

Abstract
Cholestasis is a common pathological component of numerous liver diseases. The initiating event during cholestatic liver injury is widely believed to be the accumulation of bile acids in hepatocytes and the hepatic parenchyma. As bile acids are considered the primary toxic compounds in the injury, numerous in vitro models of bile acid-induced injury and bile acid-induced changes in gene expression have been developed to attempt to better define cholestasis at a cellular level. This chapter focuses on the establishment of a system for determining the effects of cholestatic concentrations of bile acids on hepatocytes using primary hepatocytes or hepatoma cell lines. Moreover, this chapter addresses significant differences in the response of different species to bile acid exposure and novel information on the relevance of treating hepatocytes with concentrations of specific bile acids.
Keywords: Apoptosis, Necrosis, In vitro model, Bile acid, Primary hepatocyte, Cholestasis, Inflammation, Liver, Bile salt, Human hepatocytes, Rodent models, Sodium taurocholate, Co-transporting polypeptide, Bile salt export pump, HepG2, Huh7, Transfected hepatocytes, Neutrophils, Methods
1 Introduction
Cholestasis results from either blockade of any portion of the biliary tracts or inhibition of bile acid export from hepatocytes. When left untreated, chronic cholestasis proceeds to significant liver damage and cirrhosis. Common in vivo models for study include the bile duct ligation (BDL) model [1], which results in extrahepatic cholestasis, administration of lithocholic acid (LCA) [2] or alpha-naphthylisothiocyanate [3], which result in intrahepatic cholestasis, or use of the multi-drug resistance two transporter knockout mouse [4] or inhibition of the bile salt export pump (BSEP) [5], which mimic inhibition of export of bile acids from hepatocytes. As the accumulation of bile acids in hepatocytes and the hepatic parenchyma is considered to be the initiator of the injury process [6], considerable effort has gone into the establishment of a suitable in vitro model for directly studying the effects of increased bile acid levels on hepatocytes. In general, the administration of bile acids to cultured hepatocytes is a simple process. However, recent advances have illustrated numerous potential pitfalls that can be encountered during otherwise routine cell culture experiments. As a preface, current models for the study of cholestatic liver injury are discussed in this chapter, along with potential pitfalls that must be considered before initiation of experimentation.
1.1 Differential Effects of Specific Bile Acids on Hepatocytes
Bile acids are the major constituents of bile. Numerous species are present in the body and are derived from the two primary bile acids, namely cholic acid (CA) and chenodeoxycholic acid (CDCA). Dehydroxylation reactions result in secondary bile acids, namely deoxycholic acid (DCA) and lithocholic acid (LCA) from cholic acid and chenodeoxycholic acid, respectively. A majority of these bile acids are then conjugated to either of the amino acids taurine or glycine before they enter enterohepatic circulation. Recent methodology has allowed for a quantitative assessment of individual bile acid concentrations in multiple species and in multiple tissue compartments during cholestasis [7–11]. Perhaps the most important aspect of these new data is the elucidation of vast differences in individual bile acid concentrations in rodents and humans, both in control patients and during cholestatic liver injury [1]. While numerous studies [6, 12, 13] have focused on administering micromolar levels of toxic bile acids or their conjugate salt, such as glycochenodeoxycholate (GCDC) or taurolithocholate (TLC), to hepatocytes in vitro, newer data based on mass spectrometric bile acid analysis questions the use of micromolar concentrations of these bile acids in rodent cultures, as they likely do not accumulate to similar values in any compartment in vivo [8, 10]. Moreover, murine hepatocytes are highly resistant to TLC-induced injury in vitro, suggesting mechanisms of toxicity specific to the rat [10, 14]. Additionally, the use of unconjugated bile acids to investigate pathophysiology seems unwarranted as more than 99 % of bile acids in liver, plasma and bile are conjugated to taurine or glycine in multiple species [7–9, 11]. While intrahepatic concentrations of bile acids are considered to be integral to the injury, recent reports have asserted that the characteristic areas of necrosis seen after BDL or after administration of 1 % LCA via the diet are due to infarction of the small biliary tracts and the ensuing release of bile into the hepatic parenchyma, suggesting that biliary levels of bile acids may be driving the hepatic accumulation of bile acids during some models of cholestatic liver injury [2, 10]. Cholestasis models, such as BDL, display a lack of zonation in the area of injury. Instead, the necrosis correlates specifically with areas of biliary leakage, implicating leakage of biliary constituents onto the hepatic parenchyma as an initiator of injury [2, 4, 10, 11]. However, even in bile, a majority of the rodent bile acid milieu is largely composed of hydrophilic bile acids, such as taurocholic acid (TCA), that remain non-toxic up to millimolar concentrations [8, 10]. While the hydrophilic bile acids, like TCA, do not cause direct hepatotoxicity [8, 10], exposing these bile acids to hepatocytes results in dramatic increases in pro-inflammatory genes, such as macrophage inflammatory protein 2, mouse keratinocyte-derived chemokine, intercellular adhesion molecule 1, early growth response protein 1 and others [15, 16]. Many of these genes play a significant role in the pathophysiology of cholestatic liver injury, as intercellular adhesion molecule 1 and early growth response protein 1 knock-out mice are protected against BDL injury [17, 18]. This reduction in injury is due to attenuation of neutrophil function and recruitment, as BDL injury is mediated by cytotoxic neutrophils [17, 19, 20]. In contrast to the BDL model, only in models where the bile is toxified, such as through administration of LCA via the diet, direct hepatotoxicity can be seen when hepatocytes are exposed to rodent bile [2, 10]. As such, experiments where