

Abstract
The ribosome is the quintessential antibacterial drug target, with many structurally and mechanistically distinct classes of antibacterial agents acting by inhibiting ribosome function. Detecting and quantifying ribosome inhibition by small molecules and investigating their binding modes and mechanisms of action are critical to antibacterial drug discovery and development efforts. To develop a ribosome inhibition assay that is operationally simple, yet provides direct infonnation on drug target and mechanism of action, we have developed engineered E. coli strains harboring an orthogonal ribosome-controlled green fluorescent protein (GFP) reporter that produce fluorescent signal when the orthogonal ribosome is inhibited. As a proof of concept, we demonstrate that these strains, when co-expressing homogenous populations of aminoglycoside resistant ribosomes, act as sensitive and quantitative detectors of ribosome inhibition by a set of twelve structurally diverse aminoglycoside antibiotics. We suggest that this strategy can be extended to quantifying ribosome inhibition by other drug classes.
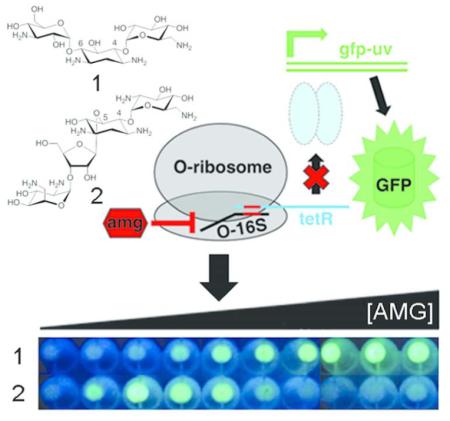
The ribosome is a complex, highly conserved biomolecular machine essential for the biosynthesis of cellular proteins and peptides. The essentiality and ancient origin of the ribosome have made it one of the most frequent targets of antibacterial natural products.1 Of the many known classes of ribosome inhibitors, the aminoglycosides (Figure 1a) are perhaps the best-studied with regard to their mechanisms of action2-4, drug resistance2-4, and biosynthesis.5-7 Aminoglycosides are a clinically useful class of ribosome inhibitors with broad spectrum activity towards a variety of microbial pathogens. However their widespread clinical use has been hampered by low-level toxicity to human mitochondrial ribosomes.2-4 Aminoglycosides can impair ribosome function by affecting the efficiency of intersubunit rotations.8-10, translocation2,3,11,12, and ribosome recycling8 and by inducing translational miscoding2-4,13 through specific interactions with the decoding site (A-site) of the 16S ribosomal RNA (rRNA) in helix 44 (h44) of the small (30S) ribosomal subunit14-17 (Figure 1b), and in the case of some aminoglycosides, with helix 69 (H69) of the large (50S) ribosomal subunit.8,10,12,15,18 Aminoglycosides also show promise as treatments for other diseases, including HIV2; human genetic disorders, where their ability to induce miscoding has been used to suppress disease-associated premature termination codons2,19; and fungal infection, where amphiphilic aminoglycoside analogs have been shown to perturb the function of the fungal plasma membrane.20
Figure 1.

A) Structures of aminoglycosides used in this study. B) Overlayed X-ray crystal structures of the decoding site (A-site) of the E.coli 16S ribosomal RNA (rRNA) with select aminoglycosides bound. Residues 1403-1411, 1489-1498 of 16S rRNA are shown in grey. Aminoglycosides are color-coded as follows; and the source PDB file for each is given in parentheses: kanamycin A - maroon (2ESI23), gentamicin - purple (2QB915), geneticin (G418) - pink (1MWL22, neomycin B - dark green (2QAL15), paromomycin-light green (2Z4K15), apramycin - orange (4AQY17), hygromycin - blue (3DF116).
Much of our current understanding of aminoglycoside/rRNA interactions, potency as ribosome inhibitors, and mechanisms of action comes from in vitro studies. The target sites of several aminoglycosides were first identified using RNA footprinting14 and were later examined in detail by X-ray crystallography.15-7,21,22 Affinities of arninoglycosides for rRNA hairpins mimicking the A-site have been assessed using mass spectrometry23, surface plasmon resonance (SPR)24, NMR18, and competition assays.25 However, it has been observed that binding affinities of some aminoglycosides to A-site rRNA mimics do not correlate linearly with inhibition of translation in vitro or antibacterial potency in vivo26, indicating that the results of assays using A-site mimics are not fully equivalent to those obtained when employing intact ribosomes in vitro or in vivo. Ribosome inhibition is also commonly measured directly using in vitro translation systems. These studies have revealed that individual aminoglycosides differ significantly with respect to their potencies as ribosome inhibitors.26-30 Recently such systems have been combined with powerful single molecule techniques such as smFRET to probe the effects of aminoglycosides on the kinetics and dynamics of the individual steps of translation, revealing that individual aminoglycosides differ markedly with respect their effects on these steps.8-10,12 However, in vitro translation experiments designed to investigate drug binding mode or mechanism of action are technically challenging to carry out; and in vitro translation assays in general either require expensive reagents or specialized preparation methods and cannot provide information on in vivo properties such as compound membrane permeability or efilux.
