

Abstract
The histone H3K27 demethylase, UTX, is a known component of the H3K4 methyltransferase MLL complex, but its functional association with H3K4 methylation in human cancers remains largely unknown. Here we demonstrate that UTX loss induces epithelial–mesenchymal transition (EMT)‐mediated breast cancer stem cell (CSC) properties by increasing the expression of the SNAIL, ZEB1 and ZEB2 EMT transcription factors (EMT‐TFs) and of the transcriptional repressor CDH1. UTX facilitates the epigenetic silencing of EMT‐TFs by inducing competition between MLL4 and the H3K4 demethylase LSD1. EMT‐TF promoters are occupied by c‐Myc and MLL4, and UTX recognizes these proteins, interrupting their transcriptional activation function. UTX decreases H3K4me2 and H3 acetylation at these promoters by forming a transcriptional repressive complex with LSD1, HDAC1 and DNMT1. Taken together, our findings indicate that UTX is a prominent tumour suppressor that functions as a negative regulator of EMT‐induced CSC‐like properties by epigenetically repressing EMT‐TFs.
Keywords: breast CSC, EMT, UTX
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Cancer
Introduction
A ubiquitously transcribed tetratricopeptide repeat X chromosome (UTX) functions as a histone demethylase towards di‐ and tri‐methylated histone H3 on lysine 27 (H3K27me2/H3K27me3) 1. This demethylase is also a known component of the mixed‐lineage leukaemia (MLL) 2/3 H3K4 methyltransferase complex, although its catalytic effect on H3K4me remains unclear 2, 3, 4. Polycomb group (PcG)‐dependent H3K27 methylation has redundant functions in many biological processes, including stem cell regulation and tumour development 5, 6, 7, although less evidence supports the functional role of UTX in these cellular processes. Several studies suggest that UTX regulates somatic cell reprogramming and embryonic development 8, 9, as well as tumour suppression in leukaemia 10, 11. Furthermore, genomewide analyses have identified various somatic mutations and deletions of UTX in several human cancers, including breast, bladder, and renal cancers and leukaemia 12, 13, 14. However, the demethylation‐dependent or demethylation‐independent molecular function of UTX in human cancers has not been clearly explained, and it remains controversial whether UTX has tumour‐suppressive activity 15, 16, 17.
Accumulating evidence shows that many histone methyltransferases and demethylases are involved in the regulation of cancer stem cells (CSCs), a small population of stem‐like cancer cells possessing tumorigenic and metastatic capacities 18, 19. Moreover, recent evidence suggests that regulators of the epithelial–mesenchymal transition (EMT), which repress E‐cadherin (encoded by CDH1) transcriptionally, such as SNAIL, SLUG, TWIST, ZEB1 and ZEB2, play a key role in inducing CSC self‐renewal 20, 21, 22, 23. Notably, these EMT transcription factors (EMT‐TFs) cooperate with various chromatin‐modifying enzymes (including the histone methylases G9a and Set8 and the H3K4/K9 demethylase LSD1) as well as histone deacetylases and DNMTs to form a repressive complex for CDH1 24, 25, 26. UTX is also known to interact physically with transcriptional and epigenetic regulators, such as MLL 2/3 H3K4 methyltransferase 2, 3, 4, histone acetyltransferase and the SWI/SNF complex 27, 28, during the assembly of multi‐protein complexes. However, the molecular relationship between UTX and EMT‐TFs in the epigenetic regulation of EMT‐associated CSC properties remains unknown.
Herein, we found that UTX is critical for inhibiting EMT‐induced breast CSC properties by suppressing the c‐Myc‐dependent transcription of SNAIL, ZEB1 and ZEB2 in cooperation with H3K4 demethylase LSD1, HDAC1 and DNMT1. These findings suggest a novel epigenetic mechanism for UTX during its inhibition of EMT‐TFs as a tumour suppressor in human breast cancer.
Results and Discussion
UTX loss enhances CSC‐like properties and the EMT in human breast cancer
Given the evidence that Drosophila and murine Utx antagonize Notch signalling and activate tumour suppressor Rb proteins, previous studies have suggested a putative tumour‐suppressive function for UTX in human cancer 15, 16. Consistently, in human breast cancer, UTX somatic mutations have been discovered that might be associated with the loss of UTX function 12. In contrast, a recent study suggested that UTX has oncogenic potential in human breast cancer cell lines 17. To clearly determine the role of UTX in human cancer, we evaluated the expression of UTX in several cell lines derived from several stages of breast tumour development. UTX expression was downregulated in the cancer cell lines compared with normal or immortalized cells (Fig 1A). Notably, UTX mRNA was expressed at lower levels in CD44+/CD24−/ESA+ cells with stem cell‐like characteristics (Fig 1B). Thus, we investigated whether UTX affects stem‐like phenotypes in normal and malignant breast epithelial cells. The percentage of CD44+/CD24−/ESA+ cells in the cell population was increased by UTX knockdown in the immortalized MCF10A cells and in breast cancer cell lines highly expressing UTX; however, this population declined in response to UTX overexpression in MDA‐MB‐231 cells having a low UTX expression (Figs 1C and EV1A and B). UTX also negatively regulated mammosphere formation and anchorage‐independent growth (Fig 1D and E). Consistently, in vivo breast tumour xenograft assays indicated that mice bearing UTX‐overexpressing MDA‐MB‐231 tumours exhibited retarded tumour initiation and growth (Table EV1, Fig EV1C), whereas mice injected with UTX‐deficient SK‐BR‐3 cells presented more rapid tumour formation and growth than control mice (Table EV1, Fig EV1D). Moreover, UTX knockdown induced the neoplastic transformation of the non‐tumorigenic MCF10A cell lines in vivo (Fig EV1E). Considering the ability of CD44+/CD24−/ESA+ cells to transform mammary epithelial cells 22, 23, these data suggest that UTX loss might promote breast tumorigenesis by expanding stem‐like cells and enhancing their self‐renewal and tumour‐initiating capacities.