mouse or rat hepatocytes are exposed to 50–100 μM GCDC or TLC may not be relevant to the pathophysiology of cholestatic liver injury in mice or man. Furthermore, as hepatocytes are exposed to multiple bile acids simultaneously during cholestasis, exposure of hepatocytes to single bile acids may obscure the anti-apoptotic effects that hydrophilic bile acids can induce through activation of nuclear factor kappa beta via upregulation of cytokines [21]. Thus, while the simplicity of adding a single bile acid to hepatocytes can produce interesting and repeatable results, the relevance of exposure of hepatocytes to a single toxic bile acid may bear little resemblance to the in vivo pathophysiology. An increased focus is instead called for on how a complete bile acid milieu derived from pathophysiological assessment of levels in the appropriate species affects hepatocytes both in vitro and in vivo. Recent studies using complete mixtures of serum or biliary bile acid values suggest that hepatocytes may be less susceptible on a mole to mole basis to a total bile acid mixture than to direct exposure to a single bile acid [11, 22]. It is therefore imperative to carefully assess what bile acids and what models are being used to investigate biological effects of bile acids to ensure pathophysiological relevance.
1.2 Species Differences in In Vitro Bile Acid Toxicity
Bile acid levels in serum, liver, and bile, and the subsequent cellular response vary substantially between commonly used laboratory species and man. These differences have resulted in significant controversy in the field with regard to which method of cell death, apoptosis or necrosis occurs during cholestasis [2, 6, 23–25]. Part of this discussion revolves around differences seen during in vitro exposure to bile acids, particularly pro-apoptotic bile acids, such as GCDC. Humans produce a relatively larger amount of the more toxic glycine-conjugated bile acids, particularly GCDC, whereas mice and rats produce larger quantities of the less toxic taurine-conjugated bile acids [7–9, 11, 26]. Numerous studies expose rat hepatocytes or transfected human hepatoma lines to about 50 μM GCDC, which then respond with prototypical apoptosis including nuclear condensation, cellular shrinkage, nuclear fragmentation, and activation of caspases [27]. Complete rescue is possible by pretreatment with the pan-caspase inhibitor z-VAD-fmk, indicating the injury is predominately apoptosis [28]. Even given this well-defined and established response, the validity of applying bile acids commonly observed in humans to rat hepatocytes does not accurately model the pathophysiology, especially as levels of these bile acids either do not reach or far exceed 50 μM during cholestasis [7, 12]. In contrast, human hepatocytes show little evidence for apoptosis at doses consistent with the pathophysiology and die primarily through necrosis, although only when exposed to higher concentrations of GCDC or a complete biliary bile salt mixture [11]. Interestingly, primary human hepatocytes are particularly resistant to GCDC-induced injury requiring near millimolar levels of specific toxic bile acids before the onset of toxicity [11, 29, 30]. Species differences also exist between mice and humans, as humans do not exhibit the same increases in cytokine production that mice do when stimulated with millimolar concentrations of TCA [11, 15]. As human bile is considerably more concentrated with glycine-conjugated toxic bile acids, this may be a by-product of the direct toxicity produced by human bile in comparison to the relatively less harmful murine bile. While inflammation is a noted part of multiple human cholestatic disorders, cytokine production likely results from another source. A final important note is the marked difference in transporter expression levels over time between primary human and primary rodent hepatocytes. While human hepatocytes retain a majority of their transporter activity (e.g. BSEP) over the first 24 h of culture [31], rodent hepatocytes rapidly dedifferentiate and lose transporter expression [32, 33]. Loss of transporter activity rapidly decreases bile acid function in vitro. Thus, primary hepatocytes from rodents need to be used immediately after adherence for them to be useful. Bile salt efflux is considerably different between humans and rats as well. While humans have essentially similar levels of basolateral and canalicular efflux, rats rely predominantly on basolateral transport, increasing their resistance to canalicular BSEP inhibition-induced cholestasis [34]. Thus, critical differences are present between species in regard to bile acid toxicity and metabolism that merit consideration before experimentation in this field.
1.3 BSEP Inhibition In Vitro
BSEP is responsible for export of bile salts out of the hepatocytes into the biliary tracts [35]. Troglitazone, a peroxisome proliferator-activated receptor activator, and bosentan, an endothelin receptor antagonist, are currently the most widely recognized BSEP inhibitors [36]. Troglitazone was removed from the market due to hepatotoxicity and bosentan currently carries a black box warning label that requires monthly monitoring for liver damage associated with taking the drug. Drug-induced BSEP inhibition results in severe cholestasis, making it a pharmacological and a clinical issue [37]. While not directly cytotoxic, drugs that inhibit BSEP result in increases in serum transaminases and serum bile acid levels, suggesting they may kill cells through loss of homeostasis of native bile acid levels and subsequent liver injury [38]. Long-term drug-induced cholestasis also causes both a ductular reaction and hepatitis that may worsen the injury [36]. The mechanisms behind how BSEP inhibition produces hepatotoxicity are not well understood, although intrahepatic accumulation of bile acids and oxidative stress are implicated [39]. Both functional [40] and gene expression assays [41] have some predictive value in determining the degree by which a drug can inhibit BSEP. Thus, while these drugs are not directly cytotoxic, drug-induced cholestasis is a separate yet important issue. Multiple efforts are underway currently to establish a reproducible and appropriate model for drug-induced cholestasis, as it relates to liver injury in vitro [25].
