

Abstract
Cardiomyopathy is the leading cause of death worldwide. Despite progress in medical treatments, heart transplantation is one of the only current options for those with infarcted heart muscle. Stem cell differentiation technology may afford cell-based therapeutics that may lead to the generation of new, healthy heart muscle cells from undifferentiated stem cells. Our approach is to use small molecules to stimulate stem cell differentiation. Herein, we describe a novel class of 1,5-disubstituted benzimidazoles that induce differentiation of stem cells into cardiac cells. We report on the evaluation in vitro for cardiomyocyte differentiation and describe structure–activity relationship results that led to molecules with drug-like properties. The results of this study show the promise of small molecules to direct stem cell lineage commitment, to probe signaling pathways and to develop compounds for the stimulation of stem cells to repair damaged heart tissue.
Keywords: Cardiomyogenesis, Embryonic stem cells, Benzimidazoles, Cardiomyocytes, High content screen
1. Introduction
More than 5 million Americans are estimated to suffer from heart failure.1 Costs due to hospitalization, therapies and loss of productivity are estimated to be more than $37.2 billion/year.1 Current therapies for heart disease do little to regenerate damaged heart muscle. The adult human heart has a limited regenerative capability but regeneration is too low to compensate for loss due to ischemic or non-ischemic heart disease.2–5 Administration of a small molecule stem cell (SC) differentiation agent to stimulate the generation of heart cells may prove useful for heart repair.6 For example, we have reported on the ability of a class of 1,5-dihydropyridines that inhibit TGF-β signaling and a class of novel Wnt inhibitors to stimulate cardiomyogenesis (CM).7–10 In addition, other groups have reported on small molecule inducers of cardiomyogenesis.11–13 Herein, we describe potent and selective 1,5-disubstituted benzimidazoles that represent a new class of small molecules that instruct stem cell differentiation to cardiomyocytes.
A commercially available library of 13,360 chemically diverse compounds14 was screened in a high content cell-based assay employing mouse embryonic stem cells (mESCs) to identify small molecules that stimulated SC differentiation. The cell-based screen identified four chemically distinct ‘hits’. Of these ‘hits’, two compounds were robust and reproducible inducers of cardiomyogenesis. One of the ‘hits’ was reported previously.8,9 Herein, we focus on the second of these ‘hits’, (i.e., 1-phenyl-5-(4-methylphthalazinyl) aminobenzimidazole, 1) (Fig. 1). The ‘hit’ from the screen (i.e., 1) had poor drug-like properties including large predicted lipophilicity and an unfavorable PSA value (i.e., predicted cLog P = 5.44, PSA = 52.35 Å2)15, unfavorable water solubility (i.e., less than 1 mg/ml) that mitigated against 1 as a drug candidate. Accordingly, elaboration of a more potent and drug-like candidate from the ‘hit’ to improve upon pharmaceutical properties of ‘hit’ 1 was a goal of this work. As shown in Figure 1, molecular dissection of 1 showed that the molecule could be divided into three regions including: a benzimidazole core, (region A), aryl substitution at the 1-position of the benzimidazole core (i.e., region B), and a phthalazine group at the 5-position of the benzimidazole core (i.e., region C). Structure–activity relationship (SAR) studies of derivatives of 1 using Stem Cell Dynamic Medicinal Chemistry8 afforded a series of substituted benzimidazoles that stimulated differentiation of cardiomyocytes from mESCs.
Figure 1.

Screening ‘hit’, compound 1.
2. Results and discussion
2.1. Chemistry
Benzimidazoles 1, 30–48, and 75 were prepared in a sequence of four steps from commercially available 2,4-dinitro-haloarene 2 (Scheme 1). Using a modified procedure from the literature16–18, one equivalent of a desired amine, R1NH2, was combined with 2 at room temperature in tetrahydrofuran to give substituted anilines 3–15. Anilines 3–15 were hydrogenated via palladium on carbon to afford the diamino-aniline intermediate that was used without further purification or characterization. Cyclization to the benzimidazole core 16–29 was accomplished using formic acid in hydrochloric acid19 at 110 °C. The substituent at the 5-amino group was installed either via N-arylation (1, 30–48, 75), reductive amination (49–58) or acylation (59, 60) of intermediates 16–29 (Scheme 1). Benzimidazole 65 incorporated a pyridine in the benzimidazole core and was prepared following similar protocols starting with commercially available 2,4-dinitro-haloarene 61 (Scheme 2). The desired amine was combined with 61 at room temperature in tetrahydrofuran to give intermediate 62. Compound 62 was reduced using sodium dithionite in aqueous ethanol to give the triamine 63.20 Cyclization to the benzimidazole core 64 was accomplished using formic acid in hydrochloric acid19 at 110 °C. Finally, 1-chloro-4-methyl-phthalazine was condensed with 64 to give 65.
Scheme 1.

Synthesis of benzimidazoles 1, 30–48, and 75. Reagents and conditions: (a) R1NH2, THF, rt, 15 h. (b) H2, Pd/C, ethyl acetate, 15 h; (c) formic acid 110 °C, 1 h; (d) R2Cl, iPrOH, 150 °C, 1 h; (e) R3CHO, borane-pyridine complex, ethanol, 25 °C, overnight; (f) R4COCl, TEA, DCM, 25 °C, overnight.
Scheme 2.

Synthesis of 7-N-benzimidazole 65. Reagents and conditions: (a) o-anisidine, THF, 25 °C, 15 h; (b) sodium dithionite, Ethanol/H2O, 70 °C, 1 h; (c) formic acid 110 °C, 1 h; (d) 1-chloro-4-methylphthalazine, iPrOH, 150 °C, 1 h.
The use of cross coupling reactions were used for benzimidazoles that incorporated various 5-amino-pyrimidine (69), -pyrazine (70), -pyridine (71, 72) or -aryl substituents (73, 74, 68), starting with 5-bromobenzimidazoles 67a or 67b (Scheme 3). Compounds 67a and 67b were obtained in three steps starting from 4-bromo-1-fluoro-2-nitrobenzene, 66. Nucleophilic aromatic substitution of 66 with a substituted aniline gave a nitroaniline intermediate that was reduced with sodium dithionite and cyclized to give benzimidazoles 67a and 67b. Aryl and heteroaryl groups (i.e., R7) in compounds 68–74 were installed in good yield using Hartwig–Buchwald cross-coupling conditions.21
Scheme 3.

Synthesis of compounds 68–74 via Hartwig–Buchwald coupling. Reagents and conditions: (a) R1PhNH2, THF, 25 °C, 15 h; (b) sodium dithionite, Ethanol/H2O, 70 °C, 1 h; (c) trimethylorthoformate, cat. p-toluenesulfonic acid, 110 °C, 1 h; (d) R6NH2, Pd(OAc)2, Cs2 CO3, rac-BINAP in toluene or Xantphos in dioxane, 100 °C, 15 h.
In addition to benzimidazoles, we investigated the synthesis and testing of substituted indoles (Scheme 4). The synthesis of substituted indole 78 began with commercially available 5-nitroindole 76 that was coupled with phenylboronic acid under Hartwig–Buchwald conditions22,23 to afford 1-phenyl-indole 77. Compound 78 was prepared in two steps, first reduction of the nitro group of 77 using sodium dithionite, followed by nucleophilic aromatic substitution with 1-chloro-4-methylphthalazine. Other modifications to the benzimidazole core included modifications to the 2-position. This included substitution or replacement of a benzimidazole moiety with a benzotriazole group (Scheme 5). To prepare 2-amino-benzimidazoles, compound 79 was treated with either phosgene iminium chloride to give the 2-dimethylamino-benzimidazole 80 or cyanogen bromide to give the 2-amino-benzimidazole 81.24,25 Hydrogenation of 80 or 81 followed by introduction of the methylphthalazine ring system as above afforded 82 and 83. The benzotriazole analog 84 was obtained when 79 was treated with isoamyl nitrite.26 Hydrogenation of 84 followed by introduction of the methylphthalazine ring system gave 85.
Scheme 4.

Synthesis of indole 78. Reagents and conditions: (a) PhB(OH)2, Cu(OAc)2, DIPEA, DCM, 25 °C, 15 h; (b) sodium dithionite, Ethanol/H2O, 70 °C, 1 h; (c) 1-chloro-4-methylphthalazine, iPrOH, 90 °C, 2 h.
Scheme 5.

Synthesis of 2-aminobenzimidazoles 82 and 83 and benzotriazole 85. Reagents and conditions: (a) phosgene iminium chloride, TEA, DCM, 25 °C, 15 h; (b) cyanogen bromide, TEA, DCM, 25 °C, 15 h; (c) H2, Pd/C, methanol/THF, 25 °C, 4 h; (d) 1-chloro-4-methylphthalazine, iPrOH, 90 °C, 2 h; (e) isoamyl nitrite, chloroform, 25 °C, 1 h.
2.2. Biology
The high content screen used herein was based on an assay previously described.12 mESCs were stably transfected with an enhanced green fluorescent protein (eGFP) construct regulated by a myosin heavy-chain alpha gene promoter (i.e., αMHC-eGFP).12 Early stage mouse cardiomyocytes express high amounts of this myosin subunit necessary for normal function of the cardiac contractile apparatus. Thus, αMHC (Myh6) expressed in mesoderm-derived cardiac muscle from day 10-differentiated mESC cultures was used as a biomarker of differentiated cardiomyocytes (i.e., see Supporting Fig. S1 for a cell fate map). Other biological markers along the cardiomyocyte differentiation pathway were examined. These included; Bra and GSC for the mesendoderm linage; Cer1 and Sox17 for the endoderm linage; and Cdx2 and MesP1 for the mesoderm linage.6,27,28 For the initial screen, compounds were administered to mESCs in fortified media during days 2–6 (see Methods). After day 6, differentiated cells were reseeded and expanded in normal media (Fig. 2A). Fold-change in cardiomyogenesis was determined on day 10 by quantification of αMHC-eGFP fluorescence levels in cell culture. Along with the quantification of αMHC-eGFP fluorescence, additional biological markers were examined with compound treatment from days 1–10.29 After the initial primary screen, the time window of compound administration was restricted to days 2–4 for the SAR studies. This time window corresponded to the time point where ‘hit’ 1 was most potent. Treatment of mESCs with functionally potent benzimidazoles resulted in widespread αMHC-driven eGFP expression within the cell culture well (Fig. 2B). An automated image capture and custom image quantification algorithm was employed in high-content screens to score lineage bias as an indicator of eGFP expression30 (see Methods).
