

Abstract
Dihydrofolate reductase (DHFR) from Escherichia coli has long served as a model enzyme with which to elucidate possible links between protein dynamics and the catalyzed reaction. Such physical properties of its human counterpart have not been rigorously studied so far, but recent computer-based simulations suggest that these two DHFRs differ significantly in how closely coupled the protein dynamics and the catalyzed C-H→C hydride transfer step are. To test this prediction, two contemporary probes for studying the effect of protein dynamics on catalysis were combined here: temperature dependence of intrinsic kinetic isotope effects (KIEs) that are sensitive to the physical nature of the chemical step, and protein mass-modulation that slows down fast dynamics (femto- to picosecond timescale) throughout the protein. The intrinsic H/T KIEs of human DHFR, like those of E. coli DHFR, are shown to be temperature-independent in the range from 5–45 °C, indicating fast sampling of donor and acceptor distances (DADs) at the reaction’s transition state (or tunneling ready state – TRS). Mass modulation of these enzymes through isotopic labeling with 13C, 15N, and 2H at nonexchangeable hydrogens yield an 11% heavier enzyme. The additional mass has no effect on the intrinsic KIEs of the human enzyme. This finding indicates that the mass-modulation of the human DHFR affects neither DAD distribution nor the DAD’s conformational sampling dynamics. Furthermore, reduction in the enzymatic turnover number and the dissociation rate constant for the product indicate that the isotopic substitution affects kinetic steps that are not the catalyzed C-H→C hydride transfer. The findings are discussed in terms of fast dynamics and their role in catalysis, the comparison of calculations and experiments, and the interpretation of isotopically-modulated heavy enzymes in general.
Keywords: dihydrofolate reductase, kinetic isotope effects, enzyme dynamics, tunneling, isotopically enriched enzymes
A contemporary debate seems to exist over the possible roles of fast femto- to picosecond-timescale protein motions in enzymatic reactions.1–4 Several computational and experimental studies have suggested that such motions may play a role in the hydride transfer reaction catalyzed by Escherichia coli dihydrofolate reductase (ecDHFR; for recent reviews see refs 5,6). DHFR catalyzes the stereospecific transfer of the pro-R hydride from reduced nicotinamide adenine diphosphate (NADPH) to the C6 position of the dihydropterin ring of 7,8-dihydrofolate (DHF) to generate 5,6,7,8-tetrahydrofolate (THF). The reaction catalyzed by DHFR is essential in maintaining the intercellular pool of THF, which is required for the anabolism of purine nucleotides and some amino acids, thus making the enzyme an important drug target. The DHFR catalyzed chemistry (a C-H→C hydride transfer) occurs at the picosecond-femtosecond time scale.7,8 This step is much faster than any measurable rate reported for this enzyme, which makes probing the nature of that step quite challenging. One of the experimental probes that can be used to examine that step is the temperature dependence of the intrinsic kinetic isotope effects (KIEs). This probe has been applied to the wild type (WT)9 and mutant10–15 forms of ecDHFR. Theoretical studies have suggested that the dominant factor in determining the temperature dependence of the KIEs is the distribution of the hydride donor and acceptor distance (DAD).16–20 These calculations supported a theme by which motions of the protein scaffold reorganize the active site to achieve a DAD conductive to hydride tunneling. In WT ecDHFR,9 this reorganization results in a short and narrow distribution of DADs, associated with very fast sampling of all DADs at the tunneling ready state (TRS).4 In such a case, no thermal activation is required to bring the system to a DAD short enough to enable transfer of the heavier isotope, resulting in temperature-independent intrinsic KIEs.4,9 Disruption of the optimized TRS in some ecDHFR mutants results in poor reorganization with broadly distributed DADs resulting in temperature-dependent KIEs.10–15
Recently, Schramm and coworkers have developed an experimental strategy utilizing mass-modulated “heavy enzymes” in which all amino acids are labeled with 13C, 15N, and nonexchangeable 2H.21 They suggested that these heavy enzymes are “Born-Oppenheimer enzymes” because they expected that the mass modulation would not alter the protein’s electronic potential surface or its structure; it would only result in slower vibrations. If those motions in the native (light) enzyme were coupled to the bond activation, the coupling should be disturbed in the heavy enzyme, leading to altered chemical step relative to the native enzyme. This methodology was subsequently used to study several enzymes including ecDHFR.22–27 Steady state and single turnover measurements of native and heavy ecDHFR show reduced rate constants for the heavy enzyme, but had no effect on the intrinsic KIEs at the physiological temperature range.23,27 In ecDHFR these two rate constants do not reflect the chemical step but steps that precede or succeed the chemistry.9,28 Consequently, it became apparent that increasing the mass of the protein affects other protein functions not related to the chemical step (e.g., ligand binding and release).
Quantum mechanical/molecular mechanical (QM/MM) calculations suggested equal barrier heights and tunneling contributions towards the chemical step in both native and heavy ecDHFRs.23 Subsequent studies of the temperature dependence of the intrinsic KIEs of heavy ecDHFR showed that isotopic substitution of the protein does not affect the intrinsic KIEs for the hydride transfer at physiological temperatures (25–45 °C), but does lead to temperature dependent intrinsic KIEs at lower temperatures.27 In addition, the altered mass of the heavy ecDHFR affected the ground state conformational ensemble that determines protein-ligand interactions, as was evident in the larger Kd values for substrate binding of the heavy enzyme measured by stopped-flow fluorescence spectroscopy.27
Comparison of bacterial and human DHFRs drew the attention of the community, as phylogenetic analysis of DHFR sequences in organisms ranging from bacteria to human identified two key insertion sites along the evolutionary path of DHFR that were predicted to influence the motions of the protein. These sites are highlighted in yellow in the human enzyme (hsDHFR), Figure 1.10,29 The first is a four-amino acid-insert (62-PEKN-65) that appeared 499 million years ago and is conserved in higher organisms.29 A second evolutionarily significant divergence occurred 361 million years ago in the M20 loop of the enzyme and consists of a polyproline sequence (24-PWPP-27).29 The roles of these two insertions have been studied extensively by both computations and experiments,10,25,29–31 and it has been concluded that only the second insertion alters the chemical step of the reaction; however, this disturbance is alleviated by the first evolutionary insertion.
