

Abstract
Increased intracellular iron levels can both promote cell proliferation and death, as such; iron has a “two-sided effect” in the delicate balance of human health. Though the role of iron in the development of cancer remains unclear, investigations of iron chelators as anti-tumor agents have revealed promising results. Here, we investigated the influence of iron and desferrioxamine (DFO), the iron chelating agent on intracellular calcium in a human leukemia cell line, K562. Iron uptake is associated with increased reactive oxygen species (ROS) generation. Therefore, we showed that iron also caused dose-dependent ROS generation in K562 cells. The measurement of intracellular calcium was determined using Furo-2 with a fluorescence spectrophotometer. The iron delivery process to the cytoplasmic iron pool was examined by monitoring the fluorescence of cells loaded with calcein-acetoxymethyl. Our data showed that iron increased intracellular calcium, and this response was 8 times higher when cells were incubated with DFO. K562 cells with DFO caused a 3.5 times increase of intracellular calcium in the presence of doxorubicin (DOX). In conclusion, DFO induces intracellular calcium and increases their sensitivity to DOX, a chemotherapeutic agent.
KEY WORDS: Intracellular calcium, iron uptake, doxorubicin, desferrioxamine
INTRODUCTION
Calcium ion (Ca2+) is referred as a universal signaling molecule and known to regulate many aspects of cellular function, of which apoptotic signaling pathways are some of the most important [1]. Calcium stores are mobilized by some agents, including the endoplasmic reticulum (ER) Ca2+-ATPase inhibitor thapsigargin (TG), glucocorticoids, and various cancer therapeutic drugs, and early transient elevation of intracellular free calcium ([Ca2+]i) triggers to apoptosis. Although the pharmacology of most chemotherapy is still effective on them, the therapeutic success in cancer treatment today is to trigger tumor-specific cell death. Intracellular calciumsignals may have a major effect on the initiation of cell death. An intracellular calcium concentration overload and a disturbance in calcium homeostasis may cause cytotoxicity and consequently also trigger apoptosis [1].
It is well-known that iron is involved in many metabolic processes in cells such as synthesis of hemoglobin and DNA [2], cell cycling, and some important enzymes [3]. Iron overload in the human body can thereby lead to cell death and tissue damage by iron-catalyzed Fenton chemistry [4]. Cellular iron metabolism and its epigenetic regulation are well-balanced and tightly controlled in normal cells [5]. However, iron is involved in a redox reaction, oxidation-reduction reactions, and causes the formation of free radicals [6-8]. Therefore, iron considerably contributes to the onset and growth of cancer. Torti and Torti showed that iron has an effective role in the microenvironmental regulation of tumor progression and metastasis [8]. Changing the pathways of iron uptake, storage, efflux, and metabolic regulation in cancer cells shows us that reprogramming of iron metabolism is an important target for the viability of cancer cells [8].
Targeted studies on the metabolic pathways of iron may provide new tools for prognosis and treatment of cancer [9]. In the previous studies, it has been reported that effect of chelating agents on iron metabolism as anti-tumor agents in cancer cells [9,10]. This anti-tumor effect is associated with G1/S cell cycle arrest, induction of apoptosis [10,11], initiation of DNA damage and inhibition of DNA synthesis. Desferrioxamine (DFO) used in numerous in vivo and in vitro investigations has also been used clinical trials. The action of anti-tumor of DFO is still being studied.
In this study, we hypothesized that in addition to the well-investigated molecular mechanisms for iron and the action of an iron chelator, DFO, their activity may be associated with regulating intracellular calcium. We used K562 cells to study the effect of iron addition or iron chelation on intracellular Ca2+ changes. Our results indicate that iron is essential to elicit a Ca2+ signal and that intracellular iron concentrations determine the Ca2+ response. K562 cells with DFO causes [Ca2+]i to rise with much smaller doses following the administration of iron. The treatment with DFO can enhance the sensitivity of cancer cells to the chemotherapeutic agent, doxorubicin (DOX).
MATERIALS AND METHODS
Cell line and chemicals
K562 (human erytroleukemia cell line) cells were obtained from the Istanbul Medical Faculty Biophysics Department and maintained in Dulbecco’s Modified Eagles medium F-12 supplemented with 10% heat-inactivated fetal calf serum and 100 IU/mL penicillin-streptomycin. Incubation conditions at 37°C in a humidified atmosphere of 5% CO2 were maintained in a Heraeus incubator (Hanau, Germany). Calcein acetoxymethyl ester (CA-AM) was purchased from Setareh Biotech. TG, ethylene glycol-bis(β-aminoethyl ether)-N,N,N’,N’-tetraacetic acid (EGTA), dihydroethidium, Furo-2 AM, ryanodine, 2-ABP, DOX, and DFO were from Sigma.
