 4H), 2.87 – 2.65 (m, 1H). 13C NMR (75 MHz, CDCl3) δ 157.0, 109.04, 73.6, 72.9, 69.9, 55.1, 52.4, 51.5
t-Butyl (3R,3aS,4R,6aR)-4-hydroxyhexahydrofuro[2,3-b]furan-3-yl(methyl)carbamate (19)
Following the general procedure outlined above, alcohol 19 was prepared in 90% yield. Rf = 0.22 (60 % ethyl acetate/hexanes). 1H NMR (300 MHz, CDCl3) δ 5.77 (d, J = 5.1 Hz, 1H), 5.14 – 4.95 (m, 1H), 4.52 (d, J = 2.6 Hz, 1H), 4.29 – 3.84 (m, 4H), 3.57 (t, J = 8.7 Hz, 1H), 2.80 (s, 3H), 2.70 (bs, 1H), 1.46 (s, 9H). 13C NMR (75 MHz, CDCl3) δ 155.7, 109.0, 80.7, 72.6, 71.0, 70.2, 54.7, 53.9, 29.5, 28.4
t-Butyl ethyl((3R,3aS,4R,6aR)-4-hydroxyhexahydrofuro[2,3-b]furan-3-yl)carbamate (20)
Following the general procedure outlined above, alcohol 20 was prepared in 81 % yield. Rf = 0.25 (40 % ethyl acetate/hexanes). 1H NMR (400 MHz, CDCl3) δ 5.75 (d, J = 4.4 Hz, 1H), 4.77 (bs, 1H), 4.43 – 4.37 (m, 1H), 4.02 (dd, J = 10.2, 7.2 Hz, 1H), 3.94 – 3.85 (m, 2H), 3.51 – 3.45 (m, 1H), 3.18 (dd, J = 14.4, 7.1 Hz, 1H), 3.30 (m, 1H), 3.03 (bs, 1H), 2.72 (bs, 1H) 1.37 (s, 9H), 1.14 (t, J = 7.0, 3H). 13C NMR (100 MHz, CDCl3) δ 154.0, 108.9, 80.5, 72.5, 71.1, 70.1, 55.1, 54.6, 38.8, 28.3, 15.5
t-Butyl ((3R,3aS,4R,6aR)-4-hydroxyhexahydrofuro[2,3-b]furan-3-yl)(isopropyl)carbamate (21)
Following the general procedure outlined above, alcohol 21 was prepared in 95% yield. 1H NMR (400 MHz, CDCl3) δ 5.81 (d, J = 5.1 Hz, 1H), 4.88–4.64 (bs, 1H), 4.44 (quin, J = 6.8, 13.8 Hz, 1H), 4.17–4.09 (m, 1H), 4.01 (dd, J = 6.4, 9.0 Hz, 1H), 3.91 (dd, J = 4.4, 9.7 Hz, 1H), 3.65 (t, J = 8.4 Hz, 2H), 2.87–2.77 (bs, 1H), 1.49 (s, 9H), 1.26 (d, J = 6.8 Hz, 3H), 1.22 (d, J = 6.8 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 109.3, 80.7, 72.8, 71.3, 70.4, 53.7, 46.7, 28.5, 21.8, 21.4; LRMS (ESI), m/z 310 (M + Na)+.
Preparation of activated carbonates 22a–f from polycyclic P2-ligands
To a solution of the corresponding alcohol (11, 17 – 21) in dry CH2Cl2 was added pyridine (2.3 eq). The resulting mixture was cooled to 0 °C under argon and 4-nitrophenylchloroformate (2.2 eq) was added in a single portion. The resulting mixture was warmed to 23 °C and stirred until the starting material was consumed. The reaction mixture was evaporated to dryness and the residue was purified by flash column chromatography on silica gel using a gradient of 20–40% ethyl acetate/hexanes to afford the desired ligand-activated carbonate 22a–f.
(3R,3aS,4R,6aR)-4-Azidohexahydrofuro[2,3-b]furan-3-yl 4-nitrophenyl carbonate (22a)
Follow ): Following the general procedure outlined above for the activation of the bis-THF alcohol, carbonate 22a was obtained in 65% yield. Rf = 0.53, (40% ethyl acetate/hexanes). 1H NMR (400 MHz, CDCl3) δ 8.31 (d, J = 8.0 Hz, 2H), 7.40 (d, J = 12.0 Hz, 2H), 5.89 (d, J = 5.2 Hz, 1H), 5.39 – 5.34 (m, 1H), 4.32 (bs, 1H), 4.25 – 4.12 (m, 2H), 4.08 (d, J = 10.3 Hz, 1H), 3.96 (dd, J = 10.3 Hz, 5.6 Hz, 1H), 3.18 – 3.15 (m, 1H). 13C NMR (100 MHz, CDCl3) δ 154.9, 151.7, 145.6, 125.4, 121.6, 108.6, 76.1, 73.6, 70.6, 61.2, 51.7
t-Butyl(3R,3aS,4R,6aR)-4-((4-nitrophenoxy)carbonyloxy)hexahydrofuro[2,3-b]furan-3-ylcarbamate (22b)
Following the general procedure outlined above for the activation of the bis-THF alcohol, carbonate 22b was obtained in 86% yield. Rf = 0.38, (40% ethyl acetate/hexanes). 1H NMR (300 MHz, CDCl3) δ 8.20 (d, J = 12.2 Hz, 2H), 7.45 (d, J = 12.0 Hz 2H), 5.83 (d, J = 5.2 Hz, 1H), 5.37 – 5.32 (m, 1H), 4.92 (d, J = 8.2 Hz, 1H), 4.54 (bs, 1H), 4.19 – 4.09 (m, 2H), 4.02 – 3.75 (m, 2H), 3.07 – 3.03 (m, 1H), 1.44 (s, 9H). 13C NMR (75 MHz, CDCl3) δ 155.2, 154.9, 151.8, 145.6, 125.3, 122.0, 108.5, 80.2, 76.3, 74.5, 70.7, 53.3, 51.9, 28.3. LRMS (ESI), m/z 433 (M + Na)+.