Figure 1. Loss of UTX enhances CSC‐like properties in human breast cancer.

-
AScreening of protein expression levels by immunoblotting in normal and cancerous breast cell lines.
-
BUTX mRNA expression in CD44+/CD24−/ESA + cells (CSC) and other cells (non‐CSC) was sorted using flow cytometry from the indicated cell lines and analysed using qRT‐PCR. ***P < 0.001 versus Non‐CSC; two‐tailed unpaired t‐test.
-
CFlow cytometry analysis was used to measure CD44+/CD24−/ESA + cell populations in the indicated cell lines. The lower right quadrant indicates the CD44+/CD24−/ESA + cells (x‐axis, APC‐conjugated CD44; y‐axis, PE‐conjugated CD24). CON si, non‐targeting siRNA; UTX si, UTX siRNA; CON, pLVX‐puro empty vector; UTX, pLVX‐puro‐UTX. *P < 0.05, **P < 0.01, ***P < 0.001 versus controls; two‐tailed unpaired t‐test.
-
D, EMammosphere formation (D) and soft agar colony formation (E) in cells that underexpress or overexpress UTX. After 21 days (E) or as indicated (D), the numbers of spheres (D) or colonies (E) (> 100 μm diameter) were counted. shCON, control shRNA; shUTX, UTX shRNA. *P < 0.05, **P < 0.01, ***P < 0.001 versus controls; two‐tailed unpaired t‐test.
Figure EV1. Effect of UTX on breast tumorigenesis and on survival in breast cancer patients.
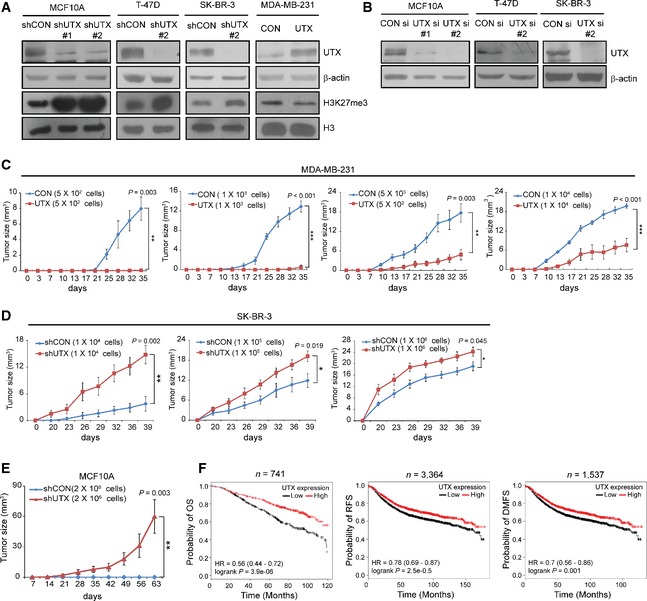
-
A, BGeneration of UTX‐knockdown or UTX‐overexpressing cells. Lysates from the indicated stable UTX‐knockdown (shUTX) and overexpressing cells (A) and UTX siRNA (si)‐transfected cells (B) were analysed for UTX and H3K27me3 expression using immunoblotting. β‐actin and histone H3 (H3) were used as loading controls for whole‐cell lysate and for histone proteins, respectively.
-
C, DUTX retards breast tumour growth in vivo. The indicated numbers of UTX‐overexpressing MDA‐MB‐231 (C) or UTX‐knockdown SK‐BR‐3 (D) cells were injected into the mammary fat pads of NOD/SCID mice, and xenograft tumour growth rates were monitored at the indicated time points for approximately 5 weeks. The data shown represent the means ± SE of n = 6 mice. *P < 0.05, **P < 0.01, ***P < 0.001 versus shCON or CON at last days, two‐tailed unpaired t‐test.
-
ETo determine whether UTX loss affects the neoplastic transformation of breast epithelial cells, NOD/SCID mice were injected with UTX‐deficient MCF10A cells and monitored at the indicated time points for 9 weeks. The data shown represent the means ± SE of n = 6 mice. **P < 0.01, two‐tailed unpaired t‐test.
-
FA meta‐analysis of the overall survival (OS), relapse‐free survival (RFS) and distant metastasis‐free survival (DMFS) of patients with breast cancer, as stratified according to UTX expression level using the Kaplan–Meier Plotter (http://kmplot.com/analysis). The high and low UTX expression groups were separated by median expression values of UTX that were based on the mean of all UTX probes (203990_s_at, 203991_s_at and 203992_s_at). P‐value was calculated based on the log‐rank test.
