

Abstract
In most vertebrates, mitotic spindles and primary cilia arise from a common origin, the centrosome. In non‐cycling cells, the centrosome is the template for primary cilia assembly and, thus, is crucial for their associated sensory and signaling functions. During mitosis, the duplicated centrosomes mature into spindle poles, which orchestrate mitotic spindle assembly, chromosome segregation, and orientation of the cell division axis. Intriguingly, both cilia and spindle poles are centrosome‐based, functionally distinct structures that require the action of microtubule‐mediated, motor‐driven transport for their assembly. Cilia proteins have been found at non‐cilia sites, where they have distinct functions, illustrating a diverse and growing list of cellular processes and structures that utilize cilia proteins for crucial functions. In this review, we discuss cilia‐independent functions of cilia proteins and re‐evaluate their potential contributions to “cilia” disorders.
Keywords: Centrosome, Cilia, Ciliopathies, IFT, MTOC
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Molecular Biology of Disease; Signal Transduction
Glossary
- APC
antigen‐presenting cell
- APC/C
anaphase‐promoting complex/cyclosome
- ATM
ataxia telangiectasia mutated
- ATMIN
ATM interactor
- ATR
ataxia telangiectasia and Rad3 related
- BBS
Bardet–Biedl syndrome
- Cc2d2A
coiled‐coil and C2 domain‐containing protein 2A
- Ccdc13
coiled‐coil domain‐containing 13
- Cdc20
cell division cycle protein 20
- Cep164
centrosomal protein of 164 kDa
- Che‐1
abnormal chemotaxis
- CHK1
checkpoint kinase 1
- Cspp1
centrosome and spindle pole‐associated protein 1
- CYLD
cylindromatosis
- DYNLL1
dynein light chain LC8‐type1
- E2F1
E2F transcription factor 1
- GMAP210
Golgi microtubule‐associated protein of 210 kDa
- Hh
Hedgehog
- HNF1β
hepatocyte nuclear factor‐1beta
- IFT
intraflagellar transport
- MTs
microtubules
- MTOC
microtubule‐organizing center
- MVA
mosaic variegated aneuploidy
- Nek8
never in mitosis A‐related kinase 8
- Nphp9
nephrocystin protein 9
- Odf1
oral‐facial‐digital syndrome 1
- PCM
pericentriolar material
- PCP
planar cell polarity
- PKD
polycystic kidney disease
- Pkhd
polycystic kidney and hepatic disease
- Rb
retinoblastoma protein
- SAC
spindle assembly checkpoint
- Sdccag8
serologically defined colon cancer antigen 8
- Shh
Sonic Hedgehog pathway
- TCR
T‐cell receptor
- TG737
gene symbol for IFT88
Cilia proteins and cilia disorders—is there more?
Primary cilia (henceforth called “cilia”) are present in almost all cell types, lymphoid cells being one exception. Thus, cilia proteins have the potential to adversely affect numerous organs and tissues when disrupted. Over the past 10–15 years, cilia loss and/or dysfunction have been linked to numerous human disorders, collectively termed ciliopathies. Phenotypes associated with cilia dysfunction are often syndromic and include cystic kidneys, polydactyly, situs inversus, obesity, and encephalocele 1, to name a few. For example, disruption of cilia‐based Hedgehog (Hh) signaling has been implicated in polydactyly 2, 3 and disruption of cilia‐based signaling through Ca2+ or planar cell polarity (PCP) has been linked to cystogenesis 4, 5. However, a growing body of evidence suggests that cilia proteins are, in fact, multifunctional. In addition to their localization to cilia, many localize to and are bona fide components of centrosomes, as well as other cellular organelles and structures. At these non‐cilia sites, cilia proteins perform functions distinct from their ciliary roles. Thus, we contend that disruption of cilia proteins—historically named for their localization to cilia, as well as their association with cilia defects and cilia‐related disorders—can additionally disrupt spindle poles and an expanding list of cellular structures that impact numerous cellular functions. As a result, it has become increasingly difficult to determine which of these cellular functions and which of the these non‐cilia organelles truly contribute to ciliopathies when cilia proteins are disrupted 6, 7. In this review, we first explore the diversity of cilia‐independent processes that involve cilia proteins and then discuss alternative hypotheses for the etiology of “ciliopathies”.
The centrosome, a common thread between cilia and mitosis
Most vertebrate centrosomes are comprised of two centriole barrels surrounded by pericentriolar material (PCM). A major function of the centrosome is to serve as the primary microtubule‐organizing center 8, 9, 10 (MTOC) of the cell. The duplicated centrosomes undergo “centrosome maturation” during mitosis by increasing their microtubule nucleation capacity through recruitment of pericentriolar material and associated microtubule‐nucleating proteins (such as γ‐tubulin). This facilitates chromosome capture and the construction of the mitotic spindle 10. During interphase (G1/Go), the mother/older centriole (basal body) docks at the plasma membrane through its distal appendages and then elongates preexisting microtubules of the centriolar barrel—rather than nucleating microtubules from the PCM as in mitosis—to form the primary cilium 11. The dual role of centrosomes in the assembly of both cilia and spindle poles provides a common link between the two structures 12 (Fig 1). Thus, centrosome defects have the potential to disrupt structural and functional aspects of both cilia and mitotic spindles, which could co‐contribute to ciliopathy phenotypes.
Figure 1. The two lives of the centrosome during the cell cycle.
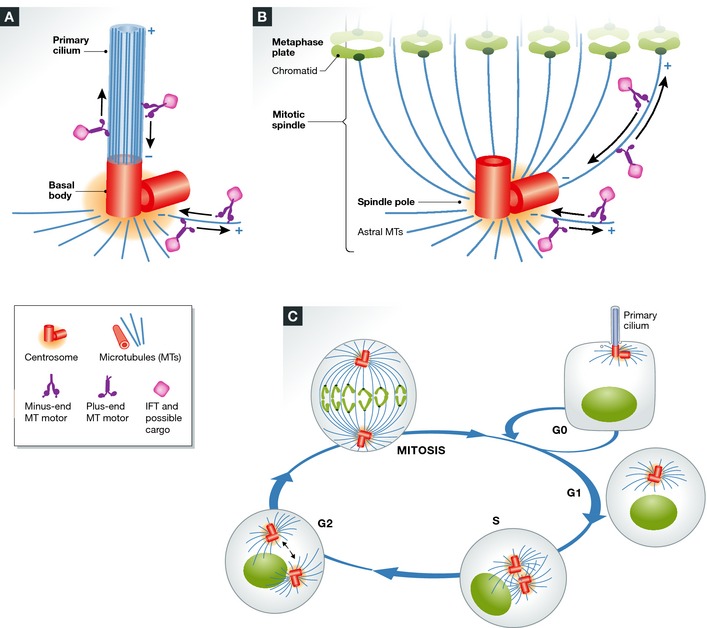
(A) In interphase, the centrosome functions as a basal body for primary cilia formation. (B) The basal body matures into the spindle poles during mitosis. (C) The centrosome duplicates once per cell cycle, during S phase, and moves to opposite sides of the cell starting at G2 and throughout mitosis. In G0, the centrosome docks at the membrane and templates cilia formation once again.
Many centrosomal protein are required for cilia integrity (see Sidebar A). Pericentrin was originally characterized as a centrosomal protein based on its localization and its role in microtubule nucleation and spindle assembly 13. Pericentrin was subsequently identified in mouse testes as a component of protein complexes involved in intraflagellar transport (IFT)—the process of transporting material within the cilium—and was shown to be required for ciliogenesis 11, 14. This triggered a second study, which revealed an additional eight centrosomal protein required for cilia integrity, and a third study identified a dozen more 15, 16. These studies identify centrosomal protein control of cilia integrity as a fundamental and conserved feature of centrosomes. Because centrosomes function throughout the cell cycle, centrosome defects could contribute to ciliopathy phenotypes at different cell cycle stages. In contrast, cilia defects occur only in G1/G0, when cilia are present (Fig 1).
Sidebar A: Centrosomes and cilia.
Not all centrosomal protein are positive contributors to ciliogenesis. For example, CP110 and Cep97 are highly expressed in proliferating cells during mitosis, but are not expressed during cilia formation 137, 138. Their depletion results in cilia formation during multiple stages of the cell cycle, thus disrupting cell cycle control of cilia assembly specifically at G1/G0 138. Conversely, when CP110 and Cep97 are ectopically expressed during interphase, cilia can no longer form 138, suggesting that CP110 and Cep97 negatively regulate ciliogenesis. These two proteins may act as a cell cycle switch that controls centrosome participation on either cell cycle progression or in cilia construction and maintenance.
The protein composition of centrosomal satellites can be altered by the autophagy machinery, contributing to ciliogenesis 139, 140. An interesting connection between centrosome satellites and the BBSome complex—one of the main contributors to ciliopathy phenotypes—was recently proposed. Bbs4, a member of the BBSome, was shown to interact with the centrosome/satellite proteins Cep290 and Azi1(Cep131) 141. Moreover, the ciliopathy protein Ofd1 is a satellite protein that might be responsible for the loss of PCM1 and Cep290 from satellites 142. Although the mechanisms connecting the components of centrosome satellites to cilia function remain to be elucidated, multiple satellite proteins are essential contributors to ciliopathies.
Defective centrosomes are an appealing alternative explanation for ciliopathy phenotypes that appear during development, especially because many cells are actively proliferating at this time and, thus, do not have cilia.
Cilia‐independent roles of cilia proteins
Orientation of the mitotic spindle and the axis of cell division
We envision two potential mechanisms for mis‐orientation of cell division caused by dysfunction of “cilia proteins”. (i) Certain cilia proteins have direct and essential roles in cell division orientation independent from their roles in cilia, and/or (ii) dysfunctional non‐canonical Wnt signaling (also known as the PCP signaling pathway) induces a downstream defect in spindle orientation, possibly through both cilia‐dependent and cilia‐independent mechanisms.