Figure 1.
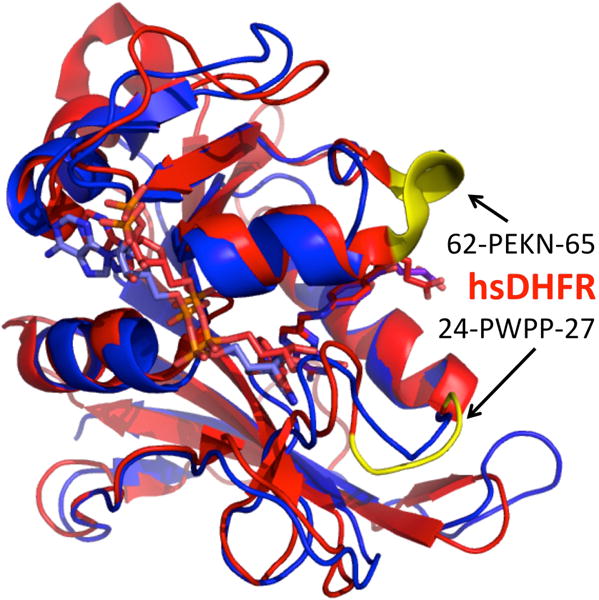
Superposition of hsDHFR (blue; PDB ID 4M6K) and ecDHFR (red; PDB ID 1RX2) bound to NADP+ and folate. The ligands are shown as sticks; hsDHFR residues 24-PWPP-27 and 62-PEKN-65 are highlighted in yellow.
The main goal of the current work is to test computational studies that predicted significant differences between ecDHFR and hsDHFR in terms of in the functional dynamics.7,8 Those QM/MM studies used transition path sampling (TPS), and suggested that fast dynamics (protein promoting vibrations, PPV, in the language of those researchers) are coupled to the CH→C hydride transfer in the human enzyme (hsDHFR), but not to the same transfer in the E. coli enzyme (ecDHFR). The TPS calculation for hsDHFR suggested that compressive motion between the active site residues I17 and P24, results in a decrease in the DAD for hydride transfer, and that this motion occurs on the same time scale as barrier crossing does (354–361 femtoseconds). Similar calculations for ecDHFR predicted that the active site is too loosely packed to support a PPV between the analogous residues I14 and P21.7,8 Those studies suggested that comparative examination of native and heavy ecDHFR and hsDHFR would serve as a critical test of these predictions. Consequently, the current study was designed to test the proposed role of fast dynamics in hsDHFR and the implication that heavy hsDHFR would alter these dynamics, rendering its coupling to the reorganization of the activated state (TRS). Steady-state kinetic parameters, product association and dissociation rate constants, and intrinsic KIEs were measured for hsDHFR and its isotopically-enriched heavy form (heavy hsDHFR). The findings suggest that the heavy hsDHFR affects steps other than the chemical step of hydride transfer, and the altered femto- to picosecond dynamics of hsDHFR do not in fact alter the chemical step under study. The findings and their comparison to studies of the ecDHFR are discussed below.
MATERIALS AND METHODS
Materials
All chemicals were reagent grade and used as purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise indicated. [Carbonyl-14C]-nicotinamide was purchased from Moravek, while [1-3H]-glucose was purchased from Perkin-Elmer. Glucose dehydrogenase from Bacillus megaterium was purchased from Affymetrix/USB. [Carbonyl-14C]-NADPH, 4R-[carbonyl-14C, 4-2H]-NADPH, 4R-3H-NADPH and DHF were synthesized and stored as per previously published protocols.32–38
Preparation of Native and Heavy hsDHFRs
The plasmid pET22a harboring the gene encoding for hsDHFR was a generous gift from Peter E. Wright of the Scripps Institute (La Jolla, CA). The enzyme was expressed and purified as described for ecDHFR39 with the following modifications. The culture medium contained 2 mM folate and a final concentration of 0.5 mM folate was added to the buffer used to lyse the cells based on a previous report showing that this improves the stability of hsDHFR.40 The isotopically-enriched heavy hsDHFR was expressed in M9 minimal medium in 2H2O supplemented with [U-13C6,1,2,3,4,5,6,6-2H7]-glucose and [15N]NH4Cl and purified as described above in buffered solutions prepared in 1H2O. Mass modulation was confirmed by MALDI-TOF mass spectrometry in 20 mM ammonium acetate. The enzymes were flash frozen in liquid N2 after purification and stored at −80 °C until use.
Steady-State Kinetics
The steady state kinetic parameters of the enzymes were determined in 50 mM MTEN buffer (50 mM MES, 25 mM Tris, 25 mM ethanolamine, and 100 mM sodium chloride) at pH 7.65 and 25 °C as described previously.41 Briefly, initial rates were determined with enzyme concentrations between 50 pM and 140 pM (from at least two independent enzyme preparations) by following the decrease in absorbance of NADPH at 340 nm. The kinetic parameters were determined by varying the concentration of DHF (0.1–50 μM) or NADPH (0.3–50 μM) at 100 μM of the other substrate. The initial rate vs. the substrate concentration was fitted to the Michaelis-Menten equation to determine the kinetic parameters. The results are reported as the average of two independent experiments carried out on different preparations of the enzyme to ensure that the differences observed are not artifacts resulting from prep-to-prep variations.