Trypan blue cytotoxicity test
We tested the sensibility of the cytotoxicity test using Trypan blue exclusion. Non-confluent (70-80% confluency) cell monolayer cells were exposed to iron and DFO for 24 hours for the determination of cell viability [12]. The cells were counted at 24 hours, and the cytotoxic dose of iron and DFO was calculated. Cell viability (%) was calculated using the equation:
Number of viable cells ÷ total number of cells × 100
Flow cytometry analysis
To determine iron uptake, cells were loaded with 0.5 µM CA-AM at 37°C for 30 minutes, in accordance with the literature [13]. Excess CA-AM on the surface was removed by washing with Hank’s balanced salt solution (HBSS: 20 mM Hepes, 10 mM glucose, 150 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1 mM Na2HPO4, pH 7.4); the cells were then resuspended in HBSS medium at a density of 106 cells/mL. CA fluorescence (excitation, 488 nm; emission, 517 nm) in CA-AM loaded cells was analyzed using a BD Aria flow cytometer (BD Bioscience, San Jose, CA, USA). Gates were set up to exclude non-viable cells and debris. The negative fraction was determined using appropriate isotype controls.
Apoptotic cell death was examined using the Annexin V-FITC Apoptosis Detection Kit (BD Bioscience, CA, USA). K562 cells were incubated with iron DFO (10, 25, 50, 75, and 100 µM) for 24 hours. All cells were washed with cold phosphate-buffered saline (PBS). The pellet was resuspended in 100 µL of ×1 binding buffer at a density of 1 × 105 cells/mL. The cells were incubated with 5 µL of FITC-conjugated Annexin-V and 5 µg/mL of PI for 15 minutes in darkness. Then, 400 µL of ×1 binding buffer was added to each sample tube, and the samples were analyzed using FACS (Becton Dickinson, CA, USA) and quantified using Cell Quest software (Becton Dickinson, San Jose, CA).
Reactive oxygen species (ROS) detection
Cellular ROS was measured using the fluorescence spectrophotometer (Perkin Elmer, LS45; λex = 392, λem = 410) with dihydroethidium (DHE) as a fluorescent probe. DHE, by virtue of its ability to freely permeate cell membranes, was used extensively to monitor ROS production [14,15]. The cells were incubated with 10 µM DHE for 15 minutes at 37°C, washed, resuspended in PBS, and then analyzed using the fluorescence spectrophotometer.
Measurement of intracellular calcium with Fura-2
To determine [Ca2+]i, 2.106 cells were rinsed with HBSS. They were then loaded with 0.5 mL of 5 µM Furo-2AM, dissolved in HBSS from a stock solution of 1.5 mM in DMSO containing 20% pluronic acid F-127 at room temperature for 30 minutes, and subsequently rinsed twice with HBSS. A Perkin-Elmer fluorescence spectrophotometer was used for the fluorometric measurement of Ca2+ with LS45 (λex = 340 and 380, λem = 510). Rmax and Rmin values of the fluorescence ratio were obtained with the addition of 10 µM ionomycin and 4 mM EGTA. We calibrated the fluorescence signal using the Grynkiewicz’s equation [16].
Statistical methods
Data are shown as means with standard deviation; at least three replicates were done for each experimental setup. Statistical significance between control conditions and each of the exposure samples was calculated with Student’s t-test by SPSS for Windows.
RESULTS
Cytotoxic effect of iron, iron uptake, and ROS generation
We had previously tested the sensibility of cytotoxicity test using Trypan blue exclusion. Compared with untreated cells, the cells’ viability was not significantly affected by each iron dose (Figure 1A). Flow cytometry analysis was used for measurements of iron uptake. Figure 1B depicts fluorescence intensity histograms of a representative experiment where K562 cells were first treated with iron for 1 hour and then loaded with CA-AM. After each dose of iron, melt flow index (MFI) value was reduced in a dose-dependent manner due to quenching (Figure 1B). The data for each dose were reported as the percent change in the MFI following incubation with iron, calculated as a percentage of the basal fluorescence (100 × [MFI−Fe]–[MFI+Fe]/[MFI−Fe]) [16]. After the treatment of iron, the graph data showed that iron uptake at the concentrations of 5, 10, and 20 µΜ significantly increased (Figure 1B). We showed that iron caused dose-dependent ROS generation in K562 cells because iron uptake is associated with increased ROS generation (Figure 1C).
FIGURE 1.