Proteins involved in the intraflagellar transport of molecules into and out of the cilium have been recently shown to also transport microtubule‐nucleating/anchoring proteins to centrosomes during their maturation into spindle poles in mitotic cells 17. Spindle poles are key players in the organization of mitotic spindles and the orientation of the cell division axis 18, 19. In fact, work from our laboratory and others indicates that centrosome protein dysfunction can induce spindle orientation defects 17, 20, 21, 22, 23, 24, 25, 26. Of note, depletion of the cilia protein IFT88 27, 28 induces spindle mis‐orientation independently of its known roles in cilia formation and maintenance 17, 29 (Fig 2). During mitosis, IFT88 is part of a dynein1‐driven complex 17 (not ciliary dynein) that transports microtubule‐nucleating proteins to spindle poles. IFT88 depletion disrupts this trafficking pathway and inhibits centrosome maturation. This, in turn, decreases the subset of astral microtubules required for spindle positioning 17 and induces spindle mis‐positioning.
Figure 2. Primary cilia‐independent functions of cilia proteins.
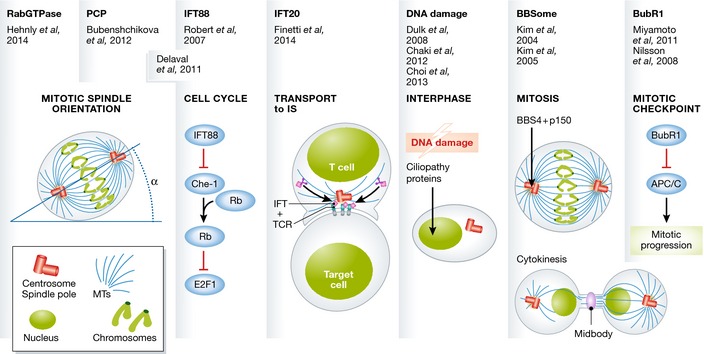
Cilia proteins participate in non‐ciliarly events, such as mitosis, immunological synapse (IS) formation, DNA damage, cytokinesis, and chromosome segregation.
The non‐canonical Wnt/PCP pathway (for canonical Wnt pathway, see Sidebar B) is responsible for ensuring the orientation of the spindle and cell division axis (Fig 2). Inactivation of the renal‐specific, PCP‐associated transcription factor Tcf2 (also known as HNF1β)—which is essential for the expression of Pkhd and Pkd2 5, two genes involved in the hallmark ciliopathy polycystic kidney disease (PKD)—causes spindle mis‐orientation in cells of kidney tubules 5. This was the first evidence linking PCP to PKD and, thus, ciliopathies. Moreover, depletion of the core PCP protein, Vangl2, induces a similar spindle mis‐orientation phenotype 30 as well as cilia mis‐positioning 31.
Sidebar B: Canonical Wnt signaling, cilia, and asymmetric division.
Mouse embryos that cannot form cilia have been shown to be hyper‐responsive to activation by canonical Wnt ligands 143, expressing increased levels of active β‐catenin 118, 144. However, mouse embryos lacking IFT88, IFT172, Kif3a, or dynein heavy chain—all of which inhibit cilia formation—express canonical Wnt target genes in the appropriate tissues as well as control embryos 145. Strikingly, all these mutant embryos that lack cilia have intact canonical Wnt signaling pathways. Moreover, there is no defect in switching between canonical and non‐canonical Wnt signaling under these conditions 145.
Exposure to exogenous canonical Wnt during mitosis was recently shown to orient the spindle for asymmetric cell division, leading to the asymmetric distribution of stem cell properties to the daughter cells 146. The lack of cilia in mitosis argues for cilia‐independent mechanisms of Wnt/centrosome interconnection. For example, Axin, a negative regulator of Wnt signaling, and β‐catenin, a major downstream target of canonical Wnt signaling, can be found at the centrosome 147. GSK3β—an inhibitor of β‐catenin signaling—regulates SMAD proteins, which are components of the BMP signaling pathway. The GSK3β‐mediated degradation of SMAD occurs at the centrosome 148, leading to the idea that β‐catenin stabilization at the centrosome occurs through Plk1‐Nek2‐dependent phosphorylation of β‐catenin at multiple sites 149. Destabilized β‐catenin is targeted for proteasomal degradation, and the ciliopathy proteins Bbs and Ofd1—which localize to the centrosome satellites—bind the proteasome lid and control degradation‐related signaling pathways 150.
Although these examples suggest that canonical Wnt signaling depends on the centrosome rather than on cilia, the older centrosome may carry membrane from a previously assembled cilium 151, which might be recognized by the Wnt pathway. Therefore, cilia could indirectly influence the centrosome, an intriguing concept that requires further investigation. Collectively, these studies underscore the idea that canonical Wnt signaling might depend on centrosomes rather than cilia.
The PCP pathway includes the cilia protein inversin, which is linked to the ciliopathy nephronophthisis type 2 32. Inversin interacts with disheveled 33, a protein involved in transducing both canonical Wnt and non‐canonical Wnt signaling pathways, and acts as a molecular switch between them. Inversin induces the degradation of cytoplasmic disheveled 33, leaving only plasma membrane‐localized disheveled and, thus, activating the PCP pathway. Importantly, inversin is localized not only to cilia 32, but also to adherens junctions 34 and the nucleus 34. Thus, it is unclear which pool of inversin mediates disheveled degradation and modulation of PCP. In addition to the aforementioned cilia‐ and PCP‐independent modulation of cell division orientation 29, it is likely that these ciliopathy proteins contribute to PCP modulation through both ciliary and non‐ciliary mechanisms.
Cytokinesis
Several cilia proteins that are linked to Bardet–Biedl syndrome (BBS) were originally shown to form a protein complex called the BBSome. These cilia components (such as Bbs4, Bbs6) also localize to centrosomes/spindle poles and retain their centrosomal localization throughout mitosis, when the cilium is resorbed and the centrosomes mature into spindle poles 35, 36. Bbs4 interacts with the dynactin subunit p150, which is important for dynein1 function in interphase and mitosis 35. Notably, p150 resides outside the cilium, suggesting that the p150‐Bbs4 complex has a cilia‐independent role. In fact, Bbs4 depletion leads to an increase in binucleated and aneuploid cells, consistent with defects in cell division 35. Another BBS member, Bbs6, localizes to the midbody, an organelle that controls abscission, which is the final step of cytokinesis 36. Like Bbs4, depletion of Bbs6 causes cell cycle defects that include binucleated cells, supernumerary centrosomes, and elongated cytokinetic bridges, all of which are hallmarks of defective cytokinesis 36. Moreover, several IFT proteins localize to the cleavage furrow 37. Among these, IFT27 seems to have an active role in regulating cytokinesis, as its depletion increases cytokinesis failure and multinucleated cell formation 37, 38. These observations indicate that the BBSome and IFT proteins likely act in concert to resolve the cytokinetic bridge during abscission.
Membrane trafficking during spindle pole maturation
Two small GTPases, Rab8 and Rab11, have been implicated in cilia formation. For instance, active Rab11 recruits the Rab8 GEF, Rabin8, to activate Rab8 and induce cilia assembly 39, 40. Rab11 and Rab8 also interact with several well‐defined proteins required for ciliogenesis, such as the BBSome, the vesicle tethering complex (exocyst), and the mother centriole appendage protein cenexin 39, 41, 42, 43, 44, 45, 46, 47, 48. However, the molecular mechanisms and interplay of these molecular components during ciliogenesis require further investigation.
The same GTPases that function in ciliogenesis are also bona fide endosome components that play important roles in endosome recycling 39, 49. The centrosome has recently been shown to be involved in organizing and mediating the recycling branch of endosomal trafficking 50. More specifically, the recycling endosome interacts with the subdistal appendages of the mother (older) centriole. These appendages are also required for assembly of the ciliary vesicle, a structure that forms at the mother centriole before the centriole docks at the plasma membrane to form the cilium 51, 52, 53, 54, 55. Rab8 and Rab11 move into and out of the centrosome area, a process possibly mediated by an interaction with the subdistal appendage protein cenexin 50. Thus, the two GTPases function in very different processes (cilia formation and endosome recycling), making it difficult to discern which contributes to cilia disorders. We speculate that the interaction between the recycling endosome and its endocytic machinery on the mother centriole appendages may facilitate the organization of the Rab11–Rab8 GTPase cascade, ensuring the initiation of cilia assembly at the right time during the cell cycle. Moreover, Rab11 depletion leads to defects in membrane trafficking toward spindle poles, which contributes to defects in spindle mis‐orientation 20 (Fig 2). Other appendage proteins have been implicated in this membrane trafficking process. For example, Cc2d2A 56 modulates Rab8 localization and function in photoreceptor cells 42, suggesting a centrosome–endosome interaction. Importantly, Cc2d2A mutations are associated with the Meckel and Joubert syndromes, which are well‐known ciliopathies 57, 58.
Polarized transport toward the immune synapse
Intraflagellar transport protein IFT20—a member of ciliary IFT complex B—also has cilia‐independent roles. IFT20 is expressed in lymphoid cells, one of the few cell types that does not form cilia 59. T lymphocytes contact antigen‐presenting cells (APCs) through the formation of a specialized, TCR‐rich membrane region at the APC contact site, known as the immunological synapse. TCRs are targeted to the immune synapse through a combination of polarized membrane recycling, lateral diffusion, and cytoskeleton‐driven movement 60. In response to T‐cell activation, IFT20 interacts with components of the TCR/CD3 complex and is required for clustering these complexes at the synapse through polarized recycling (Fig 2). More specifically, IFT20 is involved in Rab GTPase‐mediated recycling of the TCR 61. In this context, IFT20 interacts with two other IFT‐B complex members, IFT88 and IFT57 59, suggesting that the trimeric complex, rather than IFT20 alone, participates in TCR/CD3 trafficking to the immune synapse 61. Together, these data define a conserved role for IFT20 in intracellular trafficking in both ciliated and non‐ciliated cells.