1.4 Primary Cells Versus Hepatoma Cell Lines
Due to the difficulty in acquiring primary cells from animals or humans, hepatoma cell lines are sometimes used [42, 43]. Most commonly used hepatoma lines, such as the Huh7 line and HepG2 line, do not natively express the sodium taurocholate co-transporting polypeptide (NTCP), the primary bile acid uptake transporter, and must be transfected to express the protein, where-upon they will actively take up bile acids. Thus, non-transfected HepG2 or Huh7 cells make a poor model for bile acid-induced liver injury or cholestatic liver injury. Multiple reports have used HepG2 cells transfected with NTCP to assess the effects of bile acids [43–45]. In general, these cells recapitulate responses seen in the rat and not the human, which suggests that they may not be a suitable surrogate for primary human hepatocytes. In contrast, the recently isolated HepaRG cell line functionally expresses both canalicular and basolateral transporters [31, 46] and may serve as a novel human cell line for in vitro bile acid-induced toxicity. While hepatocytes from this line do not express transporters to the same degree primary human hepatocytes do, some functional activity is retained [31, 39]. As this is a bipotential cell line that actively differentiates into both cholangiocytes and hepatocytes, it is imperative to establish bile acid toxicity without cholangiocyte toxicity when assessing cell death in this model.
Given these potential pitfalls, and the current state of knowledge of the field, a model for in vitro assessment of cholestatic liver injury is proposed herein.
2 Materials
Hepatocyte seeding and culture medium. William’s medium E containing 10 % fetal bovine serum (FBS), suitable antibiotics and 2 ng/mL insulin. Media should be warmed to 37 °C before use (see Note 1).
Phosphate-buffered saline (PBS). 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4·2H2O, 1.8 mM KH2PO4 in deionized water. Adjust to pH 7.4 and store for maximum 6 months at 4 °C.
Depending on the animal model used, cell death is best measured by lactate dehydrogenase (LDH) release into media and caspase-3 activation. For the LDH assay, potassium phosphate buffer is composed of 60 mM KH2PO4 and 60 mM K2HPO4 along with 100 mM pyruvate and 250 μM nicotinamide adenine dinucleotide (NADH), pH 7.5, in deionized water. This solution can be stored for up to 1 month at 4 °C. The fluorogenic substrate for caspase-3 assay, namely acetyl-aspartic acid-glutamic acid-valine-aspartic acid-7-aminotrifluoromethylcoumarin (Ac-DEVD-AMC) is commercially available (Enzo Life Sciences, United States of America).
Depending on the animal model used, reverse transcriptase polymerase chain reaction (RT-PCR) analysis and immunoblotting can be used to assess changes in expression of pro-inflammatory genes and proteins, such as adhesion molecules, after bile acid exposure. Reagents and protocols for these assays are widely available from relevant manufacturers (Life Technologies, United States of America).
Incubator (see Note 2).
Spectrophotometer capable of reading at 340 nm.
Plate reader capable of exciting at 380 nm and reading emission at 460 nm.
Bicinchoninic acid (BCA) protein quantification reagents (Sigma-Aldrich, United States of America).
Kits and reagents for RNA extraction and RT-PCR analysis (Invitrogen, United States of America).
3 Methods
3.1 Cell Culture and Bile Acid Administration
Use freshly isolated primary human, rat, or mouse hepatocytes (see Note 3). Cell viability is assessed by means of trypan blue exclusion [47].
Plate the hepatocytes on 6-well plastic culture dishes at a density of 0.5–1.0 × 106 cells/well (see Note 4). Hepatocytes should be allowed to adhere for 3–4 h before the onset of experimentation (see Note 5).
Wash cells with 1 mL PBS and replace 1 mL culture medium.
Add the bile acid of interest to the appropriate concentration and incubate as desired (see Notes 6 and 7).
3.2 Measurement of LDH and Alanine Aminotransferase Activity (See Note 8)
Treat cells with bile acid of interest.
Remove 1 mL medium and store in a microcentrifuge tube at −80 °C. Upon thawing, centrifuge at 14,000 × g and room temperature for 3 min. Wash with 1 mL PBS.
Lyze cells in 300 μL appropriate protein buffer, scrape and store in a microcentrifuge tube at −80 °C. Upon beginning the assay, sonicate cellular mixture 3 times for 3 s at a low level to ensure complete disruption of membranes. Centrifuge at 14,000 × g and room temperature for 10 min.