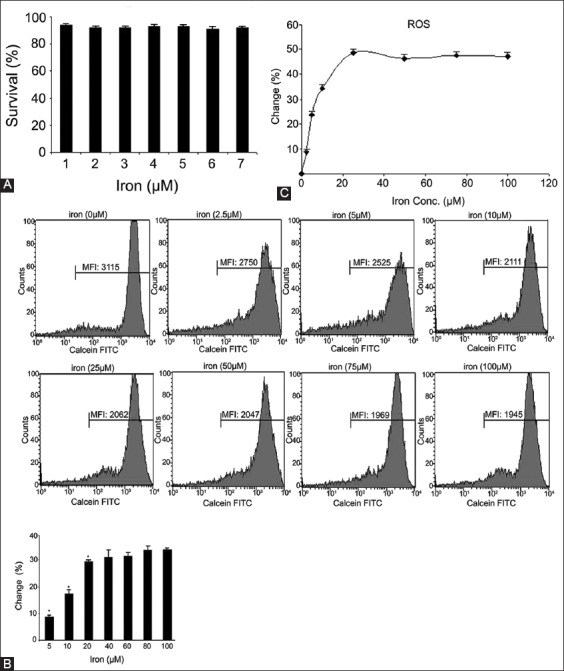
(A) Cytotoxic effect of iron at different concentrations in K562 cells. The cells were treated with iron (0, 5, 10, 20, 60, 80, and 100 µM) for 24 hours. Results are expressed as the mean percentage of control (mean ± standard deviation [SD], n = 3), (B) Flow cytometry measurements of iron uptake in K562 cells. The cells were loaded with 0.5 µM calcein-acetoxymethyl (CA-AM) at 37°C for 30 minutes. Excess CA-AM on the surface was removed by washes with Hank’s balanced salt solution; the cells were resuspended and incubated with iron at different concentrations for 1 hour. The data for each dose were calculated as the percent change in the melt flow index following incubation with iron. Iron uptake at concentrations of 5, 10, 20 µΜ significantly increased (*p < 0.05), (C) Flow cytometry measurements of reactive oxygen species in K562 after adding iron at above concentrations (means ± SD, n = 3).
Cytotoxic effect of DFO and detection of intracellular iron using DFO
Compared with untreated cells, the cell viability was not significantly affected by the iron chelating agent DFO, at the doses used in this study (Figure 2A). We speculated whether DFO would induce apoptosis of K562 cells at the doses used (Figure 2A). Therefore, we analyzed apoptosis using Annexin V after DFO treatment and showed that there was no significant apoptotic effect on the cells. DFO was also detected to chelate intracellular iron in the dose-dependent manner (Figure 2B). DFO was used to regenerate the fluorescence quenched by iron. Therefore, the differences in fluorescence reading with or without the addition of DFO positively increased due to chelation of intracellular iron.
FIGURE 2.
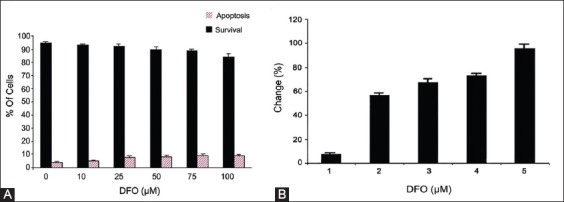
(A) Dose response of desferrioxamine (DFO) after 24 hours treatment in K562 cells. The trypan blue exclusion method and Annexin V Apoptosis Detection Kit were used as described in Materials and Methods. The data are shown as mean with standard deviations, where 100% represents the total number cells for each tested sample, (B) The chelation of intracelluar iron by DFO. K562 cells were incubated 24 hours with DFO at different concentrations (10, 25, 50, 75, 100 µΜ). The cells were loaded with 0.5 µM calcein-acetoxymethyl (CA-AM) at 37°C for 30 minutes. Excess CA-AM on the surface was removed by washes and resuspended with Hank’s balanced salt solution.
Effect of iron on intracellular Ca2+
To study, how dose-response iron uptake regulates intracellular Ca2+ levels, K562 cells without and with DFO were loaded with Furo-2. Figure 3A shows that the addition of iron (5-20 µM) to the bath solution did not induce Ca2+ elevations. Ca2+ elevation was observed above 40 µM iron concentration for Furo-2-loaded K562 cells without DFO. However, the addition of iron (5-60 µM) to Furo-2-loaded DFO-treated (100 µM) cells showed dose-dependent [Ca2+]i increase (Figure 3B). DFO-treated cells in calcium elevation appeared to be about 8-fold more sensitive in their response to iron. TG is a sarco endoplasmic reticulum calcium transport ATPase (SERCA) pump inhibitor whose action releases internal calcium from the sarcoplasmic reticulum. Figure 3C and D show [Ca2+]i of Furo-2 loaded K562 cells in the presence of TG (1 µΜ) (indicated by an arrow) of various concentrations of iron. As shown in Figure 3C and D, the dose-dependent increase of intracellular calcium was observed after adding increased concentrations iron to K562 cells with and without DFO, respectively. To examine, the role of [Ca2+]i in response to iron, the effect of EGTA, an extracellular Ca2+ chelator, was investigated. After treatment with EGTA to Furo-2-loaded K562 cells, [Ca2+]i dropped with the addition of iron (Figure 3E). Intracellular calcium started to increase with the addition of DFO (Figure 3E).
