

Abstract
Light inducible dimers are powerful tools for cellular optogenetics as they can be used to control the localization and activity of proteins with high spatial and temporal resolution. Despite the generality of the approach, application of light inducible dimers is not always straightforward as it is frequently necessary to test alternative dimer systems and fusion strategies before the desired biological activity is achieved. This process is further hindered by an incomplete understanding of the biophysical/biochemical mechanisms by which available dimers behave and how this correlates to in vivo function. To better inform the engineering process we examined the biophysical and biochemical properties of three blue-light inducible dimer variants (Cryptochrome2 (CRY2)/CIB1, iLID/SspB, and LOVpep/ePDZ) and correlated these characteristics to in vivo co-localization and functional assays. We find that the switches vary dramatically in their dark-state and lit-state binding affinities, and that these affinities correlate with activity changes in a variety of in vivo assays including transcription control, intra-cellular localization studies and control of GTPase signaling. Additionally, for CRY2 we observe that light induced changes in homo-oligomerization can have large effects on activity that are sensitive to alternative fusion strategies.
Keywords: optogenetics, dimerization, cell signaling, iLID, Cryptochrome2 (Cry2), TULIP
TOC Image
Optogenetics originally described the use of the light sensitive cation channel, Channelrhodopsin-2, to manipulate the action potential of neurons 1,2. More recently, optogenetics has extended into the realm of cell biology with the development of cellular optogenetic tools. These tools are not limited to the manipulation of action potentials, but encompass any genetically encoded and light dependent system that can be used to manipulate cellular processes. Particularly successful has been the use of light induced dimerization to control a variety of processes such as gene transcription, GTPase signaling, protein degradation, and organelle transport 3–10. For example, by fusing one half of an inducible dimer to a protein anchored in the plasma membrane and the other half to a guanine nucleotide exchange factor (GEF) it is possible to localize the GEF to the membrane with light and activate GTPase signaling.
While light inducible dimerization has proven to be a general approach for regulating biological processes, it is frequently necessary to test alternative dimer systems and fusion strategies to determine which approach will be most robust and appropriate for a given application 11. Part of the challenge is that there are a variety of light inducible dimers that have been described in the literature, but few studies have compared switches side-by-side or characterized their intrinsic biophysical properties. Here, we establish correlations between the in vitro and in vivo activities of three blue-light inducible dimers: Cryptochrome2 (CRY2)/CIB1, iLID/SspB, and LOVpep/ePDZb 12–14 (Fig. 1A). These results provide valuable input for future efforts to control biological pathways with light inducible dimerization.
FIGURE 1. Binding affinities of lit and dark states highlight difference in photoswitch dynamic range.
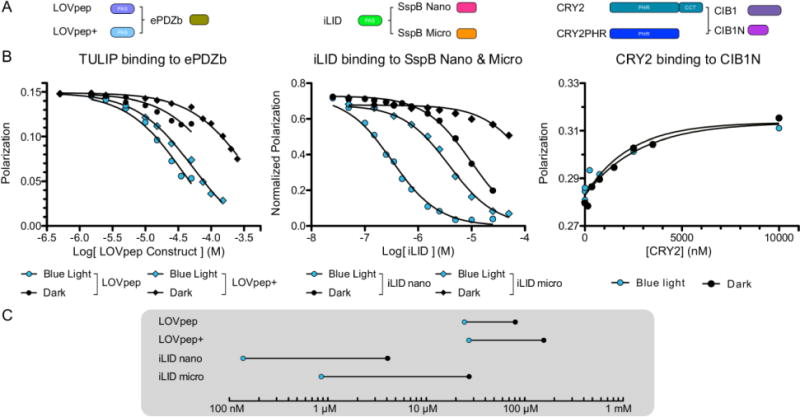
Fluorescence polarization binding plots for A) LOVpep constructs and ePDZb (left) iLID nano and micro (middle) and CRY2 and CIB1N (right). B) Fluorescence polarization of each complex was measured under blue light (blue) or darkness (black) to determine binding affinity. C) Affinity values from binding data plotted on a Dynagram highlight the dynamic range of each tool.