Products’ Association and Dissociation Rate Constants
Product association and dissociation rate constants were determined as described in refs 28 and 42. In short, the quenching of protein fluorescence that occurs upon formation of the ternary complex of hsDHFR with both products was monitored. The enzyme was pre-incubated with a saturating concentration of one ligand and mixed with varying concentrations of the second ligand in an Applied Photophysics SX20 stopped-flow spectrophotometer. To prevent oxidation of THF by atmospheric oxygen all solutions were made anaerobic through incubation with glucose/glucose oxidase as described in ref 43. The enzyme samples were made anaerobic in a tonometer through 25 cycles of vacuuming and flushing with ultra-pure argon before mounting onto the stopped flow and ligand solutions were made anaerobic by flushing the samples with argon for at least 20 minutes. Experiments were carried out using 0.2 μM enzyme pre-incubated with 1.15 mM NADPH or 25 μM THF product (i.e., 500 × Kd42), and initiated by mixing 1–32 μM of the other product (all concentrations are final, i.e., after mixing). Samples were excited at 280 nm and the protein fluorescence at 330 nm was measured. The fluorescence quenching was fitted to a single exponential function and the slope of these rates as a function of concentration yielded the rate constant.42 All measurements were carried out in triplicate, in 50 mM MTEN buffer pH 7.65 and 25 °C.
Determination of Intrinsic KIEs
The intrinsic KIEs of native and heavy hsDHFR were measured using the competitive method as described previously for ecDHFR.9,15 Briefly, KIEs were measured using mixtures of NADPH labeled with either H or T at the 4R position and unlabeled DHF at the desired temperature in 50 mM MTEN buffer at pH 9.0. The reaction was initiated with the addition of either native or heavy hsDHFR. A remote carbon at the carbonyl position of the nicotinamide ring of the cofactor was labeled by 14C to serve as a tracer for the conversion of protiated or deuterated NADPH (for H/T and D/T measurements, respectively) to product (NADP+). Reactions were quenched at different time points by the addition of methotrexate and the depletion of tritium in the product was monitored as a function of fraction conversion in order to calculate the observed KIE on the second order rate constant kcat/KM. The observed H/T and D/T KIEs were used to calculate the intrinsic KIEs using a numerical solution to the modified Northrop equation (Eq 1):38
| (1) |
where T(V/K)Hobs and T(V/K)Dobs are the observed H/T and D/T KIEs (also known as T(kcat/KM)obs), respectively, and kH/kT is the intrinsic H/T KIE. The isotope effect on the activation parameters for the intrinsic KIEs was calculated by a non-linear fit of the data to the Arrhenius equation for intrinsic KIEs (Eq 2):
| (2) |
where kl and kh are the rate constants for light and heavy isotopes, respectively, Al/Ah is the isotope effect on the Arrhenius pre-exponential factor, ΔEa is the difference in energy of activation between the two isotopologues, R is the gas constant and T is the absolute temperature.
Calculation of the Kinetic Commitment Factor (Cf)
The observed KIE measured in competitive experiments is usually smaller than the intrinsic value; this is due to the kinetic commitment factors on V/K resulting from steps other than the isotopically sensitive bond cleavage. As described in detail elsewhere44,45 Eq 3 relates the observed KIE to the intrinsic value on bond cleavage:
| (3) |
where Tkhyd is the intrinsic KIE on bond cleavage, EIE is the equilibrium isotope effect and Cf and Cr are the forward and reverse commitment factors, respectively. For ecDHFR and hsDHFR Cr is negligible because it is practically irreversible under the experimental conditions used here, in which the rate constant for the forward reaction is more than three orders of magnitude greater than the rate constant for the reverse reaction.42 Additionally, the concentration of product THF was kept close to zero by conducting the reaction aerobically (THF is extremely air sensitive). Therefore in this case Eq 3 can be simplified to Eq 4:
| (4) |
Eq 4 can therefore be rearranged to calculate Cf from the observed and intrinsic KIEs obtained from Eq 1.
RESULTS AND DISCUSSION
QM/MM TPS calculations suggested that the protein dynamics (protein promoting vibrations, PPVs) are coupled to the C-H→C hydride transfer step in human DHFR (hsDHFR) but not in the bacterial counterpart (ecDHFR).7,8 Consequently, those calculations suggested that heavy DHFR would affect the chemical step in hsDHFR but not in ecDHFR. In accordance with the Born-Oppenheimer enzyme assumption mentioned above, those calculations also assumed that only the chemical step would be affected (due to disruption of the coupling of the PPV to the reaction coordinate), and that the ensemble of protein conformations (i.e., the protein’s structure), its affinity to ligands, and other parameters would not be affected. That last assumption is very general, and broadly used in both experiments and calculations involving heavy enzymes.22,23,25,46 To begin testing these predictions and assumptions we compared the nature of the chemical step and that of other kinetic steps in native and heavy hsDHFR.
First, native and heavy hsDHFR were produced and their molecular weights were determined to be 21.3 and 23.7 kDa, respectively, by mass spectrometry (Figure S2). This corresponds to an ~11% mass increase in the heavy enzyme, which agrees well with the theoretical mass increase of ~13% for complete labeling with 13C, 15N, and 2H considering that the enzyme was purified in H2O to exchange ionizible protons. Then various kinetic steps of both enzymes were compared to each other and to native and heavy ecDHFRs.
The chemical step was examined via measurements of the temperature dependence of the intrinsic KIEs
The heavy enzyme’s intrinsic KIEs were identical to those of the native hsDHFR within experimental error (Figure 2A and Table 1). This finding indicates that the femto- to picoscond motions of hsDHFR that were altered by the mass modulation did not alter the DAD sampling frequency, which does not accord with the prediction of the TPS calculations. A possible explanation would be that originally, the notion that heavy enzymes would alter dynamics coupled to the reaction coordinate was influenced by a mass difference close to 100 % (e.g., H/D) and vibrational frequencies at the femtosecond time scale (e.g., C-H stretch of ~3000 cm−1 going to 2,100 for C–D). However, the mass modulation for hsDHFR was only ~11 %, and the DAD sampling, the frequency is only expected to be at the picosecond time scale (40 – 200 cm−1).47–49 It thus likely that the mass modulation of hsDHFR is too small to significantly alter such low frequency (i.e., less than a few wavenumber modulation) and thus does not affect the distribution of DADs at the TRS. Also, the TPS calculations in refs 7,8 include non-statistical dynamic motions, which may not manifest in the temperature dependence of the intrinsic KIE. The apparent contradiction between the experimental results and the calculations are therefore likely to be the result of the two methods examining a different phenomena.