FIGURE 3.
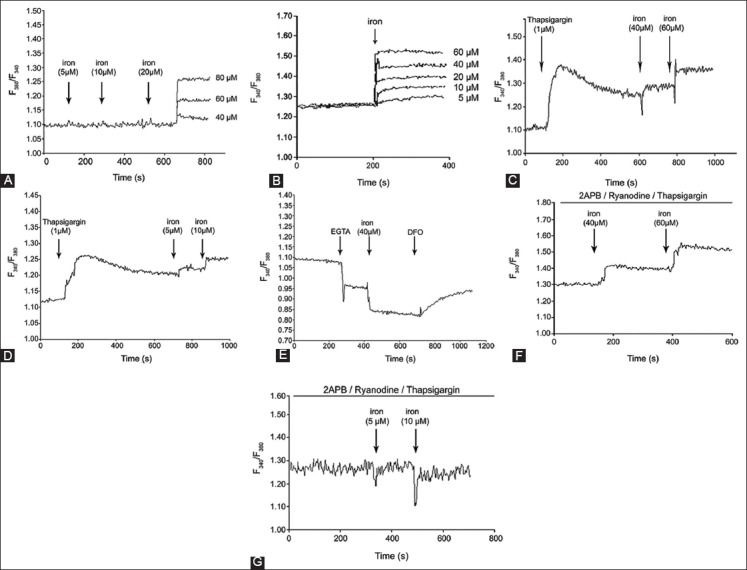
Effect of iron on the intracellular calcium of K562 cells. Iron uptake regulates intracellular Ca2+ levels, K562 cells without desferrioxamine (DFO) (A) and with DFO (100 µM) (B) were loaded with Furo-2, (C) shows [Ca2+]i of Furo-2 loaded K562 cells in presence of thapsigargin (1 µΜ) at various concentrations of iron in K562 cells without DFO and with DFO (D), (E) The effect of ethylene glycol-bis(β-aminoethyl ether)-N,N,N’,N’-tetraacetic acid (EGTA). After treatment with EGTA to Furo-2-loaded K562 cells, [Ca2+]i dropped with the addition of iron, (F) [Ca2+]i increased in K562 cells without DFO after addition of iron in presence of 2-aminoethoxydiphenyl borate (2-APB), ryanodine and thapsigargin, (G) [Ca2+]i did not increase in K562 cells treated with DFO after addition of iron in presence of 2-APB, ryanodine and thapsigargin.
2-Aminoethoxydiphenyl borate (2-APB), IP3Rs blocker, ryanodine, and TG did not inhibit Ca2+ elevation, and an increase was observed at 40 and 60 µM in K562 cells without DFO (Figure 3F). The addition of 2-APB, ryanodine, and TG into DFO-treated cells did not change intracellular Ca2+ levels at 5 and 10 µM (Figure 3G). DOX (200 nM) was applied to K562 cells without and with DFO. As shown in Figure 4, DOX significantly increased [Ca2+]i compared with cells that were not treated with DFO.
FIGURE 4.
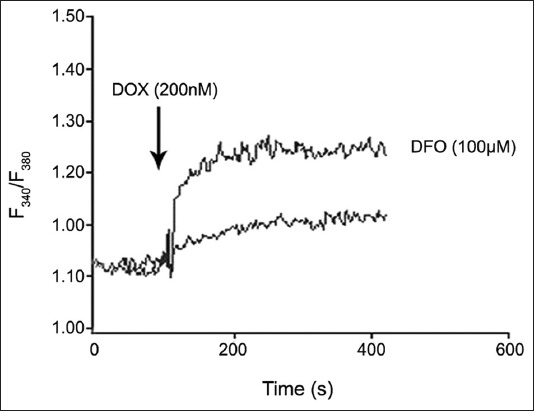
The effect of doxorubicin on intracellular calcium levels in K562 cells with and without desferrioxamine.