Figure 2.

(A) 13,360 compounds were tested at two concentrations (1 and 5 µg/mL). Mouse ESCs in growth media without Leukemia Inhibitory Factor (LIF) were applied to 384 well microtiter plates as a monolayer on day 0 and compounds were administered on days 2 and 4. On days 6 and 8 the cells were fed growth media. Differentiated cell cultures were fixed on day 10 with paraformaldehyde. (B) Nine field image mosaics from representative wells treated with DMSO (vehicle control; top row) or the benzimidazole 1 (bottom row). Intense fluorescence of eGFP expressed from the αMHC promoter was apparent in the cells treated with 1. Only sparse puncta were apparent in the DMSO-treated cells (center column). Nuclear DAPI staining (left column) indicated the relative density of the culture was similar in the 1- and DMSO-treated conditions. Non-specific red (nsRED) images indicated a measure of broad-spectrum auto fluorescence and were used for subtraction of fluorescent artifacts. There was no significant increase in this non-specific fluorescent signal in 1-treated cells compared to DMSO-treated cells (right column).
Immunohistochemistry for α-Actinin, another marker of the cardiac contractile apparatus, co-localized with αMHC-eGFP expression. This verified that eGFP expression was associated with cardiomyocyte formation. Differentiation of mESCs treated with 1 or vehicle control were monitored for 12 days of development. On day 12, the cultures were fixed in paraformaldehyde and immunostained with mouse monoclonal anti-αActinin followed by a cocktail of 2-(4-amidinophenyl)-1H-indole-6-carboxamidine (DAPI) and anti-mouse Alexa568 (Fig. 3A). DAPI, eGFP, and Alexa568 images were collected and quantified by image analysis. After administration of 6 µM compound 1 to the cell cultures at days 2 and 4, αMHC-eGFP and α-Actinin Alexa568 showed a 20-fold increase over vehicle control (integrated intensity of αMHC-eGFP and α-Actinin-Alexa568 fluorescence in control samples were 4.2% and 5.0%, respectively, of that of 1-treated samples (Fig. 3B). A few α-Actinin foci appeared to lack eGFP, possibly caused by leaching out of eGFP during cell permeabilization for immunostaining (Fig. 3A and C). For this reason, αMHC-eGFP fluorescent intensity likely represented a conservative estimate of cardiomyocyte conversion. As further evidence that the αMHC-eGFP foci contained functional cardiomyocytes, the cells were imaged live at day 10 and the image showed functionally active and coherent contractile cell beating, a property unique to cardiomyocytes (see Supporting movie). Taken together, the increase in αMHC-eGFP expression in mESC cultures by day 10 indicated that compound 1 significantly enhanced cardiomyocyte differentiation.
Figure 3.

αMHC-eGFP cardiomyocyte expression from mESCs treated with 1 was confirmed by fluorescence immunohistochemical staining of cardiomyocyte-specific α-Actinin. (A) DAPI staining (left; gray) shows the relative confluence of the culture at day 10. Widespread αMHC-eGFP expression (center; green) was apparent after treatment with compound 1. Α-Actinin immunostaining (right; red) co-localized with αMHC-eGFP fluorescence. (B) Quantification of αMHC-eGFP and α-Actinin–Alexa568 showed similar trends in response to DMSO vehicle and compound 1 with a 10-fold and 20-fold increase (at 3 µM and 6 µM concentration, respectively) in integrated intensity of eGFP signal over DMSO vehicle control. (C) An overlay of αMHC-eGFP (green) and α-Actinin (red) fluorescence showed co-localized expression of both cardiomyocyte markers in yellow.
2.3. SAR discussion
Although ‘hit’ 1 was moderately potent in the cardiomyogenesis assay, 1 had several drawbacks as a drug-like molecule: compound 1 had poor water solubility and was lipophilic with a cLog P value of 5.44 and a tPSA value of 52.4. The synthesis of analogs of 1 focused on ways to improve the overall potency and water solubility of the molecule. To identify the regions of the molecule that contributed to potency, 1 was divided into three regions for modification: region A, the benzimidazole core; region B, the benzimidazole 1-subsituent; region C, the benzimidazole 5-substituent (Fig. 1). A structure–activity relationship (SAR) of analogs of 1 was developed from results of testing and synthesis of 400 compounds. Herein, we describe the biological results of a select subset of these synthetic compounds. The results focused on the modification of the aryl groups with either electron-withdrawing groups or electron-donating groups as well as modulation of lipophilicity of benzimidazoles.
The effect of modification at the 1-position of benzimidazole 1 (region B, Fig. 1) was investigated for potency on mESC differentiation to cardiomyocytes. Compared to unsubstituted 1, substitution of electron-donating groups on the aryl ring at the 1-position of benzimidazole 1 increased cardiomyogenesis (Table 1). In contrast, substitution of electron-withdrawing groups led to compounds with diminished potency for cardiomyogenesis. For example, substitution with alkoxyphenyl groups (i.e., 30, 31 or 35), increased cardiomyogenesis by 4.3-, 18.6- and 10.0-fold, respectively. Hydroxyphenyl-substituted benzimidazole 37 increased cardiomyogenesis by 2.1-fold. By comparison, electron-withdrawing substituted benzimidazoles were generally less potent. For example, trifluoromethylphenyl-substituted benzimidazoles 33 and 36 afforded a 0.5 and 1.6 fold-change in cardiomyogenesis. Alkyl-substituted benzimidazoles such as isopropylphenyl (i.e., 34) or n-propylphenyl (i.e., 32) afforded a 4.4- and a 0.7-fold-change in cardiomyogenesis, respectively. These results suggested that addition of aryl-substituted alkyl groups at the 1-position of benzimidazoles could afford moderately potent cardiomyogenesis agents. However, these lipophilic substituents did not improve the solubility of 1 and were less potent than benzimidazoles with methoxy substituents at the 2- and 3-position of the aryl ring. Accordingly, addition of lipophilic substituents to the 1-position of benzimidazole was not explored further. Replacing the aryl group in region B with a nonaromatic moiety (i.e., cyclohexyl-substituted 40) increased cardiomyogenesis 3.3-fold. In contrast, 1-substitution of the benzimidazole with smaller alkyl groups such as methyl (i.e., 38) or cyclopropyl (i.e., 39) gave only a 1.6-fold and 0.9-fold change in cardiomyogenesis, respectively. No overall improvement in cardiomyogenesis was observed by introduction of increased polarity into region B by the addition of heteroatoms (e.g., 2-pyridine 41, 4-tetrahydropyran-substituted, 42 or N-methyl 4-piperidine, 43). Compounds 41, 42 and 43 increased cardiomyogenesis 2.2-, 1.6-, and 4.4-fold, respectively. These results showed that an aryl group with electron-donating substituents at the 2- or 3-position of region B was necessary to facilitate an improvement in cardiomyogenesis potency. In contrast, addition of polarity to the ring either by the addition of pyridine or piperidine rings decreased cardiomyogenesis. Overall, the results indicated 2- and 3-methoxyaryl groups at region B improved cardiomyogenesis and were used at the 1-position in subsequent studies.
Table 1.
Effect of R2 modification of region B of 1 on cardiomyogenesis in mESCs
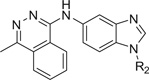 |
R2 | CM Fold-changea |
|---|---|---|
| 1 | Ph | 9.6 ± 1.1 |
| 30 | 2-OMe Ph | 4.3 ± 0.8 |
| 31 | 3-OMe Ph | 18.6 ± 7.2 |
| 32 | 4-nPr Ph | 0.7 ± 0.3 |
| 33 | 3-CF3 Ph | 0.5 ± 0.1 |
| 34 | 2-iPr Ph | 4.4 ± 0.8 |
| 35 | 4-OMe Ph | 10.0 ± 1.9 |
| 36 | 4-CF3 Ph | 1.6 ± 0.5 |
| 37 | 2-OH | 2.1 ± 0.4 |
| 38 | Me | 1.6 ± 0.6 |
| 39 | Cyclopropyl | 0.9 ± 0.3 |
| 40 | Cyclohexyl | 3.3 ± 0.8 |
| 41 | 2-Py | 2.2 ± 0.4 |
| 42 | 4-THP | 1.6 ± 0.3 |
| 43 | 4-Me piperidine | 4.4 ± 0.8 |
Fold-change in cardiomyogenesis compared to vehicle (i.e., DMSO)-treated cells.
The effect of modification of the benzimidazole at the 5-position (region C, Fig. 1) on compound potency for mESC differentiation was investigated by synthesizing and testing more than a hundred analogs. A representative number of benzimidazoles containing substituents at the 5-position were listed in Table 2 to illustrate the SAR. Modification of the phthalazine moiety was thus explored to probe the effects of heteroatoms and other ring systems in this region on mESC differentiation potency. Table 2 shows the results of the effects of substituents at region C of benzimidazole 1 on cardiomyogenesis in mESCs.
Table 2.
