

Abstract
Copy number variations at multiple chromosomal loci, including 16p11.2, have recently been implicated in the pathogenesis of autism spectrum disorder (ASD), a neurodevelopmental disease that affects 1~3% of children worldwide. The aim of this study was to investigate the roles of human genes at the 16p11.2 loci in synaptic development using Drosophila larval neuromuscular junctions (NMJ), a well-established model synapse with stereotypic innervation patterns. We conducted a preliminary genetic screen based on RNA interference in combination with the GAL4-UAS system, followed by mutational analyses. Our result indicated that disruption of klp68D, a gene closely related to human KIF22, caused ectopic innervations of axon branches forming type III boutons in muscle 13, along with less frequent re-routing of other axon branches. In addition, mutations in klp64D, of which gene product forms Kinesin-2 complex with KLP68D, led to similar targeting errors of type III axons. Mutant phenotypes were at least partially reproduced by knockdown of each gene via RNA interference. Taken together, our data suggest the roles of Kinesin-2 proteins, including KLP68D and KLP64D, in ensuring proper synaptic wiring.
Keywords: Autism, Copy number variations, 16p11.2, Kinesin-2, Drosophila
INTRODUCTION
Autism spectrum disorders (ASDs) are the heterogeneous group of childhood diseases defined by social impairments, defects in verbal and non-verbal communication, and repetitive behaviors [1]. ASD phenotypes can include co-morbidity for various neuropsychiatric disorders such as epilepsy [2]. Defining molecular pathways dysfunctional in ASD is crucial for understanding its pathophysiology. A key question is whether common molecular pathways will emerge for ASD based on defined mutations and de novo genome rearrangements that account for 5~20% of ASDs. Current evidence suggests that the disease may result from disruption in synapse formation and synaptic plasticity during development [3].
Recent progress in the genetics of ASD has revealed that the frequency of chromosomal rearrangements is strikingly high (~7%) in ASD patients [4]. Linkage analyses, genome-wide association studies (GWAS), and microarray assays have revealed rare, but strong associations between ASD and frequent microdeletions or microduplications of certain chromosomal loci defined as copy number variations (CNVs) [5]. Among the CNVs recently identified, microdeletions (as well as microduplications) of 16p11.2 have been consistently observed in several familial studies and are associated with 0.5~1% of ASD cases [6,7]. The 16p11.2 microdeletion has been sub-mapped to affect 24~27 annotated genes [8,9,10], but the loss-of-function loci responsible for the increased risk of ASD are currently unknown. Importantly, several genes at the 16p11.2 loci appear to play important roles in the neurodevelopmental processes [8,11,12,13,14]. While experimental approaches using model organisms to study the roles of these genes remain limited, a recent study using zebrafish embryos demonstrated structural changes in the brain and eyes along with formation of abnormal axonal tracts caused by defects in zebrafish homologs of human genes at the 16p11.2 loci [10], suggesting their significant contributions to development of the nervous system.
In this study, we used Drosophila melanogaster larval neuromuscular junctions (NMJs), a well-established model synapse to analyze contributions of individual genes in the 16p11.2 region to synaptic connectivity between correct partners required for normal synaptic functions. Drosophila NMJ is a glutamatergic synapse characterized with stereotypic innervation patterns of motor neurons [15] into well-defined target body-wall muscles [16], allowing to identify target selection errors with ease. Upon conducting a genetic screen using a RNA interference approach along with mutational analyses, we found that disruption of klp68D, a Kinesin-2-encoding gene closely related to human KIF22 at the 16p11.2 loci, induced ectopic targeting of motor axons. In addition, disruption of KLP64D, another component of Kinesin-2 complex assembled with KLP68D, resulted in similar morphological defects, suggesting critical roles of Kinesin-2 proteins in final target selection process during development of the nervous system.
