

SUMMARY
Intestinal Th17 cells are induced and accumulate in response to colonization with a subgroup of intestinal microbes such as segmented filamentous bacteria (SFB) and certain extracellular pathogens. Here, we show that adhesion of microbes to intestinal epithelial cells (ECs) is a critical cue for Th17 induction. Upon monocolonization of germ-free mice or rats with SFB indigenous to mice (M-SFB) or rats (R-SFB), M-SFB and R-SFB showed host-specific adhesion to small intestinal ECs, accompanied by host-specific induction of Th17 cells. Citrobacter rodentium and Escherichia coli O157 triggered similar Th17 responses, whereas adhesion-defective mutants of these microbes failed to do so. Moreover, a mixture of 20 bacterial strains, which were selected and isolated from fecal samples of a patient with ulcerative colitis on the basis of their ability to cause a robust induction of Th17 cells in the mouse colon, also exhibited EC-adhesive characteristics.
Graphical abstract
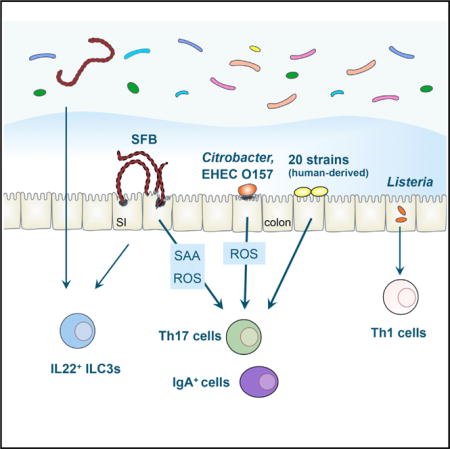
INTRODUCTION
The gut microbiota contributes to the constitutive development of Th17 cells in the intestinal lamina propria (LP) (Atarashi et al., 2008; Ivanov et al., 2008). Among commensals, segmented filamentous bacteria (SFB) are one of the most potent inducers of Th17 cells, and monocolonization of mice with SFB causes abundant accumulation of Th17 cells in the small intestinal (SI) LP (Gaboriau-Routhiau et al., 2009; Ivanov et al., 2009). Recent reports have shown that most of the intestinal Th17 cells induced by SFB have T cell receptors (TCRs) that specifically recognize SFB antigens (Goto et al., 2014; Yang et al., 2014). However, since the SFB antigens themselves do not dictate Th17 differentiation (Yang et al., 2014), and microbiota-mediated Th17 cell development occurs independently of major innate immune receptors (Atarashi et al., 2008; Ivanov et al., 2009), SFB colonization must elicit unique signaling pathways in the intestine to generate a Th17-conducive environment.
SFB are spore-forming gram-positive bacteria with a segmented and filamentous morphology, and tight adhesion to SI epithelial cells (ECs) is a remarkable characteristic feature of these bacteria (Davis and Savage, 1974). SFB are widely distributed in vertebrates (Klaasen et al., 1993). In spite of the morphological similarities of SFB isolated from various hosts, their 16S rRNA gene sequences differ, and several reports suggest that SFB have undergone host species-specific selection and adaptation (Chung et al., 2012). The complete genomic sequences of SFB colonizing the mouse and rat intestines, referred to as M-SFB and R-SFB, respectively, were determined. Although the overall genomic organization of M-SFB and R-SFB are similar, 5%–10% of the genes are specific to each strain, and the amino acid sequence identity between orthologous gene pairs is on average 80% (Prakash et al., 2011). Analysis of differences between M-SFB and R-SFB may be useful to improve understanding of the effects of SFB on the immune system.
In addition to SFB colonization, infections with several extracellular pathogens such as Candida albicans and Citrobacter rodentium are known to induce Th17 cells (Conti and Gaffen, 2010; Mangan et al., 2006). Th17 cells induce the recruitment of neutrophils and activation of ECs, leading to enhanced clearance of extracellular pathogens in concert with other immune cells such as IgA-secreting plasma cells and group 3 innate lymphoid cells (ILC3s). The induction of Th17 cells by those pathogens has been postulated to be mediated by the local cytokine milieu produced by intestinal ECs and specific subsets of myeloid cells (Weaver et al., 2013). However, it remains unclear which features of these particular microbes specifically elicit Th17 versus other types of immune cell responses at intestinal mucosal sites.
Because SFB and C. rodentium commonly adhere to ECs, we hypothesized that adhesion-mediated activation of ECs plays a pivotal role in the induction of Th17 cells. Accordingly, we examined the ability of M-SFB, R-SFB, wild-type, and mutant strains of C. rodentium and enterohemorrhagic Escherichia coli (EHEC) O157:H7 to adhere to ECs and induce Th17 cells. In addition, by combining gnotobiotic technique and anaerobic culturing of members of the intestinal microbiota from a patient with ulcerative colitis (UC), we isolated 20 strains based on their ability to induce Th17 cells in mice and examined EC-adhesive characteristics of these 20 Th17-inducing human strains. Our findings indicate that adhesion to ECs is a common mechanism used by intestinal microbes to activate host Th17 responses.
RESULTS
Host-Specific Adhesion to SI ECs and Th17 Induction by SFB
C57BL/6 (B6) or IQI germ-free (GF) mice were orally inoculated with R-SFB or M-SFB, and their intestinal colonization was monitored by qPCR analysis. The concentration of fecal and SI luminal R-SFB DNA quickly increased and reached a plateau within 1 week; the kinetics and levels were comparable to those of M-SFB (Figures 1A and S1A). Consistent with the qPCR results, Gram-stained smears of cecal luminal contents contained equivalent numbers of R-SFB and M-SFB with indistinguishable morphology (Figure S1B), indicating that R-SFB and M-SFB both colonize and grow robustly within the mouse intestinal lumen. In contrast, when SI mucosa-associated SFB DNA amounts were examined, we detected much lower levels of R-SFB than of M-SFB (Figure 1A). We also performed scanning electron microscopy (SEM) of washed SI mucosa to visualize EC-adhering SFB. Numerous M-SFB were observed adhering tightly to the mouse SI epithelium. In contrast, we did not detect any R-SFB adhering to SI ECs (Figures 1B and S1C). R-SFB also failed to adhere to SI ECs in monocolonized Rag1−/− mice or IL-2Rg−/− Rag2−/− mice (Figures S1D and S1E), making it unlikely that the impaired adhesion of R-SFB to mouse SI ECs was due to a R-SFB-specific reaction by the mouse immune system. Although we detected some EC-adhering M-SFB and R-SFB in the mouse colon, these were less frequent than in the SI (Figure 1B).
Figure 1. EC Adhesion and Th17 Induction by SFB in Mice and Rats.
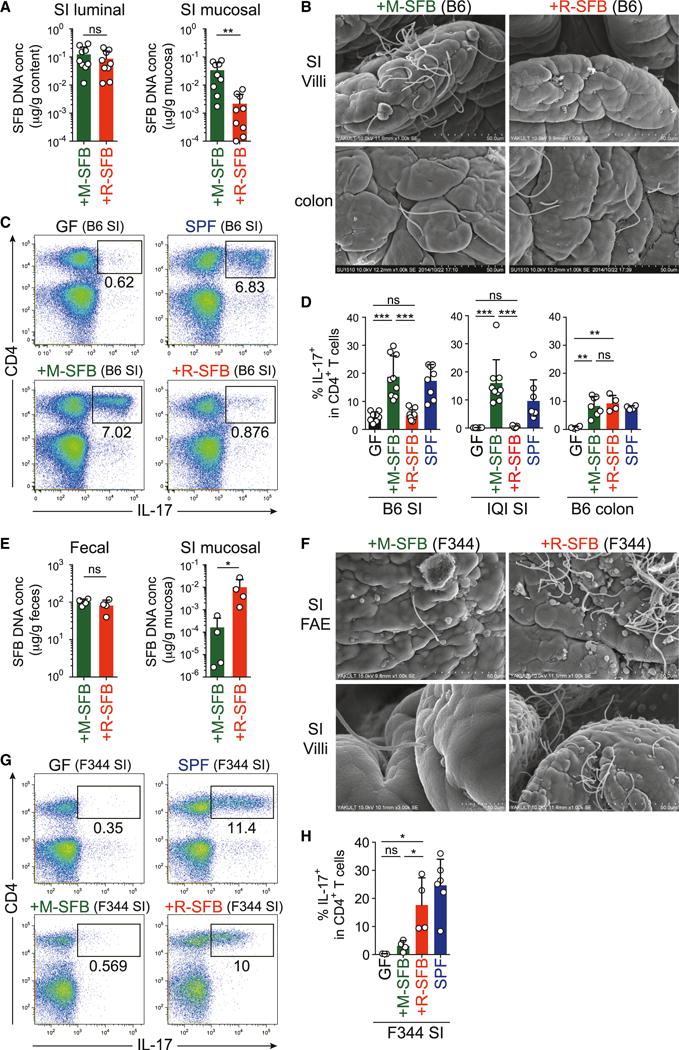
B6 or IQI mice (A–D) and F344 rats (E–H) were monocolonized with M-SFB or R-SFB for 3 weeks.
