

Abstract
This study focused on the development of flexible (i.e., deformable) multiple-unit pellets that feature (i) a prolonged drug release, (ii) drug abuse deterrence, and (iii) a minimal risk of alcohol-induced dose dumping (ADD). Deformable pellets were prepared via an advanced continuous one-step hot-melt extrusion (HME) technique, with the drug (i.e., antipyrine and codeine phosphate) fed as an aqueous solution into the molten matrix material (i.e., cornstarch, gum arabic, and xanthan). Formulations that had suitable mechanical characteristics (i.e., high compression strength) were coated with a flexible Aquacoat® ARC film to ensure prolonged release and to avoid ADD. The pellets were characterized in terms of their mechanical properties and in vitro drug release behavior in alcoholic media. All formulations were abuse deterrent: they had a high compression strength and grinding the pellets into powder was impossible. Since the pellets comprising gum arabic and xanthan as a matrix did not remain intact during dissolution testing, they had a very fast drug release rate. Cornstarch-based pellets that swelled but remained intact in the dissolution media had a slower drug release. Coated cornstarch-based pellets had a prolonged release over 8 h and resistance to dose dumping in 20 and 40% ethanol. Our results indicate that cornstarch-based pellets manufactured via the advanced HME process followed by coating are a promising formulation that makes tampering difficult due to a high compression strength combined with robustness in alcoholic media.
KEY WORDS: dose dumping, drug tampering, melt extrusion, starch
INTRODUCTION
Drug abuse of oral opioids and non-opioid drugs with a narrow therapeutic window, such as benzodiazepines (central nervous system depressants) and amphetamines (stimulants), has received increased public attention in recent years (1). Drug abuse can be defined as “the use of medication for its mind-altering effects, whether or not one also has pain or has been prescribed the medication” (2). Due to their high drug loading and euphoric effects, controlled-release opioid dosage forms that are first-choice formulations for (chronic) pain treatment (3–5) are especially popular among drug abusers (1). Drugs are either chewed or crushed to subsequently snort or dissolve them in common solvents for intravenous injections in order to achieve euphoric and mind-altering effects (1,6,7). To counteract these practices, the Food and Drug Administration (FDA) suggested several approaches described in the draft guidance document covering the evaluation and labeling of abuse-deterrent opioids (7,8), which include (i) physical and chemical barriers, (ii) agonist/antagonist combinations, (iii) aversion, (iv) certain delivery systems, (v) prodrugs, and (vi) combinations thereof (Fig. 1). Physical barriers prevent destruction of the dosage form (e.g., due to increased hardness), whereas chemical barriers impede extraction using common solvents (9). Incorporating an antagonist reduces the euphoria if the drug product is manipulated (e.g., ground). If the drug product is swallowed (i.e., used as prescribed), the antagonist is not clinically active. Aversion is created when upon manipulation, the drug product releases substances that cause unpleasant temporary side effects (e.g., warmth, flushing, itching, sweating). Furthermore, it is recommended to use unattractive delivery systems (e.g., subcutaneous implants). Prodrugs that hinder opioid activity and the associated euphoric effects until they are transformed in the gastrointestinal tract can prevent abuse via the intravenous and nasal routes (9).
Fig. 1.
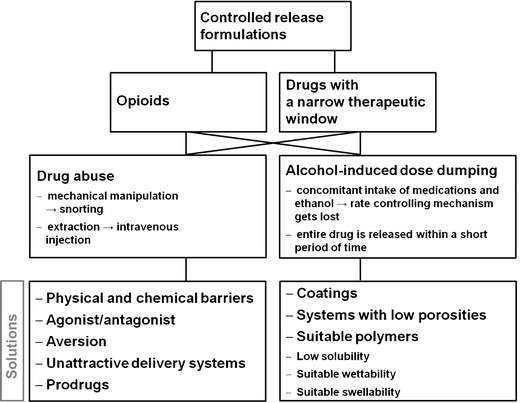
Systematic overview of drug-abuse- and alcohol-induced dose dumping
Although these are promising tools for ensuring safe and abuse-deterrent drugs, only a few proven products are available on the market and studies in this field are scarce (10–12). Hysingla™ ER tablets (Purdue Pharma) containing hydrocodone bitartrate provide abuse resistance via physical and chemical barriers (13). They are formulated using RESISTEC™, which obstructs tablet crushing by applying a unique combination of polymer and processing to ensure a high tablet hardness (i.e., physical barrier). Moreover, when dissolved in an aqueous environment, the tablet immediately forms a viscous hydrogel (i.e., chemical barrier) (13), making injecting it impossible. Another abuse-deterrent drug product is Zohydro® ER capsules comprising hydrocodone bitartrate and manufactured via BeadTek™ (14), a capsule formulation technology that incorporates well-known excipients and is designed to deter abuse. If the capsule is crushed and dissolved in liquids, a viscous gel is immediately formed (14).
Despite existing and hypothetical approaches to make affective abuse-deterrent formulations, tampering with drugs and subsequent drug abuse can never be eliminated entirely. Rather, formulators should focus on reducing the abuse potential by making tampering difficult and time consuming. Such formulations are less attractive to drug abusers, who are reported to spend no more than 10 min on tampering (15,16).
Another challenge in the development of safe drug products is minimizing the risk of alcohol-induced dose dumping (ADD) (Fig. 1). Concomitant intake of alcoholic beverages and controlled-release oral dosage forms may alter the release-rate-controlling mechanism of the formulation, resulting in an immediate and uncontrolled drug release (17) (e.g., an opioid overdose, which may lead to respiratory depression followed by hypoxia and even death (18)). Especially those patients that suffer from (chronic) pain tend to consume alcohol to cope with the pain-related stress and reduce the pain perception (17).
Overall, there is a growing demand for new flexible technologies for producing safe drug products that prevent both (1) drug abuse and (2) ADD. Recently, Gruenenthal GmbH patented a tamper-resistant oral dosage form comprising a drug with a psychotropic action and an ethylene-vinyl acetate (EVA) polymer (19). Manufactured via thermoforming technologies, such as hot-melt extrusion (HME), it is resistant to solvent extraction, grinding, and dose dumping in aqueous ethanol (19). However, the majority of the existing studies address either abuse deterrence or ADD (20) but not both topics at the same time.