- LDH activity can be measured by analyzing the oxidation of NADH in an LDH buffer via a loss in absorbance at 340 nm on a spectrophotometer. LDH activity in both medium and cells should be analyzed. Approximate LDH release can be measured using the equation:
To analyze LDH activity, dilute media 1:7 into LDH buffer and analyze the oxidation of NADH (i.e. loss of absorbance at 340 nm) in the LDH buffer with a spectrophotometer over a 2 min period. In a matched sample, dilute cellular contents 1:30 and analyze the oxidation of NADH in the LDH buffer with a spectrophotometer over a 2 min period. Use the above equation to calculate the death ratio and report as percentage LDH release. A sample figure is presented showing increasing cell death in glycocholic acid treated mouse hepatocytes in Fig. 1.
Alanine aminotransferase (ALT) activity can be measured similarly and involves a 2-step process that also measures the oxidation of NADH via a loss in absorbance at 340 nm on a spectrophotometer. In some cases, measurement of ALT can be superior to measurement of LDH, as loss of LDH activity may occur during extended cell culture. To measure ALT instead of LDH, complete the steps above, but substitute the LDH buffer with an ALT buffer (Pointe Scientific, United States of America). All calculations and steps are otherwise identical as is the equation for calculation.
Fig. 1.
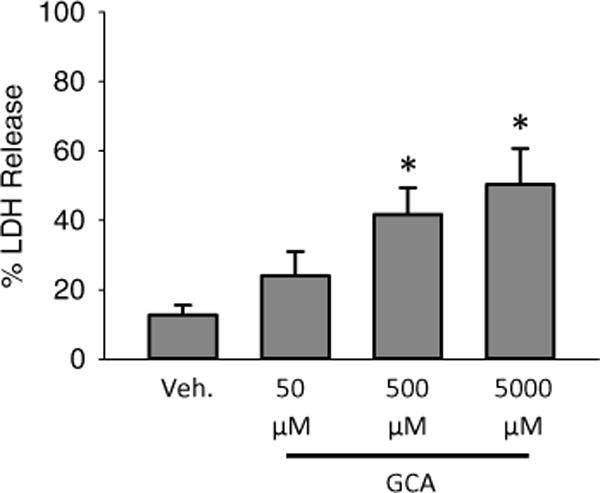
Bile acid-induced necrotic cell death in murine hepatocytes. Hepatocytes were isolated from livers of C57Bl/6 mice and exposed to various concentrations of glycocholic acid (GCA) for 6 h after an initial 3 h attachment period. LDH was measured as an indicator of cell death (n = 3; *p < 0.05 vs. control by analysis of variance)
3.3 Measurement of Caspase-3 Activity (See Note 9)
Treat cells with bile acid of interest.
Remove media and wash cells with 1 mL PBS.
Lyze cells in 150 μL of appropriate protein buffer, scrape, and store in a microcentrifuge tube at −80 °C if necessary. Upon thawing or upon starting the assay, centrifuge at 14,000 × g and room temperature for 10 min.
Dilute Ac-DEVD-AMC substrate to 2 mM. Dilute z-VAD-fmk to 100 μM (see Note 10).
- The assay is best completed in a 96-well plate on a plate reader. Generate 2 master mixes, for 1 caspase-3 activity and 1 for z-VAD-fmk inhibitable activity:
- Mix A: 50 μL protein homogenizing buffer and 10 μL diluted substrate.
- Mix B: 40 μL protein homogenizing buffer, 10 μL diluted substrate, and 10 μL diluted z-VAD-FMK.
Add 50 μL of cell lysate to each well and then pipette 50 μL of either mix A or mix B in with the samples. This yields 2 wells for each sample, namely mix A well (i.e. caspase-3 activity) and mix B well (i.e. z-VAD-inhibitable caspase activity).
Analyze activity over 1 h using an excitation of 380 nm and emission of 460 nm. Activity should be recalculated as caspase-3 activity minus z-VAD-inhibitable caspase-3 activity.
Measure protein quantity via the BCA assay as per manufacturer’s instructions and express as relative fluorescence/mg protein/min.
3.4 RNA Isolation and RT-PCR Analysis (See Note 11)
Treat cells with bile acid of interest.
Remove media and wash cells twice with PBS.
Lyse cells in Trizol buffer for RNA isolation.
Extract RNA and reverse transcribe to cDNA.
Use PCR to assess gene levels (see Note 12). Numerous protocols are available for simple quantitative PCR techniques. Following the manufacturer’s recommended instructions for the apparatus is advisable for this assay.
3.5 Immunoblot Analysis
Treat cells with bile acids of interest.
Remove media and wash cells twice with PBS.
Lyze and scrape cells in 100 μL of appropriate protein isolation buffer.
Measure protein and assess protein levels by immunoblot analysis (see Note 12). Numerous protocols are available for this process. Following the manufacturer’s recommended instructions for the apparatus and antibodies in use is advisable for this assay.
Fig. 2.