Effect of R3 modification of region C of 1 on cardiomyogenesis in mESCs
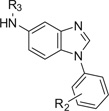 |
R2 | R3a | CM Fold-changeb |
|---|---|---|---|
| 16 | H | H | 3.0 ± 0.4 |
| 44 | 3-OMe | Phthalazin-1-yl | 13.7 ± 4.1 |
| 45 | 3-OMe | 4-Cl-Phthalazin-1-yl | 6.7 ± 1.5 |
| 46 | 3-OMe | 4-MeO-Phthalazin-1-yl | 7.1 ± 2.2 |
| 47 | 3-OMe | 4-Bn-Phthalazin-1-yl | 2.0 ± 0.9 |
| 48 | 3-OMe | 4-Ph-Phthalazin-1-yl | 1.0 ± 0.8 |
| 49 | 3-OMe | Ph-CH2CH2 | 1.4 ± 0.2 |
| 50 | 3-OMe | 2-MeO-Ph-CH2 | 1.7 ± 0.3 |
| 51 | H | 2-Furan-CH2 | 2.2 ± 0.4 |
| 52 | H | N-Me-Pyrrol-2-ylCH2 | 1.6 ± 0.3 |
| 53 | H | (2-Cl-Pyrid-3-yl)CH2 | 1.4 ± 0.3 |
| 54 | H | 5-Br-Thien-2-yl CH2 | 3.1 ± 1.0 |
| 55 | H | Thien-2-yl-CH2 | 0.75 ± 0.2 |
| 56 | H | (2-Ph-Pyrid-5-yl)CH2 | 3.0 ± 0.5 |
| 57 | H | (2-CF3-Pyrid-5-yl)CH2 | 2.9 ± 0.7 |
| 58 | 3-OMe | (N-Bn-piperidin-4yl)CH2 | 4.3 ± 0.8 |
| 59 | 3-OMe | 2-Py-CO | 1.7 ± 0.3 |
| 60 | H | Ac | 1.2 ± 0.2 |
| 68 | 2-OMe | 2-Naphthyl | 3.1 ± 0.6 |
| 69 | 2-OMe | 2-Pyrimidinyl | 1.1 ± 0.2 |
| 70 | H | 2-Pyrazinyl | 2.4 ± 0.5 |
| 71 | H | 3-Pyridyl | 7.8 ± 0.8 |
| 72 | 2-OMe | 2-Pyridyl | 1.5 ± 0.2 |
| 73 | 2-OMe | Ph | 0.7 ± 0.1 |
| 74 | 2-OMe | 3-MeO-Ph | 3.8 ± 0.7 |
| 75 | 3-Me | 4-Methyl Pyridazinyl | 7.9 ± 0.4 |
R3, substituent on the 5-amino group of the benzimidazole core.
Fold-change in cardiomyogenesis compared to vehicle (i.e., DMSO)-treated cells.
‘Hit’ 1 contained a phthalazine ring tethered through a 5-amino group to the benzimidazole core (i.e., region C, Fig. 1). Modification of the phthalazine at the 4-position was explored by synthesis and testing of compounds 44–48. The results showed that small substituents including hydrogen (i.e., 44), chloro (i.e., 45) or methoxy (i.e., 46) increased cardiomyogenesis 13.7-, 6.7- and 7.1-fold, respectively. Larger substituents (e.g., benzyl, 47 or phenyl, 48) did not improve cardiomyogenesis, because, compared to 1, they only increased cardiomyogenesis 2.0- and 1.0-fold, respectively. Replacing the phthalazine ring (region C, Fig. 1) by a naphthalene ring (i.e., 68), afforded only a 3.1-fold-increase in cardiomyogenesis. We conclude that at least one of the two nitrogen atoms in the phthalazine ring was essential for significant potency. Replacement of the phthalazine moiety with a 4-methylpyridazinyl group (i.e., 75) increased cardiomyogenesis 7.9-fold. This showed that smaller ring systems in region C of benzimidazole 1 could afford significant potency.
To decrease the lipophilicity and improve the overall drug-like properties of benzimidazole 1, additional modification of region C was undertaken. Compared to 1 (cLog P = 5.44) the absence of the fused phenyl ring in region C from the phthalazine ring in 75 decreased the overall lipophilicity by more than one cLog P unit (4.42). Several additional nitrogen-containing monocyclic analogs (i.e., 69–72) as well as the simple phenyl analog (i.e., 73) were prepared and tested to investigate the necessity and/or position of the nitrogen atom in region C of 1. Introduction of the phenyl moiety (i.e., 73) in region C slightly decreased cardiomyogenesis (i.e., 0.7 fold-change). This result suggested that one or both of the nitrogen atoms in region C might be important for cardiogenic potency. Compared to 1, introduction of a 3-pyridyl substituent (i.e., 71) increased cardiomyogenesis by 7.8-fold. The 2-pyridinyl analog (i.e., 70) was less potent with only a 2.4-fold increase in cardiomyogenesis. These data suggested that either a basic interaction or a hydrogen-bond interaction might be important in region C for optimal cardiomyogenesis potency in benzimidazole 1. For modifications in region C, the results from this series (i.e., 44–75) showed the necessity of a nitrogen atom at the 3-position of the substituent.
To investigate a possible role of a linker group (i.e., a bridge group between region A and region C of 1) in compound potency, several analogs were prepared and tested. For example, a methylene linker for compounds 49–58; a carboxy group for 59 and 60; or no moiety, compound 16 was investigated. Compounds 49–53, 55, 59 and 60 did not possess increased cardiomyogenesis potency. In contrast, compounds 54, 56, 57 and 58 that were based on thien-2-yl or pyrid-5-yl groups at the nitrogen on C-5 (Table 2) increased cardiomyogenesis by 3.1-, 3.0-, 2.9- and 4.0-fold, respectively. The results suggested that certain heteroaromatics linked through a methylene spacer at the 5-position in region C of the benzimidazole afforded compounds that increased cardiomyogenesis.
Results listed in Table 3 showed the effect of introduction of additional nitrogen atoms in region A on cardiomyogenesis potency. Replacement of carbon or nitrogen atoms at the 2, 3 and 7-positions of region A of benzimidazole 1 (i.e., compounds 65, 78, 82, 83 and 85) afforded compounds with modest increases in cardiomyogenesis, 2.5-, 2.0-, 3.7-, 2.1- and 3.1-fold, respectively. Compared to compound 65, 2-amino-benzimidazoles 82 and 83 gave small increases in potency 3.7- and 2.1-fold, respectively. Replacing N-3 of region A with a carbon atom (i.e., 78) gave slightly decreased potency (i.e., 2.0-fold). Compared to 1, replacing C-2 of region A with a nitrogen atom to elaborate a triazole (i.e., 85) gave slightly increased potency (i.e., 3.1-fold). However, compared to 1, none of these compounds substituted in region A significantly improved the overall cardiogenic potency.
Table 3.
Effect of modification of region A of 1 on cardiomyogenesis in mESCs
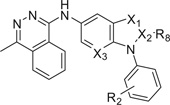 | ||||||
|---|---|---|---|---|---|---|
| R2 | R8 | X1 | X2 | X3 | CM Fold-changea | |
| 65 | 2-MeO | H | N | C | N | 2.5 ± 0.5 |
| 78 | H | H | C | C | C | 2.0 ± 0.3 |
| 82 | H | N(Me)2 | N | C | C | 3.7 ± 0.4 |
| 83 | H | NH2 | N | C | C | 2.1 ± 0.3 |
| 85 | H | H | N | N | C | 3.1 ± 0.5 |
Fold-change in cardiomyogenesis compared to vehicle (i.e., DMSO)-treated cells.
3. Conclusions
In summary, 1,5-disubstituted benzimidazole 1, discovered from a high content screen, markedly stimulated cardiomyocyte differentiation in mESC cultures. In addition to αMHC activation, 1 enhanced additional gene biomarkers for mESC differentiation to cardiomyocytes.29 Medicinal chemistry and SAR development of 1 from over 400 benzimidazole analogs yielded 31, 44 and 75. These compounds showed increased potency relative to 1 and other published small molecule inducers.11 In addition, 31, 44 and 75 trended toward improved drug-like properties, compared to 1. Thus, in the 1,5-disubstituted benzimidazole series examined herein, 1, 31, 44 and 75 constituted compounds that are useful and reliable molecular tools to direct cardiomyogenesis in mESCs.
4. Experimental
4.1. Chemistry
General: Reagents, starting materials, and solvents were purchased in the highest purity available from commercial suppliers and used as received. 1-Chloro-4-methylphthalazine, 1-chloro-4-phenylphthalazine (Enamine LLC, Kiev, Ukraine), 1-chloro-phthalazine and 1-benzyl-4-chloro-phthalazine (Frontier Scientific, Newark, DE), 1-chloro-4-methoxy-phthalazine and 2,4-dinitrofluorobenzene (Matrix Scientific, Columbia, SC). 5-nitro indole (Aldrich, St. Louis, MO), 2-chloro-3,5-dinitropyridine (Alfa Aesar, Ward Hill, MA) and 5-bromo-2-fluoronitrobenzene (TCI America, Portland, OR) were commercially available and used without further purification.
Microwave reactions were conducted using a Biotage Initiator microwave synthesizer (Biotage, Uppsala, Sweden). Reaction products were purified, when necessary, by chromatography on silica gel (Silicycle, Quebec City, Quebec) (40–63 µm) with the solvent system indicated. Nuclear magnetic resonance (NMR) data were recorded on a Varian Mercury 300 MHz Spectrometer (Agilent, Santa Clara, CA) using TMS as an internal standard and CDCl3 as a solvent except where indicated. Electrospray ionization (ESI) mass spectral data was obtained using a Hitachi M-8000 (Hitachi, Dallas, TX). Final test compounds had a purity greater than 95% on the basis LCMS analysis using UV-vis detection at 254 nM and 220 nM.
Compounds were prepared as the hydrochloride salt unless otherwise noted. Hydrochloride salts were prepared by dissolution of the benzimidazole in a minimum amount of dichloromethane and addition of excess 2 M HCl in ether. The solvent was evaporated and benzimidazole hydrochlorides were used directly for evaluation.
Aqueous solubilities were determined by preparing a DMSO stock solution of the desired compound at 10 mM then diluting the stock solution in distilled water to the desired final concentration or until turbidity was observed.
4.1.1. 2,4-Dinitro-N-phenylaniline (3)
To a flask containing 25 mL of THF was added 2 g (10.7 mmol) 2,4-dinitrofluorobenzene and 1.1 g (11.8 mmol) of aniline and stirred at 25 °C. After stirring 15 h the solvent was concentrated in vacuo to give a crude solid that was recrystallized from ethanol to afford a yellow solid (98% yield). 1H NMR δ 7.17 (d, J = 9.6 Hz, 1H), 7.31 (m, 2H), 7.39 (m, 1H), 7.51 (m, 2H), 8.17 (ddd, J = 9.6, 2.5, 0.6 Hz, 1H), 9.19 (d, J = 2.5 Hz, 1H), 9.99 (bs, 1H).