Methyl(3R,3aS,4R,6aR)-4-((4-nitrophenoxy)carbonyloxy)hexahydrofuro[2,3-b]furan-3-ylcarbamate (22c)
Following the general procedure outlined above for the activation of the bis-THF alcohol, carbonate 22c was obtained in 86% yield. Rf = 0.52, (40% ethyl acetate/hexanes). 1H NMR (300 MHz, CDCl3) δ 8.29 (d, J = 12.0 Hz, 2H), 7.45 (d, J = 9.0 Hz, 2H), 5.83 (d, J = 5.2 Hz, 1H), 5.33 (dt, J = 8.5, 6.0 Hz, 1H), 5.18 (d, J = 8.2 Hz, 1H), 4.56 (bs, 1H), 4.22 – 4.22 – 4.10 (m, 2H), 4.03 – 3.82 (m, 2H), 3.67 (s, 3H), 3.07 (t, J = 8.6, Hz, 1H). 13C NMR (75 MHz, CDCl3) δ 156.2, 155.2, 151.8, 145.6, 125.3, 121.9, 108.4, 77.2, 76.1, 74.28, 70.5, 53.1, 52.3. LRMS (ESI), m/z 391 (M + Na)+.
t-Butylmethyl((3R,3aS,4R,6aR)-4-((4-nitrophenoxy)carbonyloxy)hexahydrofuro[2,3-b]furan-3-yl)carbamate (22d)
Following the general procedure outlined above for the activation of the bis-THF alcohol, carbonate 22d was obtained in 94% yield. Rf = 0.49, (40% ethyl acetate/hexanes). 1H NMR (300 MHz, CDCl3) δ 8.25 (d, J = 12.1 Hz, 2H), 7.52 (d, J = 8.7 Hz, 2H), 5.88 (d, J = 5.3 Hz, 1H), 5.28 (bs, 2H), 4.19 – 4.10 (m, 2H), 4.01 (d, J = 10.4 Hz, 1H), 3.88 (t, J = 8.8 Hz, 1H), 3.10 (t, J = 8.0 Hz, 1H), 2.79 (s, 3H), 1.42 (s, 9H). 13C NMR (75 MHz, CDCl3) δ 155.4, 155.2, 151.8, 145.5, 125.1, 122.3, 108.9, 80.2, 76.2, 72.8, 69.5, 54.8, 51.3, 29.4, 28.3
t-Butyl ethyl((3R,3aS,4R,6aR)-4-((4-nitrophenoxy)carbonyloxy)hexahydrofuro[2,3-b]furan-3-yl)carbamate (22e)
Following the general procedure outlined above for the activation of the bis-THF alcohol, carbonate 22e was obtained in 88% yield. Rf = 0.56, (30% ethyl acetate/hexanes). 1H NMR (400 MHz, CDCl3) δ 8.25 (d, J = 8.8 Hz, 2H), 7.47 (d, J = 8.9 Hz, 2H), 5.87 (d, J = 5.2 Hz, 1H), 5.30 (q, J = 7.2 Hz, 1H), 5.00 (bs, 1H), 4.18 – 4.12 (m, 2H), 4.00 (dd, J = 10.2, 2.6 Hz, 1H), 3.89 (dd, J = 9.7, 7.4 Hz, 1H), 3.26 – 3.16 (m, 3H), 1.44 (s, 9H), 1.16 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 155.3, 151.7, 145.5, 126.0, 125.1, 121.9, 115.5, 108.9, 80.2, 76.2, 72.8, 69.5, 55.7, 51.8, 28.3, 15.5. LRMS (ESI), m/z 461 (M + Na)+.
t-Butyl isopropyl((3R,3aS,4R,6aR)-4-(((4-nitrophenoxy)carbonyl)oxy)hexahydrofuro[2,3-b]furan-3-yl)carbamate (22f)
Following the general procedure outlined above for the activation of the bis-THF alcohol, carbonate 22f was obtained in 64% yield. 1H NMR (400 MHz, CDCl3) δ 8.28 (d, J = 9.2 Hz, 2H), 7.46 (d, J = 8.4 Hz, 2H), 5.91 (d, J = 6.0 Hz, 1H), 5.28 (q, J = 7.5, 15.0 Hz, 1H), 4.27–4.15 (m, 2H), 3.98–3.92 (m, 1H), 3.91–3.85 (m, 1H), 3.31–3.21 (m, 1H), 1.45 (s, 9H), 1.26 (d, J = 6.9 Hz, 3H), 1.23 (d, J = 6.9 Hz, 3H).
General procedure for the synthesis of HIV-1-protease inhibitors
Hydroxyethylamine sulfonamide Isostere 23 or 24 was taken up in CH3CN and cooled to 0 °C. i-Pr2EtN (5eq, excess) was added, and the resulting solution was stirred for 5 min. The corresponding activated bis-THF ligand (from 22a–f) was added and the resulting yellow solution was stirred at 23 °C until the reaction was complete (18–24 h). The reaction was concentrated under vacuum and the crude residue purified by flash column chromatography on silica gel to obtain the desired inhibitor.
(3R,3aR,4R,6aR)-4-Azidohexahydrofuro[2,3-b]furan-3-yl (2S,3R)-3-hydroxy-4-(N-isobutyl-4-methoxyphenylsulfonamido)-1-phenylbutan-2-ylcarbamate (25a)
The titled inhibitor was synthesized using the general procedure outlined above. The desired inhibitor was obtained as an amorphous white solid (94% yield). Rf = 0.47 (50% ethyl acetate/hexanes). 1H NMR (300 MHz, Chloroform-d) δ 7.72 (d, J = 8.9 Hz, 2H), 7.39 – 7.10 (m, 5H), 6.99 (d, J = 8.9 Hz, 2H), 5.74 (d, J = 5.1 Hz, 1H), 5.16 – 4.91 (m, 2H), 4.02 – 3.89 (m, 2H), 3.88 (s, 3H), 3.79 (d, J = 4.0 Hz, 3H), 3.74 – 3.64 (m, 2H), 3.35 (s, 1H), 3.24 – 2.85 (m, 5H), 2.83 – 2.73 (m, 2H), 1.88 – 1.79 (m, 1H), 0.93 (d, J = 6.6 Hz, 3H), 0.89 (d, J = 6.6 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 163.1, 154.9, 137.6, 129.5, 129.3, 128.5, 126.8, 114.4, 108.6, 73.6, 72.8, 72.2, 71.0, 61.4, 58.9, 55.6, 55.2, 53.6, 52.0, 35.4, 27.3, 20.1, 19.9. Mass: HRMS (ESI), Calcd for C28H37N5O8S: m/z 626.2261 (M+Na), found m/z 626.2260 (M+Na).