Figure 2.
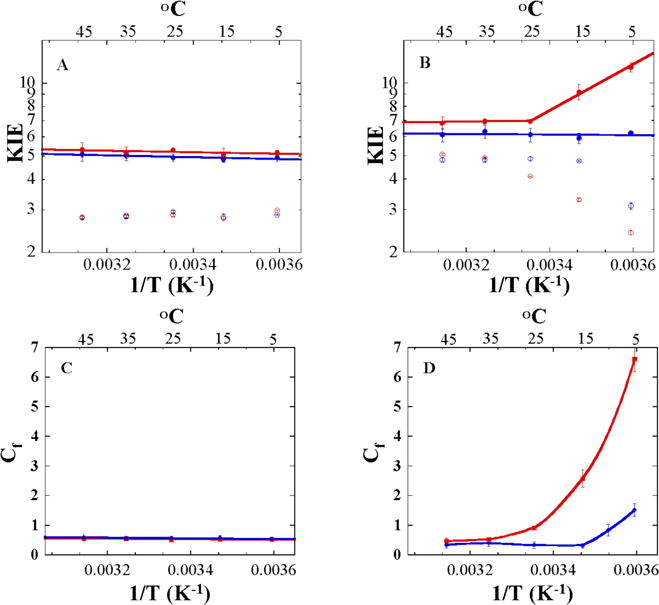
Comparison of the Arrhenius plots of the KIEs and commitment factors (Cf) of native (blue) and heavy (red) DHFRs measured in competitive experiments at pH 9.0. Shown are the observed (open circles) and intrinsic (closed circles) H/T KIEs for hsDHFR (Panel A) and ecDHFR (Panel B). The lines represent the non-linear regression to the Arrhenius equation for KIEs (Eq 2). Arrhenius plot of Cf on kcat/KM for hsDHFR (Panel C) and ecDHFR (Panel D). Cf values were calculated as described in the Materials and Methods section. The lines are an interpolation of the data and do not represent an analytical fitting. Data for ecDHFR are taken from ref 27 with permission from the American Chemical Society.
Table 1.
Comparative isotope effects for native and heavy hsDHFR at pH 9.0
| DHFR | AH/AT | ΔEa(T-H) kcal/mol |
DADavga (Å) |
|---|---|---|---|
| native | 6.6 ± 0.8 | −0.1 ± 0.1 | 3.048 ± 0.002 |
| heavy | 6.6 ± 1.1 | −0.1 ± 0.1 | 3.056 ± 0.007 |
From a fit of the data to the model described in ref 20.
It is also interesting to compare the KIEs of ecDHFR and hsDHFR. As is apparent in Figure 2A, the intrinsic KIEs of the native hsDHFR are as temperature-independent as those of ecDHFR (Figure 2B). This finding is not trivial given the fact that the enzymes share only a 26 % identity in their amino acid sequences (Figure S1 in the SI), and also given the predicted differences in the way they catalyze the isotopically sensitive C-H→C transfer.8 However, the finding accords well with earlier experiments on humanized ecDHFR, which has the two critical insertions (yellow in Figure 1);10 these experiments concluded that temperature-independent KIEs (i.e., accurate TRS) have been preserved along the evolutionary line. The KIEs of hsDHFR are slightly lower than the bacterial enzyme’s, suggesting a slightly shorter average DAD.4,18,20 The intrinsic KIEs can be fitted to a phenomenological activated-tunneling model (referred to as a activated-tunneling, environmentally-coupled-tunneling, or Marcus-like model).20 A comparison of average DADs from this fit between ecDHFR20 and hsDHFR (this work) indicates a shorter DADavg for hsDHFR (0.010 ± 0.005 Å). While small, this difference is significant within the experimental errors, as is obvious from the differences in the magnitude of the KIEs between those enzymes in Figure 2.
Rate constants and product dissociation rates were measured using steady state and pre-steady state kinetics
The steady-state kinetic parameters kcat, kcat/KM, and KM for native and heavy hsDHFR were determined at pH 7.65 and 25 °C, and are presented in Table 2. The kinetic parameters for the native enzyme were similar to those reported previously.41 The KM values for the heavy hsDHFR are similar to that of the native enzyme, but the turnover number (kcat) and the kcat/Km value for NADPH with the heavy hsDHFR are significantly slower for the heavy hsDHFR (Table 2). Early studies of hsDHFR found lower KIEs on kcat than on the burst rate, and faster product release than kcat, suggesting that conformational changes in the enzyme-product complex or product release might be partly rate-limiting for kcat.42,50 In order to test the effect of isotopically-labeled protein on product release, we directly measured the association and dissociation rate constants for the binding and release of both THF and NADP+ for both native and heavy hsDHFR, and the findings are summarized in Table 2.
Table 2.
