

Abstract
Background
Significant evidence indicates that the failing heart is “energy-starved”. During the development of heart failure, the capacity of the heart to utilize fatty acids, the chief fuel, is diminished. Identification of alternate pathways for myocardial fuel oxidation could unveil novel strategies to treat heart failure.
Methods and Results
Quantitative mitochondrial proteomics was used to identify energy metabolic derangements that occur during the development of cardiac hypertrophy and heart failure in well-defined mouse models. As expected, amounts of proteins involved in fatty acid utilization were downregulated in myocardial samples from the failing heart. Conversely, expression of β-hydroxybutyrate dehydrogenase 1 (BDH1), a key enzyme in the ketone oxidation pathway, was increased in the heart failure samples.
Studies of relative oxidation studies in an isolated heart preparation using ex vivo NMR combined with targeted quantitative myocardial metabolomic profiling using mass spectrometry revealed that the hypertrophied and failing heart shifts to oxidizing ketone bodies as a fuel source in the context of reduced capacity to oxidize fatty acids. Distinct myocardial metabolomic signatures of ketone oxidation were identified.
Conclusions
These results indicate that the hypertrophied and failing heart shifts to ketone bodies as a significant fuel source for oxidative ATP production. Specific metabolite biosignatures of in vivo cardiac ketone utilization were identified. Future studies aimed at determining whether this fuel shift is adaptive or maladaptive could unveil new therapeutic strategies for heart failure.
Keywords: heart failure, hypertrophy, metabolism, molecular biology, fatty acid
INTRODUCTION
Growing evidence indicates that derangements in myocardial fuel metabolism and bioenergetics contribute to the development of heart failure, a global health problem. The adult mammalian heart requires enormous amounts of energy to sustain contractile function. Given that cardiac myocyte energy reserves are limited, ATP must be continually generated by oxidation of carbon fuels.1–5 Over 95% of the ATP produced in the healthy adult mammalian heart comes from mitochondrial oxidative phosphorylation, with the remainder being derived from glycolysis.2–5 Genetic studies have provided evidence that alterations in mitochondrial ATP production is casually linked to the development of heart failure. Specifically, human genetic defects in mitochondrial fatty acid oxidation (FAO) and oxidative phosphorylation (OXPHOS) cause cardiomyopathy.
Studies in humans with common acquired forms of heart failure have also provided evidence that derangements in fuel and energy metabolism contribute to heart failure. Cardiac magnetic resonance spectroscopy studies have shown that myocardial “high-energy” phosphate (phosphocreatine or PCr) stores are reduced in humans with pathological ventricular hypertrophy, with further decline during the transition to heart failure.6–10 Notably, the [PCr]/[ATP] ratio correlates with heart failure severity and is a strong predictor of cardiovascular mortality.11,12 In addition, studies conducted in animal models have consistently revealed re-programming of myocardial fuel utilization in the hypertrophied and failing heart; a shift from the chief fuel metabolic pathway, fatty acid oxidation (FAO), to increased reliance on glycolysis.13–20 Cardiac positron emission tomography studies in humans with hypertensive cardiac hypertrophy or idiopathic cardiomyopathy have largely corroborated this fuel shift.21–23 The mechanisms through which the failing heart compensates for this reduced capacity for oxidizing its chief energy substrate are unknown. Delineation of such alternate fuel utilization pathways, if they exist, could unveil new therapeutic strategies for heart failure.
In this study, we undertook an unbiased proteomic approach to probe mitochondrial fuel and energy metabolic abnormalities that occur during the development of heart failure in well-defined models of compensated and de-compensated pressure overload-induced cardiac hypertrophy in mice. Our results confirmed that contents of proteins involved in fatty acid utilization are reduced in the hypertrophied and failing heart. The proteomic dataset also demonstrated that the β-hydroxybutyrate dehydrogenase 1 (BDH1), a key enzyme in the ketone oxidation pathway, is upregulated in the hypertrophied and failing heart. Metabolite profiling and labeled substrate utilization studies supported the conclusion that the hypertrophied and failing heart shifts to ketone bodies as an alternate fuel.
METHODS
Animal studies
All animal experiments and euthanasia protocols were approved by the Institutional Animal Care and Use Committee at Sanford Burnham Prebys Medical Discovery Institute at Lake Nona. Studies were performed on female C57BL/6J mice 7–12 weeks of age on either standard chow (16.4% protein, 4.0% fat and 48.5% carbohydrates; Harlan Teklad, #2016) or ketogenic diet (8.6% protein, 75.1% fat and 3.2% carbohydrates; BioServ Co, AIN-76A). Animals were fed the ketogenic diet starting at 7–8 weeks of age.
8 week old female C57BL/6J mice in the following groups were utilized: compensated hypertrophy (CH) vs sham controls; heart failure (HF) vs sham controls. CH was achieved by transverse aortic constriction (TAC). HF was achieved by TAC plus a small apical myocardial infarction as described.24–26 Mice were harvested 1 month following each procedure.
Proteomics using Stable Labeling by Amino Acids (SILAC)
Mass spectrometry-based quantitative proteomics was conducted on mitochondrial enriched fractions27 prepared from cardiac tissue of CH, HF, and sham control non-labeled (light) mice, spiked with Lys6 labeled mitochondria prepared from cardiac tissue of Lys6 (13C6-Lysine, Silantes) labeled (heavy) non-surgery mice,28 as described in the online-only Data Supplement.
The proteomics data have been deposited into the Proteome Xchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD001820.
Substrate oxidation measurements
Langendorff heart perfusions were performed as previously described.29,30 Briefly, mice received 100 units of heparin by intraperitoneal injection and 10 min later were anesthetized with an intraperitoneal injection of 390 mg/kg sodium pentobarbital.
Excised hearts were perfused with a modified Krebs-Henseliet buffer (118 mM NaCl, 25 mM NaHCO3, 4.7 mM KCl, 0.4 mM KH2PO4, 2.5 mM CaCl2, pH 7.4) supplemented with 5 mM glucose and either i) 0.6 mM [2, 4,6,8,10,12,14,16-13C8] palmitate complexed (3:1 ratio) to a 3% fatty acid free bovine serum albumin (BSA) plus unlabeled 1 mM βOHB or unlabeled 0.6 mM palmitate/BSA plus 1 mM [2, 4-13C2] βOHB, with 1 mU/ml insulin (rDNA origin; Lilly) and continuous equilibration to a 95% O2/5% CO2 gas mixture. Following each perfusion, hearts were snap frozen in LN2 cooled tongs.
In vitro NMR spectroscopy was performed on reconstituted (D2O) lyophilized samples of neutralized, acid extracts of frozen myocardium, as previously described using either direct detect proton-decoupled 13C NMR or 13C-edited, 1H-observed NMR.30,31 The relative contribution of each substrate was calculated as previously described.29,31 Briefly, glutamate enrichment was used as a reporter of carbon entry into the TCA cycle. The fractional enrichment of acetyl-coenzyme A (Fc) entering the TCA cycle and the contribution of anaplerosis relative to citrate synthase activity (Y) were determined by isotopomer analysis of the glutamate 3- and 4-carbon 13C resonance.
RNA analyses
Total RNA was isolated from mouse bi-ventricle using the RNAzol method (Tel-Test). qRT-PCR was performed as described previously32 and in the online-only Data Supplement.
Western blot
Western blotting was performed with lysates from bi-ventricle as previously described33 using the following antibodies: VDAC/porin, (Abcam #ab15895); and BDH1 (Abcam #ab93931). Detection was performed by measuring the chemiluminescent signal as assayed by SuperSignal Dura (Pierce).