Over the years, the Sonic Hedgehog pathway (Shh) 62, 63 has been linked to the presence and function of cilia. Shh receptors and downstream targets are well known to locate at the cilia 64, and the intensity of Shh signaling seems to depend on functional cilia 65. Not surprisingly, ablation of the essential centriole protein Sas4 in mice results in cilia and Shh signaling defects 66, demonstrating how interconnected centrosome and cilia function can be. Nevertheless, Shh receptors can also function outside of cilia 67. Interestingly, the Shh pathway was recently shown to function in non‐ciliated immune cells that retain centrosomes 68, suggesting that centrosomes, and not cilia, are essential for this process. Another recent study showed that cilia formation in lymphocytes could be induced if the centrosome protein CP110 was removed by another centrosome protein, centrin2 (see also Sidebar A) 69. This system may be a powerful strategy to dissect the contribution of the centrosome versus cilium in Shh signaling. For example, the centrosome can dock at the plasma membrane, which is one of the early steps in cilia assembly, but is unable to form a cilium in lymphoid cells. Whether centrosome docking at the plasma membrane is sufficient for Shh signaling remains to be elucidated. If centrosome docking is prevented, perhaps by distal appendage disruption 16, 70, is Shh signaling disabled? Additional studies are required to determine the molecular mechanism governing Shh signaling and the role of the cilium versus centrosome in this process.
Cell cycle regulation and cell proliferation
Recent studies in non‐ciliated and ciliated cells demonstrate a role for IFT proteins in cell cycle progression. For instance, depletion of IFT88 leads to a mitotic delay, whereas overabundance of IFT88 induces G1 arrest. More specifically, IFT88 over‐expression induces G1 cell cycle arrest by blocking the interaction of Che‐1 with the tumor suppressor Rb, freeing Rb to bind and repress E2F1, a transcription factor whose activation is required for the G1/S transition 71 (Fig 2). The mitotic delay and defects in spindle assembly in IFT88‐depleted cells 17 require further analysis to identify critical signaling molecules that are possibly transported by IFT88 during mitosis to ensure timely cell cycle progression. Interestingly, studies in Chlamydomonas demonstrated that an additional IFT protein, IFT27, is required for cell cycle progression 38. How and whether IFT27, IFT88, and/or other IFT proteins act in combination to regulate cell cycle progression remains an open question. It is currently difficult to delineate the complex relationship between cilia and the cell cycle. For example, the presence of cilia may delay reentry into the cell cycle 72, 73. However, we propose that the role of IFT88 in regulating mitotic progression 17 and G1 cell cycle arrest in non‐ciliated cells 71 is independent of the role of IFT88 in cilia function.
Cell proliferation defects are one of the major underlying causes of human cancer. Notably, several ciliopathy proteins are associated with different types of cancer. Loss of cilia has been linked to cancer by many independent groups 74, 75, 76, 77, 78, 79, 80, 81, 82. For instance, TG737—the gene that encodes the IFT88 protein—is required for ciliogenesis, and its mutation leads to over‐proliferation of liver progenitor cells in mice 83. Based on the high rate of TG737 mutations in human liver tumors and tumor cell lines, and the rescue of phenotypes following TG737 re‐expression, the gene has been classified as a tumor suppressor 84. These studies strongly link ciliopathy proteins to cell over‐proliferation/cancer.
DNA damage response (DDR)
The characterization of new roles of cilia and centrosomal protein in DNA damage repair is an emerging area of research. Cilia, centrosomes, and the DDR are linked in several ways: (i) the DDR pathway functions at the centrosome, (ii) ciliopathy proteins function in the nucleus during DDR, and (iii) DDR and cilia share a common regulatory protein (see below).
The ATR–CHK1 pathway is a well‐characterized mediator of the DNA damage response. CHK1 localizes to the spindle pole, where it regulates mitotic progression 85, 86. Another protein that was originally identified as a centrosome satellite protein, Ccdc13, has been implicated in both cilia formation and the DDR 87. These findings suggest that checkpoint regulation in response to DNA damage may occur through the centrosome.
Moreover, some cilia and centrosomal protein localize to the nucleus and are directly linked to DDR (Fig 2). For example, centrin2 88, 89, 90 and Cep164 91 are directly involved in DNA damage‐related events, such as excision repair. Mutations in a centrosome‐localized ciliopathy protein, Sdccag8, lead to DDR and activation of the DDR kinase ATM 92. Similarly, mutations in the centrosome protein pericentrin result in DDR defects 93. In addition, the ciliopathy protein Nek8/Nphp9 cooperates with the ATR–CHK1 DNA damage pathway to regulate CDK levels 94. Consistent with previous findings, Nek8 appears to play role in DNA stability and proliferation 95, which correlates with altered expression of Nek8 in human tumors 96, thus providing an intriguing link between DNA damage and ciliopathies 95.
Finally, both DDR and cilia might be regulated by a protein known as ATMIN. ATMIN is a co‐factor for ATM in the response to DNA damage 97 and also a transcriptional regulator of ciliary dynein, DYNLL1 98. Intriguingly, it does not regulate cytoplasmic dynein (DYNLL2) 99. Thus, the regulation of both cilia and DDR is likely tightly connected to the cell cycle.
Spindle assembly checkpoint regulation
BubR1 was recently shown to be essential for primary cilia formation in Medaka fish 100. BubR1 is an important component of the spindle assembly checkpoint (SAC, below). During the G1/G0 phase of the cell cycle, BubR1 induces ubiquitin‐mediated proteasomal degradation of Cdc20, which allows the activation of anaphase‐promoting complex/cyclosome (APC/C) by Cdh1 100. In ciliated cells, in addition to regulating cell cycle progression, APC/CCDH1 targets disheveled for destruction 101, thus enabling ciliogenesis.
However, BubR1 is best known as a kinetochore and spindle pole protein that regulates the SAC (Fig 2). In this context, BubR1 prevents progression through mitosis until all kinetochores make stable, bipolar attachments to microtubules. This occurs partly through BubR1‐induced Cdc20 degradation, which prevents the activation of the APC/C 102. When active, the APC/C catalyzes the degradation of cyclin B and securin, two proteins that are essential for maintaining metaphase 103. During both SAC maintenance and ciliogenesis, BubR1 targets Cdc20 for degradation, thereby ensuring that downstream events proceed properly, including the maintenance of essential metaphase proteins and of APC/CCDH1 activity. The interplay between BubR1 and inversin, in disheveled degradation, is unknown, as is the pool of inversin implicated in this process. These parallels again identify a single molecule that regulates two distinct processes, namely mitosis and ciliogenesis.
Additional examples
Studies showing that the cilia protein IFT20 also localizes to the cis‐Golgi complex via the centrosome/Golgi protein Gmap210 104, 105 provide further support to the notion that cilia proteins have cilia‐independent roles. Three other cilia‐localized 106, 107, 108, 109, 110 proteins that are associated with kidney disease (polycystin‐1, polycystin‐2, fibrocystin) have been implicated in the amplification of centrosomes 111, 112, 113. Moreover, the cilia proteins polycystin‐2 and fibrocystin localize to the mitotic spindle 114, although the functional relevance of this localization is unclear.
In summary, we have provided multiple examples of cilia proteins that have cilia‐independent functions in diverse cellular localizations and processes, including spindle orientation, cytokinesis, cell proliferation, cell cycle progression, checkpoint control, immune synapse integrity, cancer, Golgi organization, and endosome trafficking.
Possible cilia‐independent contributions to ciliopathies
The fact that the centrosome is linked to both ciliogenesis and mitosis can explain the function of some proteins (such as IFT, BBS, BubR1) during different cell cycle stages. It is logical to think that the cell would use similar elements to regulate both processes. Thus, future studies are required to test whether cilia‐independent pathways contribute to the pathogenesis currently attributed solely to cilia disruption. Below, we discuss possible contributions of non‐ciliary functions to the pathogenesis associated with “ciliopathies”.
Mis‐orientation of spindles and the axis of cell division
These effects have been linked to kidney disease 5, 115, 116, developmental delays, and underdeveloped cerebellum 117, the very same phenotypes associated with cilia dysfunction. To generate proper kidney tubules, spindle orientation is strictly regulated to occur in the longitudinal axis of the tubule, to ensure that tubules are elongated and not widened 5 (Fig 3). In ciliopathy models of PKD, the intrinsic polarity of kidney duct cells is lost 5, leading to cyst formation. Mis‐oriented cell divisions are observed not only in cystic kidney tubules, but also prior to cyst formation (during duct formation), suggesting that mis‐oriented cell division could be the underlying cause of cystic kidneys 5. This paradigm indicates that centrosome disruption could induce defects in structural and functional aspects of both cilia and mitotic spindle poles/centrosomes, either of which or both could contribute to ciliopathies. Improper regulation of spindle orientation has been suggested as a mechanism of kidney cyst onset in patients with PKD. This was originally proposed when mitotic spindle mis‐orientation was identified in kidney cysts that lacked IFT20 115. Counterintuitively, deletion of IFT140, which causes defects in Wnt signaling, does not affect spindle orientation but still induces cyst formation 118. Thus, cystic kidneys seem to result from cilia‐dependent as well as cilia‐independent processes 118, 119.
Figure 3. Potential primary cilia‐independent contributions to ciliopathies.
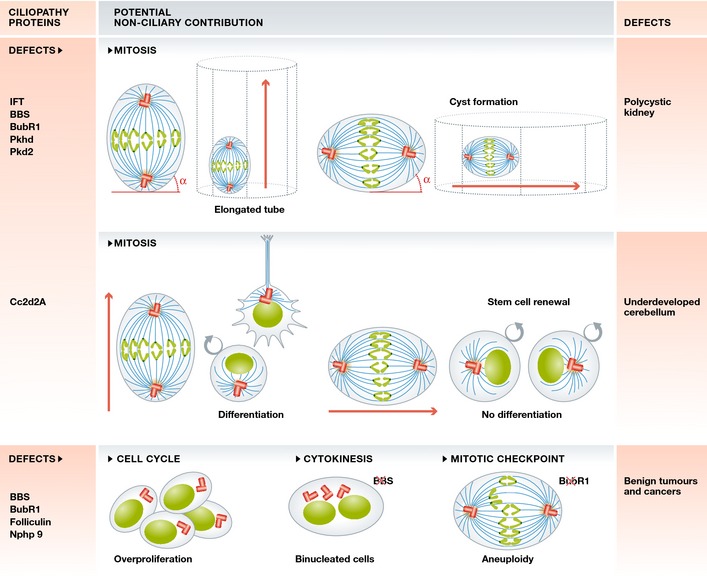
Mis‐oriented cell division can lead to kidney cyst formation and brain defects. Over‐proliferation and cytokinesis failure could result in uncontrolled cell growth and cancer.