MATERIALS AND METHODS
Fly stocks
All crosses and stocks were raised on standard media at 24℃. The w1118 strain was used as a wild-type (WT) control for mutants. The y1 v1; P{CaryP}attP2 and y1 v1; P{CaryP}attP40 stocks obtained from Bloomington Drosophila Stock Center were used as controls for RNAi experiments. The mutant lines used for our study include: Klp68DKG03849, Klp64Dk1/TM3 , and Klp64Dn123/TM3 . The elavc155-GAL4 driver combined with UAS-Dicer2 was used for expression of neuron-specific RNAi lines with the well-established GAL4-UAS system [17]. The RNAi stocks examined include: y1 v1; P{TRiP.JF03346}attP2 (UAS-Klp68D-dsRNA) and y1 v1; P{TRiP.HMS02193}attP40 (UAS-Klp64D-dsRNA) from Bloomington Drosophila Stock Center and hairpin UAS-RNAi stocks for Klp68D, rolled, Pp4-19C, CG10465, crol, CG10186, CG8421, dTao-1, rabphilin, doc1, CG9245, CG17841, CG6058, and CG15309 from Vienna Drosophila Resource Center. Whenever possible, we used multiple RNAi lines and mutant alleles for each gene to confirm the effect of mutations and RNA interference and combined the results from multiple variants since they showed similar trends, albeit slight differences in the severity of their phenotypes.
Immunohistochemistry and Imaging
Male third instar larvae were dissected in 1X Phosphate-buffered saline (PBS) and prepared for immunofluorescent staining, as previously described [18]. Alexa 594-conjugated anti-HRP antibody (Jackson ImmunoResearch Laboratories, Inc., USA) was used at 1:200 to visualize NMJs of wandering third instar larvae. The Z-stack images of ventral longitudinal bodywall muscles 12 and 13 in the abdominal segments A2 and A3 were obtained using a confocal microscope (Zeiss, LSM700; Carl Zeiss, Germany) and merged using ZEN software (Carl Zeiss). Following image acquisition, the pattern of aberrant axonal targeting was monitored in muscle 13 that is normally innervated by axon branches forming type Ib, Is and II boutons. The presence of ectopic innervation by axons other than these three types was counted for each genotype.
Statistical analysis
The 2×2 contingency tables including categorical data were constructed to represent the proportion of normal versus abnormal nerve innervation patterns. The data were then subjected to Fisher's exact text to compare the proportional differences between two groups. p-value less than 0.05 was considered significant.
RESULTS
Aberrant synaptic targeting of presynaptic axons upon knockdown of Klp68D
While copy number variations at the 16p11.2 loci were linked to a subset of familial ASD cases [6,7], the genes responsible for the pathophysiology of ASD within this region have not been clearly identified in experimental settings. The 16p11.2 loci spans 500~600 Kbps and encompasses approximately at least 24 coding genes [8] (Fig. 1A). To investigate the roles of individual human genes located within this region, we performed a genetic screen using Drosophila melanogaster, a well-established model system to study the structure and function of the nervous system. We first identified Drosophila homologs of human genes at the 16p11.2 loci (Fig. 1B). Among 24 candidates located in the 16p11.2 region, we were unable to identify Drosophila gene products homologous to human QPRT, c16orf54, MVP and HIRIP3, along with 5 more weakly related ones (Fig. 1B). Following exclusion of these candidates from the genetic screen and subsequent mutant analysis, we performed a RNAi-based genetic screen combined with the GAL4-UAS system [17] to disrupt the activity of individual gene products. The morphology of larval NMJs formed in the ventral longitudinal body-wall musculature, especially muscles 13 and 12 (Fig. 2), was then monitored in the remaining 15 candidates.
Fig. 1. Human 16p11.2 loci associated with autism spectrum disorder (ASD). (A) The approximate location of 16p11.2 loci within human chromosome 16 is indicated (red box), along with annotated genes located at the loci. (B) The 24 annotated human genes within the 16p11.2 region are matched with corresponding Drosophila homologs or most closely related genes. Human genes with Drosophila counterparts of weak homology (i.e. SPN) as well as those with no obvious homolog (i.e. MVP) were excluded from further analyses.