4.1.2. N1-Phenylbenzene-1,2,4-triamine
To a flask containing 2 g (16.9 mmol) of 3 in 75 mL of ethyl acetate, 200 mg of Pd/C was added. The flask was purged 3 times by vacuum and each time refilled with an atmosphere of H2. H2 was introduced to the flask and the mixture was stirred at 25 °C until the mixture showed no more starting material as determined by TLC (hexane/ethyl acetate, 90:10, v/v). The reaction mixture was filtered through celite and rinsed with ethyl acetate. The solvent was evaporated and the crude product was used without further purification.
4.1.3. 1-Phenyl-1H-benzo[d]imidazol-5-amine (16)
To a flask containing 1.5 g (7.5 mmol) N1-phenylbenzene-1,2,4-triamine was added 25 mL of 4 N HCl and 1 mL of formic acid and heated under reflux until no starting material was detected as determined by TLC. The reaction was then cooled to 0 °C and basicified with solid NaOH until a pH 11 was obtained. The aqueous fraction was extracted 3 times with ethyl acetate (100 mL). The organic phases were combined, dried over sodium sulfate and concentrated. The crude material was purified via column chromatography (silica gel, DCM with a gradient from 2% to 5% methanol) to give a red brown solid (80% yield). 1H NMR δ 3.73 (bs, 2H), 6.74 (dd, J = 2.0, 8.5 Hz, 1H), 7.14 (d, J = 2.0 Hz, 1H), 7.33 (dd, J = 8.5, 0.6 Hz, 1H), 7.38–7.56 (m, 5H), 8.00 (s, 1H).
4.1.4. N-(4-Methylphthalazin-1-yl)-1-phenyl-1H-benzo[d]-imidazol-5-amine·HCl (1)
To a microwave vial was added 16 mg (0.07 mmol) of 1-phenyl-1H-benzo[d]imidazol-5-amine, 27 mg (0.15 mmol) of 1-chloro-4-methylphthalazine in 0.5 mL of isopropyl alcohol. The mixture was heated to 150 °C for 1 h. The mixture was concentrated to dryness and ethyl acetate (1 mL) was added to precipitate the product. The hydrochloride salt of 1 was isolated by filtration and washing with cold ethyl acetate to give 17 mg (70% yield) as a tan solid. 1H NMR (CD3OD) δ 2.87 (s, 3H), 7.59 (dd, J = 8.5, 2.2 Hz, 2H), 7.68–7.70 (m, 4H), 7.80 (d, J = 8.5 Hz, 1H), 8.06 (d, J = 2.2 Hz, 1H), 8.23–8.27 (m, 2H), 8.38–8.42 (m, 1H), 8.60 (s, 1H), 8.74–8.77 (m, 1H). LRMS (ESI) m/z calcd 351.15; found 352.28 (M+1)+.
Compounds 6–24 were prepared using procedures similar to those described for the synthesis of 1.
4.1.5. 5-(4-Methylphthalazinyl-1-amino)-1-(2-methoxyphenyl)-benzimidazole·HCl (30)
The hydrochloride salt of 30 (60% yield) was obtained as a tan solid starting from 2-methoxyaniline. 1H NMR (CD3OD) δ 2.84 (s, 3H), 3.88 (s, 3H), 7.19–7.25 (m, 2H), 7.30 (d, J = 8.8 Hz, 1H), 7.35 (d, J = 8.3 Hz, 1H), 7.54–7.58 (m, 2H), 7.70–7.74 (m, 1H), 8.00–8.03 (m, 2H), 8.16–8.19 (m, 1H), 8.23 (s, 1H), 8.32 (s, 1H), 8.52–8.55 (m, 1H). LRMS (ESI) m/z calcd 381.43; found 382.5 (M+1)+.
4.1.6. 5-(4-Methylphthalazinyl-1-amino)-1-(3-methoxyphenyl)-benzimidazole·HCl (31)
The hydrochloride salt of 31 (75% yield) was obtained as a yellow solid starting from 3-methoxyaniline. LRMS (ESI) m/z calcd 381.43; found 382.13 (M+1)+.
4.1.7. 5-(4-Methylphthalazinyl-1-amino)-1-(4-propylphenyl)-benzimidazole·HCl (32)
The hydrochloride salt of 32 (50% yield) was obtained as a tan solid starting from 4-n-propylaniline. 1H NMR (CD3OD) δ 1.01 (t, J = 7.4 Hz, 3H), 1.67–1.80 (m, 2H), 2.73 (t, J = 7.7 Hz, 2H), 2.87 (s, 3H), 7.48 (d, J = 8.5 Hz, 2H), 7.55–7.60 (m, 3H), 7.75 (d, J = 8.8 Hz, 1H), 8.06 (d, J = 1.7 Hz, 1H), 8.21–8.28 (m, 2H), 8.37–8.41 (m, 1H), 8.56 (s, 1H), 8.75–8.81 (m, 1H). LRMS (ESI) m/z calcd 393.20; found 394.07 (M+1)+.
4.1.8. 5-(4-Methylphthalazinyl-1-amino)-1-(3-trifluoromethylphenyl)benzimidazole·HCl (33)
The hydrochloride salt of 33 (65% yield) was obtained as a tan solid starting from 3-trifluoromethylaniline. 1H NMR (CD3OD) δ 2.88 (s, 3H), 7.62 (dd, J = 1.9, 8.8 Hz, 2H), 7.8 (d, J = 8.8 Hz, 1H), 7.9–7.91 (m, 2H), 7.99–8.03 (m, 1H), 8.06 (s, 1H), 8.1 (d, J = 1.9 Hz, 1H), 8.25–8.30 (m, 1H), 8.39–8.43 (m, 1H), 8.7 (s, 1H), 8.75–8.78 (m, 1H). LRMS (ESI) m/z calcd 419.14; found 420.05 (M+1)+.
4.1.9. 5-(4-Methylphthalazinyl-1-amino)-1-(2-isopropylphenyl)-benzimidazole·HCl (34)
The hydrochloride salt of 34 (10% yield) was obtained as an orange solid starting from (4-dimethylamino) aniline. 1H NMR (CD3OD) δ 1.18 (d, J = 6.9 Hz, 3H), 1.22 (d, J = 6.6 Hz, 3H), 2.52–2.65 (m, 1H), 2.88 (s, 3H), 7.30–7.40 (m, 2H), 7.44–7.49 (m, 1H), 7.55 (d, 8.5 Hz, 1H), 7.61–7.69 (m, 2H), 8.09 (s, 1H), 8.21–8.29 (m, 2H), 8.37–8.42 (m, 1H), 8.53 (s, 1H), 8.74–8.77 (m, 1H). LRMS (ESI) m/z calcd 394.19; found 395.4 (M+1)+.
4.1.10. 5-(4-Methylphthalazinyl-1-amino)-1-(4-methoxyphenyl)-benzimidazole·HCl (35)
The hydrochloride salt of 35 (65% yield) was obtained as a tan solid starting from 4-methoxyaniline. 1H NMR (CD3OD) δ 2.88 (s, 3H), 3.91 (s, 3H), 7.21 (d, J = 8.8 Hz, 2H), 7.60 (d, J = 8.8 Hz, 3H), 7.74 (d, J = 8.8 Hz, 1H), 8.09 (s, 1H), 8.25–8.28 (m, 2H), 8.40–8.43 (m, 1H), 8.67 (s, 1H), 8.75–8.78 (m, 1H). LRMS (ESI) m/z calcd 381.43; found 382.13 (M+1)+.
4.1.11. 5-(4-Methylphthalazinyl-1-amino)-1-(4-trifluoromethylphenyl)benzimidazole·HCl (36)
The hydrochloride salt of 36 (70% yield) was obtained as a tan solid starting from 4-trifluoromethylaniline. 1H NMR (CD3OD) δ 2.90 (s, 3H), 7.67 (d, J = 8.5 Hz, 1H), 7.90 (d, J = 8.5 Hz, 1H), 7.94–8.04 (m, 4H), 8.14 (s, 1H), 8.23–8.28 (m, 2H), 8.42–8.44 (m, 1H), 8.75–8.81 (m, 1H). LRMS (ESI) m/z calcd 419.14; found 420.07 (M+1)+.
4.1.12. 5-(4-Methylphthalazinyl-1-amino)-1-(2-hydroxyphenyl)-benzimidazole·HCl, 37
The hydrochloride salt of 37 (60% yield) was obtained as a tan solid starting from 2-methoxyaniline. 1H NMR (CD3OD) δ 3.80 (s, 3H), 6.71 (dd, J = 2.0, 8.5 Hz, 1H), 7.07–7.12 (m, 3H), 7.14 (d, J = 2.0 Hz, 1H), 7.38–7.45 (m, 2H), 7.98 (s, 1H). LRMS (ESI) m/z calcd 381.43; found 382.5 (M+1)+.
4.1.13. 5-(4-Methylphthalazinyl-1-amino)-1-methylbenzimidazole·HCl (38)
The hydrochloride salt of 38 (65% yield) was obtained as a tan solid from the reaction of 1-chloro-4-methyl phthalazine and commercially available 5-amino-2-methylbenzimidazole. 1H NMR (CD3OD) δ 2.83 (s, 3H), 3.96 (s, 3H), 7.55 (dd, J = 8.8, 1.9 Hz, 1H), 7.67 (d, J = 8.5 Hz, 1H), 7.95 (d, J = 1.6 Hz, 1H), 8.13–8.17 (m, 1H), 8.25 (s, 1H), 8.27–8.31 (m, 1H), 8.40–8.43 (m, 1H), 8.63–8.66 (m, 1H). LRMS (ESI) m/z calcd 289.13; found 290.08 (M+1)+.