In this study, we focus on the development of multiple-unit dosage forms (i.e., pellets) that show a prolonged release and are designed to resist ADD and (due to their high compression strength) tampering with the most common household devices. For pellet preparation, an innovative manufacturing process based on the HME technique was applied. Thereby, the (visco)elastic properties of the matrix materials (i.e., cornstarch, gum arabic, and xanthan) can be achieved by introducing water directly into the matrix melt. Subsequent coating of the pellets ensured a prolonged release of the model drugs (i.e., antipyrine and codeine phosphate) and ADD resistance.
MATERIALS AND METHODS
Materials
Antipyrine (Fluka, Sigma Aldrich, St. Louis, USA) and codeine phosphate hemihydrate (donated by G.L. Pharma GmbH, Lannach, Austria) were used as the model drugs. Cornstarch (Carl Roth GmbH, Karlsruhe, Germany), gum arabic (ACM Herba Chemosan AG, Vienna, Austria), and xanthan (Carl Roth GmbH, Karlsruhe, Germany) were the matrix carrier systems. Purified water was used as a plasticizer.
The in vitro drug release studies were carried out with 0.1 N hydrochloric acid (HCl) and a trisodiumphosphate-dodecahydrate buffer purchased from Merck, Darmstadt, Germany. For the dose dumping studies, absolute ethanol (EP) was purchased from VWR International, Darmstadt, Germany. The mobile phase for the reversed-phase (RP) high-performance liquid chromatography (HPLC) consisted of MilliQ water, acetonitrile (VWR International, Darmstadt, Germany), phosphoric acid (85%, VWR International, Darmstadt, Germany), and methanol (LiChrosolv® Reag., EP, VWR International, Darmstadt, Germany). Aquacoat® ARC (Alcohol-Resistant Coating, consisting of ethanol-soluble Aquacoat® ECD 30 and ethanol-insoluble guar gum) from FMC BioPolymer was used for pellet coating. Talc (Carl Roth GmbH, Karlsruhe, Germany) served as an anti-tacking agent, and trieethyl citrate (TEC) (donated by G.L. Pharma GmbH, Lannach, Austria) was applied as a plasticizer for the coating dispersion.
Drug Solubility Determination
The drug solubility was determined in (a) water, (b) 0.1 N HCl, (c) 0.1 N HCl with ethanol concentrations of 20 and 40% (v/v), and (d) pure ethanol (96 v%). Saturated solutions were prepared and stored in an incubator shaker at 37 ± 0.5°C for 48 h. Samples of 1 ml were taken, filtered through a cellulose acetate filter (pore size 0.2 μm), and, if necessary, diluted. The concentration of the dissolved drug was quantified via UV/vis spectrometry at wavelengths of 244 nm (antipyrine) and 284 nm (codeine phosphate). The solubility studies were performed in triplicate.
HME Process
Extrusion was based on the NANEX process (21), which allows side feeding of aqueous drug suspensions and solutions directly into a molten polymer that is miscible with water (22). The matrix material (i.e., cornstarch, xanthan, and gum arabic) in powder form was placed into a co-rotating twin screw extruder (ZSK 18, Coperion GmbH, Stuttgart, Germany). The model drugs (i.e., codeine phosphate hemihydrates and antipyrine) were dissolved in purified water (i.e., plasticizer) at concentrations of 0.5 and 1.0 g/ml for codeine phosphate and 1.0 g/ml for antipyrine. The solutions were fed from the side into the extruder containing the matrix melt. For codeine phosphate, two concentrations were used to provide different drug loadings while keeping the water content constant. Details regarding the process parameters are summarized in Table I.
Table I.
Formulations and Process Parameters Applied During HME
| Formulation | Matrix material | Active ingredient (wt%) | Process parameters | Knife rotational speed (rpm) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Torque (%) | Barrel zone temperatures (°C) | |||||||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |||||
| 1 | Cornstarch | Without (20% H2O) | 18 | 65 | 85 | 85 | 85 | 85 | 85 | 85 | 85 | 85 | 85 | 1300 |
| 2 | Cornstarch | Antipyrine 20% (aqueous solution) | 7 | 65 | 85 | 85 | 85 | 85 | 85 | 85 | 85 | 85 | 85 | 1500 |
| 3 | Cornstarch | Codeine phosphate 10% (aqueous solution) | 22 | 65 | 85 | 85 | 85 | 85 | 85 | 88 | 88 | 88 | 87 | 1500 |
| 4a | Cornstarch | Codeine phosphate 20% (aqueous solution) | – | – | – | – | – | – | – | – | – | – | – | – |
| 5 | Xanthan | Without (20% H2O) | 16 | 70 | 85 | 85 | 85 | 85 | 85 | 85 | 85 | 85 | 85 | 1600 |
| 6 | Xanthan | Antipyrine 20% (aqueous solution) | 2 | 50 | 50 | 63 | 85 | 85 | 85 | 85 | 85 | 85 | 83 | 2000 |
| 7 | Gum arabic | Without (20% H2O) | 25 | 60 | 65 | 75 | 75 | 75 | 75 | 75 | 75 | 74 | 74 | 1600 |
| 8 | Gum arabic | Antipyrine 20% (aqueous solution) | 11 | 60 | 60 | 65 | 65 | 65 | 65 | 65 | 65 | 60 | 65 | 1600 |
aAdding 20% codeine phosphate resulted in highly viscous strands that were not suitable for hot die face pelletizing
The matrix material was fed into the extruder via a gravimetric dosing device (K-Tron, Niederlenz, Switzerland) at a feeding rate of 0.5 kg/h. The screws had a length-to-diameter ratio (L/D) of 40, and the barrel consisted of 10 individually controllable heating sections. The screw speed was 200 rpm. The drug solutions were fed via a calibrated peristaltic pump (Ismatec IP 65, IDEX Health & Science GmbH, Wertheim, Germany) into barrel 3 of the extruder. The feeding rate of the aqueous solution was adjusted to 100 g/h to yield a final drug loading of 20% for antipyrine and of 10 and 20% for codeine phosphate. The material was extruded through a die plate with die holes of 1.0 mm in diameter. The cornstarch/antipyrine formulation was also extruded through a die plate with die holes of 1.5 mm diameter. The homogeneous strands were cut directly at the die face using a hot die face pelletizer (23) (Automatik Plastics Machinery GmbH, Großostheim, Germany) with two rotating knives developed in-house. The rotational speed of the knives was adjusted manually to obtain pellets in the desired size range (i.e., 1.25–1.40 mm), which were immediately air-cooled (24). An overview of the formulations and the processing parameters is provided in Table I.