As a family, blue light inducible dimers provide a powerful experimental platform. Their photosensitive cofactor is abundant in nature making them broadly applicable to many organisms, and the single wavelength of light necessary to manipulate their dimerization makes for a simple experimental setup. CRY2/CIB1 is a naturally occurring light-dependent heterodimer from Arabidopsis thaliana. Additionally, it has been shown that CRY2 forms homooligomers when activated with light 15. Both CRY2/CIB1 dimerization and CRY2 homooligomerization have been used to control a variety of cellular processes 11,16–20. However, neither the dark state nor lit state binding affinities between CRY2 and CIB1 have been measured, and the stoichiometry of oligomerization has also not been determined.
The TULIP (LOVpep/ePDZb) 13 and iLID (iLID/SspB) 14 systems are engineered heterodimer pairs built upon the light-induced conformational change of the Avena sativa (As) phototropin LOV2 domain 21. In the TULIP system a PDZ binding motif was encoded in the Jα helix of AsLOV2, sterically caged from binding an engineered PDZ (ePDZ) domain in the dark 13. Blue light induces a conformational change within AsLOV2, relieving this occlusion and increasing affinity to ePDZ. The iLID system works in a similar fashion, caging the E. coli ssrA peptide from its binding partner, SspB 14,22. Despite the mechanistic similarities between TULIPs and iLID, the lack of molecular characterization prevents direct comparison and empirical switch selection when developing a new application.
Recently, the Tucker group began the process of benchmarking light inducible dimers by comparing CRY2/CIB1, TULIPs, and Phy/Pif in a set of standardized yeast functional assays 23. The Phy/Pif pair is a light induced dimer that rapidly forms under red light and rapidly dissociates when illuminated with far red light. The Phy/Pif system requires a cofactor, phycocyanobilin (PCB), which is not readily available in some organisms. These studies demonstrated a wide range of activities when using the switches to co-localize DNA binding and activation domains for control of reporter gene transcription in yeast. To better understand these variations and extend the results to mammalian systems, we continue the benchmarking process by measuring binding constants for the dimers in the lit and dark states, and performing a variety of activity assays including: co-localization experiments in mammalian cell culture, transcription-control assays in yeast, and the activation of small GTPases via the sub-cellular recruitment of guanine nucleotide exchange factors. In general, we find that the measurements made in vitro correspond to what we observe in cells. The switches with the largest changes in in vitro binding affinities upon light stimulation make the most effective switches for the in-cell benchmarks.
Results and Discussion
Biochemical Comparison of the Switches
Dark and Lit-state Binding Affinities
We used fluorescence polarization binding assays to measure the lit and dark state binding affinities of each pair. For the TULIPs and iLID this was performed using a competitive binding experiment. The photoactivatable domains were used to compete off fluorescently labeled peptides from the binding partners. The interaction between AtCRY2 and CIB1N (the N-Terminus of CIB1 necessary for dimerization) was not amenable to this experimental format, so CIB1N was covalently labeled with a fluorescent dye and direct binding was measured. As we previously reported, the iLID binding partner SspB comes in two variations, Nano and Micro; each with a different affinity range. The iLID Nano system has an affinity of 0.13 μM under blue light and 4.7 μM in the dark. The iLID Micro pair has an affinity of 0.8 μM under blue light and 47 μM in the dark (Fig. 1B) 14. The TULIP switches we examined function over a weaker range of affinities. The LOVpep construct binds to ePDZb with an affinity of 12 μM under blue light and 72 μM in the dark, for a 6-fold change. The presence of additional “caging” mutations, T406-7A, I532A (LOVpep+), weaken the lit state affinity to 18 μM and the dark state affinity to 150 μM, for an 8 fold change (Fig. 1B). The different affinity ranges sampled by the TULIP and iLID switches reflects the affinities of the peptides that are being caged in each case. The SsrA peptide used to create iLID binds to its partner, SspB (Nano), with an affinity of 35 nM 22. The PDZ binding peptide used in TULIP binds to ePDZb with an affinity of 14 μM.