Incontrast to the breadth of techniques available for in vitro studies, in vivo analyses of the effects of aminoglycosides have been limited to measuring the growth inhibitory effects (most commonly reported as minimum inhibitory concentrations - MICs) of the compounds on wild-type bacterial strains and on those carrying ribosomal resistance mutations.31-33 Although such assays are operationally simple, and provide direct information on a compound's ability to inhibit bacterial growth, they do not provide any information a priori regarding the biomolecular target(s) of a drug (the ribosome in the case of aminoglycosides), the target binding site(s) (the A-site, and in some cases H69 for aminoglycosides), or the mechanism(s) by which a drug exerts its effects on the target.
Considering the limitations of existing in vitro and in vivo technologies, we envisioned an alternative in vivo strategy for investigating ribosome inhibition by small molecules that combines the ability of in vitro assays to quantitatively probe the effects of binding to a specific target site with the operational ease and in vivo relevance of live cell antibacterial assays. This strategy utilizes orthogonal ribosomes (O-ribosomes) - specialized mutant ribosomes that, by virtue of a mutated 16S rRNA anti-Shine-Dalgarno (ASD) sequence, are unable to translate native mRNA, yet retain the ability to translate mRNA carrying a complementary mutant Shine-Dalgamo (SD) sequence34-36 to circumvent the limitations imposed by ribosome essentiality. We hypothesized that O-ribosomes, if used to control expression of an engineered genetic circuit that results in a quantifiable “turn-on” phenotype upon ribosome inhibition, could be used to quantify ribosome inhibition by aminoglycosides or other ribosome inhibitors, providing a means of rapidly assessing the target specificity and ribosome inhibiting potency of these compounds in live bacterial cells. Such an O-ribosome-based assay would facilitate studies of mechanisms of drug action and of the relationship between target rRNA sequence and drug activity that can lead to discovery of ribosome inhibitors with improved therapeutic properties.
Here, we have created engineered E. coli strains harboring an O-ribosome-controlled green fluorescent protein (GFP) reporter that are non-fluorescent in the absence of drug, but express GFP upon O-ribosome inhibition. We show that these strains are able to detect ribosome inhibition by a collection of twelve structurally diverse aminoglycosides in a highly sensitive, dose-dependent manner with essentially no background. The fluorescence dose response patterns we observed for engineered E. coli detection strains treated with these aminoglycosides correlate with the results of in vitro translation inhibition assays, validating the accuracy of our assay in assessing the potencies of aminoglycosides as ribosome inhibitors. Our results provide a full comparative assessment of the ribosome inhibiting potencies of the twelve aminoglycosides assayed. Thus, the O-ribosome reporter system developed here provides a powerful new tool for easily and rapidly assessing the relative potencies of aminoglycosides as ribosome inhibitors in live bacterial cells.
Our arninoglycoside responsive strains were designed to harbor an engineered plasmid-borne reporter system comprised of three elements (Figure 2a): 1) a constitutively expressed arninoglycoside-sensitive orthogonal 16S rRNA (0-16S) gene bearing a mutated anti-Shine-Dalgarno (0-ASD) sequence35, 2) the tetR gene encoding a constitutively expressed tetracycline-responsive repressor protein TetR37 with orthogonal SD (0-SD) sequence complementary to the 16S rRNA 0-ASD sequence35, and 3) the gfp-uv gene encoding green fluorescent protein variant GFPuv38 under transcriptional control of the TetR-repressed promoter PLtet0-1.39 In the absence of arninoglycoside (Figure 2a, left panel), cells bearing these three elements would produce O-ribosome-derived TetR that represses transcription of gfp-uv, resulting in no fluorescence. However inthe presence of aminoglycoside (Figure 2a, right panel), the O-ribosome is inhibited, resulting in a reduced level of TetR, de-repression of gfp-uv transcription, and production of GFP. The system is designed to be highly sensitive by substantially amplifying the arninoglycoside input signal through transcription and translation of gfp-uv, resulting in production of multiple GFP proteins per aminoglycoside molecule.