(A and E) qPCR analysis for SFB DNA in feces, SI luminal contents, and mucosal tissue specimens.
(B and F) SEM images of epithelial surfaces in the SI and colon.
(C, D, G, and H) Th17 cell frequencies in SI and colon LP. Representative dot plots gated on total lymphocytes (C and G) and summarized data gated on CD4+ T cells (D and H) are shown.
Error bars represent SD. See also Figure S1.
We then investigated the effects of monocolonization of mice with M-SFB or R-SFB on the induction of Th17 cells. In agreement with previous reports (Gaboriau-Routhiau et al., 2009; Ivanov et al., 2009), a robust accumulation of Th17 cells was observed in the SI LP of B6 or IQI mice monocolonized with M-SFB. In contrast, R-SFB colonization had no significant effect on the number of Th17 cells (Figures 1C and 1D). Notably, both M-SFB and R-SFB induced the accumulation of Th17 cells in the colonic LP, although less efficiently than that in the SI LP following M-SFB colonization (Figure 1D), correlating with the EC adhesion efficacy of the SFB.
We next performed the reciprocal experiment by monocolonizing GF F344 rats with R-SFB or M-SFB. After oral inoculation of GF rats with R-SFB, high levels of R-SFB DNA were detected in feces and SI mucosal tissues by qPCR (Figure 1E). Examination of the rat SI mucosal surface by SEM revealed the frequent presence of R-SFB adhering tightly to the follicular-associated epithelium (FAE) and villous ECs (Figure 1F). In contrast, far fewer M-SFB associated with the rat SI ECs, although the amount of M-SFB and R-SFB DNA in the feces was comparable (Figures 1E and 1F). We then investigated the effects of R-SFB and M-SFB monocolonization on Th17 cells in rats. The frequency of Th17 cells in the SI LP was markedly increased after monocolonization of GF rats with R-SFB, whereas M-SFB-monocolonization induced only a slight increase in the frequency of these cells (Figures 1G and 1H). Taken together, adhesion of SFB to intestinal ECs is a host-specific event and strongly correlates with Th17 cell induction.
Induction of Antigen-Specific Th17 Cells by EC-Adhesive SFB
To examine whether EC adhesion is required for priming SFB antigen-specific Th17 cell differentiation, we generated mice co-colonized with M-SFB and R-SFB and assessed the antigen specificity of the SI LP Th17 cells. In the feces of co-colonized mice, similar amounts of M-SFB and R-SFB DNA were detected (Figure S2A). SI LP cells, which include T cells and antigen-presenting cells (APCs), were isolated from the co-colonized mice, and stimulated ex vivo either with PMA and ionomycin (P/I) or with autoclaved cecal content from GF, M-SFB-, or R-SFB-monocolonized mice. Cytokine expression was analyzed as a readout for TCR activation. Although less efficient than P/I, the M-SFB antigens evoked IL-17 expression in a significant number of cells (Figures 2A and 2B). In contrast, the R-SFB antigens elicited no response. Therefore, a substantial fraction of Th17 cells that accumulated in the SI LP of co-colonized mice had TCRs specific to the EC-adhering M-SFB, but not to the non-adhering R-SFB. These results suggest that antigens from EC-adhering SFB are taken up and presented by APCs much more efficiently than those of non-adhering SFB.
Figure 2. Effects of SFB Adhesion on Antigen-Specific Th17 Cells, IgA+ Cells, and ILC3s.
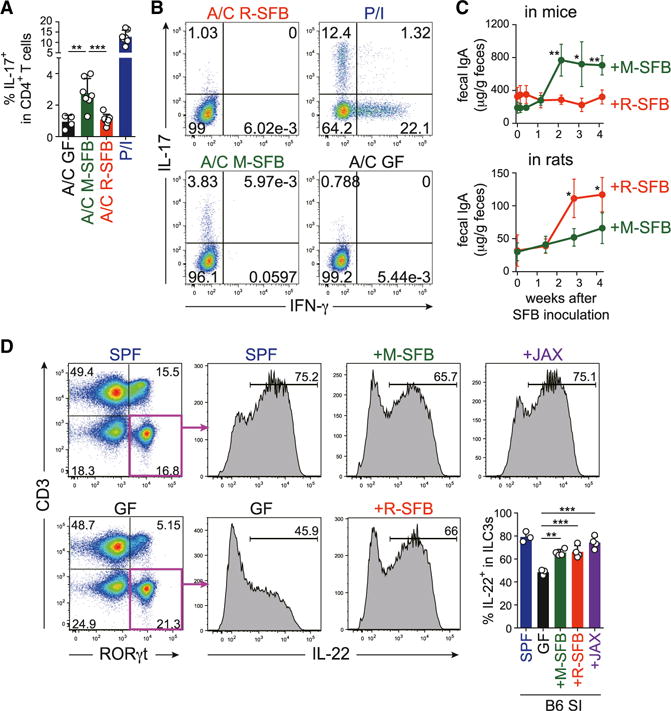
(A and B) Cytokine responses of SI LP CD4+ T cells from M-SFB and R-SFB co-colonized B6 mice to the indicated ex vivo stimulation. A/C, autoclaved cecal contents; P/I, PMA/ionomycin.
(C) Fecal IgA levels of indicated monocolonized B6 mice and F344 rats (n = 5).
(D) The frequencies of ILC3s among CD90+ cells (left dot plots) and IL22+ cells among ILC3s (right histograms and bar graphs) in SI LP cells of the indicated mice.
Error bars represent SD. See also Figure S2.
Recently, two major protein antigens responsible for M-SFB-mediated SI LP Th17 cell induction were identified (SFBNYU_003340 and SFBNYU_004990) (Yang et al., 2014). We analyzed the R-SFB genome and identified the gene encoding the orthologous protein corresponding to SFBNYU_003340, but could not identify an SFBNYU_004990 ortholog. The minimal epitope within M-SFB SFBNYU_003340 was mapped as QFSGAVPNK (Yang et al., 2014), and the corresponding epitope region in R-SFB was found to be QFNGQVPNN. The M-SFB epitope peptide efficiently induced a Th17 response when added to the ex vivo cultures of SI LP cells isolated from specific pathogen-free (SPF) mice (Figure S2B). In contrast, the same SI LP cells had no reactivity to the R-SFB peptide QFNGQVPNN. Therefore, peptides serving as dominant antigens for SI LP Th17 cell induction in mice are specifically expressed by M-SFB.
Effects of EC-Adhering SFB on IgA+ and ILC3 Cells
SFB colonization has been shown to enhance IgA production (Talham et al., 1999; Umesaki et al., 1999). Consistent with these reports, monocolonization of B6 mice with M-SFB induced a significant increase in the levels of fecal IgA and frequencies of IgA+CD138+ plasma cells in the SI LP (Figures 2C and S2C). In contrast, R-SFB monocolonization in mice resulted in a weak IgA response (Figures 2C and S2C). We also examined IgA production in SFB monocolonized rats. Fecal IgA levels were substantially increased after monocolonization of GF rats with R-SFB, whereas M-SFB-mono-colonization induced only a modest increase (Figure 2C). These findings suggest that luminal colonization is insufficient for the full induction of IgA+ cells, but instead an interaction between SFB and ECs is required, which is similar to Th17 cells.
Next, we examined the effects of SFB colonization on ILC3s, which share many features with Th17 cells (Spits et al., 2013). Thy1+CD3−RORγt+ cells corresponding to ILC3s were much more abundant in the SI LP of SPF B6 mice than in the spleen (Figure S2D), and ~70% of these cells became positive for IL-22 after ex vivo stimulation with IL-23 (Figure 2D). Although the abundance of SI LP ILC3s in GF versus SPF mice did not differ significantly, the frequency of IL-22-proficient (IL-22+) cells among ILC3s was dramatically decreased in GF mice (Figure 2D). Monocolonization of mice with M-SFB resulted in a significant increase in the frequency of IL-22+ SI ILC3s, although the level did not reach that observed in SPF mice (Figure 2D). The increase in IL-22+ cells upon monocolonization with M-SFB was observed for both NKp46+ and NKp46− ILC3 populations (Figure S2E). Interestingly, R-SFB induced a significant increase in the frequency of IL-22+ cells with a magnitude similar to that of M-SFB (Figure 2D). The induction of IL-22+ ILC3s was also observed in mice inoculated with feces from Jackson Laboratory (JAX) SPF mice, which are known to be devoid of SFB (Ivanov et al., 2009) (Figure 2D), indicating that IL-22+ ILC3s can be induced by non-adhering SFB and even by non-SFB commensals, which is in contrast to Th17 and IgA+ cells.