Bardet–Biedl Syndrome
Ciliopathy phenotypes are associated with mutations in at least 14 different BBS genes 120, 121. BBS presents with nine or more diverse ciliopathy phenotypes, including retinopathy (damage to retina), polydactyly (supernumerary digits), cystic kidneys, situs inversus (mis‐positioning of major visceral organs), hypoplastic (underdeveloped) cerebellum, hydrometrocolpos (distension of uterine cavity), obesity, and hepatic dysfunction. As described above, some of the proteins associated with ciliopathies are organized at centrosomes/spindle poles and when mutated or depleted cause cytokinetic defects, mitotic failure, multipolar spindle formation, and cell cycle perturbation 35, 36. All of these defects could lead to benign tumor formation and cancer (Fig 3), which are associated with BBS ciliopathy 122. Moreover, cell division abnormalities are associated with brain development 21, 25, 123, 124, (Fig 3), and thus, mutations in the BBSome could contribute to hypoplastic cerebellum via both ciliary and mitotic defects.
BubR1 depletion
This causes left–right asymmetry, polycystic kidney disease, and aberrant cerebral development 100, again phenotypes associated with cilia disorders (Fig 3). Strikingly, mutations in BubR1 in humans are responsible for mosaic variegated aneuploidy (MVA), a rare disorder with complex symptoms such as microcephaly, polycystic kidneys, and malignancies, among others 100, 125.
Birt–Hogg–Dube syndrome and Nephronophthisis (NPH)
These are two other ciliopathies with possible cilia‐independent underlying mechanisms (Fig 3). Birt–Hogg–Dube syndrome is caused by mutations in the tumor suppressor gene folliculin, which localizes to cilia, centrosomes, and spindle poles. Folliculin mutations disrupt the onset of ciliogenesis and cause spindle mis‐orientation, putting patients at high risk of kidney cancer, cysts, and benign skin tumors 22. Nephronophthisis (NPH) 126 results from defects in cilia‐associated nephrocystin proteins (Nphp 1–12). For example, Nphp 9 is linked to the development of breast cancer and strongly promotes proliferation through the activation of the transcription regulator Taz 81. Collectively, these studies suggest that various mechanisms lead to the human disorders, including cilia defects as one of the causes, but indicating that other pathways could co‐contribute to the etiology of these disorders.
Centrosomal proteins and disease
Perhaps the most compelling data for the role of centrosomal proteins in ciliopathies is the significant number of mutations that have been recently described in them. The fast‐growing list of ciliopathy disorders is based on newly identified protein mutations that are associated with ciliopathy phenotypes (see Sidebar C). For example, mutation of the centriolar protein Cep120 results in skeletal ciliopathy 127; mutations in the centrosome protein Poc1A result in microcephaly and cilia defects 128; and defects in Poc1B cause syndromic retinal ciliopathy 129. Depletion of Poc1B also results in photoreceptor defects 130. Cspp1 is yet another example of a centrosomal protein with mutations linked to two different ciliopathies, Joubert and Meckel–Gruber syndromes 131. Intriguingly, at least two subdistal appendage proteins of the mother (older) centriole are linked to ciliopathy disorders 56, 132. First, Sdccag8, a protein that co‐localizes with ninein at the subdistal appendages of the mother centriole, was found to be mutated in nephronophthisis‐related ciliopathies 132. Second, lack of Cc2d2A, which is also localized to subdistal appendages 56, leads to a reduction in subdistal appendage proteins—such as Odf2 and ninein—along with ciliarly defects 56. Investigation of the relationship between Cc2d2A and Sdccag8 proteins could uncover the mechanisms whereby centrosomal subdistal appendages contribute to cilia defects. A relationship between centrosome anchoring to the plasma membrane—an early step in ciliogenesis—and ciliopathies was recently demonstrated in cylindromatosis (CYDL) null mice 133. Centrosomes were unable to dock at the plasma membrane, which correlated with ciliopathy phenotypes. Earlier studies showed that distal appendages promote ciliogenesis 70, which could explain the effects of CYLD deletion. Mutations in Cep83, a major component of distal appendages, cause ciliary defects and nephronophthisis 70, further supporting a role of distal appendages in ciliopathies. Cep164 is another component of mother centriole distal appendages involved in cilia assembly 16. Overall, these data are in agreement with the previously elucidated roles of centrosomal proteins in cilia formation 15 and indicate that centrosomal defects contribute significantly to ciliopathies.
Sidebar C: Centrosome phenotypes overlap with ciliopathy phenotypes.
Defects in centrosomes are known to be a cause of ciliopathies, as well as a cause of primary microcephaly 124, 152, 153, 154, 155, 156, 157. Polydactyly seems to be an overlapping feature of both disorders. Ciliopathies and microcephaly, which involve the same organelle (the centrosome), might in fact share more than the polydactyly phenotype. Mutation of the cell division motor Kif14, which participates in different aspects of mitosis and is critical for cytokinesis 158, appears to be the cause of a lethal ciliopathy phenotype 159. It is therefore possible that “centrosomopathies” (defects in centrosome) that can affect both cilia and mitotic spindle pole function are not always compatible with life and, therefore, are not well documented. Mutations in Plk4, a protein kinase that regulates centriole biogenesis, have also been recently shown to cause a combination of ciliopathy and centrosomopathy phenotypes 160.
However, the obvious role of distal appendages in cilia formation is no longer the only centrosome–cilia connecting axis. Centrosome substructures—such as subdistal appendages, PCM, and centriolar satellites—contain proteins involved in ciliopathies. As discussed above, the centrosome is the common element between cilia and mitotic spindle poles. The fact that defects in centrosome subdistal appendage proteins—like Sdccag8 or Cc2d2A—contribute to ciliopathy phenotypes and have important roles in MT organization, rather than in the structural organization of the cilium, implies that centrosome defects could contribute to ciliopathies in both ciliary and non‐ciliary capacities. Defects in MT organization and nucleation lead to mitotic spindle mis‐orientation 17, and Cc2d2A−/− cells lack or have abnormal subdistal appendages 56, suggesting that Cc2d2A mutants can mis‐regulate cilia or lead to mitotic defects. Cc2d2A mutations are associated with Meckel–Gruber 134 and Joubert 135 syndromes, both of which present with cystic kidneys, which have been proposed to be at least partly caused by mitotic spindle mis‐orientation.
Conclusions
The discovery that cilia proteins are present at a growing number of cellular organelles and structures that collectively perform diverse cellular functions opens new frontiers in our quest to understand the etiology of ciliopathies. Moreover, their multifunctionality could account for the syndromic nature of cilia disorders. In fact, the phenotypes caused by dysfunction of cilia proteins at cilia‐independent cellular sites can often more accurately account for the generation of organ defects in cilia disorders. The eight examples provided in the previous section illustrate the potential contribution of cilia‐independent phenotypes to cilia disorders.
We can now begin to test which of these cilia‐independent functions contribute to the etiology of cilia disorders. For example, do they co‐conspire to cooperatively generate ciliopathies, or can (at least some) so‐called ciliopathies be entirely independent of cilia function? For example, mitosis, ciliogenesis, and PCP have all been implicated in spindle mis‐orientation during cyst formation. It is currently difficult to determine whether one or more of these processes dominates, or if all contribute and, if so, which pathway is upstream of the others. For example, does PCP regulate mitotic spindle orientation through cilia, as has been suggested 5? Or does PCP regulate spindle orientation through cilia proteins that function directly in mitotic spindle assembly and orientation, as shown for IFT88 17? Do cilia truly transduce PCP signals, or do they instead require PCP signals for their genesis, as has been suggested by a study in which some PCP pathway members are required for ciliogenesis 136? These and other questions (see Sidebar D) can now be addressed directly by generating “separation of function” mutations and other strategies.
Sidebar D: In need of answers.
Does Shh pathway in lymphoid cells without cilia require centrosome docking to the membrane?
Does the older centrosome that acquires ciliary membrane affect signaling events?
Do subdistal appendages directly contribute to cilia formation through transport of cargo to cilia?
Can cilia‐dependent and cilia‐independent pathologies be dissected?
With the increased recognition that many cilia proteins perform more than one function, we argue that the contradictory findings surrounding the roles of cilia, PCP signaling, and mitosis in ciliopathies are due to the multifaceted role of proteins originally defined as cilia proteins, and subsequently shown to function at different cellular sites and different stages of the cell cycle.
In closing, ciliopathies are a class of disorders originally grouped together based on the ciliary localization and function of their causative proteins and the observation that cilia are lost or dysfunctional in tissues from afflicted patients. However, different ciliopathy syndromes manifest in different organs, with varying severity, and at different stages of life, raising questions about the true etiology of ciliopathies. We believe that these differences can be explained in part by the important roles of cilia proteins at sites outside the cilium. Consistent with this idea, recent work on cilia overwhelmingly indicates that ciliopathies are caused by disruption of a number of different cellular functions alone, or in combination. It has become increasing clear that ciliopathies could be caused by a complex set of disrupted functions (such as spindle orientation, cilia assembly/signaling, cell polarity, DNA damage) mediated by cilia proteins with multiple functions and localization patterns.
Conflict of interest
The authors declare that they have no conflict of interest.
Acknowledgements
We would like to express our thanks to Prof. Gregory J Pazour and Wendy Zimmerman for critical reading and thoughtful comments. The work was supported by grants K99GM107355 to HH and GM 051994 to SJD.
EMBO Reports (2015) 16: 1275–1287
See the Glossary for abbreviations used in this article.