DISCUSSION
Recent studies have emphasized the importance of altering the iron requirement of cancer cells as a potential therapeutic strategy for overcoming cancer cell progression and increasing the efficacy of anti-cancer agents [11,17,18]. It is known that the iron requirement of cancer cells is increased, and recent evidence has demonstrated that high intracellular iron is a marker of poor prognosis in patients with breast cancer [19]. Several possible mechanisms have been reported for the anti-tumor effects of iron chelating agents, including prevention of ribonucleotide reductase activity [20], which is a rate-limiting enzyme in DNA synthesis; inhibition of cell cycle progression by inducing G1/S cell cycle arrest [21]; and inhibition of metastasis via the up-regulation of N-Myc downstream-regulated gene 1 [22]. The present study showed that the dose-dependent increase in [Ca2+]i was caused as a response to the addition of iron, both to K562 cells with and without DFO and the response was more sensitive in cells that were incubated with DFO against iron treatment. The treatment of various cancer cells with DFO has been shown in previous studies to induce apoptosis through the activation of various pro-apoptotic pathways [23-27]. Furthermore, the results of the present study demonstrate that the exposure of K562 cells to DFO induced a rise of [Ca2+]i that was more sensitive to the iron concentration than K562 cells without DFO. These results imply that this intracellular [Ca2+]i increase may promote apoptosis.
Fluorescent probes are very commonly used because of their excellent ROS sensitivity and simplicity in data collection. Of the small-molecule fluorescent ROS probes, 2’-7’-dichlorodihydrofluorescein (DCFH) and DHE are the most popular. However, recent studies show that the use of DCFH/DCFHDA in biologic systems with peroxidase activity may produce a number of misleading results because of probe-derived radicals that produce ROS [28]. Furthermore, it has been shown that cytochrome c is a powerful catalyst of DCFH oxidation [29]. Thus, because an increase in cytosolic cytochrome c levels could result in higher fluorescence without any change in cellular peroxide levels, use of DCFH-DA as an indicator of oxidative stress during apoptosis should be approached with caution. We used DHE to monitor ROS production though it still has some limitations. It has been affirmed that the DHE reaction with superoxide anions forms a red fluorescent product (ethidium), which intercalates with DNA. Hence, DHE is supposed to be a more convenient and sensitive dye to detect changes of ROS generations [30].
Iron treatment after the elevation of [Ca2+]i by preincubation with TG, a potent endomembrane Ca2+-ATPase inhibitor, also induces calcium release from internal stores. It is clear that this elevation is dependent on the concentration of iron because intracellular calcium fell with the addition 80 µM iron. Previous studies showed that iron supplementation of primary hippocampal neurons kept in Ca2+-free medium also elicited Ca2+ signals, which suggests that hippocampal neurons require Fe to generate RyR-mediated Ca2+ signals after N-methyl-D-aspartate receptor stimulation [31]. In addition, pretreatment of osteoblasts with an L-type Ca2+ channel blocker or a RyR antagonist inhibited Ca2+ release from the SR, which demonstrates that the increase of intracellular Ca2+ induced by hepcidin, a key regulator of body iron homeostasis, probably reflected Ca2+ release from the ER, triggered by the Ca2+ influx [32]. The ability of iron to deplete intracellular calcium via ryanodine, an IP3R blocker, 2-APB, which is also a blocker of TRPM2 channels and TG was examined and determined that it did not inhibit the rise in [Ca2+]i in K562 cells without DFO. Interestingly, intracellular calcium levels did not significantly change after adding iron to cells with DFO. These results show that iron does not stimulate RyR, IP3R, or SERCA, which also regulate other intracellular calcium stores.
Hanahan and Weinberg suggested that cancer is determined by six features: the ability to escape apoptosis; self-sufficiency in growth signaling; insensitivity to anti-growth signals; the capacity for metastasis; unlimited proliferation capability; and the promotion of angiogenesis [33]. Calcium signaling is either directly or indirectly associated to each of these processes [34-38]. Changes in Ca2+ signaling are not always a requirement for the initiation of cancer. However, altered calcium transport may be significant in cancer cells and thereby contribute to tumor progression. Characterizing such changes may help to identify new therapeutic targets [39,40]. It has been shown that DFO triggers apoptosis by the activation of various pro-apoptotic pathways [24-27]. Our results demonstrate that exposure of K562 a cell to DFO increases the sensitivity of iron as a calcium response and boosts the sensitivity to DOX.
CONCLUSION
The results of the present study provide evidence that DFO induces intracellular calcium and increases their sensitivity to the chemotherapeutic agent, DOX. This may render them more vulnerable to disturbances of intracellular calcium concentration because cancer cells have a relatively high rate of mitosis. Therefore, use of drugs that elevate [Ca2+]i could result in a higher apoptotic rate of cancer cells. If this effect could be controlled and used to increase [Ca2+]i to much higher levels, this may result in a much faster apoptotic rate and, therefore, the elimination of cancer cells.