Source data are available online for this figure.
Recent studies have shown that EMT is essential for generating CD44+/CD24low/− stem‐like cells that can convert breast epithelial cells into tumorigenic cells and can confer breast cancer aggressiveness 20, 21, 22, 23. Consistent with these findings, UTX knockdown in MCF10A cells resulted in a mesenchymal‐like sporadic long spindle phenotype (Fig 2A). We also observed a reduced expression of epithelial markers including E‐cadherin but an increased expression of mesenchymal markers in these cells (Fig 2B–D). Furthermore, UTX‐overexpressing MDA‐MB‐231 cells exhibited epithelial‐like morphology (Fig 2A), increased epithelial marker expression and decreased mesenchymal marker expression (Fig 2B–D). Contrary to a recent study showing decreased invasion after UTX knockdown in breast cancer 17, EMT promotion resulting from UTX loss conferred invasion and migration abilities on non‐invasive MCF10A cells, whereas UTX overexpression inhibited these effects in invasive MDA‐MB‐231 cells (Fig 2E and F). Consistently, a meta‐analysis of breast cancer patient survival performed using the Kaplan–Meier Plotter 29 showed that high UTX expression is associated with favourable overall survival (P < 0.001, log‐rank test; n = 741), relapse‐free survival (P < 0.001, log‐rank test; n = 3,364) and distant metastasis‐free (P < 0.001, log‐rank test; n = 1,537) survival (Fig EV1F). Together, these findings indicate that UTX negatively regulates CSC‐like features including EMT and tumorigenesis, the loss of which might be associated with breast cancer aggressiveness, thereby supporting evidence that suggests a tumour‐suppressive function of UTX in human breast cancer.
Figure 2. Loss of UTX induces the EMT and invasion of breast cancer cells.

-
ARepresentative bright‐field images of cells that underexpress or overexpress UTX. Scale bars: 100 μm.
-
B, CThe expression of epithelial and mesenchymal markers in the indicated cells was measured using immunoblotting (B) and qRT‐PCR (C).
-
DThe immunofluorescence staining of E‐cadherin (red) and vimentin (green) was detected using confocal microscopy and quantified. DAPI (blue) was used for nuclear staining. Scale bars: 10 μm.
-
E, FChamber transwell assays of cellular invasion or migration by the indicated cells. Invasion and migration of test cells are expressed relative to values for control cells. Scale bars: 100 μm.
UTX regulates EMT‐induced stemness by inhibiting EMT‐TFs in an EZH2‐independent manner
To further investigate the molecular mechanism whereby UTX regulates E‐cadherin transcription during the EMT, we examined the effect of UTX on CDH1 promoter activity using a luciferase (luc) reporter assay. Wild‐type CDH1 promoter activity was repressed in UTX‐deficient MCF10A cells but was increased in UTX‐overexpressing MDA‐MB‐231 cells; however, an E‐box mutation in the promoter abrogated these effects, implying that regulation of the CDH1 promoter by UTX is E‐box‐dependent (Fig 3A). We also found that UTX negatively regulates the expression of SNAIL, ZEB1 and ZEB2, which are capable of repressing CDH1 expression by binding to the E‐box motifs within the CDH1 promoter, at the transcriptional level (Fig 3B and C), thereby decreasing their nuclear accumulation (Fig 3D and E). Moreover, a chromatin immunoprecipitation (ChIP) assay showed that the binding of these TFs on the proximal CDH1 promoter E‐box was higher in UTX‐knockdown cells (Figs 3F and EV2A). Epigenetic changes occur in the CDH1 promoter during the EMT through the cooperation of various epigenetic regulators (such as histone methyltransferases, HDACs and DNMTs) with EMT‐TFs 6, 25, 26, 30. Consistent with previous data supporting EZH2‐dependent CDH1 regulation 6, H3K27me3 (but not H3K4me2 or H3K9me2) was enriched at the CDH1 promoter and gene body regions in UTX‐knockdown cells (Figs 3F and EV2A). These findings indicate that UTX enhances CDH1 transcription by inhibiting EMT‐TF expression and activity towards the CDH1 promoter, in addition to demethylating H3K27me3. These EMT‐TFs have been identified as critical regulators of mammary stem cell activity 20; therefore, we next explored whether UTX regulates breast stem‐like cells via EMT‐TF regulation. Flow cytometry of siRNA‐treated MCF10A cells showed that the increase in the CD44+/CD24−/ESA+ cell population that results from UTX deficiency was reversed by the transient knockdown of SNAIL, ZEB1 and ZEB2 (Fig 3G), implying that UTX controls EMT and stemness by modulating the expression and function of EMT‐TFs.
Figure 3. UTX depletion inhibits E‐cadherin transcription by recruiting EMT‐related transcription factors.

-
ATo measure CDH1 promoter activity, a luciferase (luc) assay was performed in UTX‐knockdown MCF10A and in UTX‐overexpressing MDA‐MB‐231 cells that were co‐transfected with either CDH1 wild‐type (WT) or E‐box mutant (Mut)‐luc and β‐galactosidase constructs for 24 h.
-
B, CThe expression of EMT‐TFs was measured in the indicated cells using immunoblotting (B) and qRT‐PCR (C).