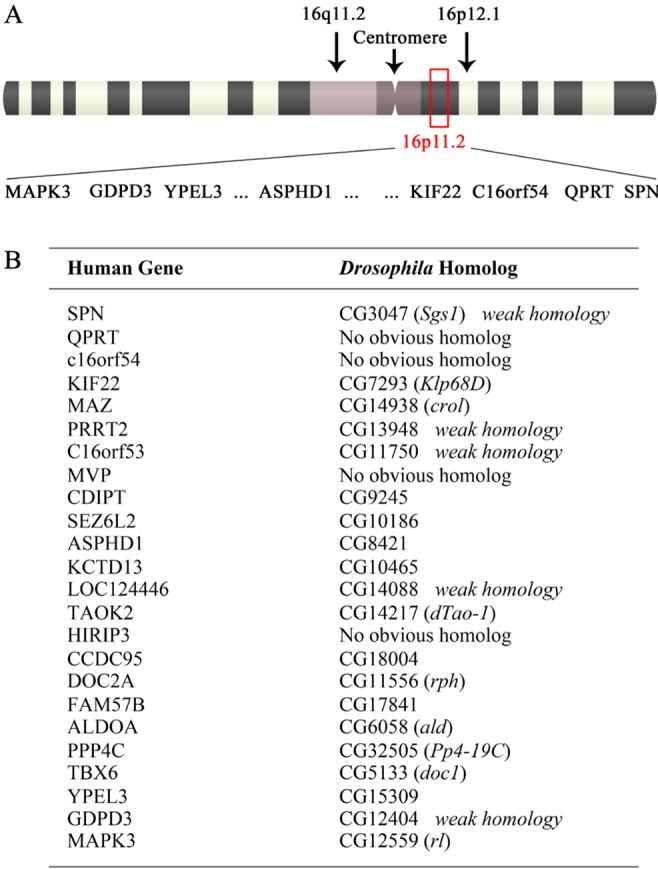
Fig. 2. Arrangements of larval neuromuscular junctions (NMJs) formed in ventral longitudinal body-wall musculature. (A) A schematic diagram is shown to represent the segmental arrangement of larval body-wall musculature. The ventral longitudinal body-wall muscles (VLMs) consisting of muscles 7, 6, 13 and 12 are enclosed in the red box. (B) A representative example of NMJs formed in the VLMs (A) is shown for WT. Three and four different innervations by presynaptic axon branches are indicated in muscles 13 and 12, respectively (arrowheads). Note a distinctive innervation of an axon forming type III boutons (hereafter type III axon) in muscle 12 (red arrowhead).
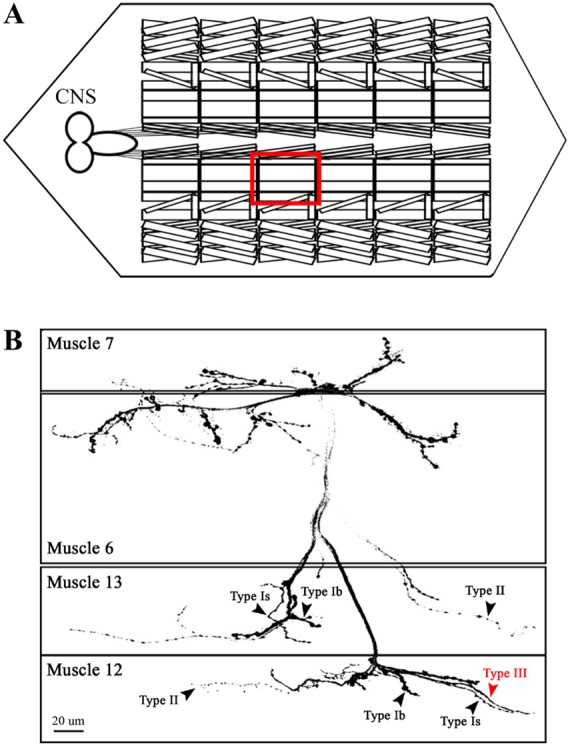
In WT, muscle 13 is normally innervated by three motor axon branches (Fig. 2B, arrowheads). These branches are originated from different motoneurons located in the ventral nerve cord, including ISN, SNa and SNb/SNd, and form type Ib, Is and II boutons that are different in size, ranging from 1~2 (type Is) to 3~6 µm in diameter (type Ib) [19,20]. In contrast, type III boutons from a SNb/SNd motoneuron [20] are observed almost exclusively in muscle 12, but rarely in muscle 13 (Fig. 2B, red arrowhead). Such distinctive innervation patterns in neighboring muscles allowed us to analyze potential errors in specific targeting of presynaptic axons. A preliminary RNAi-based genetic screen performed for the majority of candidate genes revealed NMJ morphology similar to that observed in WT. For instance, knockdown of doc1 and CG10465 did not induce any errors in targeting of different axon branches (Fig. 3A and 3B). In contrast, aberrant branching patterns were observed when KLP68D, a Drosophila gene product with high homology to human KIF22, was downregulated by RNA interference. As shown in Fig. 3, we were able to detect recurrent axon branches that were re-routed back to muscle 13 after their initial entry to muscle 12 (arrowheads, Fig. 3C). This phenotype was rarely caused by knockdown of other gene products (data not shown), suggesting a specific target selection influenced by the activity of KLP68D.