References
- 1. Gerdes JM, Davis EE, Katsanis N (2009) The vertebrate primary cilium in development, homeostasis, and disease. Cell 137: 32–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Anderson E, Peluso S, Lettice LA, Hill RE (2012) Human limb abnormalities caused by disruption of hedgehog signaling. Trends Genet 28: 364–373 [DOI] [PubMed] [Google Scholar]
- 3. Huangfu D, Anderson KV (2005) Cilia and Hedgehog responsiveness in the mouse. Proc Natl Acad Sci USA 102: 11325–11330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fischer E, Pontoglio M (2009) Planar cell polarity and cilia. Semin Cell Dev Biol 20: 998–1005 [DOI] [PubMed] [Google Scholar]
- 5. Fischer E, Legue E, Doyen A, Nato F, Nicolas J‐F, Torres V, Yaniv M, Pontoglio M (2006) Defective planar cell polarity in polycystic kidney disease. Nat Genet 38: 21–23 [DOI] [PubMed] [Google Scholar]
- 6. Yuan S, Sun Z (2013) Expanding horizons: ciliary proteins reach beyond cilia. Annu Rev Genet 47: 353–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Taulet N, Delaval B (2014) Non‐ciliary functions of cilia proteins (in French). Med Sci 30: 1040–1046 [DOI] [PubMed] [Google Scholar]
- 8. Schatten H (2008) The mammalian centrosome and its functional significance. Histochem Cell Biol 129: 667–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Doxsey S (2001) Re‐evaluating centrosome function. Nat Rev Mol Cell Biol 2: 688–698 [DOI] [PubMed] [Google Scholar]
- 10. Bettencourt‐Dias M, Glover DM (2007) Centrosome biogenesis and function: centrosomics brings new understanding. Nat Rev Mol Cell Biol 8: 451–463 [DOI] [PubMed] [Google Scholar]
- 11. Rosenbaum JL, Witman GB (2002) Intraflagellar transport. Nat Rev Mol Cell Biol 3: 813–825 [DOI] [PubMed] [Google Scholar]
- 12. Smith KR, Kieserman EK, Wang PI, Basten SG, Giles RH, Marcotte EM, Wallingford JB (2011) A role for central spindle proteins in cilia structure and function. Cytoskeleton 68: 112–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Doxsey SJ, Stein P, Evans L, Calarco PD, Kirschner M (1994) Pericentrin, a highly conserved centrosome protein involved in microtubule organization. Cell 76: 639–650 [DOI] [PubMed] [Google Scholar]
- 14. Jurczyk A, Gromley A, Redick S, San Agustin J, Witman G, Pazour GJ, Peters DJM, Doxsey S (2004) Pericentrin forms a complex with intraflagellar transport proteins and polycystin‐2 and is required for primary cilia assembly. J Cell Biol 166: 637–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mikule K, Delaval B, Kaldis P, Jurcyzk A, Hergert P, Doxsey S (2007) Loss of centrosome integrity induces p38‐p53‐p21‐dependent G1‐S arrest. Nat Cell Biol 9: 160–170 [DOI] [PubMed] [Google Scholar]
- 16. Graser S, Stierhof Y‐DD, Lavoie SB, Gassner OS, Lamla S, Le Clech M, Nigg EA (2007) Cep164, a novel centriole appendage protein required for primary cilium formation. J Cell Biol 179: 321–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Delaval B, Bright A, Lawson ND, Doxsey S (2011) The cilia protein IFT88 is required for spindle orientation in mitosis. Nat Cell Biol 13: 461–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Walczak CE, Heald R (2008) Mechanisms of mitotic spindle assembly and function. Int Rev Cytol 265: 111–158 [DOI] [PubMed] [Google Scholar]
- 19. Siller KH, Doe CQ (2009) Spindle orientation during asymmetric cell division. Nat Cell Biol 11: 365–374 [DOI] [PubMed] [Google Scholar]
- 20. Hehnly H, Doxsey S (2014) Rab11 endosomes contribute to mitotic spindle organization and orientation. Dev Cell 28: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen C‐T, Hehnly H, Yu Q, Farkas D, Zheng G, Redick SD, Hung H‐F, Samtani R, Jurczyk A, Akbarian S et al (2014) A unique set of centrosome proteins requires pericentrin for spindle‐pole localization and spindle orientation. Curr Biol 24: 2327–2334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Luijten MNH, Basten SG, Claessens T, Vernooij M, Scott CL, Janssen R, Easton JA, Kamps MA, Vreeburg M, Broers JL et al (2013) Birt‐Hogg‐Dube syndrome is a novel ciliopathy. Hum Mol Genet 22: 4383–4397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yamashita YM, Mahowald AP, Perlin JR, Fuller MT (2007) Asymmetric inheritance of mother versus daughter centrosome in stem cell division. Science 315: 518–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kitagawa D, Kohlmaier G, Keller D, Strnad P, Balestra FR, Fluckiger I, Gonczy P (2011) Spindle positioning in human cells relies on proper centriole formation and on the microcephaly proteins CPAP and STIL. J Cell Sci 124: 3884–3893 [DOI] [PubMed] [Google Scholar]
- 25. Feng Y, Walsh CA (2004) Mitotic spindle regulation by Nde1 controls cerebral cortical size. Neuron 44: 279–293 [DOI] [PubMed] [Google Scholar]
- 26. Chen J‐F, Zhang Y, Wilde J, Hansen KC, Lai F, Niswander L (2014) Microcephaly disease gene Wdr62 regulates mitotic progression of embryonic neural stem cells and brain size. Nat Commun 5: 3885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Serra R (2008) Role of intraflagellar transport and primary cilia in skeletal development. Anat Rec 291: 1049–1061 [DOI] [PubMed] [Google Scholar]
- 28. Pazour GJ, Dickert BL, Vucica Y, Seeley ES, Rosenbaum JL, Witman GB, Cole DG (2000) Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene Tg737, are required for assembly of cilia and flagella. J Cell Biol 151: 709–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Borovina A, Ciruna B (2013) IFT88 plays a cilia‐ and PCP‐independent role in controlling oriented cell divisions during vertebrate embryonic development. Cell Rep 5: 37–43 [DOI] [PubMed] [Google Scholar]
- 30. Bubenshchikova E, Ichimura K, Fukuyo Y, Powell R, Hsu C, Morrical SO, Sedor JR, Sakai T, Obara T (2012) Wtip and Vangl2 are required for mitotic spindle orientation and cloaca morphogenesis. Biol Open 1: 588–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Borovina A, Superina S, Voskas D, Ciruna B (2010) Vangl2 directs the posterior tilting and asymmetric localization of motile primary cilia. Nat Cell Biol 12: 407–412 [DOI] [PubMed] [Google Scholar]
- 32. Otto EA, Schermer B, Obara T, O'Toole JF, Hiller KS, Mueller AM, Ruf RG, Hoefele J, Beekmann F, Landau D et al (2003) Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left‐right axis determination. Nat Genet 34: 413–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Simons M, Gloy J, Ganner A, Bullerkotte A, Bashkurov M, Krönig C, Schermer B, Benzing T, Cabello OA, Jenny A et al (2005) Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat Genet 37: 537–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nürnberger J, Bacallao RL, Phillips CL (2002) Inversin forms a complex with catenins and N‐cadherin in polarized epithelial cells. Mol Biol Cell 13: 3096–3106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim JC, Badano JL, Sibold S, Esmail MA, Hill J, Hoskins BE, Leitch CC, Venner K, Ansley SJ, Ross AJ et al (2004) The Bardet‐Biedl protein BBS4 targets cargo to the pericentriolar region and is required for microtubule anchoring and cell cycle progression. Nat Genet 36: 462–470 [DOI] [PubMed] [Google Scholar]
- 36. Kim JC, Ou YY, Badano JL, Esmail MA, Leitch CC, Fiedrich E, Beales PL, Archibald JM, Katsanis N, Rattner JB et al (2005) MKKS/BBS6, a divergent chaperonin‐like protein linked to the obesity disorder Bardet‐Biedl syndrome, is a novel centrosomal component required for cytokinesis. J Cell Sci 118: 1007–1020 [DOI] [PubMed] [Google Scholar]
- 37. Wood CR, Wang Z, Diener D, Zones JM, Rosenbaum J, Umen JG (2012) IFT proteins accumulate during cell division and localize to the cleavage furrow in chlamydomonas. PLoS ONE 7: e30729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Qin H, Wang Z, Diener D, Rosenbaum J (2007) Intraflagellar transport protein 27 is a small G protein involved in cell‐cycle control. Curr Biol 17: 193–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Knödler A, Feng S, Zhang J, Zhang X, Das A, Peränen J, Guo W (2010) Coordination of Rab8 and Rab11 in primary ciliogenesis. Proc Natl Acad Sci USA 107: 6346–6351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Welz T, Wellbourne‐Wood J, Kerkhoff E (2014) Orchestration of cell surface proteins by Rab11. Trends Cell Biol 24: 407–414 [DOI] [PubMed] [Google Scholar]
- 41. Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peränen J, Merdes A, Slusarski DC, Scheller RH, Bazan JF, Sheffield VC et al (2007) A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell 129: 1201–1213 [DOI] [PubMed] [Google Scholar]