(3R,3aS,4R,6aR)-4-Aminohexahydrofuro[2,3-b]furan-3-yl (2S,3R)-3-hydroxy-4-(N-isobutyl-4-methoxyphenylsulfonamido)-1-phenylbutan-2-ylcarbamate (25b)
Inhibitor 25a was dissolved in ethyl acetate and treated with Pd/C and under a hydrogen balloon for 1 h. The reaction mixture was filtered through a plug of celite. The solution was concentrated under vacuum and the crude residue purified by flash column chromatography on silica gel to yield inhibitor 25b as an amorphous white solid (71% yield). Rf = 0.10 (10% methanol/CH2Cl2). 1H NMR (300 MHz, Chloroform-d) δ 7.72 (d, J = 8.8 Hz, 2H), 7.38 – 7.11 (m, 5H), 6.99 (d, J = 8.9 Hz, 2H), 5.76 (d, J = 5.1 Hz, 1H), 5.06 (d, J = 8.0 Hz, 2H), 3.99 – 3.88 (m, 8H), 3.75 – 3.53 (m, 2H), 3.21 – 2.95 (m, 5H), 2.83 – 2.76 (m, 2H), 2.64 (d, J = 8.7 Hz, 1H), 2.23 (bs, 2H), 1.88 – 1.79 (m, 1H), 0.91 (dd, J = 14.7, 6.6 Hz, 6H). Mass: HRMS (ESI), Calcd for C28H39N3O8S: m/z 578.2535 (M+Na), found m/z 578.2527 (M+Na).
(3R,3aS,4R,6aR)-4-((2S,3R)-3-Hydroxy-4-(N-isobutyl-4-methoxyphenylsulfonamido)-1-phenylbutan-2-ylcarbamoyloxy)hexahydrofuro[2,3-b]furan-3-yl t-butyl carbamate (25c)
The titled inhibitor was synthesized using the general procedure outlined above. The desired inhibitor 25c was obtained as an amorphous white solid (69% yield). Rf = 0.3 (50% ethyl acetate/hexanes). 1H NMR (400 MHz, CDCl3) δ 7.67 (d, J = 8.7 Hz, 2H), 7.31 – 7.19 (m, 5H), 6.97 (d, J = 8.9 Hz, 2H), 5.74 (d, J = 5.0 Hz, 1H), 5.24 – 5.02 (m, 2H), 4.86 (d, J = 6.7 Hz, 1H), 4.19 (bs, 1H), 3.98 – 3.91 (m, 2H), 3.86 (s, 3H), 3.80 – 3.69 (m, 3H), 3.20 – 2.61 (m, 7H), 1.99 – 1.70 (m, 1H), 1.42 (d, J = 2.6 Hz, 9H), 0.87 (dd, J = 15.4, 6.5 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 163.0, 155.2, 154.8, 137.5, 129.4, 128.5, 126.5, 114.3, 108.3, 79.8, 74.8, 72.2, 71.5, 65.8, 58.7, 55.6, 55.1, 53.6, 52.6, 52.0, 35.2, 30.2, 28.3, 27.2, 20.0, 19.8, 15.2. Mass: HRMS (ESI), Calcd for C33H47N3O10S: m/z 700.2880 (M+Na), found m/z 700.2865 (M+Na).
(3R,3aS,4R,6aR)-4-((2S,3R)-3-Hydroxy-4-(N-isobutyl-4-methoxyphenylsulfonamido)-1-phenylbutan-2-ylcarbamoyloxy)hexahydrofuro[2,3-b]furan-3-yl methyl carbamate (25d)
The titled inhibitor 25d was synthesized using the general procedure outlined above. The desired inhibitor 25d was obtained as an amorphous white solid (96% yield). Rf = 0.11 (50% ethyl acetate/hexanes). 1H NMR (300 MHz, Chloroform-d) δ 7.69 (d, J = 8.7 Hz, 2H), 7.43 – 7.16 (m, 5H), 6.98 (d, J = 8.9 Hz, 2H), 5.75 (d, J = 5.0 Hz, 1H), 5.11 (bs, 2H), 4.95 (bs, 1H), 3.99 (d, J = 6.1 Hz, 2H), 3.87 (s, 5H), 3.83 – 3.52 (m, 6H), 3.23 – 2.64 (m, 8H), 1.97 – 1.66 (m, 1H), 0.88 (dd, J = 12.0, 6.7 Hz, 6H). 13C NMR (100 MHz, Chloroform-d) δ 162.9, 156.1, 155.2, 137.6, 129.7, 129.4, 128.5, 126.5, 114.3, 108.3, 74.5, 72.3, 72.1, 71.2, 58.6, 55.5, 55.2, 53.5, 52.5, 52.2, 52.1, 35.2, 27.1, 20.0, 19.8. Mass: HRMS (ESI), Calcd for C30H41N3O10S: m/z 658.2410 (M+Na), found m/z 658.2429 (M+Na).
(3R,3aS,4R,6aR)-4-((2S,3R)-3-Hydroxy-4-(N-isobutyl-4-methoxyphenylsulfonamido)-1-phenylbutan-2-ylcarbamoyloxy)hexahydrofuro[2,3-b]furan-3-yl methyl t-butyl carbamate (25e)
The titled inhibitor was synthesized using the general procedure outlined above. The desired inhibitor 25e was obtained as an amorphous white solid (78% yield). Rf = 0.33 (50% ethyl acetate/hexanes). 1H NMR (300 MHz, Chloroform-d) δ 7.84 – 7.53 (m, 2H), 7.39 – 7.07 (m, 5H), 6.96 (d, J = 8.8 Hz, 2H), 5.78 (s, 1H), 5.14 (s, 2H), 4.96 – 4.34 (m, 1H), 4.13 – 4.01 (m, 3H), 3.87 (s, 6H), 3.69 – 3.37 (m, 1H), 3.16 – 2.59 (m, 9H), 1.64 – 1.52 (m, 1H), 1.45 (d, J = 9.4 Hz, 9H), 1.00 – 0.75 (m, 6H). Mass: HRMS (ESI), Calcd for C34H49N3O10S: m/z 692.3217 (M+H), found m/z 692.3205 (M+Na).