Figure 2.

A) Schematic showing the functionality of the orthogonal ribosome-controlled fluorescent reporter in the absence (left panel) and presence (right panel) of aminoglycoside. The 0-16S rRNA is shown in black, 0-SD/O-ASD pair is shown in red, tetR mRNA and TetR protein are shown in cyan, PLtet0-1 is shown in blue, the gfp-uv gene and GFP protein are shown in green, and aminoglycoside is shown as a red hexagon. B) Cell pellet fluorescence and fluorescence quantification of E. coli DHSa cells transformed with individual and combined functional elements of the orthogonal ribosome-controlled fluorescent reporter.
To protect the E. coli host itself from inhibition by aminoglycosides, the strain's native rRNA was made aminoglycoside resistant using a previously-developed host, E. coli SQ380, inwhich all seven chromosomal copies of the rRNA operon were deleted and replaced by a single rRNA operon on plasmid prmC-sacB bearing the counterselectable marker sacB. The A1408G 16S rRNA mutation, which confers resistance to many aminoglycosides31, was introduced into rRNA operon-expressing plasmid pRRSH2. The resulting plasmid, pRRSH2-A1408G, was used to replace prmC-sacB in SQ380. As expected, the resulting strain SH386 possessed a high-level of resistance to kanamycin A (up to 500 µM, the highest concentration tested, Figure S1).
To demonstrate that our reporter system functions properly we sequentially constructed plasmids harboring sets of the system's three functional elements and assayed them individually by expression in E. coli DH5a and whole cell fluorescence quantification after growth to stationary phase (18 h) (Figure 2b). First, performance of the element bearing gfp-uv under control of PLtet0-1 with the optimized 5’-untranslated region BCD240 (see Figure S2 and Supporting Information for optimization) was examined. As expected, in the absence of tetR, PLtet0-1 behaves as a strong constitutive promoter and results in high GFP expression (Figure 2b). Next, the O-SD-controlled tetR cassette was inserted into the gfp-uv-expressing construct. The resulting two element system retained robust fluorescence (~60% of that of the gfp-uv-expressing construct, Figure 2b). Finally, the 0-16S cassette was inserted into the gfp-uv/tetR-expressing construct to give final reporter construct pSH3-KF. As expected for a functional reporter system in which the O-ribosome makes TetR which in turn represses gfp-uv, addition of the 0-16S cassette completely abolished fluorescence (Figure 2b). However fluorescence could be completely recovered by addition of a saturating concentration of anhydrotetracycline (ATC), which binds to TetR causing its dissociation from PLtet0-1 (Figure 2b, Figure S3). Together, these results indicate that the reporter system as a whole, as well as its individual elements, function correctly in E. coli DH5α. We attribute the ~40% loss of fluorescence upon insertion of the tetR cassette to a polar effect on gfp-uv expression by tetR - not a lack of 0-SD orthogonality - since addition of saturating ATC to the pSH3-KF-expressing strain resulted in a fluorescence level nearly identical to (106.6% ± 1.2%) that observed in the gfp-uv/tetR-expressing strain. Thus tetR is not translated to a significant extent without co-expression of the 0-16S gene.
With the aminoglycoside resistant E. coli strain SH386 and reporter plasmid pSH3-KF in hand, we carried out an initial O-ribosome inhibition assay using kanamycin A. E. coli SH391 (SH386 transformed with pSH3-KF) grown to stationary phase (18 h) in the presence of a range of kanamycin A concentrations showed a modest dose-dependent increase in fluorescence (maximum 1.6-fold induction, Figure 3), but displayed strong background fluorescence in the absence of kanamycin A. Reasoning that the high background and low sensitivity to kanamycin A was the result of a decreased intracellular TetR concentration in the rRNA deletion mutant compared to DHSa, we generated eleven new pSH3-KF variants in which the promoter strengths of the tetR and 0-16S elements were combinatorially altered, and examined their dose-dependent responses to kanamycin A in SID86 (Figure 3, Figure S4). To our satisfaction, one variant strain, SH399, with reporter plasmid pSH6-KF carrying strong promoter BBa_J23100 controlling 0-16S expression. had essentially no background and displayed a robust dose-dependent increase in fluorescence in response to kanamycin A (Figure 3).
Figure 3.