SI EC Activation by Adhering SFB
We next examined the influence of SFB adhesion on SI EC gene expression profiles by RNA sequencing (RNA-seq). The expression of several genes was highly upregulated in SI ECs of M-SFB-colonized mice compared to GF and R-SFB-colonized mice (Figures 3A and S3A). The upregulated genes included three isoforms of serum amyloid A (SAA), regenerating islet-derived protein 3 β (Reg3β) and Reg3γ and nitric oxide synthase 2 (Nos2). The expression differences of selected genes were confirmed by qPCR analysis (Figures 3B and S3B). Elevated levels of SAA1 protein in SI ECs of M-SFB-monocolonized mice were clearly visualized by immunofluorescence microscopy (Figure 3C). Recombinant SAA1 markedly enhanced the in vitro differentiation of naive CD4+ T cells into Th17 cells mediated by CD11c+ cells, IL-6, and transforming growth factor β (TGF-β) (Figure 3D). This SAA1-mediated enhancement was severely attenuated in the absence of CD11c+ cells or of IL-6 and TGF-β, or in the presence of an anti-IL-1 receptor 1 (IL-1R1) blocking antibody (Figures 3D and 3E). In fact, SAA1 elevated the expression level of IL-1β mRNA in splenic CD11c+ cells (Figure 3F). Therefore, the current data extend previous studies reporting that SAAs are induced in SI ECs by colonization with SFB (Ivanov et al., 2009) and suggest that SAA induction requires SFB adhesion to ECs. Furthermore, the SAAs produced by this mechanism likely condition neighboring CD11c+ myeloid cells to produce IL-1β and probably other factors that act with IL-6 and TGF-β to enhance Th17 cell differentiation.
Figure 3. Adhesion-Mediated EC Activation.
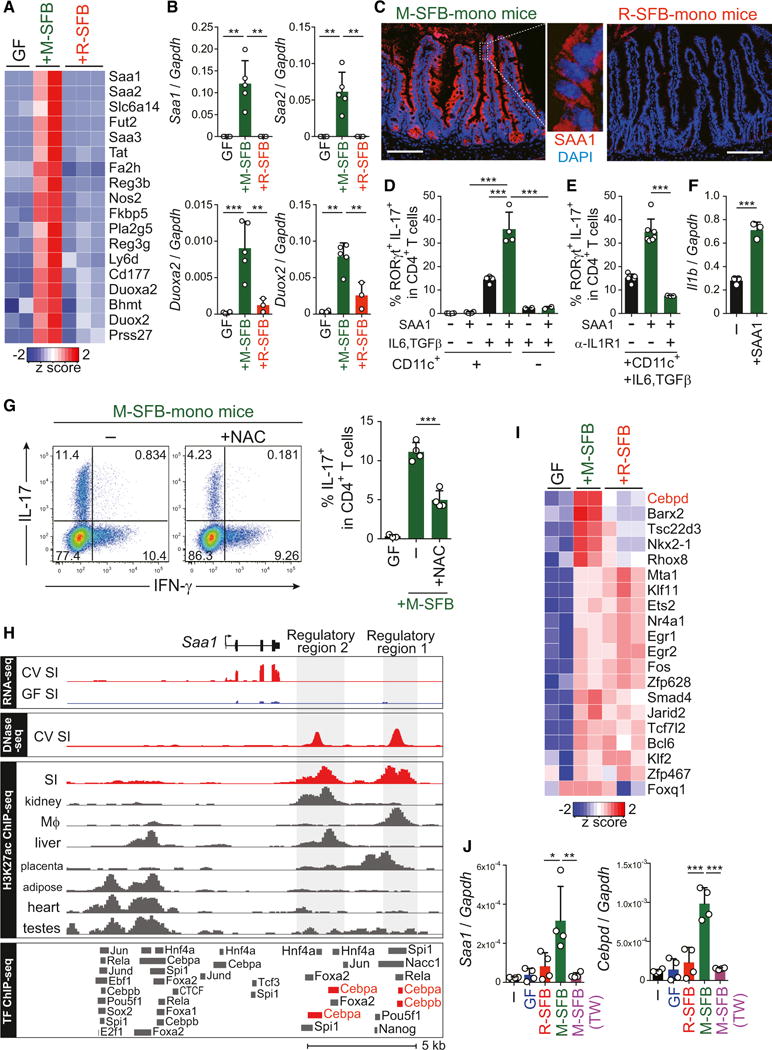
(A) Heatmap showing the relative abundance for gene transcripts upregulated in SI ECs of M-SFB-mono versus GF and R-SFB-mono IQI mice. Each column represents a single mouse.
(B) qPCR for the selected genes relative to Gapdh in SI ECs from the indicated mice.
(C) Immunostaining of SIs from the corresponding mice for SAA1 (red) and DAPI (blue). Scale bar, 100 μm.
(D and E) The percentage of RORγt+IL-17+ cells after culture of splenic naive CD4 T cells in the presence of various combinations of splenic CD11c+ cells, IL-6, TGF-β, SAA1, and/or anti-IL-1R1 antibody.
(F) Il1b expression in splenic CD11c+ cells from B6 SPF mice stimulated with recombinant SAA1.
(G) SI LP Th17 cell frequencies of IQI M-SFB-mono mice either untreated (−) or treated with NAC.
(H) RNA-seq and DNase-seq data from SI ECs of GF and conventional (CV) mice at the region surrounding the Saa1 locus. H3K27ac ChIP-seq data for SI and various other tissues obtained from (Shen et al., 2012). ChIP-seq datasets for TFs assayed in various cell and tissue types. SI EC regulatory regions are highlighted in gray.
(I) The relative abundance for TF genes that are >2-fold different between M-SFB-mono mice versus GF or R-SFB-mono mice.
(J) SAA1 and C/EBPδ genes expression in aMoS7 cells cultured with cecal content from GF mice or a Percoll-enriched M-SFB or R-SFB either in contact or separated by a Transwell membrane (TW).
Error bars represent SD. See also Figure S3.
In addition to the increase in SAAs, mRNA levels of the reactive oxygen species (ROS)-generating enzyme dual oxidase 2 (Duox2) and of its maturation factor Duoxa2 were also highly up-regulated in SI ECs in M-SFB-monocolonized mice (Figures 3A and 3B). Treatment of M-SFB-monocolonized mice with a ROS scavenger, N-acetyl-L-cysteine (NAC), in the drinking water significantly limited Th17 cell induction (Figure 3G) without affecting adhesion of M-SFB to SI ECs (Figure S3C). Therefore, EC-adhering SFB affect Th17 cell accumulation via integration of a number of different molecular mechanisms including production of SAAs and ROS by ECs.
Toward an understanding of the molecular basis of adherent SFB-mediated SAA induction, we explored the transcriptional regulatory elements of the Saa1 gene locus. We identified two DNase I hypersensitive sites downstream of the Saa1 gene, which were uniquely active in the SI and distinguished by acetylated histone 3 lysine 27 (H3K27ac) active enhancer marks (Shen et al., 2012) (Figure 3H). Investigation of publicly available datasets of chromatin immunoprecipitation sequencing (ChIP-seq) for transcription factor (TF) binding (Camp et al., 2014) suggested interactions of CCAAT-enhancer-binding protein (C/EBP) with both regulatory regions of Saa1 (Figure 3H). Our SI EC RNA-seq data and qPCR analysis showed that C/EBPδ was specifically upregulated in SI ECs of M-SFB-monocolonized mice (Figures 3I and S3B), suggesting that it may be a key TF regulating SAA1 expression. In line with this finding, we observed co-induction of SAA1 and C/EBPδ expression in a mouse SI EC line (aMos7) co-cultured with M-SFB in vitro (Figure 3J). Induction of SAA1 and C/EBPδ expression was observed only when ECs were in direct contact with M-SFB (Figure 3J); under these conditions M-SFB adhesion to the aMoS7 cells was confirmed by fluorescence in situ hybridization (FISH) and SEM (Figures S3D and S3E). The induction of SAA1 and C/EBPδ expression by M-SFB was further enhanced by the addition of F-actin inhibitory drugs, latrunculin A, or swinholide A (Figure S3F). Therefore, actin reorganization in ECs induced by SFB adhesion may lead to the elevation of C/EBPδ expression, which then contributes to SAA1 expression.