Steady-state kinetic parameters and product binding kinetics of native (l-DHFR) and heavy (h-DHFR) hsDHFR at pH 7.65 and 25 °Ca,b
| l-DHFR | h-DHFR | h-KIE | |
|---|---|---|---|
| kcatDHF, (s−1) | 14.03 ± 0.02 | 9.77 ± 0.12 | 1.44 ± 0.02 |
| KmDHF, (μM) | 0.29 ± 0.05 | 0.27 ± 0.09 | 1.07 ± 0.40 |
| (kcat/Km)DHF × 107 (M−1s−1) | 4.84 ± 0.83 | 3.62 ± 1.20 | 1.34 ± 0.50 |
| konTHF × 10−6(M−1s−1) | 38.7 ± 1.8 | 38.5 ± 0.5 | 1.01 ± 0.05 |
| koffTHF, (s−1) | 110 ± 10 | 80 ± 5 | 1.38 ± 0.13 |
|
| |||
| kcatNADPH, (s−1) | 14.10 ± 0.01 | 9.75 ± 0.11 | 1.45 ± 0.02 |
| KmNADPH, (μM) | 0.36 ± 0.05 | 0.34 ± 0.07 | 1.06 ± 0.26 |
| (kcat/Km)NADPH × 107 (M−1s−1) | 3.92 ± 0.54 | 2.87 ± 0.59 | 1.37 ± 0.34 |
| konNADP × 10−6 (M−1s−1) | 1.19 ± 0.05 | 1.26 ± 0.05 | 0.94 ± 0.06 |
| koffNADP, (s−1) | 140 ± 1 | 140 ± 1 | 1.00 ± 0.01 |
kcatDHF and KmDHF are the turnover number and Michaelis constant with DHF as substrate measured in the presence of 100 μM NADPH and varying concentrations of DHF
kcatNADPH and KmNADPH are the turnover number and Michaelis constant with NADPH as substrate, measured in the presence of 100 μM DHF and varying concentrations of NADPH.
h-KIE is the heavy enzyme KIE, calculated by taking the ratio between the kinetic parameter of l-DHFR and the same parameter of h-DHFR.
Apparently, neither the rate constants for the release of the product (koff) NADP+ nor kon for the product THF were affected by the isotopic labeling of the heavy enzyme (h-KIE close to unity). The THF release (koff), on the other hand, was slower, with h-KIE of 1.38 ± 0.13, which is similar to that measured for kcat (1.44 ± 0.02). Interestingly, the rate constant for NADP+ release from the enzyme-THF-NADP+ complex was not affected by the isotopic labeling of the enzyme (Table 2), nor was the release of the two substrates (NADPH and DHF), as is evident in the lack of heavy-enzyme KIE on commitment to catalysis on the second order rate constant V/K (Cf, Figure 2C).
Taken together, the results indicate that the isotopic substitution in heavy hsDHFR altered both the conformational changes of the enzyme-product complex prior to product release, and the protein-THF interactions affecting the release of that product. The decrease in the koffTHF and the two steady-state rate constants for the heavy enzyme could be due to changes in the geometry and electrostatics of the protein. Such effects may result from the substitution of nonexchangeable protons with deuterium, probably because C–D has a smaller dipole moment than C–H, altering the electronic potential surface of the protein.51,52 At any rate, this finding is not in accordance with the concepts behind the term “Born-Oppenheimer enzyme” coined in ref 21. The lack of any measurable effect of the protein mass-modulation on the nature of the C-H→C hydride transfer step (Figure 2A and Table 1) may suggest a more complex structure-dynamic-functional relationship than that proposed before. The current findings suggest that further investigation by both experimentalists and theoreticians is needed to obtain a model explaining both experimental and computational findings.
It also would have been interesting to examine whether the mass modulation of the hsDHFR affected its single-turnover rates. However, as has been the case in other such investigations of the native WT hsDHFR,53 our attempts to measure the pre-steady state rate of native and heavy hsDHFR were unsuccessful because the reaction was completed within the mixing time of the stopped-flow spectrophotometer (2 ms).
CONCLUSIONS
The use of heavy (isotopically-labeled) enzymes to examine protein motions that are coupled to its catalyzed chemistry is at the center of a contemporary debate. The current study, together with studies of heavy ecDHFR23,25,27 and other heavy enzymes21,22,24,26 demonstrate that isotopic enrichment of a protein can affect the catalyzed reaction in a variety of ways, not merely by altering the chemical step. For purine nucleoside phosphorylase (PNP), for instance, mass modulation resulted in differences in the intrinsic KIEs of the light and heavy enzymes,21 which the authors interpreted as a decoupling of fast femtosecond bond vibrations being part of the reaction coordinate. For PNP the steady-state kinetic parameters were not affected by isotopic enrichment, which suggest that electrostatic potential surface of the protein motions has not been altered by the mass modulation of the protein, in accordance with the “Born-Oppenheimer enzyme” suggestion. Altered KIEs and/or their temperature dependence were also reported for alanine racemase with either D- or L-alanine substrate,26 and for pentaerythritol tetranitrate reductase,24 although both reported observed rather than intrinsic KIEs.
Here we tested predictions from TPS calculations that suggested that protein dynamics on a fast time scale (which should be modulated in heavy enzymes) are coupled to the chemical step in human DHFR (hsDHFR) but not in the bacterial one (ecDHFR).7,8 Those studies suggested that the heavy enzyme could be used to test their predictions. However, the experimental findings reported above indicate that mass modulation of the hsDHFR has no detectable effect on the intrinsic KIEs or their temperature dependence. The lack of mass-modulation effect on the intrinsic KIEs suggests that fast protein dynamics either are insensitive to the mass modulation of the protein. More specifically, the PPV that ref 8 predicted would promote the C-H→C hydride transfer step might not be sensitive to isotopic substitution of the substrate; or might not be significantly affected by the protein’s mass modulation (11%). The last explanation is quite reasonable given that 11% mass difference for native DAD fluctuating at ca. 100 cm−1 47,48 may result in rather a small reduction in sampling frequency for the heavy enzyme (ca. 90 cm−1).
While the heavy hsDHFR did not affect the catalyzed C-H→C transfer step, interactions between the enzyme and THF and conformational rearrangements of the enzymatic complex appear to be altered, as is indicated by slower product release (koffTHF) and slower first and second order rate constants (kcat and kcat/KM) with h-KIEs of 1.3 – 1.5. It appears that mass modulation of enzymes have complex effects on function that depend on the specific protein architecture and dynamics. It also appears that a variety of chemical and physical properties can be altered by the isotopic labeling of the protein. In summary, the term “Born-Oppenheimer enzyme” was coined in ref 21 to suggest that heavy enzymes (i.e., isotopically labeled protein) have altered vibrations but the same electronic potential surface. The current study suggests that that term might not describe heavy hsDHFR and several other enzymes accurately, because it appears that the mass modulation also affects the protein conformational ensemble, protein-ligand interactions, and protein electrostatics.