ACKNOWLEDGMENTS
This study was supported by Scientific Research Projects Coordination Unit of the Istanbul University (Project number: 3124).
DECLARATION OF INTERESTS
The authors declare no conflict of interest.
REFERENCES
- [1].Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death:The calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4(7):552–65. doi: 10.1038/nrm1150. http://dx.doi.org/10.1038/nrm1150 . [DOI] [PubMed] [Google Scholar]
- [2].Cazzola M, Bergamaschi G, Dezza L, Arosio P. Manipulations of cellular iron metabolism for modulating normal and malignant cell proliferation:Achievements and prospects. Blood. 1990;75(10):1903–19. [PubMed] [Google Scholar]
- [3].Hoffbrand AV, Ganeshaguru K, Hooton JW, Tattersall MH. Effect of iron deficiency and desferrioxamine on DNA synthesis in human cells. Br J Haematol. 1976;33(4):517–26. doi: 10.1111/j.1365-2141.1976.tb03570.x. http://dx.doi.org/10.1111/j.1365-2141.1976.tb03570.x . [DOI] [PubMed] [Google Scholar]
- [4].Pantopoulos K, Porwal SK, Tartakoff A, Devireddy L. Mechanisms of mammalian iron homeostasis. Biochemistry. 2012;51(29):5705–24. doi: 10.1021/bi300752r. http://dx.doi.org/10.1021/bi300752r . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Pogribny IP, Tryndyak VP, Pogribna M, Shpyleva S, Surratt G, Gamboa da Costa G, et al. Modulation of intracellular iron metabolism by iron chelation affects chromatin remodeling proteins and corresponding epigenetic modifications in breast cancer cells and increases their sensitivity to chemotherapeutic agents. Int J Oncol. 2013;42(5):1822–32. doi: 10.3892/ijo.2013.1855. http://dx.doi.org/10.3892/ijo.2013.1855 . [DOI] [PubMed] [Google Scholar]
- [6].Torti SV, Torti FM. Ironing out cancer. Cancer Res. 2011;71(5):1511–4. doi: 10.1158/0008-5472.CAN-10-3614. http://dx.doi.org/10.1158/0008-5472.CAN-10-3614 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128(4):683–92. doi: 10.1016/j.cell.2007.01.029. http://dx.doi.org/10.1016/j.cell.2007.01.029 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Torti SV, Torti FM. Iron and cancer:More ore to be mined. Nat Rev Cancer. 2013;13(5):342–55. doi: 10.1038/nrc3495. http://dx.doi.org/10.1038/nrc3495 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Buss JL, Torti FM, Torti SV. The role of iron chelation in cancer therapy. Curr Med Chem. 2003;10(12):1021–34. doi: 10.2174/0929867033457638. http://dx.doi.org/10.2174/0929867033457638 . [DOI] [PubMed] [Google Scholar]
- [10].Yu Y, Gutierrez E, Kovacevic Z, Saletta F, Obeidy P, Suryo Rahmanto Y, et al. Iron chelators for the treatment of cancer. Curr Med Chem. 2012;19(17):2689–702. doi: 10.2174/092986712800609706. http://dx.doi.org/10.2174/092986712800609706 . [DOI] [PubMed] [Google Scholar]
- [11].Rao VA, Klein SR, Agama KK, Toyoda E, Adachi N, Pommier Y, et al. The iron chelator Dp44mT causes DNA damage and selective inhibition of topoisomerase IIalpha in breast cancer cells. Cancer Res. 2009;69(3):948–57. doi: 10.1158/0008-5472.CAN-08-1437. http://dx.doi.org/10.1158/0008-5472.CAN-08-1437 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Günes DA, Florea AM, Splettstoesser F, Büsselberg D. Co-application of arsenic trioxide (As2O3) and cisplatin (CDDP) on human SY-5Y neuroblastoma cells has differential effects on the intracellular calcium concentration ([Ca2]i) and cytotoxicity. Neurotoxicology. 2009;30(2):194–202. doi: 10.1016/j.neuro.2008.12.001. http://dx.doi.org/10.1016/j.neuro.2008.12.001 . [DOI] [PubMed] [Google Scholar]