Fig. 3. Innervation patterns in animals expressing double-stranded RNA (dsRNA) constructs against Drosophila homologs of human genes at the 16p11.2 loci. Two representative NMJs are shown for larvae expressing dsRNAs against doc1 (A), CG10465 (B) and Klp68D (C). Note recurrent innervations induced by downregulation of KLP68D that re-route back to muscle 13 after their initial entry to muscle 12 (C), in contrast to WT-like morphology observed in (A) and (B). Scale bar, 20 µm.

Ectopic innervation of type III axons in muscle 13 in Klp68D mutants
Upon a detailed analysis of nerve innervation patterns in the RNAi-based screen, we also found ectopic innervation of axons forming type III boutons (hereafter type III axons) in muscle 13 along with recurrent axon branches caused by downregulation of KLP68D. Such targeting error was rarely seen in WT (Fig. 2B), but more frequently observed in animals expressing a double-stranded RNA (dsRNA) construct against Klp68D. To confirm that this phenotype was specifically induced by disruption of KLP68D, we examined the NMJ morphology of Klp68D mutants. Indeed, the mutants defective in Klp68D often displayed aberrant nerve innervation patterns characterized by additional targeting of type III axons in muscle 13 (Fig. 4A, red arrowheads). When the number of muscle 13 with type III axons was counted, we found a significant increase in the fraction of such ectopic innervation (i.e. the percentage of samples with ectopic innervations) in Klp68D mutants compared to WT (Fig. 4C, p<0.01). In addition, similar phenotypes were reproduced upon downregulation of KLP68D by RNA interference (Fig. 4C, p<0.05), confirming an important contribution of KLP68D to the axon targeting process.
Fig. 4. Ectopic innervation patterns of type III axon branches caused by disruptions in KLP68D and KLP64D. (A and B) Aberrant targeting of type III axon branches (red arrowheads) is observed in Klp68D (A) and Klp64D mutants (B). Scale bar, 20 µm. (C, left) Total number of abdominal segments used in the quantification of ectopic innervations is indicated for each genotype. The segments with normal branching patterns and aberrant type III branches in muscle 13 are indicated in white and black, respectively. (C, right) The fraction of samples with ectopic type III axons is analyzed for each genotype. *p<0.05, Fisher's exact test between WT and the genotype indicated. †p<0.05, Fisher's exact test between the paired groups (UAS-dsRNA control vs. GAL4-driven expression of UAS-dsRNA).