Supplementary Material
Acknowledgments
The authors thank Professors Peter E. Wright for providing the plasmid encoding for human dihydrofolate reductase and Steven D. Schwartz for providing ref 8 before publication.
FUNDING SOURCES: This work was supported by NIH (R01GM65368) and NSF (CHE-1149023) to AK.
ABBREVIATIONS
- DHFR
dihydrofolate reductase reductase
- KIEs
kinetic isotope effects
- DAD
donor and acceptor distance
- TRS
tunneling ready state
- NADPH
nicotinamide adenine diphosphate
- DHF
7,8-dihydrofolate
- THF
5,6,7,8-tetrahydrofolate
- WT
wild-type
- ecDHFR
Escherichia coli dihydrofolate reductase
- hsDHFR
human dihydrofolate reductase
Footnotes
SUPPORTING INFORMATION AVALIBLE
Tables showing the observed and intrinsic H/T and D/T KIEs, a sequence alignment of human and E. coli DHFR, the kinetic curves for product binding to light and heavy human DHFR and the mass spectra of native and heavy hsDHFR are provided. This information is available free of charge via the Internet at http://pubs.acs.org/.
While this paper was under review a study of heavy ecDHFR was published, which questioned the validity of the Northrop method used to calculate intrinsic KIE for heavy ecDHFR at temperatures under 25 °C.54 The main error in ref 54 is that instead of using the reciprocal of the measured KIE used in the Northrop equation (Eq 1 here and S12 in ref 54), they derived normal KIEs, which lead to the commitments for hydrogen and deuterium (Eqs S2–S5 in ref 54), but not for tritium. Consequently, their derivations did not lead to the necessary isolation of the commitment for tritium (CfT) needed in the Northrop procedure with H/T and D/T measurements. Running into a dead-end, the authors had to invoke a new “first assumption” to indirectly assess C for T. However, when the Northrop equations is derived correctly, that assumption is not needed and the commitment factor for tritium can be derived directly.44 That false assumption is the basis for the criticism of the Northrop method in ref 54, although the authors fail to suggest why it is only a problem for heavy ecDHFR under 25 °C, but is correct above that temperature or for the light ecDHFR (whose intrinsic KIE calculated from the Northrop method they used to suggest their assessment of intrinsic KIEs is valid).
AUTHOR CONTRIBUTIONS
AK led and oversaw the studies, KF conducted the experiments, PS and AL expressed isotopically-labeled human dihydrofolate reductase. The manuscript was written through contributions of all authors who have given approval to the final version of the manuscript.
References
- 1.Glowacki DR, Harvey JN, Mulholland AJ. Taking Ockham’s razor to enzyme dynamics and catalysis. Nat Chem. 2012;4:169–176. doi: 10.1038/nchem.1244. [DOI] [PubMed] [Google Scholar]
- 2.Kamerlin SCL, Warshel A. At the dawn of the 21st century: Is dynamics the missing link for understanding enzyme catalysis? Proteins: Structure, Function, and Bioinformatics. 2010;78:1339–1375. doi: 10.1002/prot.22654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klinman JP. Dynamically Achieved Active Site Precision in Enzyme Catalysis. Acc Chem Res. 2015;48:449–456. doi: 10.1021/ar5003347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kohen A. Role of Dynamics in Enzyme Catalysis: Substantial vs. Semantic Controversies. Acc Chem Res. 2015;48:466–473. doi: 10.1021/ar500322s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanoian P, Liu CT, Hammes-Schiffer S, Benkovic S. Perspectives on electrostatics and conformational motions in enzyme catalysis. Acc Chem Res. 2015;48:482–489. doi: 10.1021/ar500390e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Singh P, Abeysinghe T, Kohen A. Linking protein motion to enzyme catalysis. Molecules. 2015;20:1192–1209. doi: 10.3390/molecules20011192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dametto M, Antoniou D, Schwartz SD. Barrier Crossing in Dihydrofolate Reductasedoes not involve a rate-promoting vibration. Mol Phys. 2012;110:531–536. doi: 10.1080/00268976.2012.655337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Masterson JE, Schwartz SD. Evolution alters the enzymatic reaction coordinate of dihydrofolate reductase. J Phys Chem B. 2015;119:989–996. doi: 10.1021/jp506373q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sikorski RS, Wang L, Markham KA, Rajagopalan PT, Benkovic SJ, Kohen A. Tunneling and coupled motion in the Escherichia coli dihydrofolate reductase catalysis. J Am Chem Soc. 2004;126:4778–4779. doi: 10.1021/ja031683w. [DOI] [PubMed] [Google Scholar]