- 42. Bachmann‐Gagescu R, Phelps IG, Stearns G, Link BA, Brockerhoff SE, Moens CB, Doherty D (2011) The ciliopathy gene cc2d2a controls zebrafish photoreceptor outer segment development through a role in Rab8‐dependent vesicle trafficking. Hum Mol Genet 20: 4041–4055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Westlake CJ, Baye LM, Nachury MV, Wright KJ, Ervin KE, Phu L, Chalouni C, Beck JS, Kirkpatrick DS, Slusarski DC et al (2011) Primary cilia membrane assembly is initiated by Rab11 and transport protein particle II (TRAPPII) complex‐dependent trafficking of Rabin8 to the centrosome. Proc Natl Acad Sci USA 108: 2759–2764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chang J, Seo SG, Lee KH, Nagashima K, Bang JK, Kim BY, Erikson RL, Lee K‐W, Lee HJ, Park J‐E et al (2013) Essential role of Cenexin1, but not Odf2, in ciliogenesis. Cell Cycle 12: 655–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gaillard CLB, Pallesi‐Pocachard E, Massey‐Harroche D, Richard F, Arsanto JP, Chauvin JP, Lecine P, Krämer H, Borg JP, Le Bivic A (2011) Hook2 is involved in the morphogenesis of the primary cilium. Mol Biol Cell 22: 4549–4562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yoshimura S‐I, Egerer J, Fuchs E, Haas AK, Barr FA (2007) Functional dissection of Rab GTPases involved in primary cilium formation. J Cell Biol 178: 363–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zuo X, Fogelgren B, Lipschutz JH (2011) The small GTPase Cdc42 is necessary for primary ciliogenesis in renal tubular epithelial cells. J Biol Chem 286: 22469–22477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fogelgren B, Lin S‐Y, Zuo X, Jaffe KM, Park KM, Reichert RJ, Bell PD, Burdine RD, Lipschutz JH (2011) The exocyst protein Sec10 interacts with Polycystin‐2 and knockdown causes PKD‐phenotypes. PLoS Genet 7: e1001361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Das S, Hehnly H, Doxsey S (2014) A new role for Rab GTPases during early mitotic stages. Small GTPases 5: 1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hehnly H, Chen CT, Powers CM, Liu HL, Doxsey S (2012) The centrosome regulates the Rab11‐ dependent recycling endosome pathway at appendages of the mother centriole. Curr Biol 22: 1944–1950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schmidt KN, Kuhns S, Neuner A, Hub B, Zentgraf H, Pereira G (2012) Cep164 mediates vesicular docking to the mother centriole during early steps of ciliogenesis. J Cell Biol 199: 1083–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Joo K, Kim CG, Lee M‐S, Moon H‐Y, Lee S‐H, Kim MJ, Kweon H‐S, Park W‐Y, Kim C‐H, Gleeson JG et al (2013) CCDC41 is required for ciliary vesicle docking to the mother centriole. Proc Natl Acad Sci USA 110: 5987–5992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ye X, Zeng H, Ning G, Reiter JF, Liu A (2014) C2 cd3 is critical for centriolar distal appendage assembly and ciliary vesicle docking in mammals. Proc Natl Acad Sci USA 111: 2164–2169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sorokin S (1962) Centrioles rudimentary and smooth and the formation of cilia muscle by fibroblasts. J Cell Biol 15: 363–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lu Q, Insinna C, Ott C, Stauffer J, Pintado PA, Rahajeng J, Baxa U, Walia V, Cuenca A, Hwang YS et al (2015) Early steps in primary cilium assembly require EHD1/EHD3‐dependent ciliary vesicle formation. Nat Cell Biol 17: 228–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Veleri S, Manjunath SH, Fariss RN, May‐Simera H, Brooks M, Foskett TA, Gao C, Longo TA, Liu P, Nagashima K et al (2014) Ciliopathy‐associated gene Cc2d2a promotes assembly of subdistal appendages on the mother centriole during cilia biogenesis. Nat Commun 5: 4207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gorden NT, Arts HH, Parisi MA, Coene KLM, Letteboer SJF, van Beersum SEC, Mans DA, Hikida A, Eckert M, Knutzen D et al (2008) CC2D2A is mutated in joubert syndrome and interacts with the ciliopathy‐associated basal body protein CEP290. Am J Hum Genet 83: 559–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tallila J, Jakkula E, Peltonen L, Salonen R, Kestilä M (2008) Identification of CC2D2A as a meckel syndrome gene adds an important piece to the ciliopathy puzzle. Am J Hum Genet 82: 1361–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Finetti F, Paccani SR, Riparbelli MG, Giacomello E, Perinetti G, Pazour GJ, Rosenbaum JL, Baldari CT (2009) Intraflagellar transport is required for polarized recycling of the TCR/CD3 complex to the immune synapse. Nat Cell Biol 11: 1332–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cemerski S, Shaw A (2006) Immune synapses in T‐cell activation. Curr Opin Immunol 18: 298–304 [DOI] [PubMed] [Google Scholar]
- 61. Finetti F, Patrussi L, Masi G, Onnis A, Galgano D, Lucherini OM, Pazour GJ, Baldari CT (2014) Specific recycling receptors are targeted to the immune synapse by the intraflagellar transport system. J Cell Sci 127: 1924–1937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rohatgi R, Milenkovic L, Scott MP (2007) Patched1 regulates hedgehog signaling at the primary cilium. Science 317: 372–376 [DOI] [PubMed] [Google Scholar]
- 63. Roy S (2012) Cilia and Hedgehog: when and how was their marriage solemnized? Differentiation 83: S43–S48 [DOI] [PubMed] [Google Scholar]
- 64. Dorn KV, Hughes CE, Rohatgi R (2012) A smoothened‐Evc2 complex transduces the hedgehog signal at primary cilia. Dev Cell 23: 823–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Keady BT, Samtani R, Tobita K, Tsuchya M, San Agustin JT, Follit JA, Jonassen JA, Subramanian R, Lo CW, Pazour GJ (2012) IFT25 links the signal‐dependent movement of hedgehog components to intraflagellar transport. Dev Cell 22: 940–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bazzi H, Anderson KV (2014) Acentriolar mitosis activates a p53‐dependent apoptosis pathway in the mouse embryo. Proc Natl Acad Sci USA 2014: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bijlsma MF, Damhofer H, Roelink H (2012) Hedgehog‐stimulated chemotaxis is mediated by smoothened located outside the primary cilium. Sci Signal 5: ra60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. De la Roche M, Ritter AT, Angus KL, Dinsmore C, Earnshaw CH, Reiter JF, Griffiths GM (2013) Hedgehog signaling controls T cell killing at the immunological synapse. Science 342: 1247–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Prosser S, Morrison C (2015) Centrin2 regulates CP110 removal in primary cilium formation. J Cell Biol 208: 693–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Tanos BE, Yang HJ, Soni R, Wang WJ, Macaluso FP, Asara JM, Tsou MFB (2013) Centriole distal appendages promote membrane docking, leading to cilia initiation. Genes Dev 27: 163–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Robert A, Margall‐Ducos G, Guidotti J‐E, Brégerie O, Celati C, Bréchot C, Desdouets C (2007) The intraflagellar transport component IFT88/polaris is a centrosomal protein regulating G1‐S transition in non‐ciliated cells. J Cell Sci 120: 628–637 [DOI] [PubMed] [Google Scholar]
- 72. Jackson PK (2011) Do cilia put brakes on the cell cycle? Nat Cell Biol 13: 340–342 [DOI] [PubMed] [Google Scholar]
- 73. Kim S, Zaghloul NA, Bubenshchikova E, Oh EC, Rankin S, Katsanis N, Obara T, Tsiokas L (2011) Nde1‐mediated inhibition of ciliogenesis affects cell cycle re‐entry. Nat Cell Biol 13: 351–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hanks S, Coleman K, Summersgill B, Messahel B, Williamson D, Pritchard‐Jones K, Strefford J, Swansbury J, Plaja A, Shipley J et al (2006) Comparative genomic hybridization and BUB1B mutation analyses in childhood cancers associated with mosaic variegated aneuploidy syndrome. Cancer Lett 239: 234–238 [DOI] [PubMed] [Google Scholar]
- 75. Egeberg DL, Lethan M, Manguso R, Schneider L, Awan A, Jørgensen TS, Byskov AG, Pedersen LB, Christensen ST (2012) Primary cilia and aberrant cell signaling in epithelial ovarian cancer. Cilia 1: 15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Toftgård R (2009) Two sides to cilia in cancer. Nat Med 15: 994–996 [DOI] [PubMed] [Google Scholar]
- 77. Hassounah NB, Bunch TA, McDermott KM (2012) Molecular pathways: the role of primary cilia in cancer progression and therapeutics with a focus on hedgehog signaling. Clin Cancer Res 18: 2429–2435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Han Y‐G, Alvarez‐Buylla A (2010) Role of primary cilia in brain development and cancer. Curr Opin Neuroviol 20: 58–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hassounah NB, Nagle R, Saboda K, Roe DJ, Dalkin BL, McDermott KM (2013) Primary cilia are lost in preinvasive and invasive prostate cancer. PLoS ONE 8: e68521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Basten SG, Willekers S, Vermaat JS, Slaats GG, Voest EE, van Diest PJ, Giles RH (2013) Reduced cilia frequencies in human renal cell carcinomas versus neighboring parenchymal tissue. Cilia 2: 2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Habbig S, Bartram MP, Sägmüller JG, Griessmann A, Franke M, Müller RU, Schwarz R, Hoehne M, Bergmann C, Tessmer C et al (2012) The ciliopathy disease protein NPHP9 promotes nuclear delivery and activation of the oncogenic transcriptional regulator TAZ. Hum Mol Genet 21: 5528–5538 [DOI] [PubMed] [Google Scholar]
- 82. Palmirotta R, Savonarola A, Ludovici G, Donah P, Cavaliere F, De Marchis ML, Ferroni P, Guadagni F (2010) Association between Birt Hogg Dubé syndrome and cancer predisposition. Anticancer Res 30: 751–758 [PubMed] [Google Scholar]
- 83. You N, Liu W, Zhong X, Ji R, Zhang M, You H, Dou K, Tao K (2012) Tg737 inhibition results in malignant transformation in fetal liver stem/progenitor cells by promoting cell‐cycle progression and differentiation arrest. Mol Carcinog 51: 659–673 [DOI] [PubMed] [Google Scholar]
- 84. Isfort RJ, Cody DB, Doersen CJ, Richards WG, Yoder BK, Wilkinson JE, Kier LD, Jirtle RL, Isenberg JS, Klounig JE et al (1997) The tetratricopeptide repeat containing Tg737 gene is a liver neoplasia tumor suppressor gene. Oncogene 15: 1797–1803 [DOI] [PubMed] [Google Scholar]