To biophysically characterize the binding properties of CRY2 we purified full-length protein from insect cells. We were not able to observe a light-dependent change in binding affinity between CRY2 and CIB1N. In our direct binding assay, we observed low micromolar binding (~4μM) with and without blue-light stimulation (Fig. 1B). This result is consistent with the previous observation that CRY2 purified from insect cells did not show differential affinity for CIB1 in pull-down assays performed in the light and the dark 27. It has been hypothesized that insect cell purified CRY2 is missing an important chromophore, 5,10-methylenetetrahydrofolate (MTHF) 27. However, even in the presence of saturating MTHF concentrations, we did not observe a significant change in binding affinity due to light (Fig. S1). We ran an electrophoretic mobility shift assay with CRY2 and CIB1N and found the same result. CRY2 binds CIB1N similarly under blue light as in the dark, both in the low micromolar range (Fig. S2). We were unable to express and purify a shorter variant of CRY2, CRY2PHR, that has also been shown to exhibit light-dependent binding to CIB1 in cells.
Light-dependent CRY2 Homo-oligomerization
Although purified CRY2 did not show light-dependent changes in CIB1N binding, we did observe robust homo-oligomerization of CRY2 with light stimulation as probed by multi-angle light scattering (SEC-MALS). A single symmetric peak was observed both in the light and the dark, with the retention time being delayed in the dark (Fig. 2A). The light scattering indicated a species with a molecular weight of 75 kD in the dark and 301 kD in the light. The expected molecular weight of monomeric CRY2 is 71 kD, so these results are consistent with the formation of a monomer in the dark and a tetramer in the light. Saturating amounts of MTHF did not change the elution times or molecular weight fits (Fig. S3). Using dynamic light scattering (DLS), we were able to measure the kinetics of the lit state oligomer to dark state monomer transition. In this assay, there is a 60 second delay between removal of blue light and the first DLS reading, as shown in grey (Fig. 2B). Factoring in this dead time, the reversion to dark state has a half-life of 90 ± 20 seconds. We also ran co-elution experiments of CRY2 and CIB1N in gel filtration experiments (Fig. S4). Samples of CRY2 and CIB1N (2:1 molar ratio) were run in the light and dark, however in both states CIB1N did not co-elute with CRY2. This is consistent with the micromolar binding affinities that we observed in the fluorescence experiments.
FIGURE 2. Light induces CRY2 oligomerization.
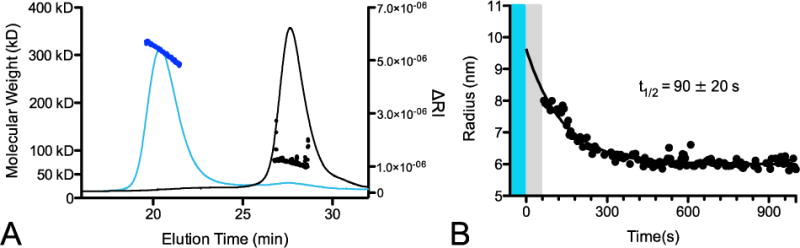
A) Size exclusion chromatography multi-angle light scattering traces for full length CRY2 run under blue light (blue line) or darkness (black line). Fit molecular weight from MALS data for each peak is shown for lit (blue dots) and dark (black dots) peaks. B) Reversion of light induced oligomer to monomer by dynamic light scattering. Blue bar represents blue light irradiation of sample; grey bar represents instrument dead time before initial measurement
Reversion Kinetics
We used our purified protein samples to also measure the reversion kinetics of the photoactivated states. Using an absorbance recovery after photoactivation assay, we determined the lit state half-life for each of our photosensitive domains. In order of fastest to slowest, the half-lives were measured to be 18 ± 2 s for iLID, 23 ± 1 s for LOVpep, 51 ± 2 s for LOVpep+, and 92 ± 10 s for CRY2 (Fig. 3).
FIGURE 3. Photoreceptor reversion kinetics.