(3R,3aR,4R,6aR)-4-(Methylamino)hexahydrofuro[2,3-b]furan-3-yl(2S,3R)-3-hydroxy-4-(N-isobutyl-4-methoxyphenylsulfonamido)-1-phenylbutan-2-ylcarbamate (25f)
Inhibitor 25e was dissolved in CH2Cl2/TFA (4:1) at 0 °C and stirred for 2 h and then warmed to 23 °C. The reaction mixture was concentrated under vacuum and purified by flash column chromatography on silica gel. The desired inhibitor 25f was obtained as an amorphous white solid (65% yield). Rf = 0.20 (10% methanol/CH2Cl2). 1H NMR (300 MHz, Chloroform-d) δ 7.71 (d, J = 8.9 Hz, 2H), 7.41 – 7.13 (m, 5H), 6.98 (d, J = 8.9 Hz, 2H), 5.72 (d, J = 5.2 Hz, 1H), 5.22 – 4.90 (m, 2H), 3.96 (dd, J = 9.7, 6.2 Hz, 1H), 3.87 (s, 5H), 3.84 – 3.62 (m, 3H), 3.22 – 2.89 (m, 5H), 2.79 (dt, J = 21.8, 8.1 Hz, 4H), 2.17 (s, 3H), 2.16 (s, 1H), 1.91 – 1.75 (m, 1H), 0.90 (dd, J = 12.8, 6.6 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 163.1, 155.3, 137.7, 129.6, 129.5, 129.3, 128.5, 126.6, 114.4, 108.8, 74.1, 72.7, 71.0, 60.4, 58.8, 55.6, 55.2, 53.6, 51.6, 35.4, 33.9, 27.3, 20.1, 19.9. Mass: HRMS (ESI), Calcd for C29H41N3O8S: m/z 592.2692 (M+Na), found m/z 592.2690 (M+Na).
(3R,3aS,4R,6aR)-4-(Methylamino)hexahydrofuro[2,3-b]furan-3-yl(2S,3R)-4-(4-amino-N-isobutylphenylsulfonamido)-3-hydroxy-1-phenylbutan-2-ylcarbamate (25g)
Inhibitor 25g was obtained over two steps. Amine 24 was reacted with carbonate 22d and the resulting Boc protected intermediate was dissolved in CH2Cl2/TFA (4:1) at 0 °C and stirred for 2 h at 23 °C. The reaction mixture was concentrated under vacuum and purified by flash column chromatography on silica gel. The desired inhibitor 25g was obtained as an amorphous white solid (65% yield). Rf = 0.10 (10% methanol/CH2Cl2). 1H NMR (300 MHz, Chloroform-d) δ 7.54 (d, J = 8.6 Hz, 2H), 7.36 – 7.16 (m, 5H), 6.69 (d, J = 8.6 Hz, 2H), 5.75 (d, J = 5.1 Hz, 1H), 5.30 – 5.05 (m, 2H), 4.19 (s, 2H), 3.97 (dd, J = 9.7, 6.2 Hz, 1H), 3.86 (d, J = 3.6 Hz, 4H), 3.71 (dq, J = 11.1, 4.7 Hz, 2H), 3.22 – 2.87 (m, 5H), 2.87 – 2.68 (m, 4H), 2.27 (s, 3H), 1.92 – 1.71 (m, 1H), 0.91 (dd, J = 12.1, 6.6 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 155.2, 150.7, 137.8, 129.5, 129.3, 128.5, 126.6, 125.9, 114.0, 108.8, 73.5, 72.7, 72.6, 71.0, 67.9, 60.3, 58.8, 55.3, 53.7, 51.1, 35.5, found m/z 577.2679 (M+H).
t-Butylethyl((3R,3aS,4R,6aR)-4-((((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl) sulfonamido)-1-phenylbutan-2-yl)carbamoyl)oxy)hexahydrofuro[2,3-b]furan-3-yl) carbamate (25h)
The titled inhibitor was synthesized using the general procedure outlined above. The desired inhibitor 25h was obtained as an oil (56% yield). Rf = 0.38 (50% ethyl acetate/hexanes). 1H NMR (500 MHz, CDCl3) δ 7.72 (m, 2H), 7.29 (m, 3H), 7.22 (m, 2H), 6.97 (d, J = 8.8 Hz, 2H), 5.80 (m, 1H), 5.13 (m, 1H), 4.04 (m, 1H), 3.91 (d, J = 9.7 Hz, 2H), 3.87 (s, 3H), 3.84 (bs, 2H), 3.69 (m, 1H), 3.19–2.87 (m, 8H), 2.76 (dd, J = 13.3 Hz, J = 6.6 Hz, 2H), 1.80 (m, 1H), 1.45 (s, 9H), 0.90 (d, J = 6.6 Hz, 3H), 0.86 (d, J = 6.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 163.2, 155.5, 155.0, 137.7, 129.9, 129.7, 129.6, 128.7, 126.8, 114.5, 109.1, 80.3, 73.6, 72.6, 72.4, 70.8, 58.9, 55.8, 55.2, 53.8, 35.4, 28.6, 27.4, 20.3, 20.0, 15.8; LRMS-ESI (m/z) 728.6 [M + Na]+; HRMS-ESI (m/z) [M + Na]+ calcd for (C35H51N3O10S), 728.3193; found, 728.3186.