Influence of Mouse Genetic Background on SFB-Mediated Th17 Cell Induction
All of the above experiments were performed using B6 or IQI mice. We next examined the influence of mouse genetic background on intestinal Th17 cell induction. The frequency of Th17 cells in SPF C57BL/10 and B10.D2 mice was similar to that in B6 and IQI (ICR) mice (Figure 4A). The only exception was BALB/c mice, which had a much lower frequency of Th17 cells even under SPF conditions or after monocolonization with M-SFB (Figures 4A, 4B, and S4A). It is noteworthy that BALB/c mice had substantial numbers of RORγt+ single-positive cells but had severe reduction of RORγt+IL-17+ double-positive cells (Figure S4A). M-SFB, but not R-SFB, were found to adhere to SI EC surfaces in BALB/c mice, similar to the results in B6 mice (Figures 4C and S4B) and splenic naive CD4+ T cells from BALB/c mice gave rise to a normal frequency of RORγt+IL-17+ cells when differentiated under Th17 conditions (Figure S4C), suggesting that SFB adhesion to SI ECs occurs normally and that CD4+ T cells do not have an intrinsic differentiation defect, but instead that EC activation and/or subsequent LP cell signaling required for RORγt+IL-17+ Th17 induction may be defective in BALB/c mice.
Figure 4. Influences of Mouse Genetic Background.
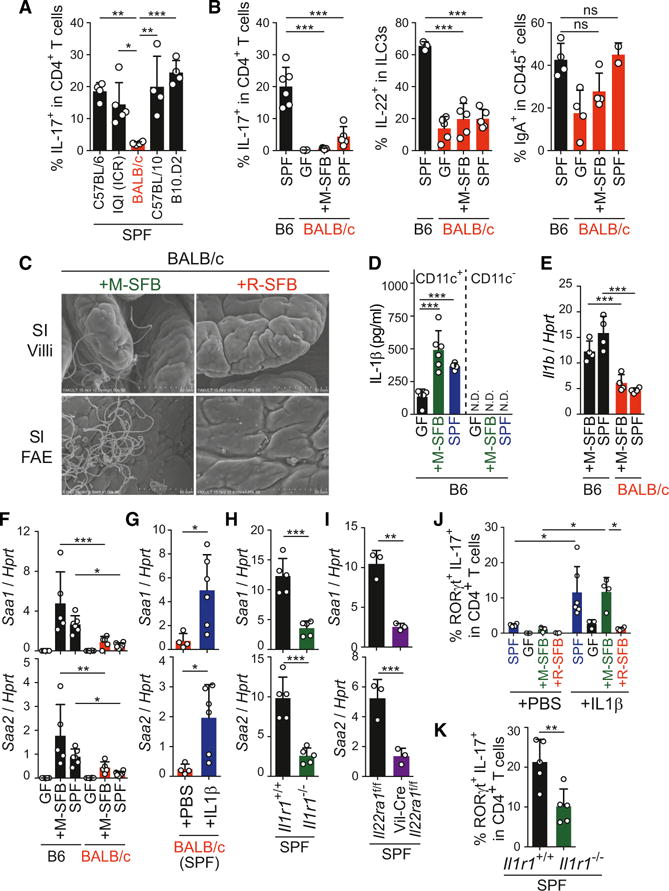
(A and B) The percentages of Th17, IL-22+ ILC3, and CD138+IgA+ cells in SI LP of the indicated mice.
(C) SEM images of SI epithelial surface of the indicated BALB/c mice.
(D and E) IL-1β protein production (D) and mRNA expression (E) from SI LP CD11c+ and CD11c− cells from the indicated mice.
(F–I) Saa1/2 mRNA expression in SI ECs from the indicated mice.
(J and K) The percentage of RORγt+IL-17+ cells among SI LP CD4+ T cells in IL-1β-injected BALB/c mice (J) or Il1r1−/− B6 mice (K).
Error bars represent SD. See also Figure S4.
Among Th17-conducting cytokines, microbiota-induced constitutive and high-level expression of IL-1β was observed in SI LP CD11c+ cells of B6 mice (Figure 4D), which is in line with previous reports (Mortha et al., 2014; Shaw et al., 2012). IL-1β expression was significantly reduced in M-SFB-monocolonized and SPF BALB/c mice, compared with that of B6 mice (Figure 4E). Besides the reduced expression of IL-1β in CD11c+ cells, expression of SAA1/2 was significantly decreased in SI ECs of BALB/c mice (Figures 4F and S4D). Injection of exogenous IL-1β into SPF BALB/c mice elevated expression of SAAs in SI ECs (Figure 4G) and this was accompanied by an increase in SI LP RORγt+IL-17+ Th17 cells (Figures 4J and S4E), suggesting that insufficient IL-1β production by LP CD11c+ cells is responsible, at least in part, for the reduction in SAA expression and Th17 cell accumulation in BALB/c mice. Consistent with this interpretation, B6 Il1r1−/− mice phenocopied BALB/c mice, displaying decreased expression of SAAs in SI ECs (Figure 4H) and a significant reduction in the number of SI LP RORγt+IL-17+ Th17 cells (Figures 4K and S4F). It is noteworthy that IL-1β administration induced Th17 cell accumulation in M-SFB-colonized mice, but not in R-SFB-colonized or GF BALB/c mice, revealing a requirement for the presence of EC-adhering bacteria for the IL-1β effect (Figures 4J and S4E).
BALB/c mice had reduced frequencies of IL-22+ cells among ILC3s compared with B6 mice under both SPF and M-SFB-monocolonized conditions, whereas the frequency of IgA+ cells was normal (Figures 4B and S4G). An EC-specific deficiency of the IL-22 receptor a1 gene (Vil-Cre crossed with Il22ra1f/f) on an SPF B6 background resulted in reduced expression of SAAs by SI ECs (Figure 4I), suggesting that IL-22 from ILC3s, together with IL-1β from CD11c+ cells, act on SI ECs to potentiate their expression of SAA. Vil-Cre crossed with Il22ra1f/f mice showed a tendency toward reduction of RORγt+IL-17+ Th17 cells in the SI LP (Figure S4H). Therefore, adhesive SFB may create a complex circuitry of interactions between ECs, DCs and ILC3s mediated by SAA, IL-1β, IL-22, ROS, and probably other factors, to amplify and maintain signaling for constitutive accumulation of Th17 cells (Figure S4I).
Extracellular Pathogens Induce Identical Th17 Responses through EC Adhesion
To further test the link between bacterial EC adhesion and Th17 induction, we examined Citrobacter rodentium, an attaching/effacing enteropathogen known to induce Th17 cells in mice (Mangan et al., 2006). B6 GF mice were mono-colonized with wild-type or a mutant C. rodentium strain lacking the eae gene, which encodes Intimin, a protein essential for EC adhesion (Kamada et al., 2012). The Δeae and wild-type C. rodentium expanded equivalently in the intestinal lumen, as demonstrated by recovery of similar amounts of bacterial DNA from the colon luminal contents of both groups of mice after 5 days of monocolonization (Figure 5A). SEM and immunofluorescence microscopy revealed that the wild-type strain had a strong tendency to adhere to colonic ECs, whereas the Δeae strain did not adhere to ECs and the inner mucus layer of the colon remained intact (Figures 5B and 5C). These results were further confirmed by qPCR quantification of mucosa-associated C. rodentium (Figure 5A). Colonization with wild-type C. rodentium resulted in potent induction of Th17 cells in the colon, whereas Δeae C. rodentium elicited a far weaker Th17 response (Figure 5D). Colonization with wild-type C. rodentium induced a greater increase in fecal IgA levels than the mutant strain (Figure 5E). Therefore, similarly to commensal SFB, pathogenic C. rodentium induces Th17 and IgA responses, most likely through adhesion to ECs. Of note, during the early phase (5 days after infection), when the epithelial layer was relatively intact, infection with wild-type C. rodentium caused a more conspicuous increase in Th17 cells than IFN-γ-producing Th1 cells. However, in the late phase (14 days after infection), when the epithelial layer was severely disrupted and bacterial invasion had occurred, there was a massive increase in Th1 cells rather than Th17 cells (Figures S5A and S5B), supporting the requirement of intact ECs for Th17 induction in response to C. rodentium, without which the same microbe would elicit a different type of immune response. We also examined IL-22+ ILC3 induction by C. rodentium colonization. The Δeae strain induced a robust increase in the frequency of colonic IL-22+ ILC3 cells comparable to the response observed in mice colonized with the wild-type strain (Figure S5C), indicating that immune stimulatory activity is not completely abrogated in the Δeae strain, and the induction of IL-22+ ILC3 cells occurs independently of bacterial EC-adhesion.