- 85. Löffler H, Bochtler T, Fritz B, Tews B, Ho AD, Lukas J, Bartek J, Krämer A (2007) DNA damage‐induced accumulation of centrosomal Chk1 contributes to its checkpoint function. Cell Cycle 6: 2541–2548 [DOI] [PubMed] [Google Scholar]
- 86. Krämer A, Mailand N, Lukas C, Syljuåsen RG, Wilkinson CJ, Nigg EA, Bartek J, Lukas J (2004) Centrosome‐associated Chk1 prevents premature activation of cyclin‐B‐Cdk1 kinase. Nat Cell Biol 6: 884–891 [DOI] [PubMed] [Google Scholar]
- 87. Staples CJ, Myers KN, Beveridge RDD, Patil AA, Howard AE, Barone G, Lee AJX, Swanton C, Howell M, Maslen S et al (2014) Ccdc13; a novel human centriolar satellite protein required for ciliogenesis and genome stability. J Cell Sci 127: 2910–2919 [DOI] [PubMed] [Google Scholar]
- 88. Den Dulk B, van Eijk P, de Ruijter M, Brandsma JA, Brouwer J (2008) The NER protein Rad33 shows functional homology to human Centrin2 and is involved in modification of Rad4. DNA Repair (Amst) 7: 858–868 [DOI] [PubMed] [Google Scholar]
- 89. Molinier J, Ramos C, Fritsch O, Hohn B (2004) CENTRIN2 modulates homologous recombination and nucleotide excision repair in Arabidopsis. Plant Cell 16: 1633–1643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Acu ID, Liu T, Suino‐Powell K, Mooney SM, D'Assoro AB, Rowland N, Muotri AR, Correa RG, Niu Y, Kumar R et al (2010) Coordination of centrosome homeostasis and DNA repair is intact in MCF‐7 and disrupted in MDA‐MB 231 breast cancer cells. Cancer Res 70: 3320–3328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Chaki M, Airik R, Ghosh AK, Giles RH, Chen R, Slaats GG, Wang H, Hurd TW, Zhou W, Cluckey A et al (2012) Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell 150: 533–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Airik R, Slaats GG, Guo Z, Weiss A‐C, Khan N, Ghosh A, Hurd TW, Bekker‐Jensen S, Schrøder JM, Elledge SJ et al (2014) Renal‐retinal ciliopathy gene Sdccag8 regulates DNA damage response signaling. J Am Soc Nephrol 25: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Griffith E, Walker S, Martin C‐A, Vagnarelli P, Stiff T, Vernay B, Al Sanna N, Saggar A, Hamel B, Earnshaw WC et al (2008) Mutations in pericentrin cause Seckel syndrome with defective ATR‐dependent DNA damage signaling. Nat Genet 40: 232–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Choi H, Lin JR, Vannier JB, Slaats G, Kile A, Paulsen R, Manning D, Beier D, Giles R, Boulton S et al (2013) NEK8 links the ATR‐regulated replication stress response and S phase CDK activity to renal ciliopathies. Mol Cell 51: 423–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Frank V, Habbig S, Bartram MP, Eisenberger T, Veenstra‐Knol HE, Decker C, Boorsma RAC, Göbel H, Nürnberg G, Griessmann A et al (2013) Mutations in NEK8 link multiple organ dysplasia with altered Hippo signalling and increased c‐MYC expression. Hum Mol Genet 22: 2177–2185 [DOI] [PubMed] [Google Scholar]
- 96. Bowers AJ, Boylan JF (2004) Nek8, a NIMA family kinase member, is overexpressed in primary human breast tumors. Gene 328: 135–142 [DOI] [PubMed] [Google Scholar]
- 97. Kanu N, Behrens A (2008) ATMINistrating ATM signalling: regulation of ATM by ATMIN. Cell Cycle 7: 3483–3486 [DOI] [PubMed] [Google Scholar]
- 98. Jurado S, Conlan LA, Baker EK, Ng JL, Tenis N, Hoch NC, Gleeson K, Smeets M, Izon D, Heierhorst J (2012) ATM substrate Chk2‐interacting Zn2+ finger (ASCIZ) is a bi‐functional transcriptional activator and feedback sensor in the regulation of dynein light chain (DYNLL1) expression. J Biol Chem 287: 3156–3164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Goggolidou P, Stevens JL, Agueci F, Keynton J, Wheway G, Grimes DT, Patel SH, Hilton H, Morthorst SK, DiPaolo A et al (2014) ATMIN is a transcriptional regulator of both lung morphogenesis and ciliogenesis. Development 141: 3966–3977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Miyamoto T, Porazinski S, Wang H, Borovina A, Ciruna B, Shimizu A, Kajii T, Kikuchi A, Furutani‐Seiki M, Matsuura S (2011) Insufficiency of BUBR1, a mitotic spindle checkpoint regulator, causes impaired ciliogenesis in vertebrates. Hum Mol Genet 20: 2058–2070 [DOI] [PubMed] [Google Scholar]
- 101. Ganner A, Lienkamp S, Schäfer T, Romaker D, Wegierski T, Park TJ, Spreitzer S, Simons M, Gloy J, Kim E et al (2009) Regulation of ciliary polarity by the APC/C. Proc Natl Acad Sci USA 106: 17799–17804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Nilsson J, Yekezare M, Minshull J, Pines J (2008) The APC/C maintains the spindle assembly checkpoint by targeting Cdc20 for destruction. Nat Cell Biol 10: 1411–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Sullivan M, Morgan DO (2007) Finishing mitosis, one step at a time. Nat Rev Mol Cell Biol 8: 894–903 [DOI] [PubMed] [Google Scholar]
- 104. Follit JA, Tuft RA, Fogarty KE, Pazour GJ (2006) The intraflagellar transport protein IFT20 is associated with the Golgi complex and is required for cilia assembly. Mol Biol Cell 17: 3781–3792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Follit JA, San Agustin JT, Xu F, Jonassen JA, Samtani R, Lo CW, Pazour GJ (2008) The Golgin GMAP210/TRIP11 anchors IFT20 to the Golgi complex. PLoS Genet 4: e1000315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Barr MM, Sternberg PW (1999) A polycystic kidney‐disease gene homologue required for male mating behaviour in C. elegans . Nature 401: 386–389 [DOI] [PubMed] [Google Scholar]
- 107. Yoder BK (2002) The polycystic kidney disease proteins, polycystin‐1, polycystin‐2, polaris, and cystin, are co‐localized in renal cilia. J Am Soc Nephrol 13: 2508–2516 [DOI] [PubMed] [Google Scholar]
- 108. Wang S (2004) The autosomal recessive polycystic kidney disease protein is localized to primary cilia, with concentration in the basal body area. J Am Soc Nephrol 15: 592–602 [DOI] [PubMed] [Google Scholar]
- 109. Ward CJ, Yuan D, Masyuk TV, Wang X, Punyashthiti R, Whelan S, Bacallao R, Torra R, LaRusso NF, Torres VE et al (2003) Cellular and subcellular localization of the ARPKD protein; fibrocystin is expressed on primary cilia. Hum Mol Genet 12: 2703–2710 [DOI] [PubMed] [Google Scholar]
- 110. Pazour GJ, San Agustin JT, Follit JA, Rosenbaum JL, Witman GB (2002) Polycystin‐2 localizes to kidney cilia and the ciliary level is elevated in orpk mice with polycystic kidney disease. Curr Biol 12: R378–R380 [DOI] [PubMed] [Google Scholar]
- 111. Zhang J, Wu M, Wang S, Shah JV, Wilson PD, Zhou J (2010) Polycystic kidney disease protein fibrocystin localizes to the mitotic spindle and regulates spindle bipolarity. Hum Mol Genet 19: 3306–3319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Burtey S, Riera M, Ribe É, Pennenkamp P, Rance R, Luciani J, Dworniczak B, Mattei MG, Fontés M (2008) Centrosome overduplication and mitotic instability in PKD2 transgenic lines. Cell Biol Int 32: 1193–1198 [DOI] [PubMed] [Google Scholar]
- 113. Battini L, Macip S, Fedorova E, Dikman S, Somlo S, Montagna C, Gusella GL (2008) Loss of polycystin‐1 causes centrosome amplification and genomic instability. Hum Mol Genet 17: 2819–2833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Rundle DR, Gorbsky G, Tsiokas L (2004) PKD2 interacts and co‐localizes with mDia1 to mitotic spindles of dividing cells: role of mDia1 in PKD2 localization to mitotic spindles. J Biol Chem 279: 29728–29739 [DOI] [PubMed] [Google Scholar]
- 115. Jonassen JA, Agustin JS, Follit JA, Pazour GJ (2008) Deletion of IFT20 in the mouse kidney causes misorientation of the mitotic spindle and cystic kidney disease. J Cell Biol 183: 377–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Patel V, Li L, Cobo‐stark P, Shao X, Somlo S, Lin F, Igarashi P (2008) Acute kidney injury and aberrant planar cell polarity induce cyst formation in mice lacking renal cilia. Hum Mol Genet 17: 1578–1590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Lancaster MA, Knoblich JA (2012) Spindle orientation in mammalian cerebral cortical development. Curr Opin Neurobiol 22: 737–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Jonassen JA, SanAgustin J, Baker SP, Pazour GJ (2012) Disruption of IFT complex a causes cystic kidneys without mitotic spindle misorientation. J Am Soc Nephrol 23: 641–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Delaval B, Covassin L, Lawson ND, Doxsey S (2011) Centrin depletion causes cyst formation and other ciliopathy‐related phenotypes in zebrafish. Cell Cycle 10: 3964–3972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Zaghloul NA, Katsanis N (2009) Mechanistic insights into Bardet‐Biedl syndrome, a model ciliopathy. J Clin Invest 119: 428–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Fliegauf M, Benzing T, Omran H (2007) When cilia go bad: cilia defects and ciliopathies. Nat Rev Mol Cell Biol 8: 880–893 [DOI] [PubMed] [Google Scholar]
- 122. Beales PL, Reid HA, Griffiths MH, Maher ER, Flinter FA, Woolf AS (2000) Renal cancer and malformations in relatives of patients with Bardet‐Biedl syndrome. Nephrol Dial Transplant 15: 1977–1985 [DOI] [PubMed] [Google Scholar]
- 123. Arquint C, Nigg EA (2014) STIL microcephaly mutations interfere with APC/C‐mediated degradation and cause centriole amplification. Curr Biol 24: 351–360 [DOI] [PubMed] [Google Scholar]