Cell pellet fluorescence of initial detector strain E. coli SH391 (boxed inred), eleven additional strains with tetR and 0-16S promoter strengths combinatorially altered (promoter strengths are show on the left: + - very weak, ++ - weak, +++ - medium, ++++ - strong), and E. coli SH399, the strain with the best detection performance (boxed in green), in response to increasing concentrations of kanamycin. Fluorescence quantification of E. coli SH391 and E. coli SH399 at each of the ten kanamycin concentrations tested is shown inthe bar graphs to the right.
Next we tested the ability of SH399 to detect ribosome inhibition by a panel of eleven additional structurally diverse aminoglycosides, including other 4,6-disubstituted 2-deoxystreptamines (2-DOS) gentamicins, G418, sisomicin, tobramycin, and amikacin; 4,S-disubstituted 2-DOS paromomycin, neomycin B, and ribostamycin; and atypical 2-DOS apramycin, hygromycin B, and neamine (Figure 1a). In addition to kanamycin A, SH399 grown to stationary phase (18 h) in the presence of a range of concentrations of each aminoglycoside was able to detect O-ribosome inhibition by nine of these compounds (Figure 4a). The two compounds that failed to give signal, G418 and hygromycin B, also caused significant growth inhibition of SH399 (Figure S5), indicating that the A1408G 16S rRNA mutation does not confer sufficient resistance to these compounds to allow survival of the detector strain. This observation is consistent with previously reported levels of resistance conferred by the A1408G mutation to a subset of the compounds examined31; and, for those compounds that were detectable the A1408G mutation confers resistance well above the detection threshold (Figure S5).
Figure 4.

Cell pellet fluorescence of A) E. coli SH399 and B) E. coli SH431 in response to increasing concentrations of aminoglycosides. Kan - kanamycin A, Apr - apramycin, Neo - neomycin B, Paro - paromomycin, Gen - gentamicins, Ami - amikacin, Nea -neamine, Rib -ribostamycin, Sis - sisomicin, Toh - tobramycin, G418 - geneticin (G418), Hyg - hygromycin B.
The fluorescence dose-response patterns observed for SH399 treated with these ten aminoglycosides were different for each compound (Figure 4a, Figure S5) and appeared upon qualitative inspection to correlate with previously reported compound potencies determined by both in vitro translation assays2-3 and E. coli growth inhibition assays26,28,30, suggesting that the O-ribosome reporter system may be useful for comparing aminoglycoside potencies. To examine this possibility, we first compared IC50 values calculated from fluorescence data obtained from aminoglycoside-treated SH399 (see Supporting Information for calculations) with previously detennined IC50 values of a subset of six aminoglycosides (kanamycin A, neomycin B, paromomycin, gentamicins, ribostamycin, and tobramycin) measured through inhibition of translation in vitro.28 We found a strong correlation (R2 = 0.97, Figure S6) between the two datasets, suggesting that IC50 values determined using the O-ribosome reporter assay are comparably accurate to those determined using in vitro translation assays. Next, to test whether the fluorescence dose-response patterns also correlate with inhibition of E. coli growth, we compared dose-dependent growth inhibition (represented as LD50 values) of the parent aminoglycoside sensitive E. coli strain SH434 with IC50 values calculated from fluorescence data obtained from aminoglycoside-treated SH399. While data obtained by the two methods correlated for a subset of the compounds (apramycin, gentamicins, amikacin, ribostamycin, sisomicin, and tobramycin; R2 = 0.97, Figure S6), there was a lack of correlation between the two datasets for kanamycin A, neomycin B, and paromomycin, and therefore between the two datasets as a whole (R2 = 0.40, Figure S6). While the reason for the incomplete correlation between inhibition of E. coli growth and fluorescence-derived IC50 values is unclear, we suggest that these inconsistencies are the result of differences between the pleiotropic effects of differentially inhibiting the ribosome, whose activity is required for synthesis of the entire E. coli proteome, on cell viability; and the effects of differentially inhibiting the O-ribosome, which are restricted to the TetR-GFP output system. Taken together, these results are consistent with the ability of the O-ribosome reporter system to compare the potencies of aminoglycosides as ribosome inhibitors. Of the ten compounds examined, sisomicin was found to have the strongest ribosome inhibition activity, followed in order of decreasing activity by gentamicins, neomycin B, paromomycin, tobramycin, amikacin, ribostamycin, apramycin, kanamycin A, and neamine (Figure S5, S6). These results provide a complete comparative assessment of the ribosome inhibiting potencies of these ten compounds that is consistent with previous reports of aminoglycoside potencies and structure-activity relationships obtained through in vitro translation inhibition assays using subsets of these compounds.26-30