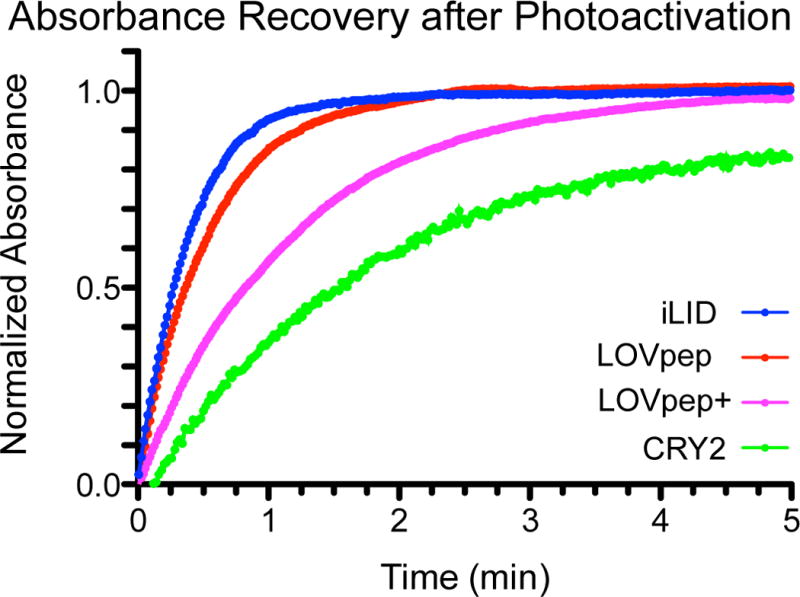
Thermal reversion kinetics of the excited state for each photoreceptor show differences in timescale of deactivation. Reversions were measured at room temperature in Tris-HCl buffer.
Comparison of Switch Behavior in the Cytoplasm of Live Cells
Controlling Sub-cellular Localization
Next, we examined how effective the photoswitches were at recruiting proteins to a specified region of the cell. In particular, we were curious if the in vitro binding properties of the dimers would correlate with in-cell behavior. Each half of the switches were fused to a fluorescent protein (Venus or tgRFPt) with spectral properties distinct from the excitation wavelength of the photoactive domain. The Venus labeled half of the switch was also fused to a membrane-anchoring domain (N-Myristoylation (Myr) or C-Farnesylation (CAAX)). The two proteins were then co-expressed in mouse fibroblasts and continuously imaged with a confocal laser-scanning microscope. During imaging a region of interest (ROI) was activated with a 488 nm laser, and changes in protein localization were quantified as a function of time by measuring the ratio of tgRFPt fluorescence intensity inside the activated ROI to the intensity in a ROI of the same size outside the area of activation. The analysis produces a maximum intensity ratio as well as the half-life of activation and reversion (Table 1).
Table 1.
| In Vitro | |||||
|---|---|---|---|---|---|
| In Vitro Binding | |||||
| Switch | Binder | Method | Lit Affinity | Dark Affinity | Reversion Halflife (s) |
| iLID | Nano | FP | 0.132 ± 0.005 μM | 4.7 ± 0.7 μM | 18 ± 2 |
| iLID | Micro | FP | 0.8 ± 0.1 μM | 47 ± 13 μM | 18 ± 2 |
| LOVpep | ePDZb | FP | 12 ± 3 μM | 72 ± 3 μM | 23 ± 1 |
| LOVpep+ | ePDZb | FP | 18 ± 1 μM | 150 ± 60 μM | 51 ± 2 |
| CRY2 | CIB1N | FP,SEC | 4 ± 2 μM | 92 ± 10 | |
| Cell Localization | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Membrane Localization | Mitochondrial Localization | ||||||||||
| Membrane Bound | Cyto | N | Max Δ Intensity | Activation Halflife (s) | Reversion Halflife (s) | N | Starting Value | Max Value | Fold Change | Activation Halflife (s) | Reversion Halflife (s) |
| iLID | Nano | 7 | 6.36 ± 0.21 | 95.4 ± 18.1 | 63.8 ± 6.7 | 5 | 1.72 ± 0.5 | 9.23 ± 2.18 | 5.4 | 21.2 ± 1.4 | 32.0 ± 3.7 |
| iLID | Micro | 7 | 4.88 ± 0.18 | 72.0 ± 10.5 | 52.5 ± 2.5 | 5 | 1.45 ± 0.07 | 7.58 ±1.49 | 5.2 | 26.7 ± 6.0 | 23.2 ± 1.5 |
| LOVpep+ | ePDZb | 13 | 1.34 ± 0.03 | 39.4 ± 15.7 | 53.8 ± 12.6 | 7 | 0.59 ± 0.09 | 1.4 ± 0.20 | 2.4 | 25.0 ± 6.7 | 19.5 ± 0.9 |
| CIB1N | CRY2PHR | 12 | 4.98 ± 0.47 | 186.4 ± 30.9 | 380.7 ± 73.3 | 6 | 2.88 ± 0.67 | 8.84 ± 1.39 | 3.1 | 58.8 ± 9.4 | 317.6 ± 85.25 |