Figure 5. EC Adhesion-Mediated Th17 Induction by C. rodentium.
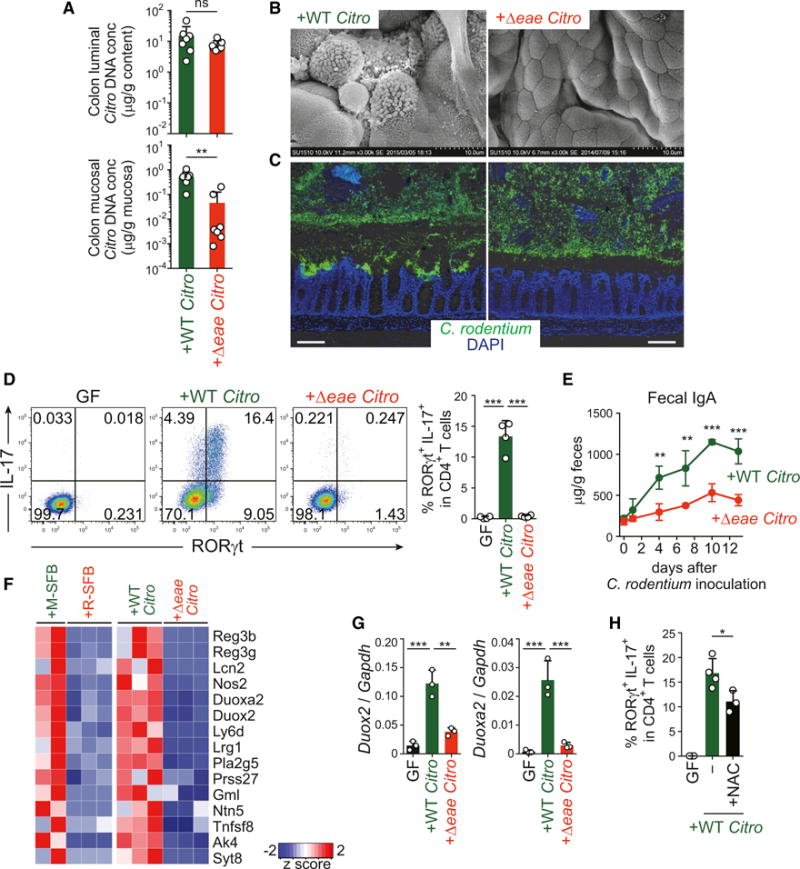
IQI mice were monocolonized with wild-type (WT) or Δeae mutant of C. rodentium for 5 days.
(A) DNA of C. rodentium in colonic luminal contents and mucosal tissues.
(B) SEM images of colonic villi of the indicated mice.
(C) C. rodentium localization visualized by O antigen antisera (green) and DAPI (blue) staining. Scale bar, 100 μm.
(D) The percentage of RORγt+IL-17+ colonic CD4+ T cells.
(E) Fecal IgA levels of the indicated mice (n = 4).
(F) The relative abundance of gene transcripts that were commonly upregulated in M-SFB-mono mice versus R-SFB-mono mice and WT C. rodentium-mono mice versus Δeae C. rodentium-mono mice.
(G) Duox2 and Duoxa2 mRNA expression in colonic ECs of the indicated mice.
(H) Th17 cell frequencies of WT C. rodentium-mono IQI mice either untreated (−) or treated with NAC.
Error bars represent SD. See also Figure S5.
Next, we performed gene expression analysis by RNA-seq of colon ECs from mice monocolonized with the Δeae or wild-type C. rodentium strain. In comparison to the Δeae strain, the wild-type strain specifically induced the expression of genes, such as Duox2, Duoxa2, RegIIIb, RegIIIg, and Nos2, which overlap with genes induced in SI ECs by colonization with EC-adherent SFB (Figures 5F, 5G, S5D, and S5E), although the expression of SAAs in the colon was not significantly enhanced by C. rodentium monocolonization (Figure S5D). Treatment of wild-type C. rodentium-monocolonized mice with NAC in the drinking water significantly limited Th17 cell induction (Figure 5H), without affecting adhesion of C. rodentium to ECs (Figure S5F). Therefore, EC adhesion and subsequent ROS production is a potential mechanism underlying C. rodentium-mediated Th17 cell induction.
We next examined EHEC O157:H7, another EC-adherent bacterium that can cause hemolytic uremic syndrome in humans and is a global public health concern (Croxen and Finlay, 2010). Because the wild-type O157 strain was lethal in GF mice shortly after the monocolonization (data not shown), we used an O157 strain deficient in both Shiga toxin 1 and 2 genes (Δstx1Δstx2) (Yokoyama et al., 2001). This double mutant strain expanded in the intestine upon monocolonization without killing the mice and adhered to colonic EC surfaces (Figures 6A and 6B). The adhesion was accompanied by induction of colonic Th17 cells (Figure 6C). We subsequently constructed a Δstx1Δstx2Δeae triple mutant strain, which expanded comparably to the Δstx1Δstx2 double mutant strain in the intestinal lumen but did not adhere to colonic ECs (Figures 6A and 6B), and this was accompanied by loss of the ability to induce Th17 cells (Figure 6C).
Figure 6. EC Adhesion-Mediated Th17 Induction by Extracellular Pathogens.
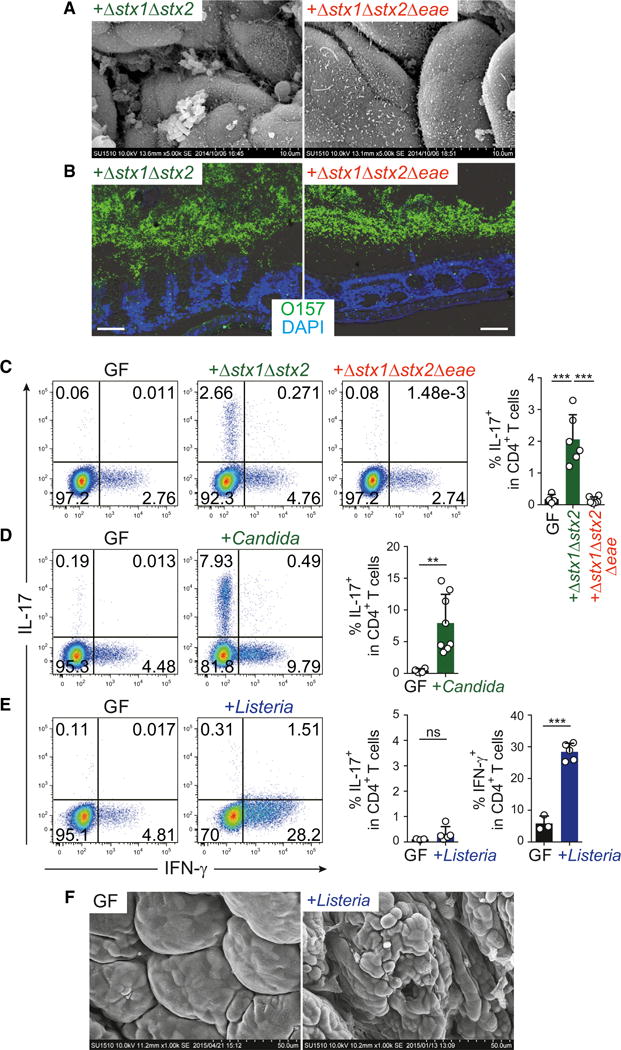
(A) SEM images of colonic villi from IQI GF mice monocolonized with Δstx1Δstx2 or Δstx1Δstx2Δeae EHEC O157:H7 for 3 weeks.
(B) O antigen antisera (green) and DAPI (blue) staining for EHEC O157 visualization. Scale bar, 50 μm.
(C–E) Th17 and Th1 cell proportions in the colonic LP CD4+ T cells from IQI mice inoculated with EHEC O157:H7 (C), C. albicans (D), or L. monocytogenes (E).
(F) SEM images of the colon of IQI GF mice and mice infected orally with L. monocytogenes.
Error bars represent SD. See also Figure S6.
Candida albicans is also an EC-adhesive microbe (Figure S6) and was found to increase the frequency of Th17 cells in the colon LP of monocolonized mice (Figure 6D). In contrast, monocolonization of mice with Listeria monocytogenes mainly induced Th1 cells, rather than Th17 cells (Figure 6E). The mice colonized with L. monocytogenes displayed disrupted structure of colon epithelial surface, whereas adhering bacteria were rarely observed (Figure 6F).