Metabolomic analysis of organic acids and acylcarnitines
Measurements of succinate, C4OH-carnitine, and acetylcarnitine (C2-carnitine) in mouse heart were conducted as described25,34 and in the online-only Data Supplement.
Plasma biochemistry measurements
Ketone bodies (total and β-hydroxybutyrate) were measured in blood serum using assays from Wako (Wako Diagnostics) according to the manufacturer’s instructions or on a Beckman-Coulter UniCel DxC 600 Analyzer. Plasma glucose and free fatty acids were measured using assays from Cayman (Cayman Chemical). Plasma triglyceride levels were determined using the Stanbio (Stanbio Laboratory) assay. The assays were conducted according to the manufacturer’s instructions.
Statistical Analyses
All data were analyzed with a 2-tailed Mann-Whitney or Student’s T-test (GraphPad Prism 6), where noted. The level of significance was set at p < 0.05 in all cases. The Pearson’s correlation coefficient was used to define the relationship between the amounts of CH and HF proteins.
RESULTS
Mitochondrial proteomic profiling reveals evidence for altered fuel utilization in the hypertrophied and early stage failing mouse heart
As an initial step towards defining energy metabolic alterations in the hypertrophied and early stage failing heart, quantitative mitochondrial proteomics was conducted on two well-defined mouse surgical models that exhibit distinct cardiac functional manifestations over a 4 week period: i) Left ventricular (LV) hypertrophy with preserved ventricular function (compensated hypertrophy or CH) achieved via surgically-placed transverse aortic constriction (TAC);24 and ii) decompensated cardiac hypertrophy (heart failure or HF) caused by combining TAC with a small apical myocardial infarction (TAC/MI) leading to reduced LV systolic function and global chamber dilatation.25,26 To identify regulated proteins, mitochondrial-enriched samples prepared from cardiac ventricles of CH and HF mice, and corresponding sham-operated control mice were subjected to quantitative proteomics using Stable Isotope Labeling by Amino Acids (SILAC) in mouse. Heavy isotope-tagged mitochondrial-enriched proteins were prepared from the hearts of control mice fed a diet containing 13C6-Lysine (Lys6) for 3 generations35 (Figure 1A). 516 mitochondrial proteins were identified in all samples (Table 1 in the online-only Data Supplement). The levels of 55 of these proteins were determined to be regulated in CH (23), HF (10) or both (22) groups compared to corresponding controls, using a cutoff of < −1.25 or > 1.5-fold change (Figure 1B, and in the online-only Data Supplement Table 2). Notably, the majority of dysregulated proteins in the HF group were similarly impacted in the CH group. In addition, changes in protein amounts in CH and HF are significantly correlated (Pearson correlation coefficient r=0.82; Figure 1C).
Figure 1.
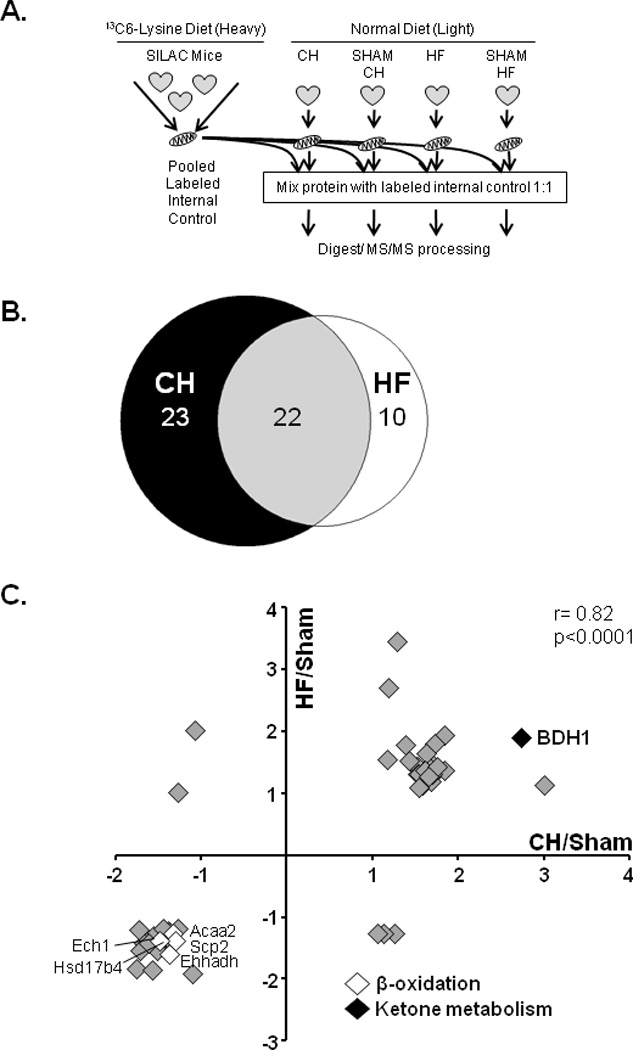
Mitochondrial proteomic profiling in the hypertrophied and failing mouse heart. (A) Schematic of the experimental design for quantitative proteomic analysis using Stable Isotope Labeling by amino ACids (SILAC), in mitochondrial enriched fractions, from the ventricles of sham-operated, compensated hypertrophy (CH), and heart failure (HF) animals. (B) Venn diagram displaying the number of regulated proteins identified in the CH, HF or both groups using a cut-off of < −1.25 or > 1.5 fold change (FC) as compared to sham-operated controls (n=2 per group). (C) The graph denotes fold change in levels of proteins that meet the defined cut-offs; HF/sham (ordinate) and CH/sham (abscissa). The key denotes regulated proteins involved in two fuel utilization pathways of interest as described in the text: fatty acid β-oxidation (white) and ketone catabolism (black). Spearman’s correlation coefficient (r) was calculated to determine the relationship between the CH and HF protein changes.
Rather surprisingly, the proteomic data revealed that few proteins involved in the electron transport chain (ETC) or mitochondrial OXPHOS were downregulated in CH or HF mice, in contrast to the results from other studies using models of chronic heart failure.36–39 These results are consistent, however, with the results of our previous transcriptomic profiling of the same samples demonstrating that very few genes involved in ETC/OXPHOS were regulated in CH or HF samples.25 Many of the regulated proteins detected in our study were involved in myocyte fuel metabolism. First, proteins needed for cellular fatty acid utilization were reduced in both the CH and HF groups [enoyl-CoA, hydratase/3-hydroxyacyl CoA dehydrogenase (EHHADH), enoyl CoA hydratase 1 (ECH1), acetyl-CoA acyltransferase 2 (ACAA2), and hydroxysteroid (17-beta) dehydrogenase 4 (HSD17B4), non-specific lipid transfer protein (SCP2); Figure 1C]. These results are concordant with many published studies showing that expression of genes involved in FAO are downregulated in the hypertrophied and failing hearts.15,16,20,40–42 Secondly, BDH1, an enzyme involved in ketone body metabolism, was increased in both CH and HF samples (2.8 and 1.9 fold, respectively; Figure 1C, and in the online-only Data Supplement Table 2). The induction of BDH1 protein expression was among the highest in the dataset. Quantitative real-time PCR (qRT-PCR) and immunoblotting confirmed a significant increase in Bdh1 mRNA and protein expression in CH and HF hearts harvested under both fed and fasted conditions (Figures 2A, B).