4.1.14. 5-(4-Methylphthalazinyl-1-amino)-1-cyclopropylbenzimidazole·HCl (39)
The hydrochloride salt of 39 (70% yield) was obtained as a yellow solid starting from cyclopropylamine. 1H NMR (CD3OD) δ 1.10–1.17 (m, 2H), 1.21–1.28 (m, 2H), 2.84 (s, 3H), 3.56–3.61 (m, 1H), 4.45–4.55 (m, 1H), 7.56 (dd, J = 8.5, 1.6 Hz, 1H), 7.87 (d, J = 8.5 Hz, 1H), 7.95 (d, J = 1.6 Hz, 1H), 8.19–8.24 (m, 2H), 8.33–8.35 (m, 1H), 8.36 (s, 1H), 8.69–8.72 (m, 1H). LRMS (ESI) m/z calcd 315.15; found 316.20 (M+1)+.
4.1.15. 5-(4-Methylphthalazinyl-1-amino)-1-cyclohexylbenzimidazole·HCl (40)
The hydrochloride salt of 40 (70% yield) was obtained as a yellow solid starting from cyclohexylamine. 1H NMR (CD3OD) δ 1.38–2.22 (m, 10H), 2.86 (s, 3H), 4.45–4.55 (m, 1H), 7.58 (dd, J = 8.5, 1.6 Hz, 1H), 7.89 (d, J = 8.5 Hz, 1H), 8.02 (d, J = 1.6 Hz, 1H), 8.13–8.17 (m, 1H), 8.21–8.25 (m, 1H), 8.37–8.40 (m, 1H), 8.61 (s, 1H), 8.71–8.74 (m, 1H). LRMS (ESI) m/z calcd 357.20; found 358.17 (M+1)+.
4.1.16. 5-(4-Methylphthalazinyl-1-amino)-1-(2-pyridyl)benzimidazole·HCl (41)
The hydrochloride salt of 41 (76% yield) was obtained as a yellow solid starting from 2-aminopyridine. 1H NMR (CD3OD) δ 2.90 (s, 3H), 7.45–7.49 (m, 1H), 7.66 (d, J = 8.8 Hz, 1H), 8.02 (d, J = 8.3 Hz, 1H), 8.10 (d, J = 8.3 Hz, 1H), 8.15 (s, 1H), 8.21–8.32 (m, 2H), 8.42 (d, J = 8.8 Hz, 2H), 8.66 (s, 1H), 9.09–9.14 (m, 2H). LRMS (ESI) m/z calcd 352.14; found 353.22 (M+1)+.
4.1.17. 5-(4-Methylphthalazinyl-1-amino)-1-(4-tetrahydropyranyl) benzimidazole·HCl (42)
The hydrochloride salt of 42 (63% yield) was obtained as a yellow solid starting from 4-aminotetrahydropyran. 1H NMR (CD3OD) δ 2.16–2.34 (m, 5H), 2.88 (s, 3H), 3.67–3.76 (m, 2H), 4.12–4.20 (m, 2H), 7.62 (d, J = 1.9 Hz, 1H), 7.95 (d, J = 8.8 Hz, 1H), 8.05 (d, J = 1.9 Hz, 1H), 8.12–8.16 (m, 1H), 8.23–8.26 (m, 1H), 8.37–8.42 (m, 1H), 8.71 (s, 1H), 8.74–8.77 (m, 1H). LRMS (ESI) m/z calcd 359.17; found 360.20 (M+1)+.
4.1.18. 5-(4-Methylphthalazinyl-1-amino)-1-(1-methylpiperidin-4-yl)-1H-benzo[d]imidazol-5-amine·HCl (43)
The hydrochloride salt of 43 (76% yield) was obtained as a red solid starting from 1-methylpiperidin-4-amine. 1H NMR (DMSO-d6) δ 2.20–2.26 (m, 4H), 2.75 (s, 3H), 2.77 (s, 3H), 3.04–3.18 (m, 4H), 4.61–4.73 (m, 1H), 7.65 (d, J = 8.0 Hz, 1H), 7.74 (d, J = 9.6 Hz, 1H), 7.98–8.00 (m, 2H), 8.07–8.10 (m, 1H), 8.23–8.28 (m, 2H), 8.59–8.63 (m, 1H). LRMS (ESI) m/z calcd 372.47; found 373.5 (M+1)+.
4.1.19. 5-(Phthalazinyl-1-amino)-1-(3-methoxyphenyl)benzimidazole·HCl (44)
The hydrochloride salt of 44 (65%% yield) was obtained as a yellow solid starting from 1-chlorophtalazine. 1H NMR (CD3OD) δ 3.92 (s, 3H), 7.25–7.44 (m, 4H), 7.59–7.70 (m, 2H), 7.90–7.99 (m, 2H), 8.27–8.42 (m, 2H), 8.88–8.91 (m, 1H), 9.32 (s, 1H), 9.71 (s, 1H). LRMS (ESI) m/z calcd 367.14; found 368.27 (M+1)+.
4.1.20. 5-(4-Chlorophthalazinyl-1-amino)-1-(3-methoxyphenyl)-benzimidazole·HCl (45)
The hydrochloride salt of 45 (55% yield) was obtained as a yellow solid starting from 1,4-dichlorophtalazine. 1H NMR (CD3OD) δ 3.93 (s, 3H), 7.22–7.25 (m, 1H), 7.35 (bs, 2H), 7.61–7.66 (m, 1H), 7.80 (d, J = 8.8 Hz, 1H), 7.93 (d, J = 8.8 Hz, 1H), 8.24–8.30 (m, 2H), 8.33 (s, 1H), 8.45 (d, J = 7.2 Hz, 1H), 8.81 (d, J = 7.2 Hz, 1H), 9.41 (s, 1H). LRMS (ESI) m/z calcd 401.10; found 402.13 (M+1)+.
4.1.21. 5-(4-Methoxyphthalazinyl-1-amino)-1-(3-methoxyphenyl)benzimidazole·HCl (46)
The hydrochloride salt of 46 (62% yield) was obtained as a yellow solid starting from 1-chloro-4-methoxyphtalazine. 1H NMR (CD3OD) δ 3.93 (s, 3H), 4.24 (s, 3H), 7.15–7.17 (m, 1H), 7.25 (bs, 2H), 7.32–7.66 (m, 5H), 8.33–8.45 (m, 2H), 8.51–8.65 (m, 2H).
4.1.22. 5-(4-Benzylphthalazinyl-1-amino)-1-(3-methoxyphenyl)-benzimidazole·HCl (47)
The hydrochloride salt of 47 (65% yield) was obtained as a tan solid starting from 1-chloro-4-benzylphtalazine. 1H NMR (CD3OD) δ 3.93 (s, 3H), 7.26–7.40 (m, 6H), 7.67 (apparent t, 1H), 7.97 (bs, 2H), 8.17–8.33 (m, 4H), 8.40 (s, 1H), 8.47 (d, J = 8.8 Hz, 1H), 8.85 (d, J = 7.2 Hz, 1H), 9.76 (s, 1H).
4.1.23. 5-(4-Phenylphthalazinyl-1-amino)-1-(3-methoxyphenyl)-benzimidazole·HCl (48)
The hydrochloride salt of 48 (58% yield) was obtained as a yellow solid starting from 1-chloro-4-phenylphtalazine. 1H NMR (CD3OD) δ 3.93 (s, 3H), 7.14–7.18 (m, 1H), 7.26–7.29 (m, 2H), 7.51–7.69 (m, 6H), 7.67 (apparent t, 1H), 7.87 (d, J = 8.8 Hz, 1H), 8.17–8.33 (m, 4H), 8.80 (s, 1H), 8.86 (d, J = 8.8 Hz, 1H).
4.1.24. 5-(Phenethylamino)-1-(3-methoxyphenyl)benzimidazole·HCl (49)
5 mg (0.03 mmol) of 18 (prepared using the method described in Scheme 1) was stirred with 5.4 mg (0.05 mmol) of phenylacetaldehyde and 15 µL of borane·pyridine complex in 0.1 mL ethanol for 15 h at 25 °C in a sealed vial. Solvent was evaporated and the crude reaction mixture purified by PTLC (hexane/ethyl acetate, 50:50, v/v). 5.0 mg of 5-(phenethylamino)-1-(3-methoxyphenyl)-benzimidazole was obtained as a free base (69% yield). Treatment of the free base (solution in DCM) with excess HCl (2.0 Msolution in ether) at room temperature, followed by concentration, yielded 49. 1H NMR δ 3.88 (s, 3H), 4.28 (s, 2H), 6.72 (dd, J = 9, 3 Hz, 1H), 7.10–6.79 (m, 11H), 7.36 (dd, J = 9 Hz, 1H), 7.44 (t, J = 9 Hz, 1H), 8.06 (s, 1H), 8.63 (t, J = 6 Hz, 1H).
Compounds 50–58 were prepared using a similar procedure to that described for the synthesis of 49.
4.1.25. N-(2-Methoxybenzyl)-1-(3-methoxyphenyl)-1H-benzo-[d]imidazol-5-amine·HCl (50)
The hydrochloride salt of 50 (49% yield) was obtained as a yellow oil from 2-methoxycinnamaldehyde and 18 using the procedure described for 25. 1H NMR δ 3.82 (s, 3H), 3.87 (s, 3H), 6.34 (bs, 1H), 6.81–7.48 (m, 13H).
4.1.26. N-(Furan-2-ylmethylamino)-1-phenyl-1H-benzimidazole·HCl (51)
The hydrochloride salt of 51 (75% yield) was obtained as an orange oil from furan-2-carboxaldehyde and 18 using the procedure described for 25. LRMS (ESI) m/z calcd 289.1; found 290.30 (M+1)+.
4.1.27. N-((1-Methyl-1H-pyrrol-2-yl)methylamino)-1-phenyl-1H-benzimidazole·HCl (52)
The hydrochloride salt of 52 (82% yield) was obtained as an orange oil from 1-methyl-1H-pyrrole-2-carboxaldehyde and 18 using the procedure described for 49. LRMS (ESI) m/z calcd 302.1; found 303.30 (M+1)+.