Th17-Inducing Human Microbiota
Finally, we attempted to identify members of the human gut microbiota that could exert immunological effects equivalent to those of SFB. To this end, we collected human fecal samples from patients with UC (UC4-2 and UC5-1) and healthy donors (H11, H17, H23) and inoculated them into GF IQI mice. All groups of mice exhibited a significant increase in Th17 cells in the colon (Figure 7A). The Th17 cell induction by H23 microbiota was suppressed by treatment with ampicillin (Amp) or other antibiotics (Figure 7B). In contrast, the UC5-1 microbiota-mediated Th17 cell induction was further enhanced when the mice were given Amp in the drinking water, although reduced by metronidazole (MNZ) or vancomycin (VCM) treatment (Figure 7B). Therefore, different subsets of bacterial species were likely to be responsible for the induction of Th17 cells in mice inoculated with H23 versus UC5-1 samples. Cecal microbiota analysis by 16S rRNA gene sequencing revealed several operational taxonomic units (OTUs) that were enriched in mice inoculated with UC5-1 sample treated with Amp and correlated with the number of colonic Th17 cells (Figure 7C, marked in red). To isolate Th17-inducing bacterial species, we anaerobically cultured cecal contents from Amp-treated UC5-1 mice using various culture media and picked 192 colonies. BLAST searches of 16S rRNA gene sequences revealed that the isolates contained 20 strains that broadly covered the bacterial species colonizing Amp-treated UC5-1 mice (Figure 7C, marked in green). Their genomes were sequenced and a phylogenetic comparison was carried out using 19 ribosomal protein genes predicted from the assembled draft genome of each strain. This analysis revealed that the 20 strains included diverse species, such as Clostridium, Bifidobacterium, Ruminococcus, and Bacteroides (Figures 7C and S7A; Table S1).
Figure 7. Th17 Induction and EC Adhesion by 20 Bacterial Strains Derived from the Human Intestine.
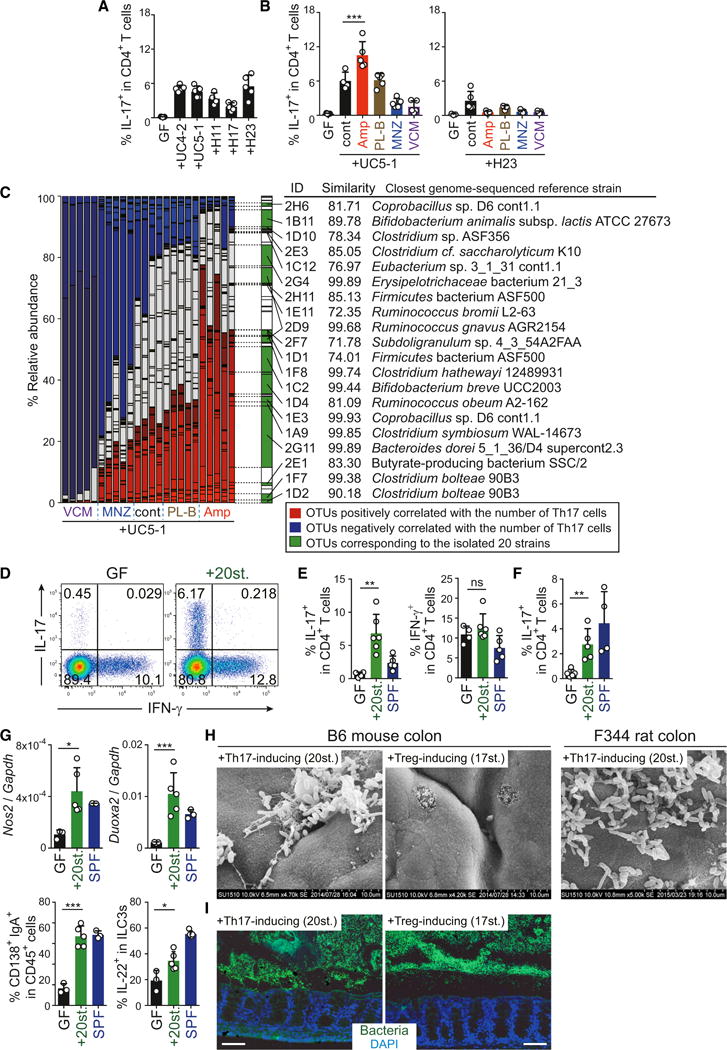
(A) Th17 cell frequencies in IQI GF mice colonized with stool from patients with UC or healthy adults.
(B) Th17 cell frequencies in UC5-1 or H23-colonized IQI mice either untreated (cont) or treated with the indicated antibiotics.
(C) Cecal microbiota compositions of each mouse (n = 4 or 5 per group). OTUs positively correlated with the frequency of Th17 cells are marked in red and those negatively correlated marked in blue. OTUs corresponding to the isolated 20 strains are marked in green. ID, isolated strain ID. See also Table S1.
(D–F) The percentage of Th17 and Th1 cells in the colonic LP of B6 mice (D and E) and F344 rats (F) colonized with the mixture of 20 strains (20st.).
(G) qPCR for Nos2 and Duoxa2 in colonic EC (upper panels) and FACS for LP IgA+ cells and IL-22+ ILC3s (lower panels) of B6 mice colonized with the 20 strains.
(H and I) SEM images and FISH staining with EUB338 of the proximal colon of mice and rats colonized with the 20 Th17-inducing strains versus the 17 Treg-inducing strains. Scale bar, 100 μm.
Error bars represent SD. See also Figure S7.
To investigate whether the isolated 20 strains have the ability to induce Th17 cells, we introduced a mixture of 20 strains into GF IQI mice. In mice inoculated with the 20 strains, we observed a robust accumulation of Th17 cells in the colon (Figures 7D and 7E). The 20-strain mix was also effective in Th17 induction in rats (Figure 7F). Similarly to SFB, the 20 strains enhanced expression of Nos2 and Duoxa2 in ECs and IgA and ILC3 responses (Figure 7G), whereas no significant effect was observed for Th1 cells in the colon (Figures 7D and 7E). To examine whether these 20 Th17-inducing strains contained EC-adherent bacteria, we performed SEM and FISH. As a control, we examined mice colonized with 17 strains of human-derived Clostridia that have the capacity to induce regulatory T cells (Tregs) (Atarashi et al., 2013). The 17 Treg-inducing strains of Clostridia were not adhering and disappeared after washing. In contrast, we observed numerous bacteria adhering to the colonic ECs in mice and rats colonized with the 20 Th17-inducing strains (Figures 7H and 7I), which is in line with our hypothesis that EC adhesion is an important signal for Th17 cell induction.
DISCUSSION
In the present study, we demonstrate that SFB, C. rodentium and EHEC O157 promote the induction of intestinal Th17 cells through adhesion to intestinal ECs. Moreover, we isolated 20 bacterial strains from human intestine that can induce Th17 cells in the mouse and rat intestine, and these 20 strains displayed similar EC adhesive characteristics. Therefore, the intestinal immune system mounts Th17 cell responses through recognition of a physical interaction with the microbes, rather than by recognition of released microbial components or soluble metabolites. EC adhesion by microbes was also associated with increases in intestinal IgA+ cells. Consistent with our observation, it has been shown that potentially colitogenic bacteria that can invade the inner mucus layer and colonize near the epithelium induce high-affinity IgA responses and become highly coated with IgA (Palm et al., 2014). In contrast to Th17 cells and IgA+ cells, IL-22+ ILC3s can be generated by nonadhering commensals. Therefore, Th17 and IgA+ cells control EC-adhering microbes, whereas IL-22+ ILC3s may play a more general role in regulation of gut microbiota. The vast majority of gut microbiota are physically separated by the mucus layer (Johansson et al., 2008; Vaishnava et al., 2011), whereas microbes that cross the mucus barrier and attach to the epithelial lining are in many cases pathogenic (SFB and the 20 human strains are exceptional in that they can cross the mucus layer and attach to ECs, but do not penetrate the EC layer). Therefore, it seems reasonable to propose that the host immune system has evolved to discriminate the biogeographical distributions of intestinal microbes.
Several reports have demonstrated that adhering SFB induce actin reorganization in SI ECs (Davis and Savage, 1974; Jepson et al., 1993). In this context, morphologically similar cytoplasmic alterations in intestinal ECs have been described in infections with EHEC O157 and C. rodentium (Croxen and Finlay, 2010). It is noteworthy that F-actin inhibition resulted in potentiated C/EBPδ and SAA1 expression in aMoS7 cells cocultured with M-SFB (Figure S3F). It is interesting to note that SFB have putative ADP-ribosyl transferases that have sequence similarities to the Clostridium perfringens iota toxin, which ribosylates G-actin and thereby inhibits its polymerization (Pamp et al., 2012). Furthermore, because ROS suppresses the activity of certain Rho GTPase family members (Stanley et al., 2014), ROS produced by the Duox2/Duoxa2 system in ECs may, in addition or instead, affect actin reorganization. In any case, there may be a link between actin cytoskeletal modulation by adherent SFB and subsequent gene induction in ECs, possibly through mechanosensing mechanisms (Dupont et al., 2011).