Figure 2.
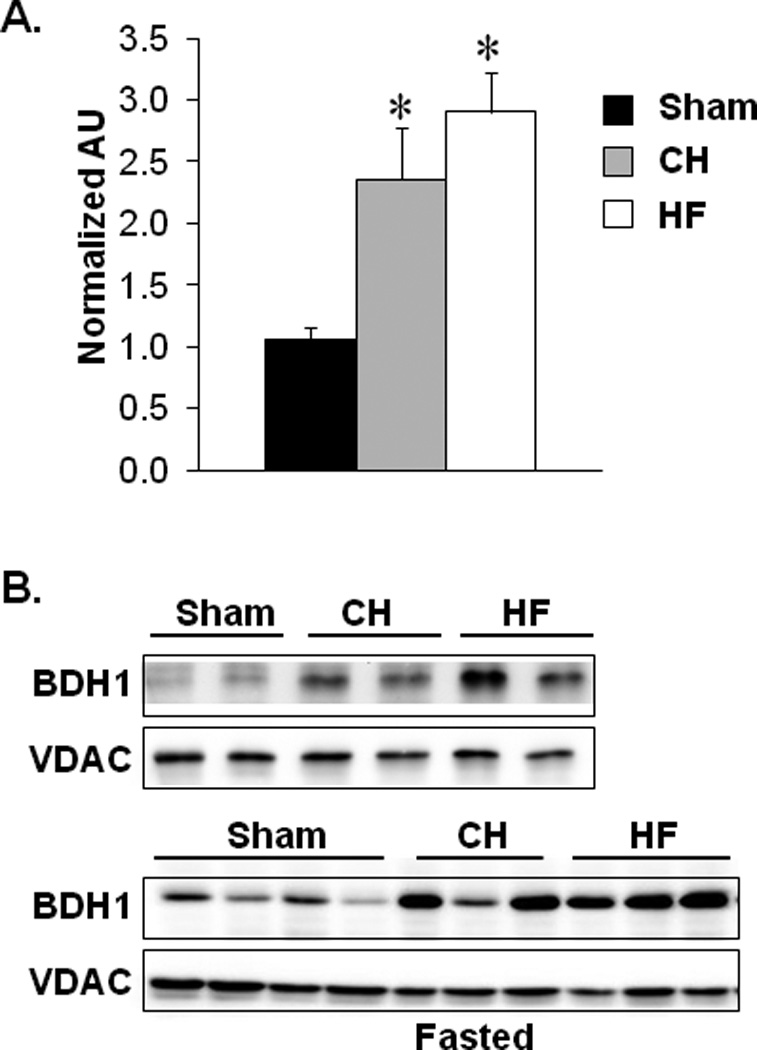
Bdh1 expression is induced in the hypertrophied and failing mouse heart. (A) Bdh1 mRNA levels in cardiac ventricular tissue from mice 4 weeks after sham, TAC (CH), or TAC/MI (HF) surgeries. Expression is normalized to Rplp0 (36B4). Bars represent mean ±SEM values (n=9–11 per group). *p<0.05. (B) Representative immunoblot analyses performed using protein extracts prepared from mouse cardiac ventricular tissue homogenates 4 weeks post-sham, -CH, or -HF surgeries collected in the fed state (4h after feeding) or following a 24 hour fast. Antibodies used are shown on the left. Anti-VDAC was used as a mitochondrial protein loading control.
The hypertrophied heart re-programs to utilize ketone bodies as an alternate fuel source
We next conducted studies to determine whether the hypertrophied heart shifts to using ketone bodies as suggested by the results of the proteomic profiling. 13C-NMR studies were performed to measure the relative contribution of fatty acids and ketone bodies to tricarboxylic acid (TCA) cycle flux. For these studies, hearts isolated from CH or sham-operated control groups were perfused in the Langendorff mode with 13C-labeled palmitate in the presence of unlabeled R-β-hydroxybutyrate (R-βOHB), or 13C-labeled R-βOHB in the presence of unlabeled palmitate. Consistent with findings described in numerous published studies15,16,18,31,41,43,44, the contribution of 13C-labeled palmitate to oxidative intermediary metabolism was decreased by approximately 40% in the CH hearts (Figure 1 in the online-only Data Supplement). Conversely, the contribution of βOHB to carbon entry into the oxidative pathway of the TCA cycle increased significantly in hearts from CH mice compared to control mice (Figure 3, left). These data indicate a 25% increase in the contribution of βOHB to oxidative production of ATP from carbon flux through the TCA cycle. In addition, the entry of anaplerotic carbon flux into the TCA cycle was increased in the CH heart, consistent with previous reports31,43,45 (Figure 3, right).
Figure 3.
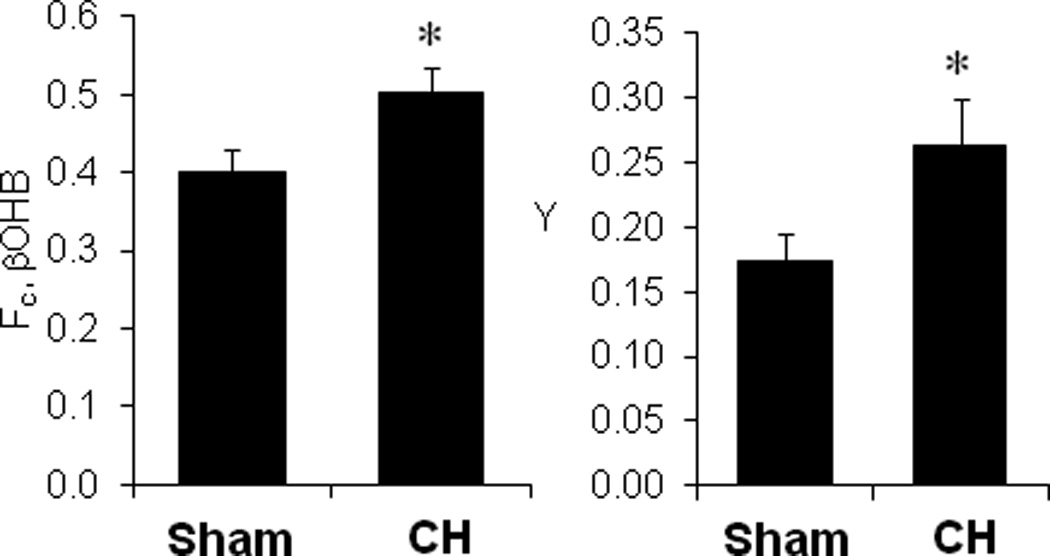
Increased βOHB oxidation in the hypertrophied heart. Left Panel: The fraction of 13C-enriched acetyl-CoA entering the TCA cycle from 13C-labeled βOHB (Fc, βOHB) is shown. Right panel: The fraction of carbon entering the TCA cycle via anaplerosis relative to that entering via citrate synthase (Y) is shown for CH and sham-operated controls. Data are shown as mean ±SEM (n=10, Sham and 11, CH). *p<0.05.
Identification of metabolite signatures of ketone utilization in the myocardium of the failing heart
We next sought to determine whether cardiac ketone utilization was increased in vivo in the failing heart. To this end, we measured myocardial levels of metabolites that reflect ketone body oxidation. Targeted quantitative metabolomic datasets generated previously from heart samples of the CH and HF groups and corresponding controls25 were analyzed for changes in metabolites that can be produced during ketone body metabolism including hydroxybutyrylcarnitine (C4OH-carnitine), acetylcarnitine (C2-carnitine), and succinate (Figure 4A). Levels of C4OH-carnitine and C2-carnitine have been shown to rise in the context of increased ketone body utilization in human and mouse skeletal muscle, and in human subcutaneous interstitial fluid.46–48 Concentrations of C4OH-carnitine, C2-carnitine, and succinate were increased in hearts of the HF group, but not the CH samples, compared to corresponding controls (Figure 4B), consistent with increased flux through the reaction catalyzed by BDH1.