-
DImmunoblotting with cytoplasmic and nuclear fractions to analyse EMT‐TF expression. SP1 and α‐tubulin were used for loading controls for nuclear and cytoplasmic proteins, respectively.
-
EImmunofluorescence analysis showing the expression level and cellular localization of SNAIL, ZEB1 and ZEB2 in UTX‐knockdown MCF10A cells. Nuclei were stained with DAPI. Scale bar: 10 μm.
-
FChIP analysis showing the indicated histone methylation status and gene recruitment to the CDH1 promoter in UTX‐deficient MCF10A cells. IgG, control IgG for negative control.
-
GFlow cytometry analysis of the CD44+/CD24−/ESA + population in MCF10A cells that were transfected with non‐targeting siRNA (CON si) or with UTX‐targeting siRNA (UTX si), together with the indicated EMT‐TF siRNAs for 48 h.
Figure EV2. UTX deficiency‐mediated regulation of stem cell‐like properties is independent of EZH2 expression.

-
AChIP analysis showing the indicated histone methylation status and gene recruitment to the CDH1 gene body (left) and promoter upstream (right) in UTX‐deficient MCF10A cells. IgG, control IgG for negative control. Error bars indicate the SD (n = 3 independent experiments). *P < 0.05, **P < 0.01 versus shCON; two‐tailed unpaired t‐test.
-
BTo determine the effect of EZH2 on UTX deficiency‐induced stemness, UTX/EZH2 double‐knockdown MCF10A cells were obtained. Tet‐inducible shCon (shTet) and shEZH2 (shTet‐shEZH2) cell lines were treated with 1 μg/ml doxycycline (DOX) for 48 h. The CD44+/CD24−/ESA + population in the indicated cells was measured using flow cytometry.
-
C, DMammosphere formation (C) and colony‐forming assays (D) were performed in UTX/EZH2 double‐knockdown MCF10A cells.
-
E, FThe protein (E) and mRNA (F) expression of the indicated genes in UTX/EZH2 double‐knockdown MCF10A cells was measured using immunoblotting and qRT‐PCR.
-
GChIP analysis showing the recruitment of the indicated proteins and histone marks at the proximal CDH1 promoter.
We further examined whether the negative effect of UTX on EMT and stemness is dependent on H3K27 methyltransferase EZH2, a known critical regulator of CSC self‐renewal and CDH1 transcription 6, 7. Under EZH2‐deficient conditions using tetracycline (Tet)‐inducible shRNA, the CD44+/CD24−/ESA+ population expansion and self‐renewal properties induced by UTX‐knockdown cells were also sustained (Fig EV2B–D). Moreover, E‐cadherin was still repressed, and SNAIL, ZEB1 and ZEB2 were continuously upregulated under these conditions (Fig EV2E and F). Although H3K27me3 was decreased by EZH2 knockdown in the CDH1 promoter, recruitment of EMT‐TFs to this region was maintained continuously in the UTX‐knockdown cells (Fig EV2G). Therefore, these data imply that UTX regulates EMT and stemness in an EZH2 activity‐independent manner; this finding supports UTX as a crucial inhibitor of EMT‐associated breast tumorigenesis that might overcome the function of EZH2 in breast cancer.
UTX transcriptionally represses EMT‐TF expression by altering c‐Myc‐dependent and c‐Myc‐independent epigenetic modifications
Little is known about the epigenetic and transcriptional mechanisms that occur in the genomic regions of EMT‐TFs; in contrast, the CDH1 regulatory mechanism has been well established. Therefore, we next investigated the molecular mechanism by which UTX transcriptionally regulates EMT‐TFs to inhibit the properties of EMT‐induced CSCs. SNAIL, ZEB1 and ZEB2 were found to have E‐box motifs that bind c‐Myc in their proximal promoters; therefore, we examined the effect of UTX on c‐Myc activity towards EMT‐TFs. Although the expression of c‐Myc was not altered by UTX (Fig EV3A), the recruitment of c‐Myc to these regions was enhanced by UTX knockdown in MCF10A cells and was inhibited by UTX overexpression in MDA‐MB‐231 cells, as assessed using a ChIP assay (Figs 4A and EV3B–D). In addition, UTX bound directly to these regions. Previous studies have shown that many UTX‐occupied genes not only exhibit altered H3K27 methylation states but also altered H3K4 methylation states 2, 3, 31, although the mechanism by which H3K4 methylation is regulated via UTX remains unclear. To identify whether UTX‐associated histone modifications participate in the transcriptional regulation of EMT‐TFs, we examined the modification status of histone proteins including H3K27me3 and H3K4me2 at these promoters. Although the level of H3K27me3 remained unchanged, the H3K4me2 and H3 acetylation (ac) active histone markers were enriched in the SNAIL, ZEB1 and ZEB2 promoters in UTX‐deficient MCF10A cells. Moreover, the recruitment of p300 histone acetyltransferase (HAT) and the dissociation of HDAC1 and DNMT1 transcriptional repressive markers were observed in these regions. Consistent results were found in UTX‐overexpressing MDA‐MB‐231 cells. Furthermore, co‐immunoprecipitation (co‐IP) of exogenous and endogenous proteins showed that UTX physically interacts with c‐Myc, HDAC1 and DNMT1 (Fig 4B and C). To determine whether c‐Myc acts as a bridge for the recognition of UTX by the target promoter and consequent epigenetic changes, we analysed epigenetic changes at the EMT‐TF promoters following siRNA‐mediated c‐Myc depletion. Although c‐Myc was required for changes in H3ac by UTX, it did not affect the enrichment of UTX and H3K4me2 in these promoters (Fig EV4A and B). However, flow cytometry following c‐Myc siRNA treatment showed that c‐Myc depletion reverses UTX‐knockdown‐induced stem cell expansion (Fig 4D). Taken together, these results show that UTX represses the transcription of EMT‐TFs by inhibiting c‐Myc‐dependent histone acetylation and inducing c‐Myc‐independent H3K4 demethylation. Together with the results of a previous study showing the regulation of SNAIL by c‐Myc 32, our data suggest that c‐Myc is a common transcription factor for the induction of SNAIL, ZEB1 and ZEB2 expression through its role in coordinating with epigenetic modifiers that involve an active gene state. Furthermore, UTX might be a key component for the epigenetic silencing of these EMT‐TFs to antagonize c‐Myc function.