((3R,3aS,4R,6aR)-4-(Ethylamino)hexahydrofuro[2,3-b]furan-3-yl (2S,3R)-3-hydroxy-4-(N-isobutyl-4-methoxyphenylsulfonamido)-1-phenylbutan-2-ylcarbamate (25i)
The titled inhibitor was obtained over two steps. First the N-Boc protected intermediate was obtained following the general procedure outlined above. The N-Boc protected intermediate to inhibitor 25i was dissolved in a solution of CH2Cl2/TFA (4:1) and stirred at 23 °C for 2 h. The reaction mixture was concentrated and the residue was purified by flash chromatography. Inhibitor 25i was obtained as an amorphous white solid (62% yield, 2 steps). 1H NMR (400 MHz, CD4OD) δ 7.77 (d, J = 8.9 Hz, 2H), 7.28 (bs, 4H), 7.23 – 7.20 (m, 1H), 7.08 (d, J = 8.9 Hz, 2H), 5.73 (d, J = 5.2 Hz, 1H), 5.09 – 5.04 (m, 1H), 4.08 (d, J = 11.5 Hz, 1H), 4.00 – 3.93 (m, 3H), 3.87 (s, 3H), 3.85 – 3.82 (m, 1H), 3.81 – 3.71 (m, 2H), 3.50 (dd, J = 15.0, 3.8 Hz, 1H), 3.20 – 3.18 (m, 1H), 3.16 – 3.06 (m, 4H), 2.91 – 2.78 (m, 3H), 2.60 – 2.52 (m, 2H), 2.03 – 1.99 (m, 1H), 1.19 (t, J = 7.2 Hz, 3H), 0.90 (d, J = 5.5 Hz, 4H), 0.85 (d, J = 6.6 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 163.1, 155.3, 137.9, 129.4, 128.5, 126.7, 114.3, 108.5, 72.1, 71.1, 60.3, 58.7, 58.4, 55.6, 53.2, 48.7, 41.4, 35.1, 29.6, 27.1, 21.0, 20.0, 19.8, 14.1. Mass: HRMS (ESI), Calcd for: C30H43N3O8S m/z 606.2849 (M+H), found m/z 606.2849 (M+H).
(3R,3aS,4R,6aR)-4-(Isopropylamino)hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)carbamate (25j)
The titled inhibitor was synthesized using the general procedure outlined above. The desired inhibitor 25j was obtained as an amorphous white solid (73% yield).). Rf = 0.4 (4% MeOH/CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.69 (d, J = 9.0 Hz, 2H), 7.32–7.26 (m, 2H), 7.25–7.19 (m, 3H), 6.98 (d, J = 9.0 Hz, 2H), 5.73 (d, J = 5.6, 1H), 5.11 (q, J = 14.0, 7.1 Hz, 1H), 5.0 (d, J = 8.1 Hz, 1H), 4.0–3.93 (m, 1H), 3.92–3.82 (m, 3H), 3.87 (s, 3H), 3.79–3.68 (m, 3H), 3.27–3.23 (m, 1H), 3.17–3.08 (m, 1H), 3.05–2.87 (m, 5H), 2.82–2.69 (m, 3H), 1.87–1.76 (m, 1H), 0.99 (d, J = 6.2 Hz, 3H), 0.96 (d, J = 6.2 Hz, 3H), 0.91 (d, J = 6.6 Hz, 3H), 0.87 (d, J = 6.6 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 163.3, 155.7, 137.6, 129.9, 129.7, 129.6, 128.8, 126.9, 114.6, 108.9, 75.1, 73.1, 72.8, 71.3, 59.0, 56.1, 55.8, 55.4, 53.8, 52.3, 46.3, 35,5, 27.5, 23.3, 23.1, 20.3, 20.1; HRMS-ESI (m/z) [M + H]+ calcd for (C31H45N3O8S), 620.3006; found, 620.2996.
(3R,3aS,4R,6aR)-4-(Dimethylamino)hexahydrofuro[2,3-b]furan-3-yl (2S,3R)-3-hydroxy-4-(N-isobutyl-4-methoxyphenylsulfonamido)-1-phenylbutan-2-ylcarbamate (25k)
Inhibitor 25k (27 mg, 0.05 mmol) was dissolved in dichloroethane (2 mL) and paraformaldehyde (powder, 60 mg, excess), NaBH3CN (50 mg, 0.23 mmol) and AcOH (20 μL) were added sequentially. The reaction mixture was stirred at 23 °C for 24 h. The reaction was diluted with CH2Cl2 and washed with sat. NaHCO3. The organic layer was combined dried over magnesium sulfate and concentrated under vacuum and purified over silica gel. The desired inhibitor 25k was obtained as an amorphous white solid (50% yield). Rf = 0.33 (10% methanol/CH2Cl2). 1H NMR (300 MHz, Chloroform-d) δ 7.71 (d, J = 8.8 Hz, 2H), 7.38 – 7.12 (m, 5H), 6.99 (d, J = 8.8 Hz, 2H), 5.68 (d, J = 5.3 Hz, 1H), 5.18 – 5.13 (m, 1H), 5.04 (d, J = 8.4 Hz, 1H), 4.06 – 3.78 (m, 8H), 3.78 – 3.57 (m, 1H), 3.18 – 2.76 (m, 8H), 2.07 (s, 6H), 1.93 – 1.69 (m, 1H), 0.89 (dd, J = 12.8, 6.6 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 163.1, 155.2, 137.5, 129.6, 129.4, 129.3, 128.6, 126.7, 114.4, 109.2, 77.2, 72.8, 72.7, 72.6, 70.5, 64.7, 58.8, 55.6, 55.2, 53.7, 45.4, 41.4, 35.3, 27.3, 20.1, 19.8. Mass: HRMS (ESI), Calcd for C30H43N3O8S: m/z 606.2849 (M+Na), found m/z 606.2849 (M+Na).
(3R,3aS,4R,6aR)-4-(Diethylamino)hexahydrofuro[2,3-b]furan-3-yl (2S,3R)-3-hydroxy-4-(N-isobutyl-4-methoxyphenylsulfonamido)-1-phenylbutan-2-ylcarbamate (25l)
Inhibitor 25b (27 mg, 0.05 mmol) was dissolved in dichloroethane (2 mL) and acetaldehyde (10 μL, excess), NaBH3CN (50 mg, 0.23 mmol) and AcOH (20 μL) were added sequentially. The reaction mixture was stirred at 23 °C for 24 h. The reaction was diluted with CH2Cl2 and washed with sat. NaHCO3. The organic layer was combined dried over magnesium sulfate and concentrated under vacuum and purified over silica gel. The desired inhibitor 25l was obtained as an amorphous white solid (53% yield). Rf = 0.38 (10% methanol/CH2Cl2). 1H NMR (300 MHz, Chloroform-d) δ 7.69 (d, J = 8.8 Hz, 2H), 7.39 – 7.13 (m, 5H), 6.98 (d, J = 8.9 Hz, 2H), 5.69 (d, J = 5.3 Hz, 1H), 5.17 (d, J = 8.3 Hz, 1H), 5.02 (d, J = 8.2 Hz, 1H), 4.01 – 3.91 (m, 3H), 3.87 (s, 5H), 3.65 (dd, J = 9.5, 6.4 Hz, 1H), 3.56 (d, J = 6.0 Hz, 1H), 3.23 – 3.04 (m, 1H), 3.04 – 2.71 (m, 7H), 2.54 -2.45 (m, 2H), 2.28 (m, 2H), 1.94 – 1.72 (m, 1H), 0.99 (t, J = 7.1 Hz, 6H), 0.88 (dd, J = 11.3, 6.6 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 163.0, 155.3, 137.3, 129.6, 129.4, 129.3, 128.6, 126.7, 114.3, 109.3, 73.4, 73.0, 72.4, 70.7, 60.7, 58.7, 55.6, 55.0, 53.6, 46.7, 43.9, 35.1, 27.2, 20.1, 19.9, 13.5. Mass: HRMS (ESI), Calcd for C32H47N3O8S: m/z 634.3162 (M+Na), found m/z 634.3152 (M+Na).