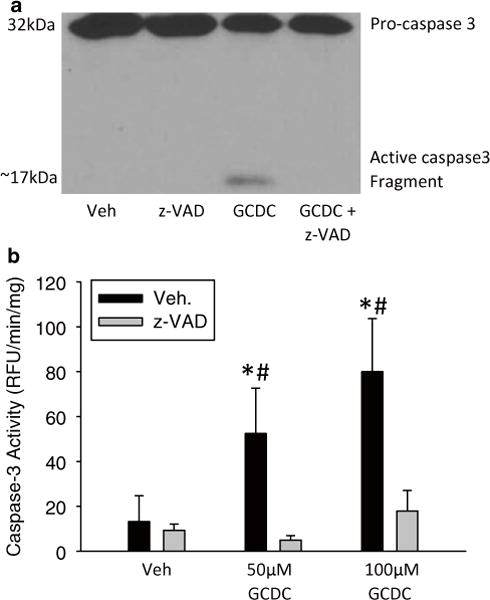
Bile acid-induced apoptosis in rat hepatocytes. Hepatocytes were isolated from rats and exposed to vehicle (i.e. 0.1 % dimethyl sulfoxide), 50 μM, or 100 μM GCDC for 6 h with and without a 30 min pretreatment with 10 μM of the pan-caspase inhibitor z-VAD-FMK. Caspase-3 activation after 50 μM GCDC was measured by immunoblot analysis (a) and through a fluorogenic caspase activity assay (b) (*p < 0.05 vs. control; #p < 0.05 vs. matched z-VAD-fmk treated sample)
Acknowledgments
Work in this laboratory was supported in part by a CTSA grant from NCRR and NCATS awarded to the University of Kansas Medical Center for Frontiers: The Heartland Institute for Clinical and Translational Research # UL1RR033179 which is now at NCATS # UL1TR000001, grants from the National Institutes of Health (R01 DK070195 and R01 AA12916) (to H.J.), and from the National Center for Research Resources (5P20RR021940) and the National Institute of General Medical Sciences (8 P20 GM103549) of the National Institutes of Health. B.L.W. was supported by the “Training Program in Environmental Toxicology” T32 ES007079-26A2 from the National Institute of Environmental Health Sciences.
Footnotes
Fetal bovine serum can be omitted, but may affect results in some assays. As cells are exposed to serum constantly in vivo, use of fetal bovine serum is recommended in all assays. If fetal bovine serum is not used, a direct comparison between cells given fetal bovine serum and cells not given fetal bovine serum should be done for the indicated assay.
Use of room air can have profound effects on hepatocyte culture [48, 49]. Physiological levels of oxygen are protective against GCDC-induced apoptosis in rat hepatocytes [49]. Primary hepatocytes are typically incubated in ambient levels of oxygen (i.e. about 20 %), whereas hepatic oxygen levels can be as low as 4 %, which results in substantial increases in available oxygen for reactive oxygen species generation in GCDC-treated cultured hepatocytes. Reactive oxygen species production likely mediates the apoptosis, although the source of the reactive oxygen species is not well established. Experiments may want to be repeated under conditions of both atmospheric oxygen and physiological hypoxia to verify changes observed during in vitro culture.
The animal model used has profound effects on the results obtained.
Typically, assays done in our laboratory are done in a 6-well plate. It is recommended to consult commercial literature on cell density numbers for other sizes of plates. About 80 % confluence is typical for these assays. For all assays listed, 1 well of a 6-well plate at the given cell density will be sufficient to complete the assay.
NTCP is rapidly downregulated after culture in murine and rat hepatocytes [32]. To avoid loss of effects, experiments should be carried out immediately at this point. Avoid cultures that do not express NTCP, as there is a serious deficit of bile acid internalization and thus quite likely, bile acid function. These cell lines may be prone to generating results with limited relevance for the human pathophysiology.
Longer incubation times will result in loss of NTCP function and alternations in read-out. Assays are best run over 4–6 h to avoid these problems. If longer time periods are needed, stably transfected cells or primary human hepatocytes, which do not lose NTCP activity as quickly, can be used [11, 31]. While the shorter timeframe likely does not mimic the consequences of long-term cholestasis, the loss of functional transporter activity is a more serious concern, as observed effects may be secondary to bile acid function.
Bile acids have varied effects. Toxicity and gene induction can be acquired through multiple different bile acids or administration of a single bile acid. In general, the more hydrophobic bile acids are more toxic and also require hydrophobic solvents, such as dimethyl sulfoxide or methanol. Concentrations of solvent below 1 % are suggested to reduce solvent effects. Vehicle solvents should always be added to experiments as a control for the solvent effects [50].
LDH is best used to measure necrosis, which may be secondary to apoptosis. The simultaneous use of LDH and caspase-3 is attainable in a single well from a 6-well plate and provides the best overall image of cell death using enzyme kinetics. Additional potential assays include staining necrotic nuclei with the cell permeable reagent propidium iodide.
Caspase-3 activity is a specific and direct marker of apoptosis. As caspase-7 generally has the same substrate specificity, some groups will label assays as caspase-3/7 activity, which is also correct. Confirmation of caspase-3 activity can be obtained via immunoblotting for pro-caspase-3 and its cleaved active form. The antibody used in our laboratory is from Cell Signaling (United States of America). While antibodies specific for cleaved caspase-3 are available, acknowledgement of the proband in immunoblot imaging is encouraged, as its compensatory decrease can be seen as a confirmation of antibody specificity. Figure 2 depicts the proper representation of an immunoblot for caspase-3 that contains both the proform band and its cleaved form. Multiple cleavage products are sometimes formed from the 32 kDa precursor between 19 and 14 kDa.