| CRY2PHR | CIB1N | 10 | 1.52 ± 0.02 | 132.7 ± 53.4 | 164.3 ± 30.3 | ||||||
| Function Manipulation | ||||||||
|---|---|---|---|---|---|---|---|---|
| Yeast Transcription | ||||||||
| Gal4-DBD | Gal4-AD | β-Gal Light | β-Gal Dark | Fold Change | His Dropout Light | His Dropout Dark | His/Ade Dropout Light | His/Ade Dropout Dark |
| Nano | iLID | 154.7 | 7.95 | 19.5 | +++ | +++ | +++ | ++ |
| Micro | iLID | 94.8 | 10.1 | 9.4 | ++ | ++ | ++ | ++ |
| ePDZb | LOVpep | ND | ND | ND | ++ | − | − | − |
| ePDZb | LovPep+ | ND | ND | ND | − | − | − | − |
| Cry2PHR | CIB1N | ND | ND | ND | +++ | − | − | − |
| CIB1N | Cry2PHR | 47.1 | 5.4 | 8.7 | +++ | +++ | +++ | ++ |
| Tiam DH/PH Induced Protrusion | ||
|---|---|---|
| Membrane Bound | Tiam DH/PH fused | Mean Protrusion Distance (μm) |
| iLID | Nano | 12.0 ± 3.8 |
| iLID | Micro | 14.5 ± 3.1 |
| LOVpep | ePDZb | 1.6 ± 0.8 |
| CIB1N | CRY2PHR | 4.2 ± 2.2 |
| CRY2PHR | CIB1N | 2.2 ± 1.7 |
In vitro characterization – Affinities and kinetics of each switch were measured in vitro by the method noted. FP – Fluorescent Polarization, SEC – Size Exclusion Chromatography. Standard deviation is shown for each value
Cell Localization – The noted half of each switch was bound to the plasma membrane or Mitochondrial membrane while the other was expressed in the cytoplasm. Localization of the cytoplasmic half of the switch was measured by fluorescence intensity and used to calculate the parameters above. Standard Error is shown for each value
+++ = Very Robust Growth
++ = Significant Growth
+ = Little Growth
− = No Growth
Function Manipulation – Each switch was used to manipulate yeast transcription or mammalian cell protrusion. The above parameters were measured for each switch. Standard Error is shown for each protrusion distance.
For each of these assays the iLID half of the switch was anchored to the membrane with a CAAX motif while Nano and Micro were diffuse in the cytoplasm. Upon activation the tgRFPt-Nano and micro fluorescence intensity increased to produce an average maximum ratio value of 6.36 and 4.88 respectively (Fig. 4A, B). As these constructs have been previously tested 14, we have reanalyzed all data using a standardized quantification strategy to allow comparison between switches. To establish a baseline for our recruitment assay cells were also transfected with the mismatched pair, Venus-iLID CAAX and tgRFPt-ePDZ. No localization was observed in this control experiment (Fig. 4A, B).
FIGURE 4. Targeted localization to the plasma membrane shows differences in switch dynamic range and kinetics.