4.1.28. N-((2-Chloropyridin-3-yl)methylamino)-1-phenyl-1H-benzimidazole·HCl (53)
The hydrochloride salt of 53 (85% yield) was obtained as a transparent oil from 2-chloronicotinaldehyde and 18 using the procedure described for 49. LRMS (ESI) m/z calcd 334.0; found 335.10 (M+1)+.
4.1.29. N-((5-Bromothiophen-2-yl)methylamino)-1-phenyl-1H-benzimidazole·HCl (54)
The hydrochloride salt of 54 (78% yield) was obtained as a clear oil from 5-bromothiophene-2-carboxaldehyde and 18 using the procedure described for 25. LRMS (ESI) m/z calcd 384.8; found 386.00 (M+1)+.
4.1.30. 1-Phenyl-N-(thiophen-2-ylmethylamino)-1H-benzimidazole·HCl (55)
The hydrochloride salt of 55 (43% yield) was obtained as a colorless oil from thiophene-2-carbaldehyde and 18 using the procedure described for 49. LRMS (ESI) m/z calcd 305.1; found 306.30 (M+1)+.
4.1.31. 1-Phenyl-N-((6-phenylpyridin-3-yl)methylamino)-1H-benzimidazole·HCl (56)
The hydrochloride salt of 56 (73% yield) was obtained as a pink solid from 6-phenylnicotinaldehyde and 18 using the procedure described for 49. LRMS (ESI) m/z calcd 375.9; found 377.1 (M+1)+.
4.1.32. 1-Phenyl-N-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-benzo[d]imidazol-5-amine·HCl (57)
The hydrochloride salt of 57 (78% yield) was obtained as a white solid from 6-(trifluoromethyl)nicotinaldehyde and 18 using the procedure described for 49. LRMS (ESI) m/z calcd 368.1; found 369.30 (M+1)+.
4.1.33. N-((1-Benzylpiperidin-4-yl)methyl)-1-phenyl-1H-benzo-[d]imidazol-5-amine·HCl (58)
The hydrochloride salt of 58 (75% yield) was obtained as an orange oil from 1-benzylpiperidine-4-carboxaldehyde and 18 using the procedure described for 49. LRMS (ESI) m/z calcd 395.2; found 396.4 (M+1)+.
4.1.34. N-(1-(3-Methoxyphenyl)-1H-benzo[d]imidazol-5-yl)-picolinamide·HCl (59)
To a 5 mL flask containing 50 mg (0.35 mmol) of picolinoyl chloride in 2 mL DCM was added 85 mg (0.35 mmol) of 18 and 0.1 mL of triethylamine. The mixture was stirred overnight at room temperature. The solution was concentrated, diluted with DCM, washed with 2 mL of brine, dried and purified by PTLC (DCM/methanol 9/1, v/v) to afford 95 mg (79% yield) of a clear liquid. 1H NMR δ 3.89 (s, 3H), 6.95–7.18 (m, 3H), 7.43–7.52 (m, 2H), 7.56 (d, J = 9 Hz, 1H), 7.82 (d, J = 9 Hz, 1H), 7.93 (t, J = 7.8 Hz, 1H), 8.14 (bs, 1H), 8.27 (bs, 1H), 8.34 (d, J = 8.1 Hz, 1H), 8.65 (d, J = 5 Hz, 1H).
4.1.35. N-(1-Phenyl-1H-benzo[d]imidazol-5-yl)acetamide (60)
To a 5 mL flask containing 5 µL (0.06 mmol) of acetyl chloride in 1 mL DCM was added 10 mg (0.05 mmol) of 18 and 5 µL of triethylamine. The mixture was stirred for 15 h at 25 °C. The solution was concentrated, diluted with DCM, washed with 2 mL of brine, dried and purified by PTLC (DCM/methanol, 9/1, v/v) to afford 7 mg (56% yield) of a light beige solid. LRMS (ESI) m/z calcd 251.3; found 252.2 (M+1)+.
4.1.36. N-(2-Methoxyphenyl)-3,5-dinitropyridin-2-amine (62)
2-Chloro-3,5-dinitropyridine (9.8 mmol), o-anisidine (9.8 mmol) and DIEA (19.6 mmol) were heated at reflux in 100 mL ethanol. After 3 h, the reaction was complete and the bright orange precipitate was filtered, washed with ethanol and dried to give 2.9 g of a bright red-orange solid. 1H NMR (300 MHz, CDCl3): 9.34 (s, 1H), 8.48 (d, J = 6.3 Hz, 1H), 7.30–7.20 (m, 4H), 7.15–7.00 (m, 3H), 3.99 (s, 3H).
4.1.37. N2-(2-Methoxyphenyl)pyridine-2,3,5-triamine, 63
1 g of 62 and 4 g of sodium dithionite were heated at 70 °C for 1 h in ethanol/water (50/50 v/v). The solvent was evaporated and the crude product was extracted with ethyl acetate to give 340 mg of 63 as an orange solid. The compound was used directly without further purification.
4.1.38. 3-(2-Methoxyphenyl)-3H-imidazo[4,5-b]pyridin-6-amine (64)
To a flask containing 330 mg (1.4 mmol) of 63 was added 5 mL of 4 N HCl and 0.2 mL of formic acid and heated under reflux until no starting material was present as determined by TLC. Upon completion, the reaction was cooled to 0 °C and basicified with solid NaOH until a pH 11 was reached. The aqueous fraction was extracted 3 times with ethyl acetate (100 mL). The organic phases were combined, dried over sodium sulfate and concentrated. The crude material was purified via column chromatography (silica gel, ethyl acetate with a gradient from 0% to 20% methanol) to give a brown solid (80% yield). 1H NMR δ 3.81 (s, 3H), 7.09–7.18 (m, 2H), 7.42–7.47 (m, 1H), 7.57 (d, J = 7.7 Hz, 1H), 7.69 (bs, 2H), 8.30 (d, J = 9.1 Hz, 1H), 8.44–8.47 (m, 1H), 8.51 (d, J = 2.2 Hz, 1H).
4.1.39. N-(3-(2-Methoxyphenyl)-3H-imidazo[4,5-b]pyridin-6-yl)-4-methylphthalazin-1-amine·HCl (65)
To a microwave vial was added 10 mg (0.04 mmol) of 1-phenyl-1H-benzo[d]imidazol-5-amine and 20 mg (0.15 mmol) of 1-chloro-4-methylphthalazine in 0.5 mL of isopropyl alcohol. The mixture was heated to 150 °C for 1 h. The mixture was concentrated to dryness and ethyl acetate (1 mL) was added to precipitate the product. The hydrochloride salt of 65 was isolated by filtration and washing with cold ethyl acetate to give 17 mg (70% yield) as a tan solid. LRMS (ESI) m/z calcd 382.15; found 383.6 (M+1)+.
4.1.40. 1-(2-Methoxyphenyl)-N-(naphthalen-2-yl)-1H-benzo[d]-imidazol-5-amine·HCl (68)
67a (30 mg, 0.1 mmol), naphthalen-2-amine (28 mg, 0.2 mmol), palladium(II)acetate (2 mg), rac-BINAP (2 mg), cesium carbonate (10 mg), and 1.5 mL of degassed toluene were added to a 1 g vial. The vial was flushed with argon gas, capped with a Teflon-coated liner, and heated overnight at 100 °C. After allowing the solution to cool to room temperature, the solution was filtered, concentrated, and purified by flash column chromatography (DCM with a gradient of 0–5% methanol) to give 17.6 mg (49% yield) of 68 as a red oil. LRMS (ESI) m/z calcd 365.15; found 366.4 (M+1)+ and 388.0 (M+Na)+.
Compounds 69–78 were prepared using procedures similar to those described for the synthesis of 68.
4.1.41. 1-(2-Methoxyphenyl)-N-(pyrimidin-2-yl)-1H-benzo[d]-imidazol-5-amine·HCl (69)
Compound 69 was prepared using procedures similar to those described for the synthesis of 68 starting from 67a and 2-amino pyrimidine in 47% yield. LRMS (ESI) m/z calcd 346.14; found 347.45 (M+1)+.
4.1.42. 1-Phenyl-N-(pyrazin-2-yl)-1H-benzo[d]imidazol-5-amine·HCl (70)
Compound 70 was prepared using procedures similar to those described for the synthesis of 68 starting from 67b and 2-amino pyrazine in 59% yield. LRMS (ESI) m/z calcd 287.33; found 288.53 (M+1)+ and 310.13 (M+Na)+.
4.1.43. 1-Phenyl-N-(pyridin-3-yl)-1H-benzo[d]imidazol-5-amine·HCl (71)
Compound 71 was obtained in 23% yield as a clear oil using procedures similar to those described for the synthesis of 68 starting from 67b and 3-aminopyridine. LRMS (ESI) m/z calcd 286.34; found 287.47 (M+1)+.
4.1.44. 1-(2-Methoxyphenyl)-N-(pyridin-2-yl)-1H-benzo[d]imidazol-5-amine·HCl (72)
Compound 72 was obtained in 48% yield using procedures similar to those described for the synthesis of 68 starting from 67a and 2-aminopyridine. LRMS (ESI) m/z calcd 287.33; found 317.73 (M+Na)+.
4.1.45. 1-(2-Methoxyphenyl)-N-(phenyl)-1H-benzo[d]imidazol-5-amine·HCl (73)
Compound 73 was obtained in 54% yield using procedures similar to those described for the synthesis of 68 starting from 67a and aniline. LRMS (ESI) m/z calcd 315.38; found 317.73 (M+1)+.
4.1.46. 1-(2-Methoxyphenyl)-N-(3-methoxyphenyl)-1H-benzo-[d]imidazol-5-amine·HCl (74)
Compound 74 was obtained in 40% yield using procedures similar to those described for the synthesis of 68 starting from 67a and 3-methoxyaniline. LRMS (ESI) m/z calcd 345.40; found 346.60 (M +1)+ and 368.27 (M+Na)+.