Ectopic innervation patterns reproduced by disruption in KLP64D, another Kinesin-2 complex protein assembled with KLP68D
KLP68D was first described as a kinesin-like motor protein with relatively high homology to kinesin heavy chain motor domain [19], thus presumably participating in axonal transport [21,22]. KLP68D is considered as a component of Kinesin-2 complex, along with KLP64D and DmKAP, a non-motor accessory protein [23], all of which are expressed in the central nervous system as well as a subset of peripheral nervous system [21,24]. Considering a high sequence homology between KLP68D and KLP64D [19], we investigated if disruption of KLP64D would also induce ectopic innervation patterns observed in Klp68D mutants.
When the innervation patterns were analyzed in animals expressing a double-stranded RNA construct against Klp64D, we found a significant increase in the fraction of targeting errors in the UAS-Klp64D-dsRNA control even in the absence of the GAL4-driver, suggesting its potential leaky expression at rest (Fig. 4C, p<0.05). As a result, the GAL4-driven expression of UAS-Klp64D-dsRNA led to a slight, but not significant increase in the fraction of ectopic innervation compared to its control (Fig. 4C, p=0.15). On the other hand, mutants defective in Klp64D exhibited ectopic innervations of type III axons in muscle 13, indistinguishable from those found in Klp68D mutants (Fig. 4B, red arrowheads; Fig. 4C, p<0.01, WT vs. Klp64D), raising a possibility of functional interplay between these two Kinesin-2 components. Together with mutant- and RNAi-based analyses of Klp68D, these results thus indicate two closely related Kinesin-2 complex proteins may participate in the same molecular pathway influencing proper selection of synaptic targets during development of the nervous system.
DISCUSSION
Recent clinical studies on ASD have revealed gross alterations in the structure of nervous system. For instance, total brain size and the rate of neuronal proliferation in the prefrontal cortex were found to be significantly increased in ASD patients [25]. Such structural changes may reflect altered neuronal connectivity between specific brain regions [26,27,28]. Moreover, recently proposed candidate genes responsible for ASD include various synaptic proteins that play important roles in neurite outgrowth, axonal guidance, axonal targeting and synaptogenesis [28,30,31], suggesting structural abnormalities at a synaptic level responsible for expression of ASD phenotypes. Based on these findings, we hypothesized that genetic perturbation at the 16p11.2 loci would lead to aberrant synaptic connectivity, thus underlying functional disturbances that lead to ASD. Our results demonstrate significant axon targeting errors caused by defects in KLP68D, a Drosophila Kinesin-2 protein closely related to human KIF22 at the autismlinked 16p11.2 loci.
Hetero-trimeric Kinesin-2 complex in Drosophila, consisting of KLP68D, KLP64D and DmKAP, has been implicated in microtubule organization [32,33] and axonal transport of synaptic proteins such as choline acetyltransferase [22]. However, experimental evidence is missing to support the idea that Kinesin-2 complex may participate in delivering molecules important for axon targeting. Potential cargos of KLP68D and KLP64D motors have been estimated to include Unc-51/ATG1, Fasciclin II [34], EB1 [35], Armadillo, Bazooka, and DE-cadherin [36], most of which have been well characterized for their roles in synaptogenesis. It will be important to investigate whether disruptions of any of these potential cargos lead to aberrant axon targeting phenotypes observed in Klp68D and Klp64D mutants.
It should be noted that Drosophila Nod ("no distributive disjunction"), mostly involed in chromosomal segregation [37] has been previously recognized as a homolog for human KIF22 [38]. However, similar levels of sequence homology to KIF22 were found in both Nod and KLP68D. In fact, a blast analysis in our hands resulted in higher sequence identity between KIF22 and KLP68D than Nod (41% vs. 33%). The specificity of motor protein cargos is often predicted to depend on the amino acid composition of motor proteins outside their core motor domain. Therefore, relatively lower level of homology between human KIF22 and Drosophila KLP68D may correspond to their distinct molecular functions. In contrast to KLP68D, the role of KIF22 in the mammalian nervous system has not been extensively investigated, but only limited to chromosomal segregation [39] and genomic stability [40]. Whether Drosophila KLP68D can be functionally replaced by human KIF22 in transgenic animals awaits further investigations.