Figure 4.
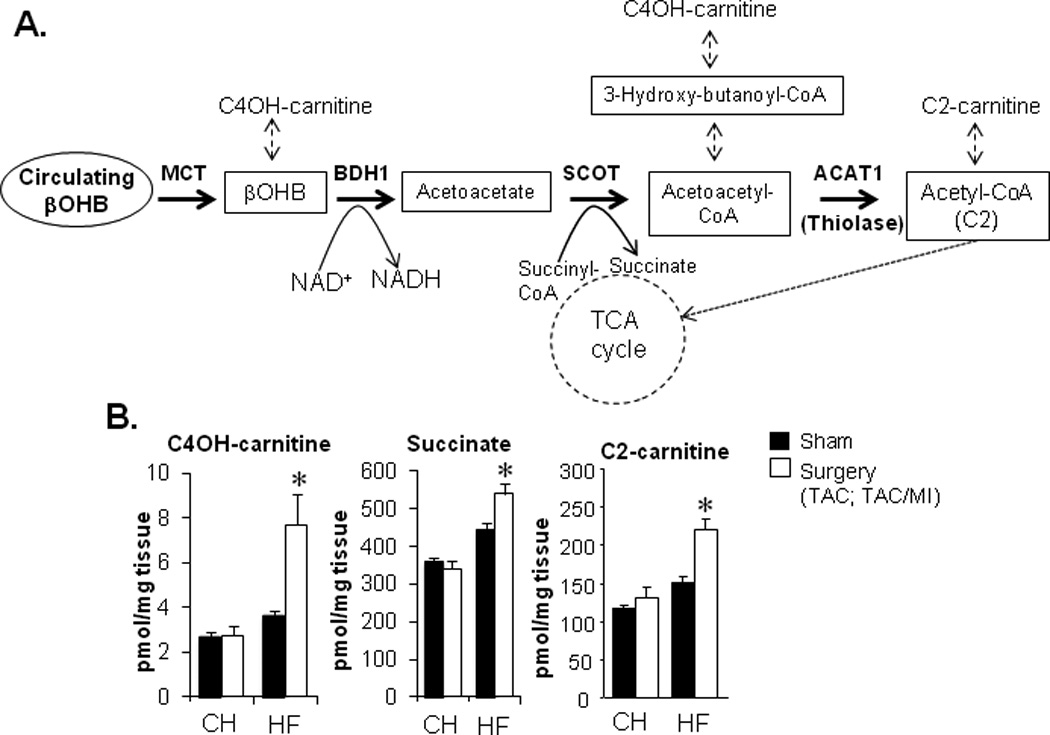
The myocardial metabolite profile of the failing heart is indicative of increased ketone utilization in the failing heart. (A) Schematic of the ketone metabolism pathway indicating relevant intermediary metabolite derivatives (dashed arrows). (B) Levels of ketone utilization pathway metabolite derivatives (C4OH-carnitine, succinate, C2-carnitine) in cardiac biventricular tissue from CH or HF mice and corresponding sham-operated controls 4 weeks post-surgery as measured previously using mass spectrometry-based quantitative metabolomics.25 Data are shown as mean ± SEM (n=6 per group). *p<0.05.
The relevance of the distinct HF metabolite signatures to myocardial ketone body metabolism was further assessed by comparison with profiles obtained from hearts of wild-type C57BL/6J mice fed a ketogenic diet for 4 weeks to increase myocardial ketone body delivery and utilization.29,49–51 As expected, the ketogenic diet resulted in a dramatic increase in concentration of plasma ketone bodies compared to controls fed a standard chow (Figure 2 in the online-only Data Supplement). Notably, this dietary intervention had no effect on ventricular function in this timeframe (echocardiographic data not shown). Importantly, the pattern of myocardial metabolite alterations observed in the mice fed a ketogenic diet was strikingly similar to that observed for the HF mice on a standard chow diet, including elevated amounts of both the R and S enantiomers of C4OH-carnitine (Figure 5A). An increase in both C4OH-carnitine enantiomers is consistent with an increase in uptake and oxidation of ketone bodies. In addition, rat ventricular myocytes cultured in fatty-acid free, ketone body-rich media also showed an elevated content of C4OH-carnitine compared to cells in control (ketone body-free) media (Figure 5B). Notably, the amount of Coenzyme A (CoASH) was not different between HF and control groups (data not shown).
Figure 5.
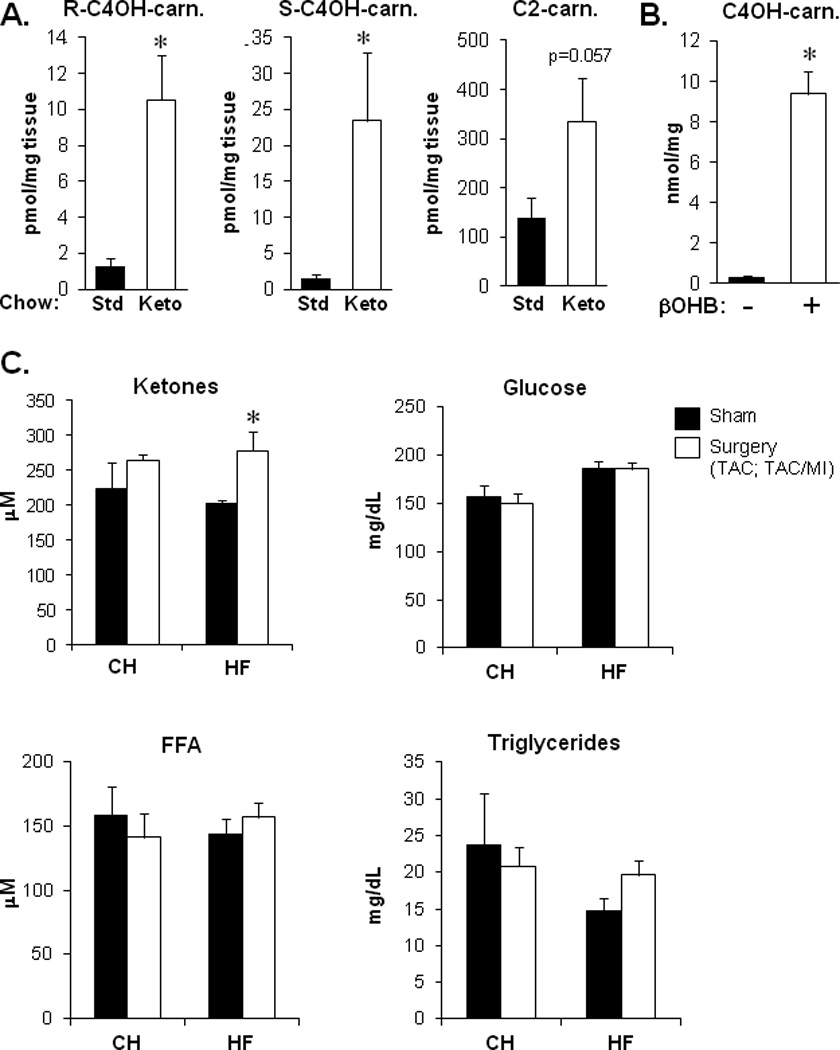
Myocardial metabolite profile on a ketogenic diet is similar to that observed for the HF mice on a standard chow diet. (A) Levels of R-C4OH-carnitine, S-C4OH-carnitine, and C2-carnitine in cardiac ventricular tissue from wild-type C57BL/6J mice fed a ketogenic (Keto) diet or standard (Std) chow diet for 4 weeks (n=5 per group). Values were determined using mass spectrometry. *p<0.05. (B) Total C4OH-carnitine levels in extracts prepared from neonatal rat ventricular myocytes (NRVMs) cultured in media +/− 8mM ketone, R-βOHB, in the presence of 1g/L glucose and 1mM carnitine, for 24h (n=3 per group, *p<0.05 by Student’s t-test). (C) Total plasma ketones (acetoacetate+βOHB), glucose, free fatty acids (FFA), and triglycerides in CH, HF and sham-operated control mice in the fed state (after a 4h morning fast; n=5–11 per group). Bars represent mean ± SEM for all panels.*p<0.05.