Figure EV3. UTX epigenetically regulates EMT‐TFs gene expression.

-
ALysates of the indicated cells were analysed by immunoblotting.
-
BSchematic illustration of the proximal promoter and transcription start site (TSS) regions of human SNAIL,ZEB1 and ZEB2 gene locus with adjacent E‐boxes. Regions #1 and #2 indicate the target sites for ChIP analysis.
-
C, DChIP analysis showing the histone methylation status and recruitment of the indicated proteins within the indicated genomic regions of EMT‐TFs in MCF10A (C) and MDA‐MB‐231 (D) cells. The data shown represent the means ± SD of three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 versus shCON or CON; two‐tailed unpaired t‐test.
Source data are available online for this figure.
Figure 4. UTX suppresses EMT‐TF expression by inducing c‐Myc‐dependent and c‐Myc‐independent epigenetic modification.

-
AChIP analysis showing the histone methylation status and recruitment of the indicated proteins within the EMT‐TF promoters. *P < 0.05, **P < 0.01, ***P < 0.001 versus controls (two‐tailed unpaired t‐test).
-
B, CThe interaction between UTX and the indicated proteins was confirmed by co‐IP in 293 T cells transfected with the indicated constructs (B) and in UTX‐overexpressing MDA‐MB‐231 cells (C).
-
DMCF10A cells were transfected with CON si or UTX si, together with c‐Myc si. The CD44+/CD24−/ESA + population was then evaluated using flow cytometry. **P < 0.01 versus CON si; †† P < 0.01 versus UTX si (one‐way ANOVA and Scheffe's post hoc test).
Figure EV4. The effect of c‐Myc and LSD1 on UTX‐mediated epigenetic alteration within the promoter regions of EMT‐TFs.

-
A, BTo transiently knock down c‐Myc, non‐targeting siRNA (CON si) or c‐Myc siRNA (c‐Myc si) was transfected into the indicated stable cells; the EMT‐TF promoters were then analysed using a ChIP assay and the indicated antibodies.
-
C, DUTX‐overexpressing MDA‐MB‐231 cells were infected with LSD1 shRNA viruses; ChIP analyses were then performed to confirm the histone methylation status and recruitment of the indicated proteins within the promoters of ZEB1 and ZEB2. The data shown represent the means ± SD of three independent experiments. **P < 0.01, ***P < 0.001 versus CON/shCON; †† P < 0.01, ††† P < 0.001 versus UTX/shCON; ns, no significance (Welch's test and Dunnett's T3 post hoc test for LSD1 and c‐Myc in C and for LSD1 in D; one‐way ANOVA and Scheffe's post hoc test for others).
Source data are available online for this figure.
UTX cooperates with LSD1 demethylase and inhibits MLL‐mediated H3K4me to epigenetically repress EMT‐TFs
To further explain how UTX negatively regulates the H3K4me2 level at the EMT‐TF promoters in a c‐Myc‐independent manner, we investigated possible histone modification enzymes targeting H3K4me that might cooperate with UTX in regulating EMT‐TFs. Consistent with previous findings 2, 4, 17, we observed that UTX physically interacts with the H3K4‐specific methyltransferase MLL4 (Fig 5A). Interestingly, UTX also bound H3K4 demethylase LSD1 and enhanced the association between LSD1 and MLL4, as confirmed by co‐IP results. Furthermore, ChIP analysis revealed that MLL4 consistently bound to the EMT‐TF promoters regardless of UTX expression, whereas LSD1 recruitment to these regions occurred only in the presence of UTX expression in both UTX‐overexpressing and UTX‐knockdown cells, thereby suggesting UTX‐dependent LSD1 recruitment and H3K4 demethylation at the EMT‐TF promoters (Fig 5B). These data also imply that UTX is not a binding partner of MLL4 but rather inhibits H3K4 methylation by facilitating competition between MLL4 and LSD1.
Figure 5. UTX cooperates with LSD1 demethylase to inhibit H3K4me in EMT‐TF promoters.

-
AThe binding of UTX to the indicated proteins was analysed using co‐IP experiments involving lysates from 293T cells transfected with the indicated constructs.
-
BChIP analysis showing the recruitment of LSD1 and MLL4 to the EMT‐TF promoters. *P < 0.05, **P < 0.01, ***P < 0.001 versus controls (two‐tailed unpaired t‐test).