Interestingly, we also observed for some aminoglycosides that at drug concentrations beyond those that give the peak response, fluorescence actually decreases, and does so to varying extents with different compounds (Figure 4, Figure S5). The effect was most pronounced with neomycin B, paromomycin, and gentarnicins; occurred to an intermediate extent with sisornicin, tobramycin, and amikacin; occurred to a slight extent with kanamycin A and apramycin; and was absent with ribostamycin and neamine. In the cases of paromomycin and gentamicins, these decreases in fluorescence were accompanied by growth inhibition. These results are consistent with previous structural and spectroscopic observations that neomycin B, paromomycin, and gentamicin bind to and inhibit the ribosome at a second, lower affinity site in helix 69 of the large ribosomal subunit.8,10,12,15,18 In our system, binding to this site on the pRRSH2-A1408G-derived ribosome would affect translation of both GFPuv and endogenous proteins, leading to both a decrease in fluorescence and loss of cell viability. When considered together with previous work, our observations suggests that the O-ribosome reporter system can identify, via dose-dependent fluorescence decrease at higher concentrations, aminoglycosides that inhibit the ribosome by interacting with a secondary site such as H69; and that other aminoglycosides in our panel that exhibit this phenomenon may also bind to H69 or to another secondary site. Examination of the X-ray crystal structures of neomycin B, paromomycin, and gentamicin C1a bound to H6915 reveals conserved contacts between the C-1 and C-3 amines of the 2-DOS core, which are present in all aminoglycoside structures, and residues 1921-1923 of the 23S rRNA, leaving open the possibility that other aminoglycosides may interact with H69. Experiments to test binding of aminoglycosides in our panel to H69 are currently underway in our laboratory.
To further explore the capabilities of the system and attempt to develop a strain that can detect O-ribosome inhibition by hygromycin B and G418, we introduced the U1406A mutation into pRRSH2. Mutations at position 1406 confer an aminoglycoside resistance spectrum distinct from that of A1408G, including resistance to G418 (U1406A)32 and hygromycin B (U1406C).33 We tested the ability of strain SH431 carrying this mutation and reporter plasmid pSH6-KF grown to stationary phase (24 h) in the presence of a range of concentrations of each aminoglycoside to detect O-ribosome inhibition by the same set of twelve aminoglycosides . As anticipated, SH431 was able to detect O-ribosome inhibition by both G418 and hygromycin B (Figure 4b) as well as kanamycin A and gentamicins (Figure S7). We observed significant growth inhibition and a lack of signal by SH431 in the presence of the remaining eight compounds, indicating that the U1406A mutation is unable to confer resistance to these compounds. Dose response patterns observed for SH431 treated with the four compounds for which fluorescence could be observed indicate that, among these, gentamicins are the most potent ribosome inhibitors, followed by G41S, hygromycin B, and with kanamycin A being the least potent (Figure S7, S8). IC50 values calculated from the O-ribosome-based fluorescence assay correlated with LD50 values of the parent aminoglycoside sensitive E.coli strain SH434 for three of the compounds (gentamicins, G41S, hygromycin B; R2 = 0.997, Figure S8); but not for kanamycin A, as was the case with SH399.
In summary, we have created engineered E. coli strains that can directly detect and quantify ribosome inhibition at the A-site by a variety of structurally distinct aminoglycosides with high sensitivity and essentially no background. The fluorescence dose-response patterns we observed for the aminoglycosides tested correlate with their reported ribosome inhibiting potencies, demonstrating that our system can be used to determine the relative potencies of aminoglycosides as ribosome inhibitors. The observation that compounds known to act by binding to H69 show dose-dependent fluorescence decrease at high concentrations suggests that our system can also report on compound secondary binding modes. We have also demonstrated that the selectivity of each strain for detection of specific aminoglycosides – even those with high structural similarity such as gentamicin and G41S – can be controlled by employing 16S rRNA mutations that confer distinct resistance profiles. We believe that the strains developed here provide a powerful new tool for assaying and studying ribosome inhibition by aminoglycosides and other ribosome inhibitors in the context of live bacterial cells; and will find broad applicability in drug discovery endeavors.