Lastly, to assess ketone delivery to the heart in the CH and HF groups, plasma substrate concentrations were measured. Plasma ketone body levels were modestly but significantly increased in HF but not CH, compared to corresponding controls (Figure 5C). Plasma glucose, free fatty acid levels, and triglycerides were unchanged in CH or HF groups (Figure 5C). The expression of the genes encoding the putative cellular ketone body transporters [Slc16a1 (MCT1) and Slc16a7 (MCT2)] were also assessed as an indirect measure of transport capacity given that Bdh1 expression was increased in CH and HF. Analysis of our published gene expression profiles25 did not reveal any differences in CH or HF compared with control myocardium following a 4h fast. However, after a 24h fast, when circulating ketone bodies are increased, we found that Slc16a7 mRNA levels were significantly increased in both CH and HF samples, compared to corresponding sham-operated controls (Figure 3 in the online-only Data Supplement). Taken together, these results provide evidence that myocardial ketone body utilization is increased in the HF mice through several potential mechanisms including increased delivery of ketone bodies and gene regulatory re-programming of ketone uptake and oxidation.
DISCUSSION
The results of this study yielded several new findings including: 1) the amounts of relatively few mitochondrial proteins involved in energy transduction and ATP production are regulated in the early stages of cardiac hypertrophy (CH) and heart failure (HF) in the mouse models studied here. Within the subset of regulated proteins in the CH and HF samples, a significant number were involved in fatty acid utilization, providing proteomic confirmation that the failing heart has reduced capacity for oxidizing fatty acids as a fuel; 2) the hypertrophied and failing rodent heart oxidizes ketone bodies as an alternate fuel for oxidative ATP production; and 3) metabolite signatures of myocardial ketone oxidation have been identified and suggest that a subset of mitochondrial ketone oxidation intermediate pools accumulate in the failing heart.
Our data support the conclusion that the shift to ketone oxidation in the failing heart occurs through several complementary mechanisms. First, ketone bodies are competitive with other substrates for heart, particularly fatty acids. The observed shift to ketone body oxidation in the hypertrophied and failing heart occurs in the context of reduced oxidation of fatty acids, the chief substrate for the normal adult heart. Downregulation of FAO gene expression is a well-characterized response in the hypertrophied and failing heart, driven at least in part, by reduced PPARα-mediated transcriptional control of genes involved in fatty acid utilization.20,52–54 Second, the delivery of ketone bodies is increased in the failing heart (increased plasma concentration). Indeed, previous studies have shown that the mammalian heart is capable of avid ketone body uptake and oxidation.55–57 We also cannot rule out the possibility that ketone body synthesis is activated in the cardiac myocyte although our gene expression data do not support this notion. Third, our results indicate that the hypertrophied and failing heart undergo gene regulatory re-programming to increase capacity for uptake and oxidation of ketone bodies. Specifically, the expression of Bdh1 and the transporter Slc16a1 were increased in CH and HF.
Our work has identified metabolite signatures of myocardial ketone utilization in the failing and normal heart (C4OH-carnitine and C2-carnitine). The metabolites were selected based on known derivatives of ketone utilization pathway intermediates (Figure 4A), and published work by others focused on tissues known to oxidize ketones.46–48 It should be noted that this set of metabolites are not unique to ketone utilization pathways, given that other metabolic pathways can generate the intermediates. However, our results demonstrate that this metabolite profile is elevated in both HF samples and normal mice fed a ketogenic diet, providing additional support for our conclusion. In addition, we found that C4OH content is increased in rat neonatal cardiac myocytes exposed to βOHB. Interestingly, the increase in C4OH-carnitine and C2-carnitine was observed in HF but not CH samples. The reason for this latter specificity is unknown, but could reflect higher ketone oxidation rates related to increased ketone body delivery (elevated plasma levels) in HF. Alternatively, capacity for flux through downstream pathways such as the TCA cycle, ETC, and OXPHOS may become reduced with progression to HF creating a mismatch with high flux rates through the ketone oxidation pathway. This latter conclusion is supported by our observation that most TCA cycle organic acid intermediates (with the exception of succinate) are reduced in HF samples,25 consistent with a “bottleneck” downstream of ketone and other fuel inputs to the TCA cycle. It will be of significant interest to explore this metabolomic signature in other experimental heart failure models, and in humans, to determine whether activation of ketone utilization is a broad paradigm relevant to energy metabolic reprogramming of the failing heart.
We speculate that the shift toward ketone body utilization in the hypertrophied heart is an early adaptive response to maintain adequate fuel supplies for oxidative ATP production in the context of reduced FAO. Consistent with this notion, a recent study demonstrated that targeted disruption of succinyl-CoA:3-oxoacid-CoA transferase (SCOT), a key enzyme in the ketone body metabolic pathway, resulted in a heart failure phenotype in mice in the context of pressure overload.30 However, it is possible that over the longer term, high rates of ketone utilization lead to maladaptive consequences. Others have shown that ketone oxidation may lead to reduced anaplerotic input in an isolated heart preparation.58 In addition, as noted above, the pools of several metabolite intermediates including succinate and C2-carnitine are expanded in the myocardial samples from the HF group. Increased availability of short-chain carbon moieties and succinate could set the stage for post-translational modifications of mitochondrial enzymes and proteins reducing oxidative flux or ATP generation.
The findings described herein raise the obvious question of relevance to human heart failure. Little is known about ketone body metabolism in the failing human heart, although studies have shown increased concentrations of ketone metabolites in urine and breath samples of patients with heart failure.59–62 In addition, increased concentrations of serum βOHB have been described in patients with severe heart failure.63
CONCLUSIONS
In summary, our findings indicate that during the development of pathologic cardiac remodeling in mouse models of heart failure, the myocardium increasingly relies on ketone bodies as a fuel. We propose that this fuel metabolic shift is triggered by reduced capacity for oxidizing fatty acids, the chief fuel for the normal adult mammalian heart. Future studies aimed at determining the relevance of these findings to human heart failure, and delineation of the impact of chronic ketone utilization on cardiac metabolism and function will be important to determine whether this response represents a new therapeutic target for the metabolic modulation of heart failure.
Supplementary Material
Clinical Perspective.