As there may be endogenous protease activity, the use of a pancaspase inhibitor (e.g. z-VAD-fmk) control ensures that the assay is specific for cellular caspase activity and excludes caspase-independent protease activities. This is a vital control, as some cell lysates will contain a great deal of endogenous protease activity.
For both RNA and protein, most changes occur within a similar timepoint as the toxicity, thus prolonged exposure results in less activity and not more, presumably due to loss of bile acid uptake.
Commonly assessed genes and proteins for assessing inflammation include macrophage inflammatory protein 2, mouse keratinocyte-derived chemokine, intercellular adhesion molecule 1, early growth response protein 1 and others [15, 16], although any gene of interest could be measured. Using RT-PCR analysis to assess caspase-3 levels is not a useful way to measure apoptosis, as caspase function is dependent upon posttranslational cleavage events largely by other caspases. Use of immunoblot analysis to assess caspase-3 cleavage is essential for demonstration of active apoptosis.
References
- 1.Woolbright BL, Antoine DJ, Jenkins R, et al. Plasma biomarkers of liver injury and inflammation demonstrate a lack of apoptosis during obstructive cholestasis in mice. Toxicol Appl Pharmacol. 2013;273:524–531. doi: 10.1016/j.taap.2013.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fickert P, Fuchsbichler A, Marschall HU, et al. Lithocholic acid feeding induces segmental bile duct obstruction and destructive cholangitis in mice. Am J Pathol. 2006;168:410–422. doi: 10.2353/ajpath.2006.050404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krell H, Metz J, Jaeschke H, et al. Drug-induced intrahepatic cholestasis: characterization of different pathomechanisms. Arch Toxicol. 1987;60:124–130. doi: 10.1007/BF00296964. [DOI] [PubMed] [Google Scholar]
- 4.Fickert P, Zollner G, Fuchsbichler A, et al. Ursodeoxycholic acid aggravates bile infarcts in bile duct-ligated and Mdr2 knock-out mice via disruption of cholangioles. Gastroenterology. 2002;123:1238–1251. doi: 10.1053/gast.2002.35948. [DOI] [PubMed] [Google Scholar]
- 5.Rodrigues AD, Lai Y, Cvijic ME. Drug-induced perturbations of the bile acid pool, cholestasis, and hepatotoxicity: mechanistic considerations beyond the direct inhibition of the bile salt export pump. Drug Metab Dispos. 2014;42:566–574. doi: 10.1124/dmd.113.054205. [DOI] [PubMed] [Google Scholar]
- 6.Malhi H, Guicciardi ME, Gores GJ. Hepatocyte death: a clear and present danger. Physiol Rev. 2010;90:1165–1194. doi: 10.1152/physrev.00061.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trottier J, Białek A, Caron P, et al. Metabolomic profiling of 17 bile acids in serum from patients with primary biliary cirrhosis and primary sclerosing cholangitis: a pilot study. Dig Liver Dis. 2012;44:303–310. doi: 10.1016/j.dld.2011.10.025. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Y, Hong JY, Rockwell CE, et al. Effect of bile duct ligation on bile acid composition in mouse serum and liver. Liver Int. 2012;32:58–69. doi: 10.1111/j.1478-3231.2011.02662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dilger K, Hohenester S, Winkler-Budenhofer U, et al. Effect of ursodeoxycholic acid on bile acid profiles and intestinal detoxification machinery in primary biliary cirrhosis and health. J Hepatol. 2012;57:133–140. doi: 10.1016/j.jhep.2012.02.014. [DOI] [PubMed] [Google Scholar]
- 10.Woolbright BL, Li F, Xie Y, et al. Lithocholic acid feeding results in direct hepatotoxicity independent of neutrophil function in mice. Toxicol Lett. 2014;228:56–66. doi: 10.1016/j.toxlet.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Woolbright BL, Li F, Gholami P, et al. Human pathophysiological concentrations of bile salts induce necrosis in primary human hepatocytes (abstract) FASEB J. 2014;28(Suppl 1):398.1. [Google Scholar]
- 12.Marrero I, Sanchez-Bueno A, Cobbold PH. Taurolithocholate and taurolithocholate 3-sulphate exert different effects on cytosolic free Ca2+ concentration in rat hepatocytes. Biochem J. 1994;300:383–386. doi: 10.1042/bj3000383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Botla R, Spivey JR, Aguilar H, et al. Ursodeoxycholate (UDCA) inhibits the mitochondrial membrane permeability transition induced by glycochenodeoxycholate: a mechanism of UDCA cytoprotection. J Pharmacol Exp Ther. 1995;272:930–938. [PubMed] [Google Scholar]