In summary, we have demonstrated ectopic innervations caused by dysfunction of Drosophila Kinesin-2 proteins, including KLP68D and KLP64D. Abnormal synaptic connectivity observed in Klp68D as well as Klp64D mutants supports the idea that a subset of ASD can at least be interpreted as clinical outcomes of structural alterations during development of the nervous system. Considering the functions of Kinesin-2 involved in molecular trafficking, future investigations on the cargos transported by these proteins will improve our understanding of the pathophysiology of CNV-related ASD, including 16p11.2, from a molecular perspective.
ACKNOWLEDGEMENTS
This work was supported by Simons Initiative on Autism and the Brain Infrastructure Grant Program, 2009 and Pusan National University Research Grant, 2013 to Ji Hye Lee.
References
- 1.Baird G, Norbury CF. Social (pragmatic) communication disorders and autism spectrum disorder. Arch Dis Child. 2015 doi: 10.1136/archdischild-2014-306944. (in press) [DOI] [PubMed] [Google Scholar]
- 2.Lee BH, Smith T, Paciorkowski AR. Autism spectrum disorder and epilepsy: Disorders with a shared biology. Epilepsy Behav. 2015;47:191–201. doi: 10.1016/j.yebeh.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walsh CA, Morrow EM, Rubenstein JL. Autism and brain development. Cell. 2008;135:396–400. doi: 10.1016/j.cell.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cook EH, Jr, Scherer SW. Copy-number variations associated with neuropsychiatric conditions. Nature. 2008;455:919–923. doi: 10.1038/nature07458. [DOI] [PubMed] [Google Scholar]
- 5.Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, Leotta A, Pai D, Zhang R, Lee YH, Hicks J, Spence SJ, Lee AT, Puura K, Lehtimäki T, Ledbetter D, Gregersen PK, Bregman J, Sutcliffe JS, Jobanputra V, Chung W, Warburton D, King MC, Skuse D, Geschwind DH, Gilliam TC, Ye K, Wigler M. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cukier HN, Perez AM, Collins AL, Zhou Z, Zoghbi HY, Botas J. Genetic modifiers of MeCP2 function in Drosophila. PLoS Genet. 2008;4:e1000179. doi: 10.1371/journal.pgen.1000179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, Saemundsen E, Stefansson H, Ferreira MA, Green T, Platt OS, Ruderfer DM, Walsh CA, Altshuler D, Chakravarti A, Tanzi RE, Stefansson K, Santangelo SL, Gusella JF, Sklar P, Wu BL, Daly MJ Autism Consortium. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. 2008;358:667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- 8.Kumar RA, KaraMohamed S, Sudi J, Conrad DF, Brune C, Badner JA, Gilliam TC, Nowak NJ, Cook EH, Jr, Dobyns WB, Christian SL. Recurrent 16p11.2 microdeletions in autism. Hum Mol Genet. 2008;17:628–638. doi: 10.1093/hmg/ddm376. [DOI] [PubMed] [Google Scholar]
- 9.Horev G, Ellegood J, Lerch JP, Son YE, Muthuswamy L, Vogel H, Krieger AM, Buja A, Henkelman RM, Wigler M, Mills AA. Dosage-dependent phenotypes in models of 16p11.2 lesions found in autism. Proc Natl Acad Sci U S A. 2011;108:17076–17081. doi: 10.1073/pnas.1114042108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blaker-Lee A, Gupta S, McCammon JM, De Rienzo G, Sive H. Zebrafish homologs of genes within 16p11.2, a genomic region associated with brain disorders, are active during brain development, and include two deletion dosage sensor genes. Dis Model Mech. 2012;5:834–851. doi: 10.1242/dmm.009944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miyazaki T, Hashimoto K, Uda A, Sakagami H, Nakamura Y, Saito SY, Nishi M, Kume H, Tohgo A, Kaneko I, Kondo H, Fukunaga K, Kano M, Watanabe M, Takeshima H. Disturbance of cerebellar synaptic maturation in mutant mice lacking BSRPs, a novel brain-specific receptor-like protein family. FEBS Lett. 2006;580:4057–4064. doi: 10.1016/j.febslet.2006.06.043. [DOI] [PubMed] [Google Scholar]