Significant evidence, based on results of pre-clinical studies and observations in humans, indicates that energy metabolic derangements contribute to the development of heart failure. A prototypical fuel shift occurs in the hypertrophied and failing heart, in which the capacity for oxidizing fatty acids, the chief substrate for the normal adult heart, becomes reduced along with an increase in reliance on glucose. It is generally believed that reduced capacity for oxidation of fatty acids leads to an “energy-starved” heart. Therefore, identification of alternate fuel utilization pathways that may compensate for this fuel shift could lead to new therapeutic strategies aimed at heart failure. In this study, using well-defined mouse models of cardiac hypertrophy and heart failure, we demonstrate that the heart begins to utilize ketone bodies en route to the development of heart failure. This shift to reliance on ketone bodies as a fuel is likely driven by multiple mechanisms, including elevation in plasma ketones, a reduction in competition with fatty acids, and gene regulatory re-programming of the heart. These findings set the stage for future studies aimed at determining whether the shift to oxidizing ketone bodies in the failing heart is adaptive or maladaptive. This fuel utilization pathway could prove to be a new candidate target for metabolic modulatory therapies aimed at early stages of heart failure.
Acknowledgments
We wish to thank Lorenzo Thomas for assistance with preparation of the manuscript and acknowledge the following Core Facilities at Sanford Burnham Prebys Medical Discovery Institute at Lake Nona: Cardiometabolic Phenotyping and Metabolomics. We wish to thank Olga Ilkayeva and the Duke University School of Medicine’s Proteomics and Metabolomics Shared Resource for metabolomics data; and Lauren Ashley Gabriel and Caron Stonebrook for assistance with the animal studies.
Funding Sources: This work was supported by NIH grants R01 HL058493 (D.P.K.), R01 HL101189 (D.P.K. and D.M.M.), R01 DK091538 (P.A.C.), R01 HL062702 (E.D.L.) and R01HL49244 (E.D.L.). G.A was supported by the Swiss National Science Foundation.
Footnotes
Disclosures: None.
References
- 1.Bing RJ. The metabolism of the heart. Harvey Lect. 1954;50:27–70. [PubMed] [Google Scholar]
- 2.Bing RJ, Siegel A, Ungar I, Gilbert M. Metabolism of the human heart. II. Studies on fat ketone and amino acid metabolism. Am J Med. 1954;16:504–515. doi: 10.1016/0002-9343(54)90365-4. [DOI] [PubMed] [Google Scholar]
- 3.Wisneski JA, Gertz EW, Neese RA, Mayr M. Myocardial metabolism of free fatty acids. Studies with 14C-labeled substrates in humans. J Clin Invest. 1987;79:359–366. doi: 10.1172/JCI112820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lopaschuk GD, Belke DD, Gamble J, Itoi T, Schonekess BO. Regulation of fatty acid oxidation in the mammalian heart in health and disease. Biochim Biophys Acta. 1994;1213:263–276. doi: 10.1016/0005-2760(94)00082-4. [DOI] [PubMed] [Google Scholar]
- 5.van der Vusse GJ, van Bilsen M, Glatz JF. Cardiac fatty acid uptake and transport in health and disease. Cardiovasc Res. 2000;45:279–293. doi: 10.1016/s0008-6363(99)00263-1. [DOI] [PubMed] [Google Scholar]
- 6.Ingwall JS, Kramer MF, Fifer MA, Lorell BH, Shemin R, Grossman W, Allen PD. The creatine kinase system in normal and diseased human myocardium. N Engl J Med. 1985;313:1050–1054. doi: 10.1056/NEJM198510243131704. [DOI] [PubMed] [Google Scholar]
- 7.de Roos A, Doornbos J, Luyten PR, Oosterwaal LJ, van der Wall EE, den Hollander JA. Cardiac metabolism in patients with dilated and hypertrophic cardiomyopathy: assessment with proton-decoupled P-31 MR spectroscopy. J Magn Reson Imaging. 1992;2:711–719. doi: 10.1002/jmri.1880020616. [DOI] [PubMed] [Google Scholar]
- 8.Tian R, Nascimben L, Kaddurah-Daouk R, Ingwall JS. Depletion of energy reserve via the creatine kinase reaction during the evolution of heart failure in cardiomyopathic hamsters. J Mol Cell Card. 1996;28:755–765. doi: 10.1006/jmcc.1996.0070. [DOI] [PubMed] [Google Scholar]
- 9.Ingwall JS, Weiss RG. Is the failing heart energy starved? On using chemical energy to support cardiac function. Circ Res. 2004;95:135–145. doi: 10.1161/01.RES.0000137170.41939.d9. [DOI] [PubMed] [Google Scholar]
- 10.Carley AN, Taegtmeyer H, Lewandowski ED. Matrix revisited: mechanisms linking energy substrate metabolism to the function of the heart. Circ Res. 2014;114:717–729. doi: 10.1161/CIRCRESAHA.114.301863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neubauer S, Horn M, Cramer M, Harre K, Newell JB, Peters W, Pabst T, Ertl G, Hahn D, Ingwall JS, Kochsiek K. Myocardial phosphocreatine-to-ATP ratio is a predictor of mortality in patients with dilated cardiomyopathy. Circulation. 1997;96:2190–2196. doi: 10.1161/01.cir.96.7.2190. [DOI] [PubMed] [Google Scholar]
- 12.Neubauer S. The failing heart--an engine out of fuel. N Engl J Med. 2007;356:1140–1151. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- 13.Bishop SP, Altschuld RA. Increased glycolytic metabolism in cardiac hypertrophy and congestive failure. Am J Physiol. 1970;218:153–159. doi: 10.1152/ajplegacy.1970.218.1.153. [DOI] [PubMed] [Google Scholar]
- 14.Taegtmeyer H, Overturf ML. Effects of moderate hypertension on cardiac function and metabolism in the rabbit. Hypertension. 1988;11:416–426. doi: 10.1161/01.hyp.11.5.416. [DOI] [PubMed] [Google Scholar]
- 15.Allard MF, Schonekess BO, Henning SL, English DR, Lopaschuk GD. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am J Physiol. 1994;267:H742–H750. doi: 10.1152/ajpheart.1994.267.2.H742. [DOI] [PubMed] [Google Scholar]
- 16.Christe ME, Rodgers RL. Altered glucose and fatty acid oxidation in hearts of the spontaneously hypertensive rat. J Mol Cell Cardiol. 1994;26:1371–1375. doi: 10.1006/jmcc.1994.1155. [DOI] [PubMed] [Google Scholar]
- 17.Paolisso G, Gambardella A, Galzerano D, D'Amore A, Rubino P, Verza M, Teasuro P, Varricchio M, D'Onofrio F. Total-body and myocardial substrate oxidation in congestive heart failure. Metabolism. 1994;43:174–179. doi: 10.1016/0026-0495(94)90241-0. [DOI] [PubMed] [Google Scholar]
- 18.Sambandam N, Lopaschuk GD, Brownsey RW, Allard MF. Energy metabolism in the hypertrophied heart. Heart Fail Rev. 2002;7:161–173. doi: 10.1023/a:1015380609464. [DOI] [PubMed] [Google Scholar]