Methods: Determination of X-ray structures of HIV-1 protease-inhibitor complexes
The optimized HIV-1 protease was expressed and purified as described.39 The protease-inhibitor complex was crystallized by the hanging drop vapor diffusion method with well solutions containing sodium chloride and sodium acetate buffer at pH 5. Diffraction data were collected on a single crystal cooled to 90 K at SER-CAT (using the 22-ID beamline for inhibitor 25f and the 22-BM beamline for inhibitor 25g, Advanced Photon Source, Argonne National Lab (Chicago, USA) with X-ray wavelengths of 0.8 and 1.0 Å, respectively. X-ray data were processed by HKL-2000 with Rmerge values of 5.7% and 7.7%, respectively.40 Using the isomorphous structure41 as an initial model, the crystal structures were solved with PHASER42 in CCP4i Suite,43,44 and refined with SHELX-97,45 to 1.34 Å and 1.29 Å resolution, respectively, for the complexes with inhibitor 25f and inhibitor 25g. COOT,46 was used for manual modification of the structures. PRODRG-2,47 was used to construct the inhibitor and the restraints for refinement. Alternative conformations were modeled when visible in the electron density maps, and isotropic atomic displacement parameters (B factors) were applied for all atoms including solvent molecules. The final refined solvent structure comprised two sodium ions, three chloride ions and 147 water molecules for the protease complex with inhibitor 25f, and one sodium ion, two chloride ions, three acetate ions and 137 water molecules for the complex with inhibitor 25g. The crystallographic statistics are listed in the supplementary material section. The coordinates and structure factors of HIV-1 protease with inhibitor 25f and inhibitor 25g structures have been deposited in Protein Data Bank with code 5BRY and 5BS4, respectively.
Acknowledgments
This research was supported by the National Institutes of Health (Grant GM53386, AKG and Grant GM62920, ITW). X-ray data were collected at the Southeast Regional Collaborative Access Team (SER-CAT) beamline 22BM at the Advanced Photon Source, Argonne National Laboratory. Use of the Advanced Photon Source was supported by the US Department of Energy, Basic Energy Sciences, Office of Science, under Contract No. W-31-109-Eng-38. This work was also supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health, and in part by a Grant-in-Aid for Scientific Research (Priority Areas) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (Monbu Kagakusho), a Grant for Promotion of AIDS Research from the Ministry of Health, Welfare, and Labor of Japan, and the Grant to the Cooperative Research Project on Clinical and Epidemiological Studies of Emerging and Reemerging Infectious Diseases (Renkei Jigyo) of Monbu-Kagakusho. The authors would like to thank the Purdue University Center for Cancer Research, which supports the shared NMR and mass spectrometry facilities.
Abbreviations
- THF
tetrahydrofuran
- bis-THF
bis-tetrahydrofuran
- tris-THF
tris-tetrahydrofuran
- PI
protease inhibitor
- CNS
central nervous system
- TFA
trifluoroacetic acid
- DRV
darunavir
- APV
amprenavir
Footnotes
Supporting Information: HPLC and HRMS data of inhibitors 25a–25l. Crystallographic data collection and refinement statistics for inhibitors 25f and 25g-bound HIVP X-ray structures.
The PDB accession codes for 25f and 25g-bound HIVP X-ray structures are 5BRY and 5BS4
References
- 1.Mitsuya H, Broder S. Strategies for antiviral therapy in AIDS. Nature. 1987;325:773–778. doi: 10.1038/325773a0. [DOI] [PubMed] [Google Scholar]
- 2.Kohl NE, Emini EA, Schleif WA, Davis LJ, Heimbach JC, Dixon RAF, Scolnick EM, Sigal IS. Active human immunodeficiency virus protease is required for viral infectivity. Proc Natl Acad Sci USA. 1988;85:4686–4690. doi: 10.1073/pnas.85.13.4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Montaner JSG, Lima VD, Barrios R, Yip B, Wood E, Kerr T, Shannon K, Harrigan PR, Hogg RS, Daly P, Kendall P. Association of highly active antiretroviral therapy coverage, population viral load, yearly new HIV diagnoses in British Columbia, Canada: a population-based study. Lancet. 2010;376:532–539. doi: 10.1016/S0140-6736(10)60936-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Braitstein P, Brinkhof MWG, Dabis F, Schechter M, Boulle A, Miotti P, Wood R, Laurent C, Sprinz E, Seyler C, Bangsberg DR, Balestre E, Sterne JAC, May M, Egger M, Grp A-L, Grp A-C. Mortality of HIV-1-infected patients in the first year of antiretroviral therapy: comparison between low-income and high-income countries. Lancet. 2006;367:817–824. doi: 10.1016/S0140-6736(06)68337-2. [DOI] [PubMed] [Google Scholar]
- 5.Este JA, Cihlar T. Current status challenges of antiretroviral research therapy. Antiviral Res. 2010;85:25–33. doi: 10.1016/j.antiviral.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 6.Waters L, Nelson M. Why do patients fail HIV therapy? Int J Clin Prac. 2007;61:983–990. doi: 10.1111/j.1742-1241.2007.01383.x. [DOI] [PubMed] [Google Scholar]
- 7.Moyle G, Back D. Principles practice of HIV-protease inhibitor pharmacoenhancement. HIV Med. 2001;2:105–113. doi: 10.1046/j.1468-1293.2001.00063.x. [DOI] [PubMed] [Google Scholar]