- [13].Tenopoulou M, Kurz T, Doulias PT, Galaris D, Brunk UT. Does the calcein-AM method assay the total cellular ‘labile iron pool’ or only a fraction of it? Biochem J. 2007;403(2):261–6. doi: 10.1042/BJ20061840. http://dx.doi.org/10.1042/BJ20061840 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bindokas VP, Jordán J, Lee CC, Miller RJ. Superoxide production in rat hippocampal neurons:Selective imaging with hydroethidine. J Neurosci. 1996;16(4):1324–36. doi: 10.1523/JNEUROSCI.16-04-01324.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, Melendez JA, et al. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J Biol Chem. 2003;278(10):8516–25. doi: 10.1074/jbc.M210432200. http://dx.doi.org/10.1074/jbc.M210432200 . [DOI] [PubMed] [Google Scholar]
- [16].Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260(6):3440–50. [PubMed] [Google Scholar]
- [17].Hoke EM, Maylock CA, Shacter E. Desferal inhibits breast tumor growth and does not interfere with the tumoricidal activity of doxorubicin. Free Radic Biol Med. 2005;39(3):403–11. doi: 10.1016/j.freeradbiomed.2005.03.029. http://dx.doi.org/10.1016/j.freeradbiomed.2005.03.029 . [DOI] [PubMed] [Google Scholar]
- [18].Whitnall M, Howard J, Ponka P, Richardson DR. A class of iron chelators with a wide spectrum of potent antitumor activity that overcomes resistance to chemotherapeutics. Proc Natl Acad Sci U S A. 2006;103(40):14901–6. doi: 10.1073/pnas.0604979103. http://dx.doi.org/10.1073/pnas.0604979103 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Pinnix ZK, Miller LD, Wang W, D’Agostino R, Jr, Kute T, Willingham MC, et al. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci Transl Med. 2010;2(43):43ra56. doi: 10.1126/scisignal.3001127. http://dx.doi.org/10.1126/scitranslmed.3001127 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Le NT, Richardson DR. The role of iron in cell cycle progression and the proliferation of neoplastic cells. Biochim Biophys Acta. 2002;1603(1):31–46. doi: 10.1016/s0304-419x(02)00068-9. http://dx.doi.org/10.1016/s0304-419x(02)00068-9 . [DOI] [PubMed] [Google Scholar]
- [21].Wilkinson MG, Millar JB. Control of the eukaryotic cell cycle by MAP kinase signaling pathways. FASEB J. 2000;14(14):2147–57. doi: 10.1096/fj.00-0102rev. http://dx.doi.org/10.1096/fj.00-0102rev . [DOI] [PubMed] [Google Scholar]
- [22].Saletta F, Suryo Rahmanto Y, Noulsri E, Richardson DR. Iron chelator-mediated alterations in gene expression:Identification of novel iron-regulated molecules that are molecular targets of hypoxia-inducible factor-1 alpha and p53. Mol Pharmacol. 2010;77(3):443–58. doi: 10.1124/mol.109.061028. http://dx.doi.org/10.1124/mol.109.061028 . [DOI] [PubMed] [Google Scholar]
- [23].Le NT, Richardson DR. Iron chelators with high antiproliferative activity up-regulate the expression of a growth inhibitory and metastasis suppressor gene:A link between iron metabolism and proliferation. Blood. 2004;104(9):2967–75. doi: 10.1182/blood-2004-05-1866. http://dx.doi.org/10.1182/blood-2004-05-1866 . [DOI] [PubMed] [Google Scholar]
- [24].Hileti D, Panayiotidis P, Hoffbrand AV. Iron chelators induce apoptosis in proliferating cells. Br J Haematol. 1995;89(1):181–7. doi: 10.1111/j.1365-2141.1995.tb08927.x. http://dx.doi.org/10.1111/j.1365-2141.1995.tb08927.x . [DOI] [PubMed] [Google Scholar]
- [25].Pan YJ, Hopkins RG, Loo G. Increased GADD153 gene expression during iron chelation-induced apoptosis in Jurkat T-lymphocytes. Biochim Biophys Acta. 2004;1691(1):41–50. doi: 10.1016/j.bbamcr.2003.12.003. http://dx.doi.org/10.1016/j.bbamcr.2003.12.003 . [DOI] [PubMed] [Google Scholar]
- [26].So EY, Ausman M, Saeki T, Ouchi T. Phosphorylation of SMC1 by ATR is required for desferrioxamine (DFO)-induced apoptosis. Cell Death Dis. 2011;2(41):e128. doi: 10.1038/cddis.2011.9. http://dx.doi.org/10.1038/cddis.2011.9 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Saletta F, Suryo Rahmanto Y, Siafakas AR, Richardson DR. Cellular iron depletion and the mechanisms involved in the iron-dependent regulation of the growth arrest and DNA damage family of genes. J Biol Chem. 2011;286(41):35396–406. doi: 10.1074/jbc.M111.273060. http://dx.doi.org/10.1074/jbc.M111.273060 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bonini MG, Rota C, Tomasi A, Mason RP. The oxidation of 2’,7’-dichlorofluorescin to reactive oxygen species:A self-fulfilling prophesy? Free Radic Biol Med. 2006;40(6):968–75. doi: 10.1016/j.freeradbiomed.2005.10.042. http://dx.doi.org/10.1016/j.freeradbiomed.2005.10.042 . [DOI] [PubMed] [Google Scholar]