- 19.Chandler MP, Kerner J, Huang H, Vazquez E, Reszko A, Martini WZ, Hoppel CL, Imai M, Rastogi S, Sabbah HN, Stanley WC. Moderate severity heart failure does not involve a downregulation of myocardial fatty acid oxidation. Am J Physiol Heart Circ Physiol. 2004;287:H1538–H1543. doi: 10.1152/ajpheart.00281.2004. [DOI] [PubMed] [Google Scholar]
- 20.Sack MN, Rader TA, Park S, Bastin J, McCune SA, Kelly DP. Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation. 1996;94:2837–2842. doi: 10.1161/01.cir.94.11.2837. [DOI] [PubMed] [Google Scholar]
- 21.Wallhaus TR, Taylor M, DeGrado TR, Russell DC, Stanko P, Nickles RJ, Stone CK. Myocardial free fatty acid and glucose use after carvedilol treatment in patients with congestive heart failure. Circulation. 2001;103:2441–2446. doi: 10.1161/01.cir.103.20.2441. [DOI] [PubMed] [Google Scholar]
- 22.Davila-Roman VG, Vedala G, Herrero P, de las Fuentes L, Rogers JG, Kelly DP, Gropler RJ. Altered myocardial fatty acid and glucose metabolism in idiopathic dilated cardiomyopathy. J Am Coll Cardiol. 2002;40:271–277. doi: 10.1016/s0735-1097(02)01967-8. [DOI] [PubMed] [Google Scholar]
- 23.de las Fuentes L, Herrero P, Peterson LR, Kelly DP, Gropler RJ, Davila-Roman VG. Myocardial fatty acid metabolism: independent predictor of left ventricular mass in hypertensive heart disease. Hypertension. 2003;41:83–87. doi: 10.1161/01.hyp.0000047668.48494.39. [DOI] [PubMed] [Google Scholar]
- 24.Huss JM, Imahashi K, Dufour CR, Weinheimer CJ, Courtois M, Kovacs A, Giguere V, Murphy E, Kelly DP. The nuclear receptor ERRalpha is required for the bioenergetic and functional adaptation to cardiac pressure overload. Cell Metab. 2007;6:25–37. doi: 10.1016/j.cmet.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 25.Lai L, Leone TC, Keller MP, Martin OJ, Broman AT, Nigro J, Kapoor K, Koves TR, Stevens R, Ilkayeva OR, Vega RB, Attie AD, Muoio DM, Kelly DP. Energy metabolic reprogramming in the hypertrophied and early stage failing heart: a multisystems approach. Circ Heart Fail. 2014;7:1022–1031. doi: 10.1161/CIRCHEARTFAILURE.114.001469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weinheimer CJ, Lai L, Kelly DP, Kovacs A. Novel mouse model of left ventricular pressure overload and infarction causing predictable ventricular remodelling and progression to heart failure. Clin Exp Pharmacol Physiol. 2015;42:33–40. doi: 10.1111/1440-1681.12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK, Hill DE, Vidal M, Evans JG, Thorburn DR, Carr SA, Mootha VK. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kruger M, Moser M, Ussar S, Thievessen I, Luber CA, Forner F, Schmidt S, Zanivan S, Fassler R, Mann M. SILAC mouse for quantitative proteomics uncovers kindlin-3 as an essential factor for red blood cell function. Cell. 2008;134:353–364. doi: 10.1016/j.cell.2008.05.033. [DOI] [PubMed] [Google Scholar]
- 29.Wentz AE, d'Avignon DA, Weber ML, Cotter DG, Doherty JM, Kerns R, Nagarajan R, Reddy N, Sambandam N, Crawford PA. Adaptation of myocardial substrate metabolism to a ketogenic nutrient environment. J Biol Chem. 2010;285:24447–24456. doi: 10.1074/jbc.M110.100651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schugar RC, Moll AR, Andre d'Avignon D, Weinheimer CJ, Kovacs A, Crawford PA. Cardiomyocyte-specific deficiency of ketone body metabolism promotes accelerated pathological remodeling. Mol Metab. 2014;3:754–769. doi: 10.1016/j.molmet.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pound KM, Sorokina N, Ballal K, Berkich DA, Fasano M, Lanoue KF, Taegtmeyer H, O'Donnell JM, Lewandowski ED. Substrate-enzyme competition attenuates upregulated anaplerotic flux through malic enzyme in hypertrophied rat heart and restores triacylglyceride content: attenuating upregulated anaplerosis in hypertrophy. Circ Res. 2009;104:805–812. doi: 10.1161/CIRCRESAHA.108.189951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huss JM, Kelly DP. Nuclear receptor signaling and cardiac energetics. Circ Res. 2004;95:568–578. doi: 10.1161/01.RES.0000141774.29937.e3. [DOI] [PubMed] [Google Scholar]
- 33.Cresci S, Wright LD, Spratt JA, Briggs FN, Kelly DP. Activation of a novel metabolic gene regulatory pathway by chronic stimulation of skeletal muscle. Am J Physiol. 1996;270:C1413–C1420. doi: 10.1152/ajpcell.1996.270.5.C1413. [DOI] [PubMed] [Google Scholar]
- 34.Minkler PE, Stoll MS, Ingalls ST, Kerner J, Hoppel CL. Validated Method for the Quantification of Free and Total Carnitine, Butyrobetaine, and Acylcarnitines in Biological Samples. Anal Chem. 2015;87:8994–9001. doi: 10.1021/acs.analchem.5b02198. [DOI] [PubMed] [Google Scholar]
- 35.Konzer A, Ruhs A, Braun T, Kruger M. Global protein quantification of mouse heart tissue based on the SILAC mouse. Methods Mol Biol. 2013;1005:39–52. doi: 10.1007/978-1-62703-386-2_4. [DOI] [PubMed] [Google Scholar]
- 36.Gao Z, Xu H, DiSilvestre D, Halperin VL, Tunin R, Tian Y, Yu W, Winslow RL, Tomaselli GF. Transcriptomic profiling of the canine tachycardia-induced heart failure model: global comparison to human and murine heart failure. J Mol Cell Cardiol. 2006;40:76–86. doi: 10.1016/j.yjmcc.2005.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bugger H, Schwarzer M, Chen D, Schrepper A, Amorim PA, Schoepe M, Nguyen TD, Mohr FW, Khalimonchuk O, Weimer BC, Doenst T. Proteomic remodelling of mitochondrial oxidative pathways in pressure overload-induced heart failure. Cardiovasc Res. 2010;85:376–384. doi: 10.1093/cvr/cvp344. [DOI] [PubMed] [Google Scholar]
- 38.Barth AS, Kumordzie A, Frangakis C, Margulies KB, Cappola TP, Tomaselli GF. Reciprocal transcriptional regulation of metabolic and signaling pathways correlates with disease severity in heart failure. Circ Cardiovasc Genet. 2011;4:475–483. doi: 10.1161/CIRCGENETICS.110.957571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu J, Nie HG, Zhang XD, Tian Y, Yu B. Down-regulated energy metabolism genes associated with mitochondria oxidative phosphorylation and fatty acid metabolism in viral cardiomyopathy mouse heart. Mol Biol Rep. 2011;38:4007–4013. doi: 10.1007/s11033-010-0519-y. [DOI] [PubMed] [Google Scholar]
- 40.Taegtmeyer H, Golfman L, Sharma S, Razeghi P, van Arsdall M. Linking gene expression to function: metabolic flexibility in the normal and diseased heart. Ann N Y Acad Sci. 2004;1015:202–213. doi: 10.1196/annals.1302.017. [DOI] [PubMed] [Google Scholar]
- 41.Akki A, Smith K, Seymour AM. Compensated cardiac hypertrophy is characterised by a decline in palmitate oxidation. Mol Cell Biochem. 2008;311:215–224. doi: 10.1007/s11010-008-9711-y. [DOI] [PubMed] [Google Scholar]