- 10.Francis K, Stojković V, Kohen A. Preservation of Protein Dynamics in Dihydrofolate Reductase Evolution. J Biol Chem. 2013;288:35961–35968. doi: 10.1074/jbc.M113.507632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu CT, Francis K, Layfield JP, Huang X, Hammes-Schiffer S, Kohen A, Benkovic SJ. Escherichia coli dihydrofolate reductase catalyzed proton and hydride transfers: Temporal order and the roles of Asp27 and Tyr100. Proc Natl Acad Sci USA. 2014;111:18231–18236. doi: 10.1073/pnas.1415940111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singh P, Francis K, Kohen A. Network of Remote and Local Protein Dynamics in Dihydrofolate Reductase Catalysis. ACS Catalysis. 2015;5:3067–3073. doi: 10.1021/acscatal.5b00331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Singh P, Sen A, Francis K, Kohen A. Extension and Limits of the Network of Coupled Motions Correlated to Hydride Transfer in Dihydrofolate Reductase. J Am Chem Soc. 2014;136:2575–2582. doi: 10.1021/ja411998h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stojković V, Perissinotti LL, Willmer D, Benkovic SJ, Kohen A. Effects of the donor-acceptor distance and dynamics on hydride tunneling in the dihydrofolate reductase catalyzed reaction. J Am Chem Soc. 2012;134:1738–1745. doi: 10.1021/ja209425w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang L, Goodey NM, Benkovic SJ, Kohen A. Coordinated effects of distal mutations on environmentally coupled tunneling in dihydrofolate reductase. Proc Natl Acad Sci USA. 2006;103:15753–15758. doi: 10.1073/pnas.0606976103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fan Y, Cembran A, Ma S, Gao J. Connecting protein conformational dynamics with catalytic function as illustrated in dihydrofolate reductase. Biochemistry. 2013;52:2036–2049. doi: 10.1021/bi301559q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hatcher E, Soudackov AV, Hammes-Schiffer S. Proton-coupled electron transfer in soybean lipoxygenase: dynamical behavior and temperature dependence of kinetic isotope effects. J Am Chem Soc. 2007;129:187–196. doi: 10.1021/ja0667211. [DOI] [PubMed] [Google Scholar]
- 18.Liu H, Warshel A. Origin of the Temperature Dependence of Isotope Effects in Enzymatic Reactions: The Case of Dihydrofolate Reductase. J Phys Chem B. 2007;111:7852–7861. doi: 10.1021/jp070938f. [DOI] [PubMed] [Google Scholar]
- 19.Pu J, Ma S, Gao J, Truhlar DG. Small Temperature Dependence of the Kinetic Isotope Effect for the Hydride Transfer Reaction Catalyzed by Escherichia coli Dihydrofolate Reductase. The Journal of Physical Chemistry B. 2005;109:8551–8556. doi: 10.1021/jp051184c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roston D, Cheatum CM, Kohen A. Hydrogen Donor-Acceptor Fluctuations from Kinetic Isotope Effects: A Phenomenological Model. Biochemistry. 2012;51:6860–6870. doi: 10.1021/bi300613e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Silva RG, Murkin AS, Schramm VL. Femtosecond dynamics coupled to chemical barrier crossing in a Born-Oppenheimer enzyme. Proc Natl Acad Sci USA. 2011;108:18661–18665. doi: 10.1073/pnas.1114900108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kipp DR, Silva RG, Schramm VL. Mass-dependent bond vibrational dynamics influence catalysis by HIV-1 protease. J Am Chem Soc. 2011;133:19358–19361. doi: 10.1021/ja209391n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luk LY, Javier Ruiz-Pernia J, Dawson WM, Roca M, Loveridge EJ, Glowacki DR, Harvey JN, Mulholland AJ, Tunon I, Moliner V, Allemann RK. Unraveling the role of protein dynamics in dihydrofolate reductase catalysis. Proc Natl Acad Sci U S A. 2013;110:16344–16349. doi: 10.1073/pnas.1312437110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pudney CR, Guerriero A, Baxter NJ, Johannissen LO, Waltho JP, Hay S, Scrutton NS. Fast protein motions are coupled to enzyme H-transfer reactions. Am Chem Soc. 2013;135:2512–2517. doi: 10.1021/ja311277k. [DOI] [PubMed] [Google Scholar]
- 25.Ruiz-Pernia JJ, Luk LY, Garcia-Meseguer R, Marti S, Loveridge EJ, Tunon I, Moliner V, Allemann RK. Increased dynamic effects in a catalytically compromised variant of Escherichia coli dihydrofolate reductase. J Am Chem Soc. 2013;135:18689–18696. doi: 10.1021/ja410519h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Toney MD, Castro JN, Addington TA. Heavy-enzyme kinetic isotope effects on proton transfer in alanine racemase. J Am Chem Soc. 2013;135:2509–2511. doi: 10.1021/ja3101243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Z, Singh P, Czekster CM, Kohen A, Schramm VL. Protein mass-modulated effects in the catalytic mechanism of dihydrofolate reductase: beyond promoting vibrations. J Am Chem Soc. 2014;136:8333–8341. doi: 10.1021/ja501936d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fierke CA, Johnson KA, Benkovic SJ. Construction and evaluation of the kinetic scheme associated with dihydrofolate reductase from Escherichia coli. Biochemistry. 1987;26:4085–4092. doi: 10.1021/bi00387a052. [DOI] [PubMed] [Google Scholar]
- 29.Liu CT, Hanoian P, French JP, Pringle TH, Hammes-Schiffer S, Benkovic SJ. Functional Significance of Evolving Protein Sequence in Dihydrofolate Reductase from Bacteria to Human. Proc Natl Acad Sci USA. 2013;110:10159–10164. doi: 10.1073/pnas.1307130110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bhabha G, Lee J, Ekiert DC, Gam J, Wilson IA, Dyson HJ, Benkovic SJ, Wright PE. A dynamic knockout reveals that conformational fluctuations influence the chemical step of enzyme catalysis. Science. 2011;332:234–238. doi: 10.1126/science.1198542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Loveridge EJ, Behiry EM, Guo J, Allemann RK. Evidence that a ‘dynamic knockout’ in Escherichia coli dihydrofolate reductase does not affect the chemical step of catalysis. Nature Chem. 2012;4:292–297. doi: 10.1038/nchem.1296. [DOI] [PubMed] [Google Scholar]