Loss on drying (LOD) of the pellets was determined using an oven at 60°C for 24 h. Pellets were sieved according to Pharm. Eu. 7.0/2.09.38.00 using analytical DIN sieves (Retzsch GmbH, Haan, Germany). The pellet fractions between 1.25 and 1.4 mm were applied for further characterization studies.
Coating Process
Selected pellet formulations were coated with Aquacoat® ARC, as proposed by Rosiaux et al. (25–27). The coating dispersion was prepared by mixing Aquacoat® ECD 30 and guar gum at a ratio of 93:7. Prior to mixing, Aquacoat® ECD 30 was plasticized with 25% TEC (w/w; based on the total polymer content) at room temperature for 30 min. Guar gum was dissolved in purified water under stirring at 65°C for 2 h. After cooling down to room temperature, the two solutions were mixed and stirred for 30 min. To reduce the stickiness of the coating formulation, talc (50% w/w; based on the total polymer content) was added as an anti-tacking agent.
The pellets were coated in a fluidized bed coater (Mycrolab H00472, Oystar Hüttlin, Schopfheim, Germany) equipped with a bottom sprayer. The coating conditions were as follows: inlet air volume 25 m3/h, inlet air temperature 38°C, spray rate 2 g/min, spray air pressure 1.2 bar, microclimate pressure 0.6 bar, nozzle diameter 1.2 mm, and coating level 20%. After coating, the pellets were dried in the fluidized bed coater for 15 min.
Characterization of Drug-Loaded Pellets
Differential Scanning Calorimetry Analysis
The solid-state properties of the hot-melt-extruded pellets were characterized using a differential scanning calorimeter (DSC 204F1 Phoenix®, Netzsch GmbH, Selb, Germany). For comparison reasons, we evaluated the thermal behavior of the model drugs, the matrix materials, and mixtures of the matrix materials with 20% purified water that acted as a plasticizer during extrusion. Samples of about 5 mg were placed into hermetically sealed aluminum crucibles and scanned with pure nitrogen as the analytical gas between 25 and 200°C at a heating rate of 50 K/min and at a flow rate of 20 ml/min. After cooling to 25°C at 10 K/min, a second heating run was performed. An empty aluminum crucible was used as a reference. The DSC data analysis was conducted with Proteus Thermal Analysis software (Netzsch GmbH, Selb, Germany). Each sample was evaluated in triplicate.
Pellet Compression Strength
The pellet compression strength was determined with a conventional tablet hardness tester (PTB 111 E, Pharma Test, Hainburg, Germany) by recording the hardness values of 10 randomly picked pellets from each batch.
Additionally, the compression force of the pellets was studied using a rheometer (Physica MCR 301, Anton Paar GmbH, Graz, Austria) equipped with a parallel plate measuring system in the non-rotational mode. A single pellet was manually placed in the center of the lower plate. The upper plate was moved down at a constant velocity of 0.5 μm/s. Based on the force displacement diagrams, the maximum force of plastic deformation upon pellet fraction (F) was determined. The crushing strength (σ) was calculated from F and the diameter of each pellet (d) according to Shipway et al. (28):
| 1 |
From each batch, 50 pellets were tested with the rheometer.
In Vitro Dissolution Studies
All in vitro dissolution tests were carried out via the USP 28 rotating basket method <711> in a USP apparatus I (Pharma Test, Hainburg, Germany). The rotational speed was 100 rpm, and the temperature was 37 ± 0.5°C. The dissolution medium consisted of 750 ml 0.1 N HCl. After 2 h, 250 ml trisodiumphosphate-dodecahydrate buffer was added to increase the pH from 1.2 to 6.8. For the ADD studies, we used 900 ml HCl (0.1 N) with ethanol concentrations of 20% (equivalent to mixed drinks) and 40% (equivalent to hard liquor) (v/v) and tested them for 2 h. The pellet sample weight was adjusted to 1 g to ensure perfect sink conditions in all dissolution media without exceeding the maximum single dose. Samples of 1 ml were withdrawn at predetermined time intervals. Each batch was investigated in triplicate.
To compare the drug release profiles in alcoholic and non-alcoholic media, the f2 similarity factor was used (29):
| 2 |
where n is the number of dissolution time points considered and Rt and Tt are the percentages of drug dissolved of the reference and test formulations at time point t. In general, an f2 value in the range of 50–100 indicates that the dissolution profiles are similar.
Drug Quantification
Drug quantification of the dissolution samples was performed via reversed-phase (RP) high-performance liquid chromatography (HPLC) for antipyrine and via UV/vis spectroscopy for codeine phosphate.
The HPLC analysis was carried out using a Merck-Hitachi system (Tokyo, Japan) at 35°C. As the stationary phase, a C18 MOS-Hypersil column (250 × 4 mm × 5 μm; 120 Ǻ pore size; VDS optilab, Berlin, Germany) was applied. The mobile phase consisted of MilliQ water, which was adjusted to pH 3.0 with phosphoric acid and a 50:50 (v/v) mixture of acetonitrile and methanol (isocratic mode 58:42 (v/v)). The injection volume was 20 μl and the flow rate was 1 ml/min. The drug was analyzed with a model series L-4250 UV-VIS detector at a wavelength 230 nm. For quantification, a single-point calibration with 100% standard solution of the active pharmaceutical ingredient (API) was used.
The codeine phosphate quantification was performed with a UV spectrophotometer (Spectronic Genesys 5, Spectronic Instruments Inc., Rochester, USA) at a wavelength of 248 nm. The samples were diluted, if necessary, and quantified via an external standard (i.e., calibration curve).
RESULTS AND DISCUSSION
Drug Solubility Determination
Since a drug’s solubility in the dissolution medium strongly affects the dissolution rate, the solubility of antipyrine and codeine phosphate was determined in water, acidic media with and without ethanol (20 and 40%), and in 96% ethanol (Fig. 2). The results indicate that antipyrine is highly soluble in all tested media. The ethanol content did not affect the solubility until its concentration reached 40%. In 96% ethanol, the solubility decreased significantly due to the different polarity/dielectric constants of the solute and the solvent. However, antipyrine was still highly soluble in 96% ethanol (30). In contrast, the solubility of codeine phosphate markedly decreased with the increasing ethanol concentrations (31) and was very low in 96% ethanol. It can be concluded that the addition of ethanol lowered the dielectric constant in relation to pure water, and hence, the solubility decreased (17,32,33).