- 42.Ardehali H, Sabbah HN, Burke MA, Sarma S, Liu PP, Cleland JG, Maggioni A, Fonarow GC, Abel ED, Campia U, Gheorghiade M. Targeting myocardial substrate metabolism in heart failure: potential for new therapies. Eur J Heart Fail. 2012;14:120–129. doi: 10.1093/eurjhf/hfr173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kolwicz SC, Jr, Olson DP, Marney LC, Garcia-Menendez L, Synovec RE, Tian R. Cardiac-specific deletion of acetyl CoA carboxylase 2 prevents metabolic remodeling during pressure-overload hypertrophy. Circ Res. 2012;111:728–738. doi: 10.1161/CIRCRESAHA.112.268128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lewandowski ED, Fischer SK, Fasano M, Banke NH, Walker LA, Huqi A, Wang X, Lopaschuk GD, O'Donnell JM. Acute liver carnitine palmitoyltransferase I overexpression recapitulates reduced palmitate oxidation of cardiac hypertrophy. Circ Res. 2013;112:57–65. doi: 10.1161/CIRCRESAHA.112.274456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sorokina N, O'Donnell JM, McKinney RD, Pound KM, Woldegiorgis G, LaNoue KF, Ballal K, Taegtmeyer H, Buttrick PM, Lewandowski ED. Recruitment of compensatory pathways to sustain oxidative flux with reduced carnitine palmitoyltransferase I activity characterizes inefficiency in energy metabolism in hypertrophied hearts. Circulation. 2007;115:2033–2041. doi: 10.1161/CIRCULATIONAHA.106.668665. [DOI] [PubMed] [Google Scholar]
- 46.Hack A, Busch V, Pascher B, Busch R, Bieger I, Gempel K, Baumeister FA. Monitoring of ketogenic diet for carnitine metabolites by subcutaneous microdialysis. Pediatr Res. 2006;60:93–96. doi: 10.1203/01.pdr.0000219479.95410.79. [DOI] [PubMed] [Google Scholar]
- 47.Soeters MR, Serlie MJ, Sauerwein HP, Duran M, Ruiter JP, Kulik W, Ackermans MT, Minkler PE, Hoppel CL, Wanders RJ, Houten SM. Characterization of D-3-hydroxybutyrylcarnitine (ketocarnitine): an identified ketosis-induced metabolite. Metabolism. 2012;61:966–973. doi: 10.1016/j.metabol.2011.11.009. [DOI] [PubMed] [Google Scholar]
- 48.DeBalsi KL, Wong KE, Koves TR, Slentz DH, Seiler SE, Wittmann AH, Ilkayeva OR, Stevens RD, Perry CG, Lark DS, Hui ST, Szweda L, Neufer PD, Muoio DM. Targeted metabolomics connects thioredoxin-interacting protein (TXNIP) to mitochondrial fuel selection and regulation of specific oxidoreductase enzymes in skeletal muscle. J Biol Chem. 2014;289:8106–8120. doi: 10.1074/jbc.M113.511535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hasselbaink DM, Glatz JF, Luiken JJ, Roemen TH, Van der Vusse GJ. Ketone bodies disturb fatty acid handling in isolated cardiomyocytes derived from control and diabetic rats. Biochem J. 2003;371:753–760. doi: 10.1042/BJ20021617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stanley WC, Meadows SR, Kivilo KM, Roth BA, Lopaschuk GD. beta-Hydroxybutyrate inhibits myocardial fatty acid oxidation in vivo independent of changes in malonyl-CoA content. Am J Physiol Heart Circ Physiol. 2003;285:H1626–H1631. doi: 10.1152/ajpheart.00332.2003. [DOI] [PubMed] [Google Scholar]
- 51.Veech RL. The therapeutic implications of ketone bodies: the effects of ketone bodies in pathological conditions: ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins Leukot Essent Fatty Acids. 2004;70:309–319. doi: 10.1016/j.plefa.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 52.Tian Q, Barger PM. Deranged energy substrate metabolism in the failing heart. Curr Hypertens Rep. 2006;8:465–471. doi: 10.1007/s11906-006-0024-9. [DOI] [PubMed] [Google Scholar]
- 53.Barger PM, Brandt JM, Leone TC, Weinheimer CJ, Kelly DP. Deactivation of peroxisome proliferator-activated receptor-alpha during cardiac hypertrophic growth. J Clin Invest. 2000;105:1723–1730. doi: 10.1172/JCI9056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lahey R, Wang X, Carley AN, Lewandowski ED. Dietary fat supply to failing hearts determines dynamic lipid signaling for nuclear receptor activation and oxidation of stored triglyceride. Circulation. 2014;130:1790–1799. doi: 10.1161/CIRCULATIONAHA.114.011687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rudolph W, Maas D, Richter J, Hasinger F, Hofmann H, Dohrn P. [on the Significance of Acetoacetate and Beta-Hydroxybutyrate in Human Myocardial Metabolism] Klin Wochenschr. 1965;43:445–451. doi: 10.1007/BF01483852. [DOI] [PubMed] [Google Scholar]
- 56.Jeffrey FM, Diczku V, Sherry AD, Malloy CR. Substrate selection in the isolated working rat heart: effects of reperfusion, afterload, and concentration. Basic Res Cardiol. 1995;90:388–396. doi: 10.1007/BF00788500. [DOI] [PubMed] [Google Scholar]
- 57.Bartelds B, van der Leij FR, Kuipers JR. Role of ketone bodies in perinatal myocardial energy metabolism. Biochem Soc Trans. 2001;29:325–330. doi: 10.1042/0300-5127:0290325. [DOI] [PubMed] [Google Scholar]
- 58.Russell RR, 3rd, Taegtmeyer H. Changes in citric acid cycle flux and anaplerosis antedate the functional decline in isolated rat hearts utilizing acetoacetate. J Clin Invest. 1991;87:384–390. doi: 10.1172/JCI115008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kupari M, Lommi J, Ventila M, Karjalainen U. Breath acetone in congestive heart failure. Am J Cardiol. 1995;76:1076–1078. doi: 10.1016/s0002-9149(99)80304-x. [DOI] [PubMed] [Google Scholar]
- 60.Chung JH, Kim JS, Kim OY, Kang SM, Hwang GS, Shin MJ. Urinary ketone is associated with the heart failure severity. Clin Biochem. 2012;45:1697–1699. doi: 10.1016/j.clinbiochem.2012.08.013. [DOI] [PubMed] [Google Scholar]
- 61.Marcondes-Braga FG, Gutz IG, Batista GL, Saldiva PH, Ayub-Ferreira SM, Issa VS, Mangini S, Bocchi EA, Bacal F. Exhaled acetone as a new biomaker of heart failure severity. Chest. 2012;142:457–466. doi: 10.1378/chest.11-2892. [DOI] [PubMed] [Google Scholar]
- 62.Samara MA, Tang WH, Cikach F, Jr, Gul Z, Tranchito L, Paschke KM, Viterna J, Wu Y, Laskowski D, Dweik RA. Single exhaled breath metabolomic analysis identifies unique breathprint in patients with acute decompensated heart failure. J Am Coll Cardiol. 2013;61:1463–1464. doi: 10.1016/j.jacc.2012.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lommi J, Kupari M, Koskinen P, Naveri H, Leinonen H, Pulkki K, Harkonen M. Blood ketone bodies in congestive heart failure. J Am Coll Cardiol. 1996;28:665–672. doi: 10.1016/0735-1097(96)00214-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.