-
C, DUTX‐overexpressing MDA‐MB‐231 cells were infected with LSD1 shRNA viruses; immunoblotting (C) and ChIP analyses (D) were then performed to confirm the expression of the indicated proteins and the epigenetic changes at the SNAIL promoters. *P < 0.05, **P < 0.01, ***P < 0.001 versus. CON/shCON; † P < 0.05, †† P < 0.01, ††† P < 0.001 versus UTX/shCON (one‐way ANOVA and Scheffe's post hoc test for c‐Myc IP; Welch's test and Dunnett's T3 post hoc test for others).
-
ESchematic summary of the regulation of EMT‐TFs by UTX.
LSD1 role as an oncogene has been well described, despite controversy regarding in its role in breast cancer 33, 34, 35. To define the potential role of LSD1 in the UTX‐mediated repression of EMT‐TFs, we inhibited LSD1 expression using lentiviral shRNA in control or UTX‐overexpressing MDA‐MB‐231 cells. Consistent with previous findings that LSD1 promotes EMT 35, 36, we found that LSD1 knockdown decreased SNAIL and ZEB1 expression and increased E‐cadherin expression in control MDA‐MB‐231 cells expressing low levels of UTX (Fig 5C). In contrast, the loss of LSD1 recovered the expression of EMT‐TFs that were inhibited by UTX and subsequently abolished E‐cadherin expression (Fig 5C), indicating that UTX overexpression causes LSD1 to function as a negative regulator of EMT‐TFs. These paradoxical results support the critical role of UTX in determining the oncogenic potential of LSD1 in human breast cancer. Because LSD1 is a component of the CoREST and NuRD transcriptional repressive complexes, which contain HDACs 33, 37, we assumed that UTX might recruit not only LSD1 but also LSD1‐containing multiple repressive complexes to its target chromatin. Indeed, the ChIP analysis results showed that the repression of H3K4me2 and H3ac by the recruitment of LSD1 and HDAC1 was recovered in UTX‐overexpressing cells by LSD1 knockdown (Figs 5D and EV4C and D), implying that LSD1 is required for the formation of a transcriptional repressive complex with UTX and HDAC1 at the EMT‐TF promoters. However, LSD1 could not alter the inhibitory effect of UTX on c‐Myc binding to these promoters. Collectively, our results indicate that UTX epigenetically inhibits the transcription of EMT‐TFs by cooperating with LSD1/HDAC1 in competition with c‐Myc/MLL4 in the regulation of EMT and CSCs. Contrary to the findings of Kim and colleagues 17, these data suggest that UTX might utilize MLL4 as a recognition signal for chromatin binding but inhibits MLL4 activity towards H3K4 by recruiting the LSD1 demethylase‐containing transcriptional repressive complex to silence the expression of EMT‐TFs in breast cancer.
In this study, we demonstrated that UTX plays an important role in breast tumour suppression as an epigenetic regulator of EMT‐TFs. UTX negatively regulated EMT‐induced breast CSC properties by transcriptionally suppressing SNAIL, ZEB1 and ZEB2 in an H3K27 demethylation activity‐independent manner (Fig 5E). In the absence of UTX, EMT‐TFs are transcriptionally activated by recruiting c‐Myc/p300 to the EMT‐TF promoters. MLL4, the H3K4 methyltransferase, was occupied and methylated H3K4 in these regions. When UTX was present, the recruitment of c‐Myc and p300 was disrupted, and LSD1/HDAC1/DNMT1 recognized the target region by promoting UTX binding to MLL4. The UTX‐containing repressive complex inhibits the H3K4me and H3ac, resulting in gene silencing of the EMT‐TFs. Collectively, these findings suggest that UTX is a novel epigenetic silencer of EMT‐TFs that represses EMT‐associated breast CSC‐like properties in an H3K27 methylation‐independent manner by cooperating with H3K4 demethylase LSD1 and HDAC1.
Materials and Methods
Flow cytometry
Breast CSC populations were isolated using flow cytometry as described previously 38, based on the expression of surface markers (CD44+/CD24−/ESA+). To evaluate UTX expression in CSC and non‐CSC populations from breast cell lines, cell populations were divided, stained using the appropriate antibodies and collected; the cells were then analysed using a FACSAria flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
Mammosphere formation
MCF10A cells (1 × 104 cells/well), T‐47D cells (5 × 103 cells/well), SK‐BR‐3 cells (1 × 104 cells/well) and MDA‐MB‐231 cells (5 × 103 cells/well) were cultured under previously described conditions 38. After 3, 5 and 7 days, mammosphere formation was analysed and quantified using an inverted microscope.
ChIP–qPCR assay
ChIP assays were performed using a ChIP assay kit according to the manufacturer's instructions (Upstate Biotechnology, Lake Placid, NY, USA). ChIP signal enrichment was determined using real‐time quantitative PCR (qPCR) (signal/input ratio) conducted using the 7300 Real‐Time PCR System and the SYBR Green Master Mix (Applied Biosystems, Foster City, CA, USA).