- 32.Agrawal N, Kohen A. Microscale synthesis of 2-tritiated isopropanol and 4R-tritiated reduced nicotinamide adenine dinucleotide phosphate. Anal Biochem. 2003;322:179–184. doi: 10.1016/j.ab.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 33.Blakley RL. Crystalline Dihydropteroylglutamic Acid. Nature. 1960;188:231–232. [Google Scholar]
- 34.Markham KA, Sikorski RS, Kohen A. Purification, analysis, and preservation of reduced nicotinamide adenine dinucleotide 2′-phosphate. Anal Biochem. 2004;322:26–32. doi: 10.1016/j.ab.2003.07.010. [DOI] [PubMed] [Google Scholar]
- 35.Markham KA, Sikorski RS, Kohen A. Synthesis and utility of 14C-labeled nicotinamide cofactors. Anal Biochem. 2004;325:62–67. doi: 10.1016/j.ab.2003.10.027. [DOI] [PubMed] [Google Scholar]
- 36.McCracken JA, Wang L, Kohen A. Synthesis of R and S tritiated reduced beta-nicotinamide adenine dinucleotide 2′ phosphate. Anal Biochem. 2003;324:131–136. doi: 10.1016/j.ab.2003.09.025. [DOI] [PubMed] [Google Scholar]
- 37.Sen A, Stojkovic V, Kohen A. Synthesis of radiolabeled nicotinamide cofactors from labeled pyridines: versatile probes for enzyme kinetics. Anal Biochem. 2012;430:123–129. doi: 10.1016/j.ab.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sen A, Yahashiri A, Kohen A. Triple isotopic labeling and kinetic isotope effects: exposing H-transfer steps in enzymatic systems. Biochemistry. 2011;50:6462–6468. doi: 10.1021/bi2003873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cameron CE, Benkovic SJ. Evidence for a functional role of the dynamics of glycine-121 of Escherichia coli dihydrofolate reductase obtained from kinetic analysis of a site-directed mutant. Biochemistry. 1997;36:15792–15800. doi: 10.1021/bi9716231. [DOI] [PubMed] [Google Scholar]
- 40.Bhabha G, Tuttle L, Martinez-Yamout MA, Wright PE. Identification of endogenous ligands bound to bacterially expressed human and E. coli dihydrofolate reductase by 2D NMR. FEBS Lett. 2011;585:3528–3532. doi: 10.1016/j.febslet.2011.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lewis WS, Cody V, Galitsky N, Luft JR, Pangborn W, Chunduru SK, Spencer HT, Appleman JR, Blakley RL. Methotrexate-resistant variants of human dihydrofolate reductase with substitutions of leucine 22. Kinetics, crystallography, and potential as selectable markers. J Biol Chem. 1995;270:5057–5064. doi: 10.1074/jbc.270.10.5057. [DOI] [PubMed] [Google Scholar]
- 42.Appleman JR, Beard WA, Delcamp TJ, Prendergast NJ, Freisheim JH, Blakley RL. Unusual transient- and steady-state kinetic behavior is predicted by the kinetic scheme operational for recombinant human dihydrofolate reductase. J Biol Chem. 1990;265:2740–2748. [PubMed] [Google Scholar]
- 43.Quaye O, Lountos GT, Fan F, Orville AM, Gadda G. Role of Glu312 in binding and positioning of the substrate for the hydride transfer reaction in choline oxidase. Biochemistry. 2008;47:243–256. doi: 10.1021/bi7017943. [DOI] [PubMed] [Google Scholar]
- 44.Northrop DB. Intrinsic isotope effects in enzyme catalyzed reactions. In: Cook PF, editor. Enzyme mechanism from isotope effects. CRC Press; Boca Raton, Fl.: 1991. pp. 181–202. [Google Scholar]
- 45.Cook PF, Cleland WW. Enzyme Kinetics and Mechanism. Taylor and Francis Group LLC; New York, NY: 2007. pp. 253–324. [Google Scholar]
- 46.Antoniou D, Ge X, Schramm VL, Schwartz SD. Mass Modulation of Protein Dynamics Associated with Barrier Crossing in Purine Nucleoside Phosphorylase. J Phys Chem Lett. 2012;3:3538–3544. doi: 10.1021/jz301670s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Basner JE, Schwartz SD. Donor-Acceptor Distance and Protein Promoting Vibration Coupling to Hydride Transfer: A Possible Mechanism for Kinetic Control in Isozymes of Human Lactate Dehydrogenase. J Phys Chem B. 2004;108:444–451. [Google Scholar]
- 48.Hatcher E, Soudackov AV, Hammes-Schiffer S. Proton-coupled electron transfer in soybean lipoxygenase. J Am Chem Soc. 2004;126:5763–5775. doi: 10.1021/ja039606o. [DOI] [PubMed] [Google Scholar]
- 49.Cheatum CM, Kohen A. Relationship between femtosecond-picosecond dynamics to enzyme catalyzed H-transfer. Top Curr Chem. 2013;337:1–39. doi: 10.1007/128_2012_407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Beard WA, Appleman JR, Delcamp TJ, Freisheim JH, Blakley RL. Hydride transfer by dihydrofolate reductase. Causes and consequences of the wide range of rates exhibited by bacterial and vertebrate enzymes. J Biol Chem. 1989;264:9391–9399. [PubMed] [Google Scholar]
- 51.Wolfsberg M, Van Hook WA, Paneth P, Rebelo LPN. Isotope Effects in the Chemical, Geological and Bio Sciences. Springer; Berlin: 2010. pp. 389–412. [Google Scholar]
- 52.Zhao C, Parrish RM, Smith MD, Pellechia PJ, Sherrill CD, Shimizu KD. Do deuteriums form stronger CH-pi interactions? J Am Chem Soc. 2012;134:14306–14309. doi: 10.1021/ja305788p. [DOI] [PubMed] [Google Scholar]
- 53.Beard WA, Appleman JR, Delcamp TJ, Freisheim JH, Blakley RL. Hydride transfer by dihydrofolate reductase. Causes and consequences of the wide range of rates exhibited by bacterial and vertebrate enzymes. J Biol Chem. 1989;264:9391–9399. [PubMed] [Google Scholar]
- 54.Wang Z, Antoniou D, Schwartz SD, Schramm VL. Hydride Transfer in DHFR by Transition Path Sampling, Kinetic Isotope Effects, and Heavy Enzyme Studies. Biochemistry. 2016;55:157–166. doi: 10.1021/acs.biochem.5b01241. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.