Fig. 2.
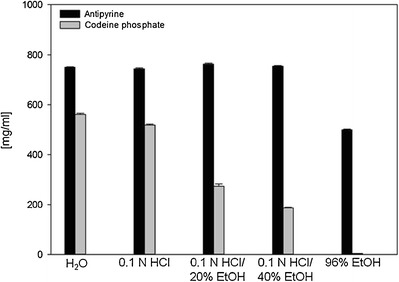
Solubility of model drugs in various media at 37 ± 0.5°C. Mean values ± SD (n = 3). As the standard deviation was very low, it is not clearly visible from the graph. Solubility of codeine phosphate is based on the data of Jedinger et al. (27)
Preparation of Pellets via HME
According to the literature, extrusion of pure native starch is not possible and adding water is required to achieve gelatinization and to transform starch into a homogeneous matrix (34). Starch is composed of α-d-glucosyl units. It comprises amorphous regions as well as crystalline domains in the short-chain clusters of amylopectin. During extrusion, the starch granules are destroyed by mechanical disruption of molecular bonds due to the intense shear forces within the extruder and loss of crystallinity is caused (i.e., gelatinization of starch) (35).
When cornstarch was extruded in our experiments with water at process temperatures between 65 and 85°C, glassy-like yellowish spherical pellets with a smooth, homogeneous surface structure were obtained. The pellets were very flexible and elastic, exhibiting mechanical properties suitable for abuse-deterrent dosage forms.
To the best of our knowledge, xanthan and gum arabic have not been used as matrix materials for HME to date. Xanthan is assigned to the polysaccharides from the chemical point of view and comprises d-glucose, d-mannose, and d-glucuronic acid. Gum arabic is a complex mixture of glycoproteins and polysaccharides. Both, xanthan and gum arabic are crystalline. During extrusion, it is assumed that crystallite softening occurs.
When gum arabic and xanthan were extruded without any plasticizers (i.e., water) at process temperatures of 60–85°C, a non-molten powdery and brittle mass was obtained. At higher process temperatures (i.e., 80–200°C), the substances failed to melt and became charred. However, when water was added as a plasticizer, smooth pellets with a yellowish coloration were produced via HME. They were slightly sticky since the amount of water added was rather high (∼20%), which was necessary to obtain suitable strands for further downstream processing (die face cutting). The gum arabic and xanthan pellets had a cylindrical shape, possibly due to faster solidification (and thus less time for forming a spherical shape), compared to cornstarch (24). Nevertheless, they were considered to be suitable for providing abuse deterrence due to their deformable character.
The next step was to incorporate drugs at various concentrations. While adding antipyrine to the starch matrix decreased the torque from 18 to 7% (Table I) due to the plasticizing effect of the drug, incorporating codeine phosphate increased the torque values (i.e., 22%; Table I). Adding 20% codeine phosphate resulted in highly viscous strands that were not suitable for hot die face pelletizing. Decreasing the drug loading to 10% yielded strands that could be cut into spherical pellets via die face pelletizing. Once again, the pellets were very flexible and elastic.
Incorporating antipyrine into the xanthan matrix decreased the extrusion temperatures from 70–85 to 50–83°C due to the plasticizing effect of the drug. This is confirmed by the torque value that decreased from 16 to 2% (Table I). Adding antipyrine to the gum arabic matrix slightly reduced the required process temperatures (i.e., from 60–75 to 60–65°C). Additionally, the torque decreased from 25 to 11% (Table I), indicating the plasticizing effect of antipyrine. Similarly to the drug-free pellets, the drug-loaded xanthan and gum arabic pellets were smooth and sticky, with a yellowish coloration. As before, the pellets were not spherical but rather cylindrical in shape. The placebo pellets exhibited approximately 20% w/w LOD, which decreased by drug addition. Incorporating antipyrine led to approximately 17% w/w LOD for all pellet formulations. The cornstarch/codeine phosphate pellets showed 15% w/w LOD. However, the added total amount of water was kept constant for all formulations. It is assumed that the lower weight loss during drying of antipyrine- and codeine-phosphate-containing pellets may be due to drug-bound water.
The pellets that were extruded with the 1.0-mm die had a yield of >50% of the sieve fraction between 1.25 and 1.40 mm.
Characterization of Pellets
Thermal Characterization
First, we evaluated the thermal behavior of the matrix materials in the presence of water (i.e., a plasticizer during extrusion) via DSC (Fig. 3a–d). For pure cornstarch and xanthan, a broad endothermic event with an onset at 107.4 and 115.0°C was recorded during the first heating cycle (data not shown) due to water loss (36). Upon cooling and reheating, no thermal events were observed. The glass transition (Tg) of pure cornstarch, which occurs at around 58°C (37), was not detected via conventional DSC with comparatively low heating rates (37,38). The thermogram of pure gum arabic showed no thermal events in the investigated temperature range (i.e., 20–200°C). Adding water to the matrix materials resulted in a broad endotherm with an onset between 100 and 120°C due to water evaporation (Fig. 3a–d). No thermal events were observed during cooling and the second heating cycle for all matrix materials. Since no degradation occurred under 200°C, the matrix materials were considered to be stable at the applied extrusion temperatures (i.e., 50–85°C).
Fig. 3.