We envision that the O-ribosome reporter strategy described here can be used to assess structure-activity relationships of synthetic aminoglycoside analogs in high-throughput; to detect and quantify aminoglycosides in natural product extracts; and to detect activity of aminoglycoside biosynthetic enzymes, as was recently done with a target-based β-lactam antibiotic detection system.41 We suggest that our strategy can be extended to specifically detect ribosome inhibition by compound classes acting at other 16S rRNA binding sites such as streptomycin, kasugamycin, spectinomycin, tetracyclines, tuberactinomycins, and pactamycins1 by employing 16S rRNA resistance mutations specific to each compound class. Furthermore, with the recent development of a functional ribosome in which the 16S and 23S rRNAs are tethered42, our strategy may be extended to detect ribosome inhibition by compounds targeting the 23S rRNA such as macrolides, thiostrepton, avilamycin, and others.1 Mutation of the O-ribosome A-site in the strains described here to mimic the A-site of other bacteria (e.g. Mycobacteria or other pathogens) or human mitochondria may also allow estimation of aminoglycoside potency against these bacteria, or toxicity to human cells, respectively.
Methods
Experimental procedures are described in detail in the Supporting Information.
Supplementary Material
Acknowledgements
We gratefully acknowledge C. Squires at Tufts University for providing E. coli SQ380 and J. Tabor at Rice University for providing plasmid pSR26_2 containing the tetR/PLtet0-1 repressor/promoter/operator system. This work was supported by NIH NM-INBRE grant P20 GM103451 (C. Melancon) and by the University of New Mexico.
Footnotes
Supporting Information
Supporting Figure S1-S5 and detailed experimental procedures can be found in the Supporting Information, which is available free of charge on the ACS Publications website at http://pubs.acs.org
Notes
The authors declare no competing financial interests.
References
- (1).Wilson DN. Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nat. Rev. Microbiol. 2014;12:35–48. doi: 10.1038/nrmicro3155. [DOI] [PubMed] [Google Scholar]
- (2).Houghton JL, Green KD, Chen W, Garneau-Tsodikova S. The future of aminoglycosides: the end or renaissance? Chembiochem. 2010;11:880–902. doi: 10.1002/cbic.200900779. [DOI] [PubMed] [Google Scholar]
- (3).Becker B, Cooper MA. Aminoglycoside antibiotics in the 21st century. ACS Chem. Biol. 2013;8:105–115. doi: 10.1021/cb3005116. [DOI] [PubMed] [Google Scholar]
- (4).Kotra LP, Haddad J, Mobashery S. Aminoglycosides: perspectives on mechanisms of action and resistance and strategies to counter resistance. Antimicrob. Agents Chemother. 2000;44:3249–3256. doi: 10.1128/aac.44.12.3249-3256.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Flatt PM, Mahmud T. Biosynthesis of aminocyclitol-aminoglycoside antibiotics and related compounds. Nat. Prod. Rep. 2007;24:358–392. doi: 10.1039/b603816f. [DOI] [PubMed] [Google Scholar]
- (6).Wehmeier UF, Piepersberg W. Enzymology of aminoglycoside antibiotics - deduction from gene clusters. In: Hopwood DA, editor. Methods in Enzymology. Vol. 459. Academic Press; New York: 2009. pp. 459–491. [DOI] [PubMed] [Google Scholar]
- (7).Park SR, Park JW, Ban YH, Sohng JK, Yoon YJ. 2-Deoxystreptamine-containing aminoglycoside antibiotics: recent advances in the characterization and manipulation of their biosynthetic pathways. Nat. Prod. Rep. 2013;30:11–20. doi: 10.1039/c2np20092a. [DOI] [PubMed] [Google Scholar]
- (8).Wang L, Pulk A, Wasserman MR, Feldman MB, Altman RB, Cate JHD, Blanchard SC. Allosteric control of the ribosome by small-molecule antibiotics. Nat. Struct. Mol. Biol. 2012;19:957–963. doi: 10.1038/nsmb.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Tsai A, Uemura S, Johansson M, Puglisi EV, Marshall RA, Aitken CE, Korlach J, Ehrenberg M, Puglisi JD. The impact of aminoglycosides on the dynamics of translation elongation. Cell Rep. 2013;3:497–508. doi: 10.1016/j.celrep.2013.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wassermann MR, Pulk A, Zhou Z, Altman RB, Zinder JC, Green KD, Gameau-Tsodikova S, Cate JHD, Blanchard SC. Chemically related 4,5-linked aminoglycoside antibiotics drive subunit rotation in opposite directions. Nat. Commun. 2015;6:7896. doi: 10.1038/ncomms8896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Cabanas MJ, Vazquez D, Modolell J. Inhibition of ribosomal translocation by aminoglycoside antibiotics. Biochem. Biophys. Res. Commun. 1978;83:991–997. doi: 10.1016/0006-291x(78)91493-6. [DOI] [PubMed] [Google Scholar]