Statistical analysis
The statistical significance of differences between the control and experimental groups was analysed using the two‐tailed unpaired t‐test after confirming that data were normally distributed based on the Shapiro–Wilk test as implemented in the SPSS (version 12.0; SPSS Inc., Chicago, IL, USA). The Levene's test was used to verify equality of variances. For multiple comparisons, one‐way ANOVA followed by Scheffe's post hoc test (for equal variances) or Welch's test with Dunnett's T3 post hoc test (for unequal variances) was performed. To estimate the frequency of the tumour‐initiating ability, limiting dilution assay results were calculated using L‐Calc software. P‐values < 0.05 were considered to indicate statistical significance.
For further methods, see Appendix Supplementary Methods.
Author contributions
GK and J‐YL designed and supervised the project. H‐JC, J‐YL and GK wrote the manuscript. H‐JC and J‐HP performed the experiments, analysed and organized the data. MP, H‐YW, H‐SJ and C‐HL conducted the experiments and analysed the data.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Review Process File
Source Data for Figure 1A
Source Data for Figure 2B
Source Data for Figure 3
Source Data for Figure 4B and C
Source Data for Figure 5A and C
Source Data for Expanded View
Acknowledgements
This study was supported by the National Research Foundation of Korea (NRF), which is funded by the Korean Government (No. 2010‐0020879).
EMBO Reports (2015) 16: 1288–1298
References
- 1. Agger K, Cloos PA, Christensen J, Pasini D, Rose S, Rappsilber J, Issaeva I, Canaani E, Salcini AE, Helin K (2007) UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 449: 731–734 [DOI] [PubMed] [Google Scholar]
- 2. Issaeva I, Zonis Y, Rozovskaia T, Orlovsky K, Croce CM, Nakamura T, Mazo A, Eisenbach L, Canaani E (2007) Knockdown of ALR (MLL2) reveals ALR target genes and leads to alterations in cell adhesion and growth. Mol Cell Biol 27: 1889–1903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lee MG, Villa R, Trojer P, Norman J, Yan KP, Reinberg D, Di Croce L, Shiekhattar R (2007) Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science 318: 447–450 [DOI] [PubMed] [Google Scholar]
- 4. Cho YW, Hong T, Hong S, Guo H, Yu H, Kim D, Guszczynski T, Dressler GR, Copeland TD, Kalkum M et al (2007) PTIP associates with MLL3‐ and MLL4‐containing histone H3 lysine 4 methyltransferase complex. J Biol Chem 282: 20395–20406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Richly H, Aloia L, Di Croce L (2011) Roles of the Polycomb group proteins in stem cells and cancer. Cell Death Dis 2: e204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cao Q, Yu J, Dhanasekaran SM, Kim JH, Mani RS, Tomlins SA, Mehra R, Laxman B, Cao X, Yu J et al (2008) Repression of E‐cadherin by the polycomb group protein EZH2 in cancer. Oncogene 27: 7274–7284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chang CJ, Yang JY, Xia W, Chen CT, Xie X, Chao CH, Woodward WA, Hsu JM , Hortobagyi GN, Hung MC (2011) EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1‐beta‐catenin signaling. Cancer Cell 19: 86–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang C, Lee JE, Cho YW, Xiao Y, Jin Q, Liu C, Ge K (2012) UTX regulates mesoderm differentiation of embryonic stem cells independent of H3K27 demethylase activity. Proc Natl Acad Sci USA 109: 15324–15329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mansour AA, Gafni O, Weinberger L, Zviran A, Ayyash M, Rais Y, Krupalnik V, Zerbib M, Amann‐Zalcenstein D, Maza I et al (2012) The H3K27 demethylase Utx regulates somatic and germ cell epigenetic reprogramming. Nature 488: 409–413 [DOI] [PubMed] [Google Scholar]
- 10. Ntziachristos P, Tsirigos A, Welstead GG, Trimarchi T, Bakogianni S, Xu L, Loizou E, Holmfeldt L, Strikoudis A, King B et al (2014) Contrasting roles of histone 3 lysine 27 demethylases in acute lymphoblastic leukaemia. Nature 514: 513–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Van der Meulen J, Sanghvi V, Mavrakis K, Durinck K, Fang F, Matthijssens F, Rondou P, Rosen M, Pieters T, Vandenberghe P et al (2015) The H3K27me3 demethylase UTX is a gender‐specific tumor suppressor in T‐cell acute lymphoblastic leukemia. Blood 125: 13–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. van Haaften G, Dalgliesh GL, Davies H, Chen L, Bignell G, Greenman C, Edkins S, Hardy C, O'Meara S, Teague J et al (2009) Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat Genet 41: 521–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gui Y, Guo G, Huang Y, Hu X, Tang A, Gao S, Wu R, Chen C, Li X, Zhou L et al (2011) Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat Genet 43: 875–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, Butler A, Davies H, Edkins S, Hardy C, Latimer C et al (2010) Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature 463: 360–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Herz HM, Madden LD, Chen Z, Bolduc C, Buff E, Gupta R, Davuluri R, Shilatifard A, Hariharan IK, Bergmann A (2010) The H3K27me3 demethylase dUTX is a suppressor of Notch‐ and Rb‐dependent tumors in Drosophila. Mol Cell Biol 30: 2485–2497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Terashima M, Ishimura A, Yoshida M, Suzuki Y, Sugano S, Suzuki T (2010) The tumor suppressor Rb and its related Rbl2 genes are regulated by Utx histone demethylase. Biochem Biophys Res Commun 399: 238–244 [DOI] [PubMed] [Google Scholar]