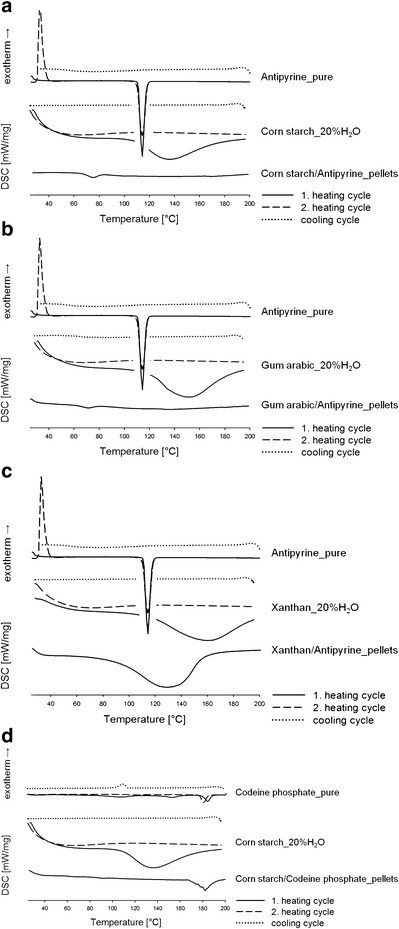
a–d DSC scans of the model drugs (i.e., antipyrine, codeine phosphate), matrix materials (i.e., cornstarch, gum arabic, xanthan) with the addition of 20% purified water (two heating cycles shown), and hot-melt-extruded pellets (one heating cycle shown)
Additionally, DSC was used to investigate the solid state of the model drugs and hot-melt-extruded pellets (Fig. 3a–d). The thermogram of the first heating cycle of antipyrine clearly indicated one sharp endotherm at 113.6°C (onset 107.6°C), which corresponds to melting. No thermal events were observed during cooling, suggesting that the drug was in its amorphous state after cooling. Upon reheating, one exothermic event and one endothermic event were recorded. The exothermic signal at 31.8°C (onset 26.5°C) was due to recrystallization of the amorphous drug, followed by melting at 113.6°C (onset 104.7°C) (39). Upon heating, codeine phosphate had three endothermic events (Fig. 3d) corresponding to water loss, structural rearrangement, and melting. For detailed interpretation, please refer to Jedinger et al. (31).
The DSC scan of the cornstarch/antipyrine pellets indicated one endothermic event at 70.8°C (onset 63.3°C) (Fig. 3a). The endotherm was attributed to the gelatinization (melting) of the starch granules, which occurs at around 70°C with high water contents (40). For the gum arabic/antipyrine pellets, one endotherm was detected at 71.0°C (onset 63.7°C), which corresponds to crystallite melting of gum arabic (Fig. 3b) (41). During the first heating of xanthan/antipyrine pellets, a broad endothermic event occurred at 129.4°C (onset 88.7°C) due to moisture evaporation (Fig. 3c). Since the melting peak of antipyrine did not show in the heating thermograms of starch/antipyrine and gum arabic/antipyrine pellets, it was assumed that the drug molecularly dissolved in the matrix, which agrees with previous studies (24,42). For the xanthan/antipyrine pellets, the solid state of antipyrine could not be determined since the melting endotherm of antipyrine appears in the same temperature range as the moisture evaporation does.
The thermogram of the cornstarch/codeine phosphate pellets suggests an endothermic event at 181.2°C (onset 176.4°C), which corresponds to the structural rearrangement (43) of codeine phosphate (Fig. 3d). The presence of crystalline codeine phosphate indicates that the drug did not molecularly dissolve in the cornstarch matrix. However, the characteristic endotherm of the drug broadened and shifted slightly towards lower temperature.
No thermal events were observed during cooling and the second heating cycle for all pellet formulations (data not shown).
Pellet Compression Strength and Physical Manipulation
The compression force of the drug-loaded pellet formulations exceeded the maximum force that can be applied by the rheometer (i.e., 50 N). All tested pellets deformed but yet did not break, suggesting that the force required for compression was higher than 50 N. Consequently, the compression force could not be determined and the compression strength could not be calculated from Eq. (1).
Additionally, the compression force was determined via a conventional tablet hardness tester with a maximum applicable force of 500 N. Again, the pellets deformed but did not recover (i.e., plastic deformation), implying that the force necessary to crush the pellets exceeds even 500 N. That means that a single pellet can be loaded with a mass of 50 kg without being crushed.
Physical (mechanical) manipulation was tested according to the FDA’s draft guidance on abuse-deterrent opioids (8): the pellet formulations were ground first with a spoon (i.e., a common household device) and then, more sophisticated, with a mortar and a pestle. Both manipulation methods resulted in large sticky fragments and no fine powders were obtained (Fig. 4). This shows that physical manipulation is impeded for all tested pellet formulations due to their high compression strengths independent upon the formulation composition.
Fig. 4.

Hot-melt-extruded xanthan/antipyrine (20%) pellets a before and b after grinding with a mortar and a pestle
In Vitro Drug Release of Hot-Melt-Extruded Pellets
Figure 5a illustrates the API release from the antipyrine pellets in non-alcoholic media. Due to the physicochemical properties of both, the pellet matrix and the drug, all pellets had an immediate drug release. High solubility of antipyrine in the dissolution media (i.e., 743.3 mg/ml) led to a fast drug release. Dissolution was fast also because the tested matrix materials (gum arabic, xanthan, and cornstarch) did not contain their shape during dissolution testing and did not retard release. Hence, diffusion of dissolved antipyrine through the matrix was not a rate-limiting step. While the gum-arabic-based pellets dissolved completely within 30 min due to physical erosion and subsequent degradation (44), the xanthan pellets swelled without dissolving, forming a highly viscous gel due to uncoiling of the structure and the formation of hydrogen bonds with the water molecules (45). Typically, a gel layer forming on the outer surface of a dosage form is considered to control drug release (46). However, the pellets produced in the process had a high specific surface area, resulting in complete hydration and dissolution of the pellets. After 30 min, individual pellets were not visible anymore and the rate-controlling effect was not observed. Also, the cornstarch pellets swelled to a great extent. Here, individual pellets were still present throughout dissolution testing (see also Bialleck et al. (24)). Again, the hydrophilic gel layer that formed upon contact with the aqueous media did not control the drug release due to the high specific surface area of the pellets.
Fig. 5.
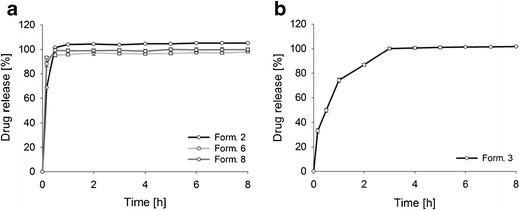
In vitro drug release profiles of the following: a formulation 2 (cornstarch/antipyrine (20%) pellets), formulation 6 (xanthan/antipyrine (20%) pellets), and formulation 8 (gum arabic/antipyrine (20%) pellets), and b formulation 3 (cornstarch/codeine phosphate (10%) pellets) in non-alcoholic media over 8 h. Mean values ± SD (n = 3). As SDs were very low, they are not clearly visible in the graph
Since starch was the most promising matrix for achieving a prolonged drug release, a different model drug, codeine phosphate, was incorporated. As illustrated in Fig. 5b, the amount of codeine phosphate released was lower compared to the starch/antipyrine pellets. Around 33% of codeine phosphate was detected in the dissolution media after 10 min. The pellets continued the drug release for 2 h, followed by an entire release of the API after 3 h. This can be explained by a lower solubility of codeine phosphate in 0.1 N HCl compared to antipyrine.