4.1.47. N-(6-Methylpyridazin-3-yl)-1-m-tolyl-1H-benzo[d]imidazol-5-amine·HCl (75)
Compound 75 was obtained in 33% yield using procedures similar to those described for the synthesis of 1 starting from 1-m-tolyl-1H-benzo[d]imidazol-5-amine and 3-chloro-6-methylpyridazine. LRMS (ESI) m/z calcd 315.15; found 317.40 (M+1)+.
4.1.48. N-Phenyl-5-nitroindole (77)
To a flame-dried sealed tube was added 5-nitroindole (486 mg, 3 mmol), phenylboronic acid (914 mg, 7.5 mmol), anhydrous copper(II) acetate (1.36 g, 7.5 mmol), diisopropylethylamine (1.3 mL, 7.5 mmol) in DCM (6 mL). The mixture was stirred at 25 °C for 4 days, concentrated in-vacuo, diluted with chloroform (50 mL) and water (50 mL), and filtered to remove particulates. The organic layer was then dried over sodium sulfate, concentrated in vacuo, and purified by flash column chromatography (hexanes/ethyl acetate, 4:1, v/v) to 77 (502 mg, 70% yield) as a yellow solid. 1H NMR δ 6.86 (d, J = 3.3 Hz, 1H), 7.43–7.6 (m, 7H), 8.11 (dd, J = 2.1, 9.0 Hz, 1H), 8.65 (d, J = 2.1 Hz, 1H).
4.1.49. N-Phenyl-5-aminoindole
N-Phenyl-5-nitroindole 77 (150 mg, 0.63 mmol) was dissolved in ethanol (15 mL). A solution of sodium dithionite (329 mg, 1.89 mmol) in water (1 mL) was added at room temperature with stirring. The solution was then heated to 70 °C until complete consumption of 77 was observed by TLC (~2 h). After the solution was cooled to 25 °C, ethanol was removed in-vacuo and the resulting crude material was diluted with ethyl acetate, washed with water, dried over sodium sulfate and concentrated in vacuo to yield crude N-phenyl-5-aminoindole (62 mg, 47%) which was used immediately in the following reaction without further purification. 1H NMR δ 3.55 (bs, 2H), 6.51 (dd, J = 0.6, 3 Hz, 1H), 6.70 (dd, J = 2.1, 8.7 Hz, 1H), 7.00 (d, J = 2.1 Hz, 1H), 7.28–7.53 (m, 7H). LRMS (ESI) m/z calcd 208.10; found 209.13 (M+1)+.
4.1.50. 4-Methyl-N-(1-phenyl-1H-indol-5-yl)phthalazin-1-amine·HCl (78)
To a microwave vial was added N-Phenyl-5-aminoindole (10 mg, 0.05 mmol) and 1-chloro-4-methylphthalzine (17 mg, 0.09 mmol) in isopropanol (1 mL). The mixture was heated at 150 °C for 1 h. The reaction mixture was concentrated to dryness and ethyl acetate was added to precipitate the hydrochloride salt of 78 as a yellow solid (14 mg, 86% yield). LRMS (ESI) m/z calcd 350.15; found 351.2 (M+1)+.
4.1.51. N,N-Dimethyl-5-nitro-1-phenyl-1H-benzo[d]imidazol-2-amine (80)
4-Nitro-N1-phenylbenzene-1,2-diamine (200 mg, 0.87 mmol) was dissolved in dichloromethane (10 mL). Phosgene imminium chloride (426 mg, 2.62 mmol) and triethylamine (0.73 mL, 5.24 mmol) were added to the solution and the mixture was stirred at room temperature overnight. The reaction mixture was carefully stopped with methanol, diluted with DCM, washed with water, washed with brine, dried over sodium sulfate, and concentrated. The crude mixture was purified by flash column chromatography (hexanes with a gradient of ethyl acetate 20–100%) to yield 160 mg (65% yield) of 80. 1H NMR δ 2.89 (s, 6H), 6.94 (d, J = 8.7 Hz, 1H), 7.42–7.62 (m, 5H), 7.95 (dd, J = 2.4, 8.4 Hz, 1H), 8.37 (d, J = 2.4 Hz, 1H). LRMS (ESI) m/z calcd 282.11; found 283.6 (M+1)+, 305.4 (M+Na)+.
4.1.52. N2,N2-Dimethyl-1-phenyl-1H-benzo[d]imidazole-2,5-diamine
To a flask containing 80 (160 mg, 0.57 mmol) in ethanol (10 mL) was added 16 mg of 10% Pd/C. The flask was purged 3 times by vacuum and each time refilled with an atmosphere of H2. H2 was introduced to the flask and the mixture was stirred at 25 °C for 15 h. The mixture was then filtered through a pad of celite and the resulting solution was concentrated to yield N2,N2-dimethyl-1-phenyl-1H-benzo[d]imidazole-2,5-diamine (130 mg, 91%). LRMS (ESI) m/z calcd 252.14; found 253.2 (M+1)+, 254.47 (M+2)+.
4.1.53. N2,N2-Dimethyl-N5-(4-methylphthalazin-1-yl)-1-phenyl-1H-benzo[d]imidazole-2,5-diamine·HCl (82)
The hydrochloride salt of 82 (72% yield) was obtained as a yellow solid using the procedure described for 1. LRMS (ESI) m/z calcd 394.19; found 395.00 (M+1)+, 396.00 (M+2)+.
4.1.54. 5-Nitro-1-phenyl-1H-benzo[d]imidazol-2-amine (81)
4-Nitro-N1-phenylbenzene-1,2-diamine 79 (200 mg, 0.87 mmol) was dissolved in DCM (10 mL). Cyanogen bromide (150 mg, 1.42 mmol) was added to the solution and the reaction mixture was stirred at 25 °C for 15 h. TLC indicated the reaction had not gone to completion. 150 mg cyanogen bromide was added and the reaction mixture was stirred at reflux for an additional 2 h. After allowing the solution to cool to room temperature a precipitate was formed. The precipitate 81 (140 mg, 63% yield) was collected by filtration and used without further purification. 1H NMR (CDCl3 + CD3OD) δ 3.52 (br s, 2H), 7.03 (d, J = 9.0 Hz, 1H), 7.43–7.46 (m, 2H), 7.63–7.68 (m, 3H), 8.12 (dd, J = 2.4, 9 Hz, 1H), 8.32 (d, J = 1.8 Hz, 1H). LRMS (ESI) m/z calcd 254.08; found 255.33 (M+1)+.
4.1.55. 1-Phenyl-1H-benzo[d]imidazole-2,5-diamine
5-Nitro-1-phenyl-1H-benzo[d]imidazol-2-amine 81 (140 mg, 0.55 mmol) was dissolved in 4:1 ethanol/water (5 mL). Excess solid sodium dithionite was added to the solution and the reaction mixture was heated to 70 °C with stirring for 1 h. The reaction mixture was diluted with ethyl acetate and washed with water. The water layer was extracted several times with ethyl acetate. The organic layers were combined, dried over sodium sulfate, and concentrated to yield 1-phenyl-1H-benzo[d]imidazole-2,5-diamine (52 mg, 42% yield). LRMS (ESI) m/z calcd 224.11; found 225.07 (M+1)+, 226.33 (M+2)+.
4.1.56. N5-(4-Methylphthalazin-1-yl)-1-phenyl-1H-benzo[d]-imidazole-2,5-diamine (83)
The hydrochloride salt of 83 (39% yield) was obtained as a yellow solid using the procedure described for 1. LRMS (ESI) m/z calcd 366.16; found 367.07 (M+1)+, 388.93 (M+Na)+.
4.1.57. 5-Nitro-1-phenyl-1H-benzo[d][1,2,3]triazole (84)
4-Nitro-N1-phenylbenzene-1,2-diamine 79 (125 mg, 0.55 mmol) was dissolved in chloroform (15 mL). Isoamyl nitrite (1 mL, 7.4 mmol) was added to the solution and the reaction mixture was stirred at room temperature for 1 h. The solution was concentrated to give 84 (131 mg, 100% yield) and used without further purification. 1H NMR δ 7.57–7.79 (m, 5H), 7.87 (dd, J = 0.6, 9 Hz, 1H), 8.47 (dd, J = 1.8, 9 Hz, 1H), 9.09 (dd, J = 0.6, 1.8 Hz, 1H).
4.1.58. 1-Phenyl-1H-benzo[d][1,2,3]triazol-5-amine
To a flask containing 84 (131 mg, 0.545 mmol) in 8:1 ethanol/tetrahydrofuran (20 mL) was added 13 mg of 10% Pd/C. The flask was purged 3 times by vacuum and each time refilled with an atmosphere of H2. H2 was introduced to the flask and the mixture was stirred at 25 °C for 15 h. The mixture was then filtered through a pad of celite and the resulting solution was concentrated to yield 1-phenyl-1H-benzo[d][1,2,3]triazol-5-amine (35 mg, 31% yield). 1H NMR δ 3.92 (br s, 2H), 7.0 (dd, J = 2, 8.7 Hz, 1H), 7.28 (dd, J = 0.6, 1.8 Hz, 1H), 7.45–7.62 (m, 5H), 7.74–7.78 (m, 1H).
4.1.59. 4-Methyl-N-(1-phenyl-1H-benzo[d][1,2,3]triazol-5-yl)-phthalazin-1-amine·HCl (85)
The hydrochloride salt of 85 (71% yield) was obtained using the procedure described for 1. LRMS (ESI) m/z calcd 352.14; found 352.93 (M+1)+, 374.87 (M+Na)+.
4.2. Biology
General: mESC normal growth media contained high glucose DMEM (prepared by supplementing it with 6% FBS and 5 µM β-mercaptoethanol). Additional supplements including sodium pyruvate, non-essential amino acids, glutamine, and pen/strep were added to 1×·from 100× stocks (Sigma, St. Louis, MO). Leukemia Inhibitory Factor (LIF) was added at 1 IU/µL (Millipore, Billerica, MA) to maintain mESC pluripotency during scale-up and maintenance. Mouse monoclonal anti-αActinin antibody (Millipore) was used at a dilution of 1:200. Donkey anti-mouse Alexa568 (Life Technologies, Carlsbad, CA) was used at a dilution of 1:400. Horse serum and Triton-X100 were used as specified (Sigma).