Since recombinant SAA1 activated CD11c+ cells to upregulate IL-1β expression (Figure 3F), and recombinant IL-1β activated ECs to potentiate expression of SAAs (Figure 4G), our results suggest a model in which the initial increase in SAAs in SI ECs caused by SFB adhesion is then augmented via an amplification loop of SAA1 and IL-1β between SI ECs and CD11c+ myeloid cells (Figure S4I). This amplification processes may be required for the maintenance of high level expression of SAAs in SI ECs and IL-1β in myeloid cells, which causes constitutive accumulation of Th17 cells. In an accompanying article published in this issue of Cell, Sano et al. (2015) show that SAAs can directly act on RORγt+ T cells to induce IL-17 expression. Derebe et al. (2014) showed that SAAs act as a transporter of retinol. Therefore, SAAs may exert their effects through multiple mechanisms. IL-22 derived from ILC3s also contributes to this loop via potentiation of SAA expression by SI ECs. IL-22 alone cannot sufficiently activate this amplification loop, as was shown that IL-22+ ILC3s induced by luminal R-SFB cannot induce Th17 cells (Figure 2D).
A substantial fraction of Th17 cells specifically recognized EC-adhesive M-SFB but not non-adhering R-SFB (Figures 2A and 2B). The simplest explanation for this observation is that antigens may be taken up by host cells only when SFB adhere to ECs. This process likely involves bacterial sampling by dendritic cells (DCs). In this context, recognition and phagocytosis of C. rodentium-infected apoptotic ECs by LP DCs were shown to preferentially trigger Th17 cell differentiation (Torchinsky et al., 2009). SFB and other EC-adhesive microbes may be engulfed by LP DCs together with their adhering apoptotic ECs, processed and loaded onto MHC class II molecules. SFB are innocuous members of the microbiota; however, SFB can be pathogenic, depending on the host genotype. For example, SFB have been implicated in T cell-mediated autoimmune diseases such as arthritis (Wu et al., 2010). In this context, we observed a striking genotype-specific difference upon monocolonization with EC-adhesive SFB in the induction of Th17 cells, with BALB/c mice having fewer Th17 cells in comparison to B6 and IQI mice. Our observation in BALB/c mice provides one remarkable example showing that the combination of genetic background and composition of the gut microbiota affects T cell status. Genotype-specific differences in the differentiation of T cells in response to a component of the gut microbiota may explain the phenotypic variations in autoimmune disease susceptibility.
Different from the host-specific Th17 induction by SFB, the 20 bacterial strains isolated from a human fecal sample were Th17-inducing in at least two different mammalian hosts (mice and rats, Figures 7E and 7F); therefore, they may function in humans and may act as pathobionts, because they were derived from UC patients. However, we did not observe any inflammatory changes in the intestines of mice inoculated with the 20 strains (data not shown). Furthermore, mining of publicly available data from the MetaHIT project (Qin et al., 2010) revealed that only three out of the 20 strains were significantly increased in the microbiomes of UC and Crohn’s disease subjects (Figure S7B). Moreover, the 20 strains were not abundant in mice inoculated with a fecal sample from subject UC4-2, in which Th17 cells were induced similarly to UC5-1 microbiota-associated mice (Figure S7C). Therefore, the 20 strains may not be essential components for Th17 induction nor contribute to the pathogenesis of IBD in humans. Our study design has some limitations in this context, including the limited number of subjects tested and the fact that robust activity in mice may not be physiologically relevant in humans and vice versa. Nevertheless, this is a clear demonstration of Th17 cell induction by human-associated bacterial species and therefore represents a significant step forward in dissecting human intestinal microbiota biology. The intestinal Th17 responses to commensal and pathogenic bacteria with shared EC-adhering characteristics have obvious clinical implications in the context of IBD, vaccine design, and probiotics.
EXPERIMENTAL PROCEDURES
Materials and experimental procedures are detailed in the Supplemental Experimental Procedures.
Animals and Microbes
GF mice and rats were purchased from CLEA and Sankyo Labo Service and kept in the GF facility of Yakult Central Institute or RIKEN Yokohama Institute. To generate SFB-monocolonized animals, cecal or fecal suspensions from SFB-monocolonized animals were orally inoculated into GF animals. The C. rodentium Δeae and E. coli O157:H7 Δstx1Δstx2 mutant strains have been described previously (Kamada et al., 2012; Nagano et al., 2003). To generate the E. coli O157:H7 Δstx1Δstx2Δeae mutant strain, the eae gene was replaced with the tetracycline-resistance gene by homologous recombination. Approximately 1 × 108 colony-forming unit (CFU) of C. rodentium, E. coli O157, or L. monocytogenes was orally administered into GF mice. C. albicans (TUA6) was provided by Dr. Takashi Umeyama (National Institute of Infectious Diseases, Japan) and orally administered (~1 × 106 CFU) into GF mice.
qPCR and 16S rRNA Gene Pyrosequencing
To quantify bacterial load, bacterial DNA was isolated from intestinal contents or mucosal tissues using a QIAamp Stool Mini Kit, and qPCR was carried out using universal or bacterial strain-specific primers for 16S rRNA genes. For 16S rRNA gene pyrosequencing, cecal contents were incubated with lysozyme and achromopeptidase. Sodium dodecyl sulfate and subsequently proteinase K was added to the suspension and high-molecular-mass DNA was purified by phenol/chloroform extraction. PCR was performed using primers to the V1–V2 region of the 16S rRNA gene, and the amplified DNA was used as template for 454 GS Junior sequencing.
Isolation of Human-Derived Th17-Inducing Bacterial Strains
Stool samples from patients with UC and from healthy donors were obtained and inoculated into IQI GF mice. The cecal contents from Amp-treated UC5-1 mice were cultured on Schaedler, BHI, GAM, CM0151, BL, or TS agar plates under strictly anaerobic conditions. Individual colonies were picked and identified by sequencing of the 16S rRNA gene fragment.
Statistical Analysis
All statistical analyses were performed using Prism software with two-tailed unpaired Student’s t test or one-way ANOVA followed by Tukey’s post hoc test. p values < 0.05 were considered significant (*p < 0.05, **p < 0.01, and ***p < 0.001).
Supplementary Material
Highlights.
A strong correlation between epithelial adhesion and Th17 induction by SFB and EHEC
Twenty Th17-inducing strains isolated from human feces show epithelial-adhesive property
Bacterial adhesion elicits a Th17-inducing gene-expression program in epithelium
Acknowledgments
This work was supported by the Japan Society for the Promotion of Science NEXT program, Health Labor Sciences Research Grant, the Waksman Foundation of Japan, Suzuken Memorial Foundation, and the Uehara Memorial Foundation. We thank the animal care staff at the Yakult Central Institute, Osamu Ohara, Mayuko Sato, and Kei Hashimoto at RIKEN for their technical supports, Hiroyuki Nobusue and Hideyuki Saya at Keio University for data discussion, and Mamoru Totsuka at the University of Tokyo for providing the aMoS7 cell line.
Footnotes
ACCESSION NUMBERS
The accession number for the RNA sequencing data reported in this paper is NCBI GEO: GSE71734. The draft genome sequences of Th17-inducing 20 strains can be retrieved under DDBJ BioProject ID: PRJDB 4119-4138 and the 16S rRNA gene sequencing data under DDBJ BioProject ID: PRJDB4113.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and one table and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2015.08.058.
AUTHOR CONTRIBUTIONS
K.H. and Y.U. planned experiments, analyzed data, and wrote the manuscript. K.A., N.K., and T.T. performed immunological analyses and bacterial cultures together with Y.Nagano, S.N., Y.Noguchi, E.W., and H. Morita. W.S. and M.H. performed bacterial sequence analyses. M.A., S.N., and S.K. performed SEM analysis. A.I., H.S., T.N., E.I., T.Shima, and T.H. prepared M-SFB- and R-SFB-monocolonized animals and performed initial experiments. S.Y., S.T., H.Mori., and T.Sugiyama generated mutant strains of EHEC O157. I.I. and G.N. provided essential materials and contributed to data discussions. T.J. and H.O. provided Il22r1f/f mice. T.K. helped with ChIP-seq experiments. K.T. instructed SEM. J.G.C. analyzed Saa1 regulatory regions.