- (12).Feldman MB, Terry DS, Altman RB, Blanchard SC. Aminoglycoside activity observed on single pre-translocation ribosome complexes. Nat. Chem. Biol. 2010;6:54–62. doi: 10.1038/nchembio.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Davies J, Gorini L, Davis BD. Misreading of RNA codewords induced by aminoglycoside antibiotics. Mo/. Pharmacol. 1965;1:93–106. [PubMed] [Google Scholar]
- (14).Moazed D, Noller HF. Interaction of antibiotics with functional sites in 16S ribosomal RNA. Nature. 1987;327:389–394. doi: 10.1038/327389a0. [DOI] [PubMed] [Google Scholar]
- (15).Borovinskaya MA, Pai RD, Zhang W, Schuwirth BS, Holton JM, Hirokawa G, Kaji H, Kaji A, Cate JHD. Structural basis for aminoglycoside inhibition of bacterial ribosome recycling. Nat. Struct. Mo/. Biol. 2007;14:727–732. doi: 10.1038/nsmb1271. [DOI] [PubMed] [Google Scholar]
- (16).Borovinskaya MA, Shoji S, Frederick K, Cate JHD. Structural basis for hygromycin B inhibition of protein biosynthesis. RNA. 2008;14:1590–1599. doi: 10.1261/rna.1076908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Matt T, Ng CL, Lang K, Sha S-H, Akbergenov R, Shcherbakov D, Meyer M, Duscha S, Dubbaka SR, Perez-Fernandez D, Vasella A, Ramakrishnan V, Schacht J, Bottger EC. Dissociation of antibacterial activity and aminoglycoside ototoxicity in the 4-rnonosubstituted 2-deoxystreptamine aprarnycin. Proc. Nat. Acad. Sci. USA. 2012;109:10984–10989. doi: 10.1073/pnas.1204073109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Scheunemann AE, Graham WD, Vendeix FAP, Agris PF. Binding of aminoglycoside antibiotics to helix 69 of 23S rRNA. Nucleic Acids Res. 2010;38:3094–3105. doi: 10.1093/nar/gkp1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Shalev M, Baasov T. When protein start to make sense: fine-tuning of aminoglycosides for PTC suppression therapy. Medchemcomm. 2014;5:1092–1105. doi: 10.1039/C4MD00081A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Chang C-WT, Takemoto JY. Anti.fungal amphiphilic aminoglycosides. Medchemcomm. 2014;5:1048–1057. doi: 10.1039/C4MD00078A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Vicens Q, Westhof E. Crystal structure of geneticin bound to a bacterial 16S ribosomal RNA A site oligonucleotide. J. Mol. Biol. 2003;326:1175–1188. doi: 10.1016/s0022-2836(02)01435-3. [DOI] [PubMed] [Google Scholar]
- (22).Fran ois B, Russell RJM, Murray JB, Aboul-ela F, Masquida B, Vicens Q, Westhof E. Crystal structures of complexes between aminoglycosides and decoding A site oligonucleotides: role of the number of rings and positive charges in the specific binding leading to miscoding. Nucleic Acids Res. 2005;33:5677–5690. doi: 10.1093/nar/gki862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Sannes-Lowry KA, Griffey RH, Hofstadler SA. Measuring dissociation constants of RNA and aminoglycoside antibiotics by electrospray ionization mass spectrometry. Anal. Biochem. 2000;280:264–271. doi: 10.1006/abio.2000.4550. [DOI] [PubMed] [Google Scholar]
- (24).Hendrix M, Priestley ES, Joyce GF, Wong C-H. Direct observation of aminoglycoside-RNA interactions by surface plasmon resonance. J. Am. Chem. Soc. 1997;119:3641–3648. doi: 10.1021/ja964290o. [DOI] [PubMed] [Google Scholar]
- (25).Watkins D, Norris FA, Ku.mer S, Arya DP. A fluorescence-based screen for ribosome binding antibiotics. Anal. Biochem. 2013;434:300–307. doi: 10.1016/j.ab.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Greenberg WA, Priestley ES, Sears PS, Alper PB, Rosenbohm C, Hendrix M, Hung S-C, Wong C-H. Design and synthesis of new aminoglycoside antibiotics containing neamine as an optimal core structure: correlation of antibiotic activity with in vitro inhibition of translation. J. Am. Chem. Soc. 1999;121:6527–6541. [Google Scholar]