- 14.Sokol RJ, Devereaux M, Khandwala R, et al. Evidence for involvement of oxygen free radicals in bile acid toxicity to isolated rat hepatocytes. Hepatology. 1993;17:869–881. [PubMed] [Google Scholar]
- 15.Allen K, Jaeschke H, Copple BL. Bile acids induce inflammatory genes in hepatocytes: a novel mechanism of inflammation during obstructive cholestasis. Am J Pathol. 2011;178:175–186. doi: 10.1016/j.ajpath.2010.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O’Brien KM, Allen KM, Rockwell CE, et al. IL-17A synergistically enhances bile acid-induced inflammation during obstructive cholestasis. Am J Pathol. 2013;183:1498–1507. doi: 10.1016/j.ajpath.2013.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gujral JS, Liu J, Farhood A, et al. Functional importance of ICAM-1 in the mechanism of neutrophil-induced liver injury in bile duct-ligated mice. Am J Physiol Gastrointest Liver Physiol. 2004;286:G499–G507. doi: 10.1152/ajpgi.00318.2003. [DOI] [PubMed] [Google Scholar]
- 18.Kim ND, Moon JO, Slitt AL, et al. Early growth response factor-1 is critical for cholestatic liver injury. Toxicol Sci. 2006;90:586–595. doi: 10.1093/toxsci/kfj111. [DOI] [PubMed] [Google Scholar]
- 19.Gujral JS, Farhood A, Bajt ML, et al. Neutrophils aggravate acute liver injury during obstructive cholestasis in bile duct-ligated mice. Hepatology. 2003;38:355–363. doi: 10.1053/jhep.2003.50341. [DOI] [PubMed] [Google Scholar]
- 20.Yang M, Ramachandran A, Yan HM, et al. Osteopontin is an initial mediator of inflammation and liver injury during obstructive cholestasis after bile duct ligation in mice. Toxicol Lett. 2014;224:186–195. doi: 10.1016/j.toxlet.2013.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schoemaker MH, Gommans WM, Conde de la Rosa L, et al. Resistance of rat hepatocytes against bile acid-induced apoptosis in cholestatic liver injury is due to nuclear factor-kappa B activation. J Hepatol. 2003;39:153–161. doi: 10.1016/s0168-8278(03)00214-9. [DOI] [PubMed] [Google Scholar]
- 22.Jang JH, Rickenbacher A, Humar B. Serotonin protects mouse liver from cholestatic injury by decreasing bile salt pool after bile duct ligation. Hepatology. 2012;56:209–218. doi: 10.1002/hep.25626. [DOI] [PubMed] [Google Scholar]
- 23.Gujral JS, Liu J, Farhood A, et al. Reduced oncotic necrosis in Fas receptor-deficient C57BL/6J-lpr mice after bile duct ligation. Hepatology. 2004;40:998–1007. doi: 10.1002/hep.20380. [DOI] [PubMed] [Google Scholar]
- 24.Fickert P, Trauner M, Fuchsbichler A, et al. Oncosis represents the main type of cell death in mouse models of cholestasis. J Hepatol. 2005;42:378–385. doi: 10.1016/j.jhep.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 25.Vinken M, Landesmann B, Goumenou M, et al. Development of an adverse outcome pathway from drug-mediated bile salt export pump inhibition to cholestatic liver injury. Toxicol Sci. 2013;136:97–106. doi: 10.1093/toxsci/kft177. [DOI] [PubMed] [Google Scholar]
- 26.Chatterjee S, Bijsmans IT, van Mil SW, et al. Toxicity and intracellular accumulation of bile acids in sandwich-cultured rat hepatocytes: role of glycine conjugates. Toxicol In Vitro. 2014;28:218–230. doi: 10.1016/j.tiv.2013.10.020. [DOI] [PubMed] [Google Scholar]
- 27.Patel T, Bronk SF, Gores GJ. Increases of intracellular magnesium promote glycodeoxycholate-induced apoptosis in rat hepatocytes. J Clin Invest. 1994;94:2183–2192. doi: 10.1172/JCI117579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones B, Roberts PJ, Faubion WA, et al. Cystatin A expression reduces bile salt-induced apoptosis in a rat hepatoma cell line. Am J Physiol. 1998;275:G723–G730. doi: 10.1152/ajpgi.1998.275.4.G723. [DOI] [PubMed] [Google Scholar]
- 29.Galle PR, Theilmann L, Raedsch R, et al. Ursodeoxycholate reduces hepatotoxicity of bile salts in primary human hepatocytes. Hepatology. 1990;12:486–491. doi: 10.1002/hep.1840120307. [DOI] [PubMed] [Google Scholar]
- 30.Gonzalez R, Cruz A, Ferrin G, et al. Nitric oxide mimics transcriptional and post-translational regulation during α-tocopherol cytoprotection against glycochenodeoxycholate-induced cell death in hepatocytes. J Hepatol. 2011;55:133–144. doi: 10.1016/j.jhep.2010.10.022. [DOI] [PubMed] [Google Scholar]
- 31.Szabo M, Veres Z, Baranyai Z, et al. Comparison of human hepatoma HepaRG cells with human and rat hepatocytes in uptake transport assays in order to predict a risk of drug induced hepatotoxicity. PLoS One. 2013;8:e59432. doi: 10.1371/journal.pone.0059432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liang D, Hagenbuch B, Stieger B, et al. Parallel decrease of Na(+)-taurocholate cotransport and its encoding mRNA in primary cultures of rat hepatocytes. Hepatology. 1993;18:1162–1166. [PubMed] [Google Scholar]