- [29].Lawrence A, Jones CM, Wardman P, Burkitt MJ. Evidence for the role of a peroxidase compound I-type intermediate in the oxidation of glutathione, NADH, ascorbate, and dichlorofluorescin by cytochrome c/H2O2. Implications for oxidative stress during apoptosis. J Biol Chem. 2003;278(32):29410–9. doi: 10.1074/jbc.M300054200. http://dx.doi.org/10.1074/jbc.M300054200 . [DOI] [PubMed] [Google Scholar]
- [30].Robinson KM, Janes MS, Pehar M, Monette JS, Ross MF, Hagen TM, et al. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc Natl Acad Sci U S A. 2006;103(41):15038–43. doi: 10.1073/pnas.0601945103. http://dx.doi.org/10.1073/pnas.0601945103 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Muñoz P, Humeres A, Elgueta C, Kirkwood A, Hidalgo C, Núñez MT. Iron mediates N-methyl-D-aspartate receptor-dependent stimulation of calcium-induced pathways and hippocampal synaptic plasticity. J Biol Chem. 2011;286(15):13382–92. doi: 10.1074/jbc.M110.213785. http://dx.doi.org/10.1074/jbc.M110.213785 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Li GF, Xu YJ, He YF, Du BC, Zhang P, Zhao DY, et al. Effect of hepcidin on intracellular calcium in human osteoblasts. Mol Cell Biochem. 2012;366(1-2):169–74. doi: 10.1007/s11010-012-1294-y. http://dx.doi.org/10.1007/s11010-012-1294-y . [DOI] [PubMed] [Google Scholar]
- [33].Hanahan D, Weinberg RA. Hallmarks of cancer:The next generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. http://dx.doi.org/10.1016/j.cell.2011.02.013 . [DOI] [PubMed] [Google Scholar]
- [34].Roderick HL, Cook SJ. Ca2+signalling checkpoints in cancer:Remodelling Ca2+for cancer cell proliferation and survival. Nat Rev Cancer. 2008;8(5):361–75. doi: 10.1038/nrc2374. http://dx.doi.org/10.1038/nrc2374 . [DOI] [PubMed] [Google Scholar]
- [35].Prevarskaya N, Skryma R, Shuba Y. Calcium in tumour metastasis:New roles for known actors. Nat Rev Cancer. 2011;11(8):609–18. doi: 10.1038/nrc3105. http://dx.doi.org/10.1038/nrc3105 . [DOI] [PubMed] [Google Scholar]
- [36].Fiorio Pla A, Avanzato D, Munaron L, Ambudkar IS. Ion channels and transporters in cancer 6. Vascularizing the tumor:TRP channels as molecular targets. Am J Physiol Cell Physiol. 2012;302(1):C9–15. doi: 10.1152/ajpcell.00280.2011. http://dx.doi.org/10.1152/ajpcell.00280.2011 . [DOI] [PubMed] [Google Scholar]
- [37].Rizzuto R, Pinton P, Ferrari D, Chami M, Szabadkai G, Magalhães PJ, et al. Calcium and apoptosis:Facts and hypotheses. Oncogene. 2003;22(53):8619–27. doi: 10.1038/sj.onc.1207105. http://dx.doi.org/10.1038/sj.onc.1207105 . [DOI] [PubMed] [Google Scholar]
- [38].Lehen’kyi V, Shapovalov G, Skryma R, Prevarskaya N. Ion channnels and transporters in cancer 5. Ion channels in control of cancer and cell apoptosis. Am J Physiol Cell Physiol. 2011;301(6):C1281–9. doi: 10.1152/ajpcell.00249.2011. http://dx.doi.org/10.1152/ajpcell.00249.2011 . [DOI] [PubMed] [Google Scholar]
- [39].Ramsey IS, Delling M, Clapham DE. An introduction to TRP channels. Annu Rev Physiol. 2006;68:619–47. doi: 10.1146/annurev.physiol.68.040204.100431. http://dx.doi.org/10.1146/annurev.physiol.68.040204.100431 . [DOI] [PubMed] [Google Scholar]
- [40].Berridge MJ. Elementary and global aspects of calcium signalling. J Exp Biol. 1997;200:315–9. doi: 10.1242/jeb.200.2.315. http://dx.doi.org/10.1113/jphysiol.1997.sp021927 . [DOI] [PubMed] [Google Scholar]