Although none of the cornstarch-based formulations had a prolonged release profile, they stayed intact throughout the dissolution testing. Together with their elastic behavior and high mechanical resistance to tampering with common household devices, this makes cornstarch-based codeine-phosphate-loaded pellets perfect candidates for further development. Thus, Aquacoat® ARC, a coating material known to retard drug release and to prevent ADD, was applied (25,27,47). Aquacoat® ARC consists of ethanol-soluble Aquacoat® ECD 30 and ethanol-insoluble guar gum. Aquacoat® ECD 30 remains intact in water and thus retards the release in aqueous media. The rate-controlling effect of the coating in the presence of ethanol is due to guar gum, which is insoluble in ethanol (47). Thereby, guar gum acts as a protective layer for the ethanol-soluble ethylcellulose, leaving the release-controlling film intact in the presence of ethanol (47). Moreover, guar gum increases the mechanical strength of the film (27), which is further enhanced by adding TEC (plasticizer), resulting in high elongation at breaking. The cornstarch pellets also showed high compression strengths and did not break but rather deformed, which means that grinding was impossible. Thus, if two highly flexible systems are combined, the possibility for abuse by mechanical destruction is decreased.
The dissolution studies of the coated pellets demonstrated that a coating level of 20% led to a significantly (p < 0.05) decreased drug release rate, compared to the non-coated pellets (Fig. 6). After 1 h, 20% of the drug was released, and after 2 h, only 40% was detected in the dissolution media. This indicates that adding Aquacoat® ARC was suitable for achieving a prolonged release from cornstarch pellets. To verify if this coating material can be alcohol resistant for 2 h, the pellets were tested in 20 and 40% ethanolic media and compared with the uncoated pellet formulation.
Fig. 6.
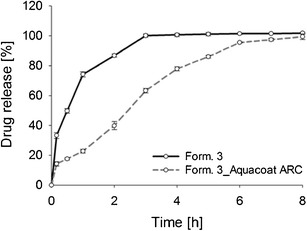
In vitro drug release profiles of formulation 3 (cornstarch/codeine phosphate (10%) pellets) and formulation 3 coated with 20% Aquacoat® ARC in non-alcoholic media over 8 h. Mean values ± SD (n = 3)
Figure 7a clearly shows that the drug release of uncoated pellets depends on the ethanol concentration. Interestingly, the pellets were robust in 40% alcoholic media (f2 = 59.77). However, adding 20% ethanol to acidic media markedly increased the drug release rate (f2 = 35.86) and led to dose dumping. This can be attributed to the effect of ethanol on the starch gel layer formation and the solubility of codeine phosphate (17,48). In general, the swelling behavior and the API release depend on the penetration of dissolution media into the pellet. Thereby, the free volume between the polymer chains increases and a gel layer that controls the drug release is formed (48). Upon contact with acidic media (without any ethanol) the cornstarch/codeine phosphate pellets immediately begin to swell. The formed gel layer acts as a protective barrier, which controls diffusion of the drug from the pellets and the drug release rate. Adding ethanol to the dissolution media inhibits the initial interaction between the ethanol-insoluble cornstarch matrix and the surrounding media and hinders the formation of a uniform and stable gel layer, leading to an uncontrolled release of the drug from the pellets. As such, the drug release rate is not controlled by the gel layer but is rather a function of the drug’s solubility in ethanol, which is lower in 40% ethanol than in 20% ethanol (i.e., 186.09 and 273.33 mg/ml, respectively). Figure 8b, c clearly indicates that swelling is impeded with increasing alcohol content.
Fig. 7.
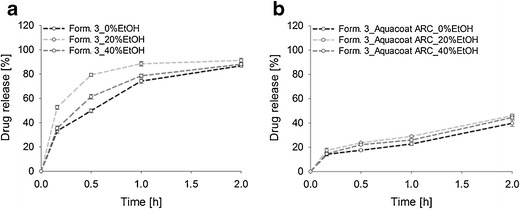
In vitro drug release profiles of a formulation 3 (cornstarch/codeine phosphate (10%) pellets) and b formulation 3 coated with 20% Aquacoat® ARC in alcoholic media over 2 h. Mean values ± SD (n = 3)
Fig. 8.

Swelling behavior of cornstarch/codeine phosphate pellets after 2 h of exposure to a 0.1 N HCl, b 0.1 N HCl/20% EtOH, and c 0.1 N HCl/40% EtOH
Figure 7b shows that drug release from the coated cornstarch/codeine phosphate pellets was not significantly affected by adding 20 and 40% ethanol (p = 0.54 and 0.72). The estimated f2 values of 62.13 and 70.96 confirmed that no dose dumping occurred. Again, the drug release was higher in 20% than in 40% alcoholic media, which was also observed in a previous study (31).
CONCLUSIONS
This paper reports the development of drug-abuse-alcohol-deterrent multi-unit dosage forms (i.e., pellets) based on matrix systems (i.e., cornstarch, gum arabic, xanthan). The pellets were manufactured via an advanced, continuous one-step HME process, during which a defined amount of water was directly fed into the matrix melt yielding deformable pellets that are tamper resistant. Formulations based on xanthan and gum arabic showed immediate drug release (i.e., complete release within less than 30 min). In contrast, cornstarch-based pellets retarded the drug release up to 3 h. To prolong the drug release and to provide resistance against ADD, cornstarch pellets were coated with Aquacoat® ARC. Overall, it was shown that processing cornstarch via advanced HME increased its resistance to common tampering practices. In combination with a coating process, this matrix system has the potential not only to diminish the abuse but also to prevent ADD.
Acknowledgments
This work was funded under the Austrian COMET Program by the Austrian Federal Ministry of Transport, Innovation and Technology (BMVIT); the Austrian Federal Ministry of Economy, Family and Youth (BMWFJ); and the State of Styria (Styrian Funding Agency SFG). The authors would like to thank the extrusion team of RCPE GmbH, Graz, Austria, for their assistance with the hot-melt extrusion process.
Conflict of interest
The authors declare that they have no competing interests.