- 33.Rippin SJ, Hagenbuch B, Meier PJ, et al. Cholestatic expression pattern of sinusoidal and canalicular organic anion transport systems in primary cultured rat hepatocytes. Hepatology. 2001;33:776–782. doi: 10.1053/jhep.2001.23433. [DOI] [PubMed] [Google Scholar]
- 34.Jemnitz K, Veres Z, Vereczkey L. Contribution of high basolateral bile salt efflux to the lack of hepatotoxicity in rat in response to drugs inducing cholestasis in human. Toxicol Sci. 2010;115:80–88. doi: 10.1093/toxsci/kfq044. [DOI] [PubMed] [Google Scholar]
- 35.Kullak-Ublick GA, Meier PJ. Mechanisms of cholestasis. Clin Liver Dis. 2000;4:357–385. doi: 10.1016/s1089-3261(05)70114-8. [DOI] [PubMed] [Google Scholar]
- 36.Padda MS, Sanchez M, Akhtar AJ, et al. Drug-induced cholestasis. Hepatology. 2011;53:1377–1387. doi: 10.1002/hep.24229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kubitz R, Droge C, Stindt J, et al. The bile salt export pump (BSEP) in health and disease. Clin Res Hepatol Gastroenterol. 2012;36:536–553. doi: 10.1016/j.clinre.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 38.Fattinger K, Funk C, Pantze M, et al. The endothelin antagonist bosentan inhibits the canalicular bile salt export pump: a potential mechanism for hepatic adverse reactions. Clin Pharmacol Ther. 2001;69:223–231. doi: 10.1067/mcp.2001.114667. [DOI] [PubMed] [Google Scholar]
- 39.Antherieu S, Bachour-El Azzi P, Dumont J, et al. Oxidative stress plays a major role in chlorpromazine-induced cholestasis in human HepaRG cells. Hepatology. 2013;57:1518–1529. doi: 10.1002/hep.26160. [DOI] [PubMed] [Google Scholar]
- 40.Stieger B, Beuers U. The canalicular bile salt export pump BSEP (ABCB11) as a potential therapeutic target. Curr Drug Targets. 2011;12:661–670. doi: 10.2174/138945011795378496. [DOI] [PubMed] [Google Scholar]
- 41.Garzel B, Yang H, Zhang L, et al. The role of bile salt export pump gene repression in drug-induced cholestatic liver toxicity. Drug Metab Dispos. 2014;42:318–322. doi: 10.1124/dmd.113.054189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rust C, Bauchmuller K, Bernt C, et al. Sulfasalazine reduces bile acid induced apoptosis in human hepatoma cells and perfused rat livers. Gut. 2006;55:719–727. doi: 10.1136/gut.2005.077461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Muhlfeld S, Domanova O, Berlage T, et al. Short-term feedback regulation of bile salt uptake by bile salts in rodent liver. Hepatology. 2012;56:2387–2397. doi: 10.1002/hep.25955. [DOI] [PubMed] [Google Scholar]
- 44.Denk GU, Maitz S, Wimmer R, et al. Conjugation is essential for the anticholestatic effect of NorUrsodeoxycholic acid in taurolithocholic acid-induced cholestasis in rat liver. Hepatology. 2010;52:1758–1768. doi: 10.1002/hep.23911. [DOI] [PubMed] [Google Scholar]
- 45.Denk GU, Kleiss CP, Wimmer R, et al. Tauro-β-muricholic acid restricts bile acid-induced hepatocellular apoptosis by preserving the mitochondrial membrane potential. Biochem Biophys Res Commun. 2012;424:758–764. doi: 10.1016/j.bbrc.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 46.Le Vee M, Jigorel E, Glaise D, et al. Functional expression of sinusoidal and canalicular hepatic drug transporters in the differentiated human hepatoma HepaRG cell line. Eur J Pharm Sci. 2006;28:109–117. doi: 10.1016/j.ejps.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 47.Bajt ML, Knight TR, Lemasters JJ, et al. Acetaminophen-induced oxidant stress and cell injury in cultured mouse hepatocytes: protection by N-acetyl cysteine. Toxicol Sci. 2004;80:343–349. doi: 10.1093/toxsci/kfh151. [DOI] [PubMed] [Google Scholar]
- 48.Yan HM, Ramachandran A, Bajt ML, et al. The oxygen tension modulates acetaminophen-induced mitochondrial oxidant stress and cell injury in cultured hepatocytes. Toxicol Sci. 2010;117:515–523. doi: 10.1093/toxsci/kfq208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hohenester S, Vennegeerts T, Wagner M, et al. Physiological hypoxia prevents bile salt-induced apoptosis in human and rat hepatocytes. Liver Int. 2014;34(8):1224–1231. doi: 10.1111/liv.12368. [DOI] [PubMed] [Google Scholar]
- 50.Woolbright BL, Ramachandran A, McGill MR, et al. Lysosomal instability and cathepsin B release during acetaminophen hepatotoxicity. Basic Clin Pharmacol Toxicol. 2012;111:417–425. doi: 10.1111/j.1742-7843.2012.00931.x. [DOI] [PMC free article] [PubMed] [Google Scholar]