- 12.Golzio C, Willer J, Talkowski ME, Oh EC, Taniguchi Y, Jacquemont S, Reymond A, Sun M, Sawa A, Gusella JF, Kamiya A, Beckmann JS, Katsanis N. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature. 2012;485:363–367. doi: 10.1038/nature11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, Shago M, Moessner R, Pinto D, Ren Y, Thiruvahindrapduram B, Fiebig A, Schreiber S, Friedman J, Ketelaars CE, Vos YJ, Ficicioglu C, Kirkpatrick S, Nicolson R, Sloman L, Summers A, Gibbons CA, Teebi A, Chitayat D, Weksberg R, Thompson A, Vardy C, Crosbie V, Luscombe S, Baatjes R, Zwaigenbaum L, Roberts W, Fernandez B, Szatmari P, Scherer SW. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82:477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Z, He X, Feng J. 16p11.2 is required for neuronal polarity. World J Neurosci. 2013;3:221–227. [Google Scholar]
- 15.Menon KP, Carrillo RA, Zinn K. Development and plasticity of the Drosophila larval neuromuscular junction. Wiley Interdiscip Rev Dev Biol. 2013;2:647–670. doi: 10.1002/wdev.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bate M, Landgraf M, Ruiz Gómez Bate M. Development of larval body wall muscles. Int Rev Neurobiol. 1999;43:25–44. doi: 10.1016/s0074-7742(08)60539-5. [DOI] [PubMed] [Google Scholar]
- 17.Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- 18.Lee J, Wu CF. Orchestration of stepwise synaptic growth by K+ and Ca2+ channels in Drosophila. J Neurosci. 2010;30:15821–15833. doi: 10.1523/JNEUROSCI.3448-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Keshishian H, Broadie K, Chiba A, Bate M. The drosophila neuromuscular junction: a model system for studying synaptic development and function. Annu Rev Neurosci. 1996;19:545–575. doi: 10.1146/annurev.ne.19.030196.002553. [DOI] [PubMed] [Google Scholar]
- 20.Hoang B, Chiba A. Single-cell analysis of Drosophila larval neuromuscular synapses. Dev Biol. 2001;229:55–70. doi: 10.1006/dbio.2000.9983. [DOI] [PubMed] [Google Scholar]
- 21.Pesavento PA, Stewart RJ, Goldstein LS. Characterization of the KLP68D kinesin-like protein in Drosophila: possible roles in axonal transport. J Cell Biol. 1994;127:1041–1048. doi: 10.1083/jcb.127.4.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ray K, Perez SE, Yang Z, Xu J, Ritchings BW, Steller H, Goldstein LS. Kinesin-II is required for axonal transport of choline acetyltransferase in Drosophila. J Cell Biol. 1999;147:507–518. doi: 10.1083/jcb.147.3.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stewart RJ, Pesavento PA, Woerpel DN, Goldstein LS. Identification and partial characterization of six members of the kinesin superfamily in Drosophila. Proc Natl Acad Sci U S A. 1991;88:8470–8474. doi: 10.1073/pnas.88.19.8470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sarpal R, Ray K. Dynamic expression pattern of kinesin accessory protein in Drosophila. J Biosci. 2002;27:479–487. doi: 10.1007/BF02705044. [DOI] [PubMed] [Google Scholar]
- 25.Courchesne E, Mouton PR, Calhoun ME, Semendeferi K, Ahrens-Barbeau C, Hallet MJ, Barnes CC, Pierce K. Neuron number and size in prefrontal cortex of children with autism. JAMA. 2011;306:2001–2010. doi: 10.1001/jama.2011.1638. [DOI] [PubMed] [Google Scholar]