REFERENCES
- 1.Mastropietro DJ, Omidian H. Current approaches in tamper-resistant and abuse-deterrent formulations. Drug Dev Ind Pharm. 2013;39(5):1–14. doi: 10.3109/03639045.2012.680468. [DOI] [PubMed] [Google Scholar]
- 2.Katz N. Abuse-deterrent opioid formulations: are they a pipe dream? Curr Rheumatol Rep. 2008;10(1):11–8. doi: 10.1007/s11926-008-0003-z. [DOI] [PubMed] [Google Scholar]
- 3.Gourlay GK. Sustained relief of chronic pain: pharmacokinetics of sustained release morphine. Clin Pharmacokinet. 1998;35(3):173–90. doi: 10.2165/00003088-199835030-00002. [DOI] [PubMed] [Google Scholar]
- 4.Skelly JP, Amidon GL, Barr WH, Benet LZ, Carter JE, Robinson JR, et al. In vitro and in vivo testing and correlation for oral controlled/modified-release dosage forms. Pharm Res. 1990;7(9):975–82. doi: 10.1023/A:1015970512368. [DOI] [PubMed] [Google Scholar]
- 5.Walden M, Nicholls FA, Smith KJ, Tucker GT. The effect of ethanol on the release of opioids from oral prolonged-release preparations. Drug Dev Ind Pharm. 2007;33(10):1101–11. doi: 10.1080/03639040701377292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Webster LR. Update on abuse-resistant and abuse-deterrent approaches to opioid formulations. Pain Med. 2009;10(S2):S124–S33. doi: 10.1111/j.1526-4637.2009.00672.x. [DOI] [PubMed] [Google Scholar]
- 7.Webster LR, Bath B, Medve RA. Opioid formulations in development designed to curtail abuse: who is the target? Expert Opin Investig Drugs. 2009;18(3):255–63. doi: 10.1517/13543780902751622. [DOI] [PubMed] [Google Scholar]
- 8.FDA guidance for industry abuse-deterrent opioids—evaluation and labeling 2015 [11 Jun 2015]. Available from: www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM334743.pdf.
- 9.Herry C, Monti A, Vauzelle-Kervroedan F, Oury P, Michel L. Reducing abuse of orally administered prescription opioids using formulation technologies. J Drug Deliv Sci Tech. 2013;23(2):103–10. doi: 10.1016/S1773-2247(13)50017-7. [DOI] [Google Scholar]
- 10.FDA approves abuse-deterrent labeling for reformulated OxyContin [12 Feb 2015]. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm348252.htm.
- 11.FDA approves Targiniq ER with abuse-deterrent properties [12 Feb 2015]. Available from: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm406290.htm.
- 12.FDA approves labeling with abuse-deterrent features for third extended-release opioid analgesic [12 Feb 2015]. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm419288.htm.
- 13.Purdue Pharma L.P. receives FDA approval for HysinglaTM ER (hydrocodone bitartrate) extended-release tablets CII, a once-daily opioid analgesic formulated with abuse-deterrent properties [04 Feb 2015]. Available from: http://www.purduepharma.com/news-media/2014/11/purdue-pharma-l-p-receives-fda-approval-for-hysinglatm-er-hydrocodone-bitartrate-extended-release-tablets-cii-a-once-daily-opioid-analgesic-formulated-with-abuse-deterrent-properties/.
- 14.Zogenix receives FDA approval for ZohydroTM ER (hydrocodone bitartrate) extended-release capsules [04 Feb 2015]. Available from: http://ir.zogenix.com/phoenix.zhtml?c=220862&p=irol-newsArticle&ID=1868848.
- 15.Vosburg SK, Jones JD, Manubay JM, Ashworth JB, Benedek IH, Comer SD. Assessment of a formulation designed to be crush-resistant in prescription opioid abusers. Drug Alcohol Depend. 2012;126(1–2):206–15. doi: 10.1016/j.drugalcdep.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sokolowska M. FDA meeting on ADF—development and evaluation of abuse deterrent opioid formulations, part 1 2014 [12 Feb 2015]. Available from: http://www.fda.gov/downloads/Drugs/NewsEvents/UCM422383.pdf.
- 17.Jedinger N, Khinast J, Roblegg E. The design of controlled-release formulations resistant to alcohol-induced dose dumping—a review. Eur J Pharm Biopharm. 2014;87(2):217–26. doi: 10.1016/j.ejpb.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 18.White JM, Irvine RJ. Mechanisms of fatal opioid overdose. Addiction. 1999;94(7):961–72. doi: 10.1046/j.1360-0443.1999.9479612.x. [DOI] [PubMed] [Google Scholar]
- 19.Wening K, Barnscheid L, Schwier S, Geißler A, inventors; Grünenthal GmbH, assignee. Tamper-resistant dosage form containing ethylene-vinyl acetate polymer patent. US 2015/0017250 A1. 2015.