4.2.1. Robotics and automation
The primary screen used a fully integrated Beckman FX fluid handler (Beckman Coulter; San Diego, CA) with a peripheral tip lift, plate hotel, incubation cytomat (ThermoFisher, Pittsburgh, PA) and rail mounted robotic arm. The microtiter plates were imaged at day 10 or day 12 using an IC100 Cell imaging system (Vala Sciences, San Diego, CA) and images were analyzed using Vala Sciences software. The confirmatory and SAR screens for cardiomyogenesis in mESCs used a fully integrated Hamilton STAR robotics system (Hamilton Robotics, Reno, NV) with incubation cytomat and plate hotel (ThermoFisher). The microtiter plates were imaged at day 10 or day 12 using an InCell 1000 (GE Healthcare; Piscataway, NJ) and images were analyzed by a custom built algorithm (see Supporting information) using Developer Toolbox (GE Healthcare, Pittsburgh, PA).
4.2.2. Compound management
The primary screen used diluted stocks in 100% DMSO (1 mg/mL daughter plate) dissolved in 20% DMSO in water. The library consisted of 13,360 compounds, from the DIVERSet collection (Chembridge; San Diego, CA), at 2 doses of 1 µg/mL and 5 µg/mL. The SAR screen was conducted at 5 concentrations for most of the compounds examined (i.e., 0.78, 1.56, 3.12, 6.25, 12.5 µM). Test compounds were reconstituted in dry DMSO as a 50 mM stock concentration (based on mass of the HCl salt). Fluid transfers were handled with automated liquid handlers with requisite peripheral devices.
4.2.3. mESC culture and differentiation
mESCs were thawed and expanded in feeder free conditions using normal growth media supplemented with LIF. Cells were passaged on poly-d-lysine coated tissue culture plastic 5 times prior to use in the assay. For differentiation experiments, cells were monodispersed and 50 µL were applied to gelatin coated, clear-bottom, black wall, 384-well microtiter plates in normal growth media without LIF. Seeding cell density was held at 92 cells/mm2 or about 1000 cells/well. Cells were grown at 37 °C and 5% CO2. After 2 days in normal growth media, compounds were added to wells by supplying an equivalent volume (50 µL) of normal growth media at 2-fold the final compound concentration. For the primary screen, compounds were refreshed on day 4 by aspirating 50 µL media from each well and replacing with 50 µL of normal growth media fortified with 1× the final compound concentration. Media was replenished with 50 µL of normal growth media on days 6 and 8. For the SAR screen, compounds were not added on day 4, but media was replenished with 50 µL of normal growth media on days 4, 6 and 8. The cell cultures were stopped on day 9 or 10 depending on the maturity of the culture as determined by control experiments. Cell cultures were terminated by adding 25 µL of 4% paraformaldehyde to each well and incubated for 40 min. Thereafter, the normal growth media was completely removed by aspiration. Microtiter plates were then washed three times with (1 mM, potassium phosphate, pH 7.4) and then incubated for 30 min with DAPI (Sigma) at 1 µg/mL. The cells were finally washed twice with PBS and then prepared for storage and imaging by adding 40 µL of PBS/Glycerol (1:1 v/v).
4.2.4. Immunostaining mESC cultures at day 10 or day 12
Cell cultures were terminated by removing media from the plate by aspiration and adding back 25 µL/well 4% paraformaldehyde (Sigma) for 40 min at room temperature. Cells were permeablized and blocked for 30 min using 1× PBS supplemented with 10% heat inactivated horse serum and 0.2% Triton X-100 (Sigma). Primary antibody (anti-αActinin, 15 µL/well) (Millipore) diluted 1:200 was incubated for 60 min, washed three times with 1 × PBS fortified with 0.2% Triton X-100. The wells were incubated at 25 °C for 10 min between washes. The secondary antibody (donkey anti-mouse Alexa568) (Life Technologies) was diluted 1:400 in 1× PBS with 0.2% Triton X-100 and 1 µg/mL DAPI and 15 µL of this solution was added to each well and incubated for 60 min. The cells were washed again 3 times as described above for the primary screen and then each well was stored in 40 µL 0.5·× PBS and 50% glycerol prior to imaging.
Supplementary Material
Acknowledgments
We are grateful to Jia J. Ding and Tara Wu for helping with the synthesis of some of the benzimidazole compounds. We thank Dr. Marcia Dawson and Dr. Zebin Xia for their intellectual contribution and support throughout the duration of this project. We are grateful to Ying Su for her contribution to the cheminformatics preceding the SAR described in this work. The financial support provided by CIRM (RC1-00132 to Mark Mercola and RS1-00169-1 to John Cashman) and NIH (R37HL059502 and R01HL108176 to Mark Mercola) is gratefully acknowledged.
Footnotes
Supplementary data (synthesis and 1H NMR of compounds 4–15, 17–29 and 67a details of the image analysis and supporting figure and movie) associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmc.2015.07.073.
References and notes
- 1.Roger VrL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Soliman EZ, Sorlie PD, Sotoodehnia N, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Circulation. 2012;125:e2. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabe-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, Jovinge S, Frisen J. Science. 2009;324:98. doi: 10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hsieh PC, Segers VF, Davis ME, MacGillivray C, Gannon J, Molkentin JD, Robbins J, Lee RT. Nat. Med. 2007;13:970. doi: 10.1038/nm1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kajstura J, Urbanek K, Perl S, Hosoda T, Zheng H, Ogorek B, Ferreira-Martins J, Goichberg P, Rondon-Clavo C, Sanada F, D’Amario D, Rota M, Del Monte F, Orlic D, Tisdale J, Leri A, Anversa P. Circ. Res. 2010;107:305. doi: 10.1161/CIRCRESAHA.110.223024. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 5.Senyo SE, Steinhauser ML, Pizzimenti CL, Yang VK, Cai L, Wang M, Wu TD, Guerquin-Kern JL, Lechene CP, Lee RT. Nature. 2013;493:433. doi: 10.1038/nature11682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Willems E, Lanier M, Forte E, Lo F, Cashman J, Mercola M. J. Cardiovasc. Transl. Res. 2011;4:340. doi: 10.1007/s12265-011-9270-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lanier M, Schade D, Willems E, Tsuda M, Spiering S, Kalisiak J, Mercola M, Cashman JR. J. Med. Chem. 2012;55:697. doi: 10.1021/jm2010223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schade D, Lanier M, Willems E, Okolotowicz K, Bushway P, Wahlquist C, Gilley C, Mercola M, Cashman JR. J. Med. Chem. 2012;55:9946. doi: 10.1021/jm301144g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Willems E, Cabral-Teixeira J, Schade D, Cai W, Reeves P, Bushway PJ, Lanier M, Walsh C, Kirchhausen T, Izpisua Belmonte JC, Cashman J, Mercola M. Cell Stem Cell. 2012;11:242. doi: 10.1016/j.stem.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Willems E, Spiering S, Davidovics H, Lanier M, Xia Z, Dawson M, Cashman J, Mercola M. Circ. Res. 2011;109:360. doi: 10.1161/CIRCRESAHA.111.249540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oh SW, Kim B, Jeon S, Go DM, Kim MK, Baek K, Oh GT, Kim DY. Life Sci. 2013;93:409. doi: 10.1016/j.lfs.2013.07.016. [DOI] [PubMed] [Google Scholar]
- 12.Takahashi T, Lord B, Schulze PC, Fryer RM, Sarang SS, Gullans SR, Lee RT. Circulation. 2003;107:1912. doi: 10.1161/01.CIR.0000064899.53876.A3. [DOI] [PubMed] [Google Scholar]
- 13.Wu X, Ding S, Ding Q, Gray NS, Schultz PG. J. Am. Chem. Soc. 2004;126:1590. doi: 10.1021/ja038950i. [DOI] [PubMed] [Google Scholar]
- 14.ChemBridge’s DIVERSettm. www.chembridge.com. [Google Scholar]
- 15.Calculated using ChemDraw Ultra version 11.0. http://www.cambridgesoft.com/Ensemble_for_Chemistry/ChemDraw/ [Google Scholar]
- 16.Bell MGW, Day M, Peters AT. J. Soc. Dyers Colour. 1966;82:410. [Google Scholar]
- 17.Day M, Peters AT. J. Soc. Dyers Colour. 1969;85:8. [Google Scholar]
- 18.Janaki WT, Rathinammal N. J. Indian Chem. Soc. 1987;64:250. [Google Scholar]
- 19.Agrawal VK, Sharma S, Iyer RN. Indian J. Chem., Sect. B. 1981;20B:398. [Google Scholar]
- 20.Yang D, Fokas D, Li J, Yu L, Baldino CM. Synthesis. 2005;2005:47. [Google Scholar]
- 21.Purandare AV, Gao A, Poss MA. Tetrahedron Lett. 2002;43:3903. [Google Scholar]
- 22.Shibata M, Kawai K, Makihara T, Yonekura N, Kawashima T, Sakai J, Muramatsu N. US6576631. 2003
- 23.Shibata M, Kawai K, Makihara T, Yonekura N, Kawashima T, Sakai J, Muramatsu N. US6872729. 2005
- 24.Queiroz M-JoRP, Calhelha RC, Kirsch G. Tetrahedron. 2007;63:13000. [Google Scholar]
- 25.Stover JS, Rizzo CJ. Org. Lett. 2004;6:4985. doi: 10.1021/ol047851m. [DOI] [PubMed] [Google Scholar]
- 26.Bekolo H. Can. J. Chem. 2007;85:42. [Google Scholar]
- 27.Shen MM. Development. 2007;134:1023. doi: 10.1242/dev.000166. [DOI] [PubMed] [Google Scholar]
- 28.Van Vliet P, Wu SM, Zaffran S, Puceat M. Cardiovasc. Res. 2012;96:352. doi: 10.1093/cvr/cvs270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bushway P. San Diego, San Diego: University of California; 2012. [Google Scholar]
- 30.Bushway PJ, Mercola M. Methods Enzymol. 2006;414:300. doi: 10.1016/S0076-6879(06)14017-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.