- 124. Guernsey DL, Jiang H, Hussin J, Arnold M, Bouyakdan K, Perry S, Babineau‐Sturk T, Beis J, Dumas N, Evans SC et al (2010) Mutations in centrosomal protein CEP152 in primary microcephaly families linked to MCPH4. Am J Hum Genet 87: 40–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. García‐Castillo H, Vásquez‐Velásquez AI, Rivera H, Barros‐Núñez P (2008) Clinical and genetic heterogeneity in patients with mosaic variegated aneuploidy: delineation of clinical subtypes. Am J Med Genet Part A 146: 1687–1695 [DOI] [PubMed] [Google Scholar]
- 126. Benzing T, Schermer B (2012) Clinical spectrum and pathogenesis of nephronophthisis. Curr Opin Nephrol Hypertens 21: 272–278 [DOI] [PubMed] [Google Scholar]
- 127. Shaheen R, Schmidts M, Faqeih E, Hashem A, Lausch E, Holder I, Superti‐Furga A, Mitchison HM, Almoisheer A, Alamro R et al (2014) A founder CEP120 mutation in Jeune asphyxiating thoracic dystrophy expands the role of centriolar proteins in skeletal ciliopathies. Hum Mol Genet 000: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Shaheen R, Faqeih E, Shamseldin HE, Noche RR, Sunker A, Alshammari MJ, Al‐Sheddi T, Adly N, Al‐Dosari MS, Megason SG et al (2012) POC1A truncation mutation causes a ciliopathy in humans characterized by primordial dwarfism. Am J Hum Genet 91: 330–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Beck BB, Phillips JB, Bartram MP, Wegner J, Thoenes M, Pannes A, Sampson J, Heller R, Göbel H, Koerber F et al (2014) Mutation of POC1B in a severe syndromic retinal ciliopathy. Hum Mutat 35: 1153–1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Roosing S, Lamers IJC, De Vrieze E, Van Den Born LI, Lambertus S, Arts HH, Peters TA, Hoyng CB, Kremer H, Hetterschijt L et al (2014) Disruption of the basal body protein poc1b results in autosomal‐recessive cone‐rod dystrophy. Am J Hum Genet 95: 131–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Shaheen R, Shamseldin HE, Loucks CM, Seidahmed MZ, Ansari S, Ibrahim Khalil M, Al‐Yacoub N, Davis EE, Mola NA, Szymanska K et al (2014) Mutations in CSPP1, encoding a core centrosomal protein, cause a range of ciliopathy phenotypes in humans. Am J Hum Genet 94: 73–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Otto EA, Hurd TW, Airik R, Chaki M, Zhou W, Stoetzel C, Patil SB, Levy S, Ghosh AK, Murga‐Zamalloa CA et al (2010) Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal‐renal ciliopathy. Nat Genet 42: 840–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Yang Y, Liu M, Li D, Ran J, Gao J, Suo S, Sun S‐C, Zhou J (2014) CYLD regulates spindle orientation by stabilizing astral microtubules and promoting dishevelled‐NuMA‐dynein/dynactin complex formation. Proc Natl Acad Sci USA 111: 2158–2163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Szymanska K, Berry I, Logan CV, Cousins SR, Lindsay H, Jafri H, Raashid Y, Malik‐Sharif S, Castle B, Ahmed M et al (2012) Founder mutations and genotype‐phenotype correlations in Meckel‐Gruber syndrome and associated ciliopathies. Cilia 1: 18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Bachmann‐Gagescu R, Ishak GE, Dempsey JC, Adkins J, O'Day D, Phelps IG, Gunay‐Aygun M, Kline AD, Szczaluba K, Martorell L et al (2012) Genotype‐phenotype correlation in CC2D2A‐related Joubert syndrome reveals an association with ventriculomegaly and seizures. J Med Genet 49: 126–137 [DOI] [PubMed] [Google Scholar]
- 136. Park TJ, Haigo SL, Wallingford JB (2006) Ciliogenesis defects in embryos lacking inturned or fuzzy function are associated with failure of planar cell polarity and Hedgehog signaling. Nat Genet 38: 303–311 [DOI] [PubMed] [Google Scholar]
- 137. Chen Z, Indjeian VB, McManus M, Wang L, Dynlacht BD (2002) CP110, a cell cycle‐dependent CDK substrate, regulates centrosome duplication in human cells. Dev Cell 3: 339–350 [DOI] [PubMed] [Google Scholar]
- 138. Spektor A, Tsang WY, Khoo D, Dynlacht BD (2007) Cep97 and CP110 suppress a cilia assembly program. Cell 130: 678–690 [DOI] [PubMed] [Google Scholar]
- 139. Tang Z, Lin MG, Stowe TR, Chen S, Zhu M, Stearns T, Franco B, Zhong Q (2013) Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature 502: 254–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Pampliega O, Orhon I, Patel B, Sridhar S, Díaz‐Carretero A, Beau I, Codogno P, Satir BH, Satir P, Cuervo AM (2013) Functional interaction between autophagy and ciliogenesis. Nature 502: 194–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Zhang Y, Seo S, Bhattarai S, Bugge K, Searby CC, Zhang Q, Drack AV, Stone EM, Sheffield VC (2014) BBS mutations modify phenotypic expression of CEP290‐related ciliopathies. Hum Mol Genet 23: 40–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Lopes CAM, Prosser SL, Romio L, Hirst RA, O'Callaghan C, Woolf AS, Fry AM (2011) Centriolar satellites are assembly points for proteins implicated in human ciliopathies, including oral‐facial‐digital syndrome 1. J Cell Sci 124: 600–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Corbit KC, Shyer AE, Dowdle WE, Gaulden J, Singla V, Chen M‐H, Chuang P‐T, Reiter JF (2008) Kif3a constrains beta‐catenin‐dependent Wnt signalling through dual ciliary and non‐ciliary mechanisms. Nat Cell Biol 10: 70–76 [DOI] [PubMed] [Google Scholar]
- 144. Lancaster MA, Schroth J, Gleeson JG (2011) Subcellular spatial regulation of canonical Wnt signalling at the primary cilium. Nat Cell Biol 13: 700–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Ocbina PJR, Tuson M, Anderson KV (2009) Primary cilia are not required for normal canonical Wnt signaling in the mouse embryo. PLoS ONE 4: e6839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Habib SJ, Chen B‐C, Tsai F‐C, Anastassiadis K, Meyer T, Betzig E, Nusse R (2013) A localized Wnt signal orients asymmetric stem cell division in vitro. Science 339: 1445–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Mbom BC, Nelson WJ, Barth A (2013) β‐catenin at the centrosome: discrete pools of β‐catenin communicate during mitosis and may co‐ordinate centrosome functions and cell cycle progression. BioEssays 35: 804–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Fuentealba LC, Eivers E, Ikeda A, Hurtado C, Kuroda H, Pera EM, De Robertis EM (2007) Integrating patterning signals: Wnt/GSK3 regulates the duration of the BMP/Smad1 signal. Cell 131: 980–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Mbom BC, Siemers KA, Ostrowski MA, Nelson WJ, Barth AIM (2014) Nek2 phosphorylates and stabilizes β‐catenin at mitotic centrosomes downstream of Plk1. Mol Biol Cell 25: 977–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Liu YP, Tsai IC, Morleo M, Oh EC, Leitch CC, Massa F, Lee BH, Parker DS, Finley D, Zaghloul NA et al (2014) Ciliopathy proteins regulate paracrine signaling by modulating proteasomal degradation of mediators. J Clin Invest 124: 2059–2070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Paridaen JTML, Wilsch‐Bräuninger M, Huttner WB (2013) Asymmetric inheritance of centrosome‐associated primary cilium membrane directs ciliogenesis after cell division. Cell 155: 333–344 [DOI] [PubMed] [Google Scholar]
- 152. Kalay E, Yigit G, Aslan Y, Brown KE, Pohl E, Bicknell LS, Kayserili H, Li Y, Tüysüz B, Nürnberg G et al (2011) CEP152 is a genome maintenance protein disrupted in Seckel syndrome. Nat Genet 43: 23–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Bond J, Roberts E, Springell K, Lizarraga SB, Scott S, Higgins J, Hampshire DJ, Morrison EE, Leal GF, Silva EO et al (2005) A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size. Nat Genet 37: 353–355 [DOI] [PubMed] [Google Scholar]
- 154. Kumar A, Girimaji SC, Duvvari MR, Blanton SH (2008) Mutations in STIL, encoding a pericentriolar and centrosomal protein, cause primary microcephaly. Am J Hum Genet 84: 286–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Sir J‐H, Barr AR, Nicholas AK, Carvalho OP, Khurshid M, Sossick A, Reichelt S, D'Santos C, Woods CG, Gergely F (2011) A primary microcephaly protein complex forms a ring around parental centrioles. Nat Genet 43: 1147–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Hussain MS, Baig SM, Neumann S, Nürnberg G, Farooq M, Ahmad I, Alef T, Hennies HC, Technau M, Altmüller J et al (2012) A truncating mutation of CEP135 causes primary microcephaly and disturbed centrosomal function. Am J Hum Genet 90: 871–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Delaval B, Doxsey SJ (2010) Pericentrin in cellular function and disease. J Cell Biol 188: 181–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Carleton M, Mao M, Biery M, Warrener P, Kim S, Buser C, Marshall CG, Fernandes C, Annis J, Linsley PS (2006) RNA interference‐mediated silencing of mitotic kinesin KIF14 disrupts cell cycle progression and induces cytokinesis failure. Mol Cell Biol 26: 3853–3863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Filges I, Nosova E, Bruder E, Tercanli S, Townsend K, Gibson W, Röthlisberger B, Heinimann K, Hall J, Gregory‐Evans C et al (2013) Exome sequencing identifies mutations in KIF14 as a novel cause of an autosomal recessive lethal fetal ciliopathy phenotype. Clin Genet 86: 220–228 [DOI] [PubMed] [Google Scholar]
- 160. Martin C‐A, Ahmad I, Klingseisen A, Hussain MS, Bicknell LS, Leitch A, Nürnberg G, Toliat MR, Murray JE, Hunt D et al (2014) Mutations in PLK4, encoding a master regulator of centriole biogenesis, cause microcephaly, growth failure and retinopathy. Nat Genet 46: 1283–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]