- 20.Bartholomaeus JH, Ashworth JB, Stahlberg H-J, Galia E, Strothmann K. Innovative formulation technology protecting intended drug action. Drug Dev Deliv. 2012;12(8):76–81. [Google Scholar]
- 21.Khinast J, Baumgartner R, Roblegg E. Nano-extrusion: a one-step process for manufacturing of solid nanoparticle formulations directly from the liquid phase. AAPS PharmSciTech. 2013;14(2):601–4. doi: 10.1208/s12249-013-9946-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baumgartner R, Eitzlmayr A, Matsko N, Tetyczka C, Khinast J, Roblegg E. Nano-extrusion: a promising tool for continuous manufacturing of solid nano-formulations. Int J Pharm. 2014;477:1–11. doi: 10.1016/j.ijpharm.2014.10.008. [DOI] [PubMed] [Google Scholar]
- 23.Treffer D, Wahl PR, Hörmann TR, Markl D, Schrank S, Jones I, et al. In-line implementation of an image-based particle size measurement tool to monitor hot-melt extruded pellets. Int J Pharm. 2014;466(1–2):181–9. doi: 10.1016/j.ijpharm.2014.03.022. [DOI] [PubMed] [Google Scholar]
- 24.Bialleck S, Rein H. Preparation of starch-based pellets by hot-melt extrusion. Eur J Pharm Biopharm. 2011;79(2):440–8. doi: 10.1016/j.ejpb.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 25.Rosiaux Y, Muschert S, Chokshi R, Leclercq B, Siepmann F, Siepmann J. Ethanol-resistant polymeric film coatings for controlled drug delivery. J Control Release. 2013;169(1–2):1–9. doi: 10.1016/j.jconrel.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 26.Rosiaux Y, Velghe C, Muschert S, Chokshi R, Leclercq B, Siepmann F, et al. Ethanol-resistant ethylcellulose/guar gum coatings—importance of formulation parameters. Eur J Pharm Biopharm. 2013;85((3, Part B)):1250–8. doi: 10.1016/j.ejpb.2013.07.014. [DOI] [PubMed] [Google Scholar]
- 27.Rosiaux Y, Velghe C, Muschert S, Chokshi R, Leclercq B, Siepmann F, et al. Mechanisms controlling theophylline release from ethanol-resistant coated pellets. Pharm Res. 2014;31(3):731–41. doi: 10.1007/s11095-013-1194-1. [DOI] [PubMed] [Google Scholar]
- 28.Shipway PH, Hutchings IM. Fracture of brittle spheres under compression and impact loading. I. Elastic stress distributions. Philos Mag A. 1993;67(6):1389–404. doi: 10.1080/01418619308225362. [DOI] [Google Scholar]
- 29.Moore JW, Flanner HH. Mathematical comparison of dissolution profiles. Pharm Techno. 1996;20:64–74. [Google Scholar]
- 30.Paruta AN. Solubility profiles for antipyrine and aminopyrine in hydroalcoholic solutions. J Pharm Sci. 1967;56(12):1565–9. doi: 10.1002/jps.2600561207. [DOI] [PubMed] [Google Scholar]
- 31.Jedinger N, Schrank S, Mohr S, Feichtinger A, Khinast J, Roblegg E. Alcohol dose dumping: the influence of ethanol on hot-melt extruded pellets comprising solid lipids. Eur J Pharm Biopharm. 2015;92:83–95. doi: 10.1016/j.ejpb.2015.02.022. [DOI] [PubMed] [Google Scholar]
- 32.Li A, Yalkowsky SH. Solubility of organic solutes in ethanol/water mixtures. J Pharm Sci. 1994;83(12):1735–40. doi: 10.1002/jps.2600831217. [DOI] [PubMed] [Google Scholar]
- 33.Seedher N, Bhatia S. Solubility enhancement of cox-2 inhibitors using various solvent systems. AAPS PharmSciTech. 2003;4(3):36–44. doi: 10.1208/pt040333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu F, Prashantha K, Soulestin J, Lacrampe M-F, Krawczak P. Plasticized-starch/poly (ethylene oxide) blends prepared by extrusion. Carbohyd Polym. 2013;91(1):253–61. doi: 10.1016/j.carbpol.2012.08.008. [DOI] [PubMed] [Google Scholar]
- 35.Liu H, Xie F, Yu L, Chen L, Li L. Thermal processing of starch-based polymers. Prog Polym Sci. 2009;34(12):1348–68. doi: 10.1016/j.progpolymsci.2009.07.001. [DOI] [Google Scholar]
- 36.Zohuriaan MJ, Shokrolahi F. Thermal studies on natural and modified gums. Polym Test. 2004;23(5):575–9. doi: 10.1016/j.polymertesting.2003.11.001. [DOI] [Google Scholar]
- 37.Kipping T, Trindade R, Rein H. The use of hot-melt extruded corn starch matrices as drug carrier systems: a thermophysical characterization. Starch - Stärke. 2014;66(9–10):923–33. doi: 10.1002/star.201400014. [DOI] [Google Scholar]
- 38.Liu P, Yu L, Liu H, Chen L, Li L. Glass transition temperature of starch studied by a high-speed DSC. Carbohyd Polym. 2009;77(2):250–3. doi: 10.1016/j.carbpol.2008.12.027. [DOI] [Google Scholar]
- 39.Bialleck S. Herstellung von Polysaccharidpellets mittels Schmelzextrusion Bonn: Rheinische Friedrich-Wilhelms-Universität 2011. Available from: http://hss.ulb.uni-bonn.de/2012/2743/2743.htm.
- 40.Liu H, Yu L, Xie F, Chen L. Gelatinization of cornstarch with different amylose/amylopectin content. Carbohyd Polym. 2006;65(3):357–63. doi: 10.1016/j.carbpol.2006.01.026. [DOI] [Google Scholar]
- 41.Mothé CG, Rao MA. Thermal behavior of gum arabic in comparison with cashew gum. Thermochim Acta. 2000;357–358:9–13. doi: 10.1016/S0040-6031(00)00358-0. [DOI] [Google Scholar]
- 42.Wauer GF, Rein H. Solid solutions of tramadol-HCl based on starch. Pharm Ind. 2010;72:1973–9. [Google Scholar]
- 43.Petruševski G, Ugarkovic S, Makreski P. Solid-state transformation of the pseudopolymorphic forms of codeine phosphate hemihydrate and codeine phosphate sesquihydrate monitored by vibrational spectroscopy and thermal analysis. J Mol Struct. 2011;993(1–3):328–35. doi: 10.1016/j.molstruc.2011.01.062. [DOI] [Google Scholar]
- 44.Toti US, Soppimath KS, Mallikarjuna NN, Aminabhavi TM. Acrylamide-grafted-acacia gum polymer matrix tablets as erosion-controlled drug delivery systems. J Appl Polym Sci. 2004;93(5):2245–53. doi: 10.1002/app.20768. [DOI] [Google Scholar]
- 45.Talukdar MM, Kinget R. Swelling and drug release behaviour of xanthan gum matrix tablets. Int J Pharm. 1995;120(1):63–72. doi: 10.1016/0378-5173(94)00410-7. [DOI] [Google Scholar]
- 46.Vendruscolo CW, Andreazza IF, Ganter JLMS, Ferrero C, Bresolin TMB. Xanthan and galactomannan (from M. scabrella) matrix tablets for oral controlled delivery of theophylline. Int J Pharm. 2005;296(1–2):1–11. doi: 10.1016/j.ijpharm.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 47.FMC. BioPolymer Aquacoat® ARC [27 May 2013]. Available from: http://www.fmcbiopolymer.com/Pharmaceutical/Products/NEWAquacoatARC.aspx.
- 48.Bialleck S, Rein H. Drug release mechanisms of hot-melt extruded starch-based pellets. Starch - Stärke. 2012;64(5):408–19. doi: 10.1002/star.201100168. [DOI] [Google Scholar]