- 17. Kim JH, Sharma A, Dhar SS, Lee SH, Gu B, Chan CH, Lin HK, Lee MG (2014) UTX and MLL4 coordinately regulate transcriptional programs for cell proliferation and invasiveness in breast cancer cells. Cancer Res 74: 1705–1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Visvader JE (2011) Cells of origin in cancer. Nature 469: 314–322 [DOI] [PubMed] [Google Scholar]
- 19. Tam WL, Weinberg RA (2013) The epigenetics of epithelial‐mesenchymal plasticity in cancer. Nat Med 19: 1438–1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M et al (2008) The epithelial‐mesenchymal transition generates cells with properties of stem cells. Cell 133: 704–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Scheel C, Weinberg RA (2012) Cancer stem cells and epithelial‐mesenchymal transition: concepts and molecular links. Semin Cancer Biol 22: 396–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lim S, Becker A, Zimmer A, Lu J, Buettner R, Kirfel J (2013) SNAI1‐mediated epithelial‐mesenchymal transition confers chemoresistance and cellular plasticity by regulating genes involved in cell death and stem cell maintenance. PLoS ONE 8: e66558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hollier BG, Tinnirello AA, Werden SJ, Evans KW, Taube JH, Sarkar TR, Sphyris N, Shariati M, Kumar SV, Battula VL et al (2013) FOXC2 expression links epithelial‐mesenchymal transition and stem cell properties in breast cancer. Cancer Res 73: 1981–1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lin Y, Wu Y, Li J, Dong C, Ye X, Chi YI, Evers BM, Zhou BP (2010) The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine‐specific demethylase 1. EMBO J 29: 1803–1816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dong C, Wu Y, Yao J, Wang Y, Yu Y, Rychahou PG, Evers BM, Zhou BP (2012) G9a interacts with Snail and is critical for Snail‐mediated E‐cadherin repression in human breast cancer. J Clin Invest 122: 1469–1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yang F, Sun L, Li Q, Han X, Lei L, Zhang H, Shang Y (2012) SET8 promotes epithelial‐mesenchymal transition and confers TWIST dual transcriptional activities. EMBO J 31: 110–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tie F, Banerjee R, Conrad PA, Scacheri PC, Harte PJ (2012) Histone demethylase UTX and chromatin remodeler BRM bind directly to CBP and modulate acetylation of histone H3 lysine 27. Mol Cell Biol 32: 2323–2334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Miller SA, Mohn SE, Weinmann AS (2010) Jmjd3 and UTX play a demethylase‐independent role in chromatin remodeling to regulate T‐box family member‐dependent gene expression. Mol Cell 40: 594–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gyorffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, Szallasi Z (2010) An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat 123: 725–731 [DOI] [PubMed] [Google Scholar]
- 30. De Craene B, Berx G (2013) Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer 13: 97–110 [DOI] [PubMed] [Google Scholar]
- 31. Wang JK, Tsai MC, Poulin G, Adler AS, Chen S, Liu H, Shi Y, Chang HY (2010) The histone demethylase UTX enables RB‐dependent cell fate control. Genes Dev 24: 327–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Smith AP, Verrecchia A, Faga G, Doni M, Perna D, Martinato F, Guccione E, Amati B (2009) A positive role for Myc in TGFbeta‐induced Snail transcription and epithelial‐to‐mesenchymal transition. Oncogene 28: 422–430 [DOI] [PubMed] [Google Scholar]
- 33. Wang Y, Zhang H, Chen Y, Sun Y, Yang F, Yu W, Liang J, Sun L, Yang X, Shi L et al (2009) LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell 138: 660–672 [DOI] [PubMed] [Google Scholar]
- 34. Lim S, Janzer A, Becker A, Zimmer A, Schule R, Buettner R, Kirfel J (2010) Lysine‐specific demethylase 1 (LSD1) is highly expressed in ER‐negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis 31: 512–520 [DOI] [PubMed] [Google Scholar]
- 35. Lin T, Ponn A, Hu X, Law BK, Lu J (2010) Requirement of the histone demethylase LSD1 in Snai1‐mediated transcriptional repression during epithelial‐mesenchymal transition. Oncogene 29: 4896–4904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ferrari‐Amorotti G, Fragliasso V, Esteki R, Prudente Z, Soliera AR, Cattelani S, Manzotti G, Grisendi G, Dominici M, Pieraccioli M et al (2013) Inhibiting interactions of lysine demethylase LSD1 with snail/slug blocks cancer cell invasion. Cancer Res 73: 235–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lee MG, Wynder C, Cooch N, Shiekhattar R (2005) An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature 437: 432–435 [DOI] [PubMed] [Google Scholar]
- 38. Won HY, Lee JY, Shin DH, Park JH, Nam JS, Kim HC, Kong G (2012) Loss of Mel‐18 enhances breast cancer stem cell activity and tumorigenicity through activating Notch signaling mediated by the Wnt/TCF pathway. FASEB J 26: 5002–5013 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Review Process File
Source Data for Figure 1A
Source Data for Figure 2B
Source Data for Figure 3
Source Data for Figure 4B and C
Source Data for Figure 5A and C
Source Data for Expanded View