- (27).Benveniste R, Davies J. Structure-activity relationships among the aminoglycoside antibiotics: role of hydroxyl and amino groups. Antimicrob. Agents Chemother. 1973;4:402–409. doi: 10.1128/aac.4.4.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Sucheck SJ, Wong AL, Koeller KM, Boehr DD, Draker K.-a., Sears P, Wright GD, Wong C-H. Design of bifunctional antibiotics that target bacterial rRNA and inhibit resistance-causing enzymes. J. Am. Chem. Soc. 2000;122:5230–5231. [Google Scholar]
- (29).Salian S, Matt T, Akbergenov R, Harish S, Meyer M, Duscha S, Shcherbakov D, Bernet BB, Vasella A, Westhof E, Bottger EC. Structure-activity relationships among the kanamycin aminoglycosides: role of ring Ihydroxyl and amino groups. Antimicrob. Agents Chemother. 2012;56:6104–6108. doi: 10.1128/AAC.01326-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Kato T, Yang G, Teo Y, Juskeviciene R, Perez-Fernandez D, Shinde HM, Salian S, Bernet B, Vasella A, Bottger EC, Crich D. Synthesis and antiribosomal activities of 4'-0-, 6'-0-, 4"-0-, 4', 6'-0-, and 4", 6"-0-derivatives in the kanamycin series indicate differing target selectivity patterns between the 4,5- and 4,6-series of disubstituted 2-deoxystreptamine aminoglycoside antibiotics. ACS Inf Dis. 2015 doi: 10.1021/acsinfecdis.5b00069. ASAP DOI: 10.1021/acsinfecdis.5b00069. [DOI] [PubMed] [Google Scholar]
- (31).Recht MI, Douthwaite S, Puglisi JD. Basis for prokaryotic specificity of action of aminoglycoside antibiotics. EMBO J. 1999;18:3133–3136. doi: 10.1093/emboj/18.11.3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Recht MI, Puglisi JD. Aminoglycoside resistance with homogeneous and heterogeneous populations of antibiotic-resistant ribosomes. Antimicrob. Agents Chemother. 2001;45:2414–2419. doi: 10.1128/AAC.45.9.2414-2419.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Pfister P, Risch M, Broderson DE, Bottger EC. Role of 16S rRNA Helix 44 in Ribosomal Resistance to Hygromycin B. Antimicrob. Agents Chemother. 2003;47:1496–1502. doi: 10.1128/AAC.47.5.1496-1502.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Hui A, De Boer HA. Specialized ribosome system: preferential translation of a single mRNA species by a subpopulation of mutated ribosomes in Escherichia coli. Proc. Nat. Acad. Sci. USA. 1987;84:4762–4766. doi: 10.1073/pnas.84.14.4762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Lee K, Holland-Staley CA, Cunningham PR. Genetic analysis of the Shine-Dalgarno interaction: selection of alternative functional mRNA-rRNA combinations. RNA. 1996;2:1270–1285. [PMC free article] [PubMed] [Google Scholar]
- (36).Rackham 0, Chin JW. A network of orthogonal ribosome•mRNA pairs. Nat. Chem. Biol. 2005;1:159–166. doi: 10.1038/nchembio719. [DOI] [PubMed] [Google Scholar]
- (37).Beck CF, Mutzel R, Barbe J, Millier W. A multifunctional gene (tetR) controls TnlO-encoded tetracycline resistance. J. Bacteriol. 1982;150:633–642. doi: 10.1128/jb.150.2.633-642.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Crameri A, Whitehorse EA, Tate E, Stemmer WPC. Improved green fluorescent protein by molecular evolution using DNA shuftling. Nat. Biotechnol. 1996;14:315–319. doi: 10.1038/nbt0396-315. [DOI] [PubMed] [Google Scholar]
- (39).Lutz R, Bujard H. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/Il-12 regulatory elements. Nucleic Acids Res. 1997;25:1203–1210. doi: 10.1093/nar/25.6.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Mutilak VK, Guimaraes JC, Cambray G, Lam C, Christoffersen MJ, Mai Q-A, Tran MA, Paull M, Keasling JD, Arkin AP, Endy D. Precise and reliable gene expression via standard transcription and translation initiation elements. Nat. Methods. 2013;10:354–360. doi: 10.1038/nmeth.2404. [DOI] [PubMed] [Google Scholar]
- (41).Phelan RM, DiPardo BJ, Townsend CA. A high-throughput screen for the engineered production of P-lactam antibiotics. ACS Chem. Biol. 2012;7:835–840. doi: 10.1021/cb200504g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Orelle C, Carlson ED, Szal T, Florin T, Jewett MC, Mankin AS. Protein synthesis by ribosomes with tethered subunits. Nature. 2015 doi: 10.1038/nature14862. Epub July 29, 2015. DOI: 10.1038/nature14862. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.