References
- Atarashi K, Nishimura J, Shima T, Umesaki Y, Yamamoto M, Onoue M, Yagita H, Ishii N, Evans R, Honda K, Takeda K. ATP drives lamina propria T(H)17 cell differentiation. Nature. 2008;455:808–812. doi: 10.1038/nature07240. [DOI] [PubMed] [Google Scholar]
- Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, Fukuda S, Saito T, Narushima S, Hase K, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013;500:232–236. doi: 10.1038/nature12331. [DOI] [PubMed] [Google Scholar]
- Camp JG, Frank CL, Lickwar CR, Guturu H, Rube T, Wenger AM, Chen J, Bejerano G, Crawford GE, Rawls JF. Microbiota modulate transcription in the intestinal epithelium without remodeling the accessible chromatin landscape. Genome Res. 2014;24:1504–1516. doi: 10.1101/gr.165845.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung H, Pamp SJ, Hill JA, Surana NK, Edelman SM, Troy EB, Reading NC, Villablanca EJ, Wang S, Mora JR, et al. Gut immune maturation depends on colonization with a host-specific microbiota. Cell. 2012;149:1578–1593. doi: 10.1016/j.cell.2012.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti HR, Gaffen SL. Host responses to Candida albicans: Th17 cells and mucosal candidiasis. Microbes Infect. 2010;12:518–527. doi: 10.1016/j.micinf.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croxen MA, Finlay BB. Molecular mechanisms of Escherichia coli pathogenicity. Nat Rev Microbiol. 2010;8:26–38. doi: 10.1038/nrmicro2265. [DOI] [PubMed] [Google Scholar]
- Davis CP, Savage DC. Habitat, succession, attachment, and morphology of segmented, filamentous microbes indigenous to the murine gastrointestinal tract. Infect Immun. 1974;10:948–956. doi: 10.1128/iai.10.4.948-956.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derebe MG, Zlatkov CM, Gattu S, Ruhn KA, Vaishnava S, Diehl GE, MacMillan JB, Williams NS, Hooper LV. Serum amyloid A is a retinol binding protein that transports retinol during bacterial infection. eLife. 2014;3:e03206. doi: 10.7554/eLife.03206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179–183. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- Gaboriau-Routhiau V, Rakotobe S, Lécuyer E, Mulder I, Lan A, Bridonneau C, Rochet V, Pisi A, De Paepe M, Brandi G, et al. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity. 2009;31:677–689. doi: 10.1016/j.immuni.2009.08.020. [DOI] [PubMed] [Google Scholar]
- Goto Y, Panea C, Nakato G, Cebula A, Lee C, Diez MG, Laufer TM, Ignatowicz L, Ivanov II. Segmented filamentous bacteria antigens presented by intestinal dendritic cells drive mucosal Th17 cell differentiation. Immunity. 2014;40:594–607. doi: 10.1016/j.immuni.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, Frutos RdeL, Manel N, Yoshinaga K, Rifkin DB, Sartor RB, Finlay BB, Littman DR. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe. 2008;4:337–349. doi: 10.1016/j.chom.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jepson MA, Clark MA, Simmons NL, Hirst BH. Actin accumulation at sites of attachment of indigenous apathogenic segmented filamentous bacteria to mouse ileal epithelial cells. Infect Immun. 1993;61:4001–4004. doi: 10.1128/iai.61.9.4001-4004.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci USA. 2008;105:15064–15069. doi: 10.1073/pnas.0803124105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada N, Kim YG, Sham HP, Vallance BA, Puente JL, Martens EC, Núñez G. Regulated virulence controls the ability of a pathogen to compete with the gut microbiota. Science. 2012;336:1325–1329. doi: 10.1126/science.1222195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaasen HL, Koopman JP, Van den Brink ME, Bakker MH, Poelma FG, Beynen AC. Intestinal, segmented, filamentous bacteria in a wide range of vertebrate species. Lab Anim. 1993;27:141–150. doi: 10.1258/002367793780810441. [DOI] [PubMed] [Google Scholar]
- Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- Mortha A, Chudnovskiy A, Hashimoto D, Bogunovic M, Spencer SP, Belkaid Y, Merad M. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science. 2014;343:1249288. doi: 10.1126/science.1249288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano K, Taguchi K, Hara T, Yokoyama S, Kawada K, Mori H. Adhesion and colonization of enterohemorrhagic Escherichia coli O157:H7 in cecum of mice. Microbiol Immunol. 2003;47:125–132. doi: 10.1111/j.1348-0421.2003.tb02795.x. [DOI] [PubMed] [Google Scholar]
- Palm NW, de Zoete MR, Cullen TW, Barry NA, Stefanowski J, Hao L, Degnan PH, Hu J, Peter I, Zhang W, et al. Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell. 2014;158:1000–1010. doi: 10.1016/j.cell.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamp SJ, Harrington ED, Quake SR, Relman DA, Blainey PC. Single-cell sequencing provides clues about the host interactions of segmented filamentous bacteria (SFB) Genome Res. 2012;22:1107–1119. doi: 10.1101/gr.131482.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash T, Oshima K, Morita H, Fukuda S, Imaoka A, Kumar N, Sharma VK, Kim SW, Takahashi M, Saitou N, et al. Complete genome sequences of rat and mouse segmented filamentous bacteria, a potent inducer of th17 cell differentiation. Cell Host Microbe. 2011;10:273–284. doi: 10.1016/j.chom.2011.08.007. [DOI] [PubMed] [Google Scholar]
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano T, Huang W, Hall JA, Yang Y, Chen A, Gavzy SJ, Lee J, Ziel J, Miraldi ER, Domingos AI, et al. An 23R/IL-22 circuit regulates epithelial serum amyloid A to promote local effector Th17 responses. Cell. 2015;163:381–393. doi: 10.1016/j.cell.2015.08.061. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw MH, Kamada N, Kim YG, Núñez G. Microbiota-induced IL-1β, but not IL-6, is critical for the development of steady-state TH17 cells in the intestine. J Exp Med. 2012;209:251–258. doi: 10.1084/jem.20111703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, Ren B. A map of the cis-regulatory sequences in the mouse genome. Nature. 2012;488:116–120. doi: 10.1038/nature11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie AN, Mebius RE, et al. Innate lymphoid cells–a proposal for uniform nomenclature. Nat Rev Immunol. 2013;13:145–149. doi: 10.1038/nri3365. [DOI] [PubMed] [Google Scholar]
- Stanley A, Thompson K, Hynes A, Brakebusch C, Quondamatteo F. NADPH oxidase complex-derived reactive oxygen species, the actin cytoskeleton, and Rho GTPases in cell migration. Antioxid Redox Signal. 2014;20:2026–2042. doi: 10.1089/ars.2013.5713. [DOI] [PubMed] [Google Scholar]
- Talham GL, Jiang HQ, Bos NA, Cebra JJ. Segmented filamentous bacteria are potent stimuli of a physiologically normal state of the murine gut mucosal immune system. Infect Immun. 1999;67:1992–2000. doi: 10.1128/iai.67.4.1992-2000.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torchinsky MB, Garaude J, Martin AP, Blander JM. Innate immune recognition of infected apoptotic cells directs T(H)17 cell differentiation. Nature. 2009;458:78–82. doi: 10.1038/nature07781. [DOI] [PubMed] [Google Scholar]
- Umesaki Y, Setoyama H, Matsumoto S, Imaoka A, Itoh K. Differential roles of segmented filamentous bacteria and clostridia in development of the intestinal immune system. Infect Immun. 1999;67:3504–3511. doi: 10.1128/iai.67.7.3504-3511.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, Yu X, Koren O, Ley R, Wakeland EK, Hooper LV. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science. 2011;334:255–258. doi: 10.1126/science.1209791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver CT, Elson CO, Fouser LA, Kolls JK. The Th17 pathway and inflammatory diseases of the intestines, lungs, and skin. Annu Rev Pathol. 2013;8:477–512. doi: 10.1146/annurev-pathol-011110-130318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HJ, Ivanov II, Darce J, Hattori K, Shima T, Umesaki Y, Littman DR, Benoist C, Mathis D. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity. 2010;32:815–827. doi: 10.1016/j.immuni.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Torchinsky MB, Gobert M, Xiong H, Xu M, Linehan JL, Alonzo F, Ng C, Chen A, Lin X, et al. Focused specificity of intestinal TH17 cells towards commensal bacterial antigens. Nature. 2014;510:152–156. doi: 10.1038/nature13279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama S, Suzuki T, Shiraishi S, Ohishi N, Yagi K, Ichihara S, Itoh S, Mori H. Construction of deletion mutants of Shiga (-Like) toxin genes (stx-1 and/or stx-2) on enterohemorrhagic Escherichia coli (O157: H7) J Clin Biochem Nutr. 2001;30:33–42. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.