- 8.Kumar GN, Dykstra J, Roberts EM, Jayanti VK, Hickman D, Uchic J, Yao Y, Surber B, Thomas S, Granneman GR. Potent inhibition of the cytochrome P-450 3A-mediated human liver microsomal metabolism of a novel HIV protease inhibitor by ritonavir: A positive drug-drug interaction. Drug Metab Dispos. 1999;27:902–908. [PubMed] [Google Scholar]
- 9.van Sighem AIvdW, Mark A, Ghani Azra C, Jambroes Mariëlle, Reiss Peter, Gyssens Inge C, Brinkman Kees, Lange Joep MA, de Wolf Frank. Mortality progression to AIDS after starting highly active antiretroviral therapy. AIDS. 2003;17:2227–2326. doi: 10.1097/00002030-200310170-00011. [DOI] [PubMed] [Google Scholar]
- 10.Staszewski S, Morales-Ramirez J, Tashima KT, Rachlis A, Skiest D, Stanford J, Stryker R, Johnson P, Labriola DF, Farina D, Manion DJ, Ruiz NM. Efavirenz plus zidovudine lamivudine, efavirenz plus indinavir, and indinavir plus zidovudine lamivudine in the treatment of HIV-1 infection in adults. Study 006 Team. N Eng J Med. 1999;341:1865–1873. doi: 10.1056/NEJM199912163412501. [DOI] [PubMed] [Google Scholar]
- 11.Wensing AM, van Maarseveen NM, Nijhuis M. Fifteen years of HIV Protease Inhibitors: raising the barrier to resistance. Antiviral Res. 2010;85:59–74. doi: 10.1016/j.antiviral.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 12.Ghosh AK, Ramu Sridhar P, Kumaragurubaran N, Koh Y, Weber IT, Mitsuya H. Bis-tetrahydrofuran: a privileged ligand for darunavir a new generation of hiv protease inhibitors that combat drug resistance. ChemMedChem. 2006;1:939–950. doi: 10.1002/cmdc.200600103. [DOI] [PubMed] [Google Scholar]
- 13.Ghosh AK, Sridhar PR, Leshchenko S, Hussain AK, Li J, Kovalevsky AY, Walters DE, Wedekind JE, Grum-Tokars V, Das D, Koh Y, Maeda K, Gatanaga H, Weber IT, Mitsuya H. Structure-based design of novel HIV-1 protease inhibitors to combat drug resistance. J Med Chem. 2006;49:5252–5261. doi: 10.1021/jm060561m. [DOI] [PubMed] [Google Scholar]
- 14.Ghosh AK, Chapsal BD. Anti-HIV protease inhibitor Darunavir. In: Ganellin CR, Jefferis R, Roberts S, editors. Introduction to Biopharmaceuticals and Small Molecule Drug Research and Development: theory and case studies. Elsevier; Waltham, MA: 2013. pp. 355–384. [Google Scholar]
- 15.Ghosh AK, Dawson ZL, Mitsuya H. Darunavir a conceptually new HIV-1 protease inhibitor for the treatment of drug-resistant HIV. Bioorg Med Chem. 2007;15:7576–7580. doi: 10.1016/j.bmc.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghosh AK, Chapsal BD, Mitsuaya H. In: Aspartic Acid Proteases as Therapeutic Targets. Ghosh AK, editor. Vol. 45. Wiley-VCH; Weinheim: 2010. p. 205. [Google Scholar]
- 17.Koh Y, Nakata H, Maeda K, Ogata H, Bilcer G, Devasamudram T, Kincaid JF, Boross P, Wang YF, Tie Y, Volarath P, Gaddis L, Harrison RW, Weber IT, Ghosh AK, Mitsuya H. Novel bis-tetrahydrofuranylurethane-containing nonpeptidic protease inhibitor (PI) UIC-94017 (TMC114) with potent activity against multi-PI-resistant human immunodeficiency virus in vitro. Antimicrob Agents Chemother. 2003;47:3123–3129. doi: 10.1128/AAC.47.10.3123-3129.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Meyer SAH, Surleraux D, Jochmans D, Tahri A, Pauwels R, Wigerinck P, de Bethune M. TMC 114, a novel human immunodeficiency virus type 1 protease inhibitor active against protease inhibitor-resistant viruses, including a broad range of clinical isolates. Antimicrob Agents Chemother. 2005;49:2314–2321. doi: 10.1128/AAC.49.6.2314-2321.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tie Y, Boross PI, Wang YF, Gaddis L, Hussain AK, Leshchenko S, Ghosh AK, Louis JM, Harrison RW, Weber IT. High resolution crystal structures of HIV-1 protease with a potent non-peptide inhibitor (UIC-94017) active against multi-drug-resistant clinical strains. J Mol Biol. 2004;338:341–352. doi: 10.1016/j.jmb.2004.02.052. [DOI] [PubMed] [Google Scholar]
- 20.Ghosh AK, Anderson DD, Weber IT, Mitsuya H. Enhancing protein backbone binding–a fruitful concept for combating drug-resistant HIV. Angew Chem Int Ed. 2012;51:1778–1802. doi: 10.1002/anie.201102762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghosh AK, Chapsal BD, Weber IT, Mitsuya H. Design of HIV protease inhibitors targeting protein backbone: an effective strategy for combating drug resistance. Acc Chem Res. 2008;41:78–86. doi: 10.1021/ar7001232. [DOI] [PubMed] [Google Scholar]
- 22.Ghosh AK, Martyr CD, Steffey M, Wang YF, Agniswamy J, Amano M, Weber IT, Mitsuya H. Design of substituted bis-tetrahydrofuran (bis-THF)-derived potent HIV-1 protease inhibitors, protein-ligand X-ray structure, and convenient syntheses of bis-THF substituted bis-THF ligands. ACS Med Chem Lett. 2011;2:298–302. doi: 10.1021/ml100289m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghosh AK, Yashchuk S, Mizuno A, Chakraborty N, Agniswamy J, Wang YF, Aoki M, Gomez PM, Amano M, Weber IT, Mitsuya H. Design of gem-difluoro-bis-tetrahydrofuran as P2 ligand for HIV-1 protease inhibitors to improve brain penetration: synthesis, X-ray studies, and biological evaluation. ChemMedChem. 2015;10:107–115. doi: 10.1002/cmdc.201402358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Salcedo Gomez PM, Amano M, Yashchuk S, Mizuno A, Das D, Ghosh AK, Mitsuya H. GRL-04810 and GRL-05010, difluoride-containing nonpeptidic HIV-1 protease inhibitors (PIs) that inhibit the replication of multi-PI-resistant HIV-1 in vitro and possess favorable lipophilicity that may allow blood-brain barrier penetration. Antimicrob Agents Chemother. 2013;57:6110–6121. doi: 10.1128/AAC.01420-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Al-Hakim AH, Haines AH, Morley C. Synthesis of 1,2-O-isopropylidene-L-threitol and its conversion to (R)-1,2-O-isopropylideneglycerol. Synthesis. 1985;1985:207–208. [Google Scholar]