- 26.Geschwind DH, Levitt P. Autism spectrum disorders: developmental disconnection syndromes. Curr Opin Neurobiol. 2007;17:103–111. doi: 10.1016/j.conb.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 27.Amaral DG, Schumann CM, Nordahl CW. Neuroanatomy of autism. Trends Neurosci. 2008;31:137–145. doi: 10.1016/j.tins.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 28.McFadden K, Minshew NJ. Evidence for dysregulation of axonal growth and guidance in the etiology of ASD. Front Hum Neurosci. 2013;7:671. doi: 10.3389/fnhum.2013.00671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Van Battum EY, Brignani S, Pasterkamp RJ. Axon guidance proteins in neurological disorders. Lancet Neurol. 2015;14:532–546. doi: 10.1016/S1474-4422(14)70257-1. [DOI] [PubMed] [Google Scholar]
- 30.Hussman JP, Chung RH, Griswold AJ, Jaworski JM, Salyakina D, Ma D, Konidari I, Whitehead PL, Vance JM, Martin ER, Cuccaro ML, Gilbert JR, Haines JL, Pericak-Vance MA. A noise-reduction GWAS analysis implicates altered regulation of neurite outgrowth and guidance in autism. Mol Autism. 2011;2:1–16. doi: 10.1186/2040-2392-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen J, Yu S, Fu Y, Li X. Synaptic proteins and receptors defects in autism spectrum disorders. Front Cell Neurosci. 2014;8:276. doi: 10.3389/fncel.2014.00276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mattie FJ, Stackpole MM, Stone MC, Clippard JR, Rudnick DA, Qiu Y, Tao J, Allender DL, Parmar M, Rolls MM. Directed microtubule growth, +TIPs, and kinesin-2 are required for uniform microtubule polarity in dendrites. Curr Biol. 2010;20:2169–2177. doi: 10.1016/j.cub.2010.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Doodhi H, Katrukha EA, Kapitein LC, Akhmanova A. Mechanical and geometrical constraints control kinesin-based microtubule guidance. Curr Biol. 2014;24:322–328. doi: 10.1016/j.cub.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 34.Mochizuki H, Toda H, Ando M, Kurusu M, Tomoda T, Furukubo-Tokunaga K. Unc-51/ATG1 controls axonal and dendritic development via kinesin-mediated vesicle transport in the Drosophila brain. PLoS One. 2011;6:e19632. doi: 10.1371/journal.pone.0019632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gu C, Zhou W, Puthenveedu MA, Xu M, Jan YN, Jan LY. The microtubule plus-end tracking protein EB1 is required for Kv1 voltage-gated K+ channel axonal targeting. Neuron. 2006;52:803–816. doi: 10.1016/j.neuron.2006.10.022. [DOI] [PubMed] [Google Scholar]
- 36.Mukhopadhyay B, Nam SC, Choi KW. Kinesin II is required for cell survival and adherens junction positioning in Drosophila photoreceptors. Genesis. 2010;48:522–530. doi: 10.1002/dvg.20642. [DOI] [PubMed] [Google Scholar]
- 37.Zhang P, Knowles BA, Goldstein LS, Hawley RS. A kinesin-like protein required for distributive chromosome segregation in Drosophila. Cell. 1990;62:1053–1062. doi: 10.1016/0092-8674(90)90383-p. [DOI] [PubMed] [Google Scholar]
- 38.Tokai N, Fujimoto-Nishiyama A, Toyoshima Y, Yonemura S, Tsukita S, Inoue J, Yamamota T. Kid, a novel kinesinlike DNA binding protein, is localized to chromosomes and the mitotic spindle. EMBO J. 1996;15:457–467. [PMC free article] [PubMed] [Google Scholar]
- 39.Vernos I, Karsenti E. Motors involved in spindle assembly and chromosome segregation. Curr Opin Cell Biol. 1996;8:4–9. doi: 10.1016/s0955-0674(96)80041-x. [DOI] [PubMed] [Google Scholar]
- 40.Maddika S, Sy SM, Chen J. Functional interaction between Chfr and Kif22 controls genomic stability. J Biol Chem. 2009;284:12998–13003. doi: 10.1074/jbc.M900333200. [DOI] [PMC free article] [PubMed] [Google Scholar]