- 26.Dueno EE, Chu F, Kim S-I, Jung KW. Cesium promoted O-alkylation of alcohols for the efficient ether synthesis. Tetrahedron Lett. 1999;40:1843–1846. [Google Scholar]
- 27.Bruckner R, Priepke H. Asymmetric induction in the [2,3]-Wittig rearrangement by chiral substituents in the allyl moiety. Angew Chem Int Ed. 1988;27:278–280. [Google Scholar]
- 28.Brückner R. Asymmetric induction and simple diastereoselectivity in the [2,3]-Wittig rearrangement of ester enolates. Chem Ber. 1989;122:703–710. [Google Scholar]
- 29.Swamy KCK, Kumar NNB, Balaraman E, Kumar KVP. Mitsunobu and related reactions: advances and applications. Chem Rev. 2009;109:2551–2651. doi: 10.1021/cr800278z. [DOI] [PubMed] [Google Scholar]
- 30.Ghosh AK, Bischoff A. A convergent synthesis of (+)-cryptophycin B, a potent antitumor macrolide from Nostoc sp. cyanobacertia. Organic Lett. 2000;2:1573–1575. doi: 10.1021/ol000058i. [DOI] [PubMed] [Google Scholar]
- 31.Toth MV, Marshall GR. A simple, continuous fluorometric assay for HIV protease. Int J Pept Protein Res. 1990;36:554–550. doi: 10.1111/j.1399-3011.1990.tb00994.x. [DOI] [PubMed] [Google Scholar]
- 32.Tie Y, Boross PI, Wang YF, Gaddis L, Hussain AK, Leshchenko S, Ghosh AK, Louis JM, Harrison RW, Weber IT. High resolution crystal structures of HIV-1 protease with a potent non-peptide inhibitor (UIC-94017) active against multi-drug-resistant clinical strains. J Mol Biol. 2004;338:341–352. doi: 10.1016/j.jmb.2004.02.052. [DOI] [PubMed] [Google Scholar]
- 33.Miller MM, Liedtke MD, Lockhart SM, Rathbun RC. The role of dolutegravir in the management of HIV infections. Infect Drug Resist. 2015;8:19–29. doi: 10.2147/IDR.S58706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mitsuya Y, Lie TF, Rhee SY, Fessel WJ, Shafter RW. Prevalence of darunavir resistance-associated mutations: patterns of occurrence and association with past treatment. J Infect Dis. 2007;196:1177–1179. doi: 10.1086/521624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koh Y, Amano M, Towata T, Danish M, Leshchenko-Yashchuck S, Das D, Nakayama M, Tojo Y, Ghosh AK, Mitsuya H. In vitro selection of highly darunavir-resistant-competent HIV-1 variants by using a mixture of clinical HIV-1 isolates resistant to multiple conventional protease inhibitors. J Virol. 2010;84:11961–11969. doi: 10.1128/JVI.00967-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gatanaga H, Suzuki Y, Tsang H, Yoshimura K, Kavlick MF, Mardy S, Gorelick RJ, Tang C, Summers MF, Mitsuya H. Amino acid substitutions in non cleavage sites of the gag region are indispensable for high level HIV-1 resistance to protease inhibitors. J Biol Chem. 2002;277:5952–5961. doi: 10.1074/jbc.M108005200. [DOI] [PubMed] [Google Scholar]
- 37.Koh Y, Aoki M, Danish ML, Aoki-Ogata H, Amano M, Das D, Shafer RW, Mitsuya H. Loss of protease dimerization inhibition activity of darunavir is associated with the acquistion of resistance to darunavir by HIV-1. J Virol. 2011;85:10079–10089. doi: 10.1128/JVI.05121-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hayashi H, Takamune N, Nirasawa T, Aoki M, Morishita Y, Das D, Koh Y, Ghosh AK, Misumi S, Mitsuya H. Dimerization of HIV-1 protease occurs through two steps relating to the mechanism of protease dimerization inhibition by darunavir. Proc Natl Acad Sci. 2014;79:5697–5709. doi: 10.1073/pnas.1400027111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mahalingam B, Louis JM, Hung J, Harrison RW, Weber IT. Structural implications of drug resistant mutants of HIV-1 protease: high resolution crystal structures of the mutant protease/substrate analog complexes. Proteins. 2001;43:455–464. doi: 10.1002/prot.1057. [DOI] [PubMed] [Google Scholar]
- 40.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Carter CW Jr, Sweet RM, editors. Methods in Enzymology. Vol. 276. Academic Press; New York: 1997. pp. 307–326. (Macromolecular Crystallography, Part A). [DOI] [PubMed] [Google Scholar]
- 41.Shen C-H, Wang Y-F, Kovalevsky AY, Harrison RW, Weber IT. Amprenavir complexes with HIV-1 protease and its drug-resistant mutants altering hydrophobic clusters. FEBS J. 2010;277:3699–3714. doi: 10.1111/j.1742-4658.2010.07771.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr Sect D: Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Potterton E, Briggs P, Turkenburg M, Dodson E. A graphical user interface to the CCP4 program suite. Acta Crystallogr Sect D: Biol Crystallogr. 2003;59:1131–1137. doi: 10.1107/s0907444903008126. [DOI] [PubMed] [Google Scholar]
- 45.Sheldrick GM. A short history of SHELX. Acta Crystallogr Sect A: Found Crystallogr. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 46.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr Sect D: Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schuettelkopf AW, van Aalten DMF. PRODRG;a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr Sect D: Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]