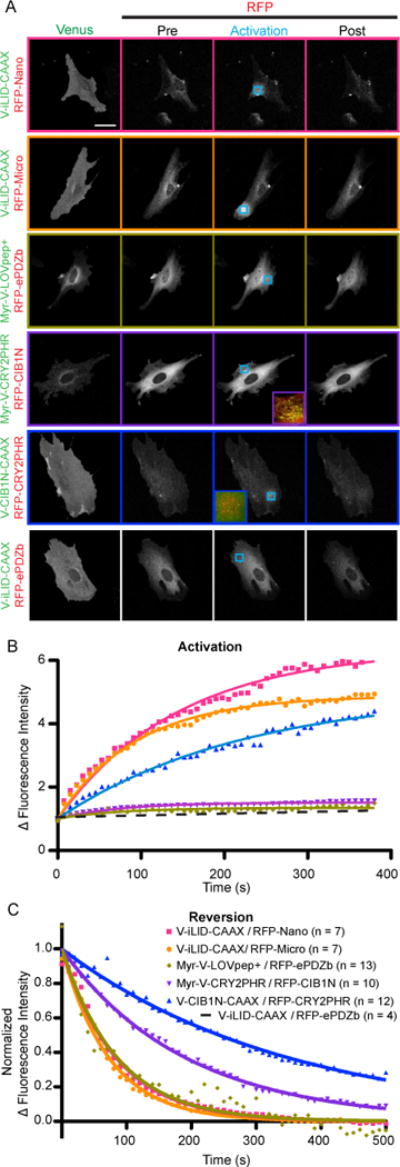
A) Representative images of the data analyzed in B and C. Cells transfected with each membrane bound switch pair were visualized and activated by confocal microscopy. Venus labeled constructs are bound to the plasma membrane while tgRFPt labeled constructs are cytoplasmic. The activated ROI is identified by the blue arrow. The activation and post activation images represent the final image of the specified time frame. (Bar = 50 μm) B) A ratio of tgRFPt fluorescence intensity inside the activated ROI to outside the activated ROI during the period of activation as shown in A. C) A normalized ratio of tgRFPt fluorescence intensity inside the activated ROI to outside the activated ROI during the period of activation as shown in A.
Our first experiments with the TULIP system used a similar approach, LOVpep+ was fused to Venus and a CAAX motif while ePDZb was fused to tgRFPt. However, with LOVpep+ fused to a CAAX motif the C-terminal PDZ binding motif was prevented from binding to ePDZb and no change in fluorescence intensity at the activated ROI was observed (Fig. S5A). Therefore, we fused the LOVpep+ to an N-Terminal myristoylation sequence, freeing the PDZ binding motif. Upon expression, we found that the myristoylated sequence localized to the plasma membrane but also localized to other membrane bound organelles. This lead to two issues; while the overall expression levels were similar to iLID a large portion of the protein was not localized to the membrane, and the portion of the protein that was in the ER could lead to background signal. To circumvent the second issue we chose ROIs that predominantly consisted of only plasma membrane bound LOVpep+ for activation and analysis. Upon light stimulation, we observed a small increase in protein localization (average maximum ratio value = 1.34) (Fig. 4A, B).
The initial CRY2 publication was unable to show functionality with CRY2 bound to the plasma membrane 12. However, Pathak et. al. recently maintained functionality in yeast by fusing CRY2PHR to the C-terminus of Mid2, a membrane anchored protein 23. We therefore tested 3 experimental approaches varying the switch positions as follows: Venus-CRY2PHR-CAAX, tgRFPt-CIB1N; Myr-Venus-CRY2PHR, tgRFPt-CIB1N; and Venus-CIB1N-CAAX, tgRFPt-CRY2PHR. While the CAAX fused CRY2PHR localized to the plasma membrane, the tgRFPt fluorescence intensity did not increase upon activation within the ROI (Fig. S5A). Myr-Venus-CRY2PHR had a similar localization pattern as Myr-Venus-LOVpep+ so we again carefully chose ROIs outside of the ER. Upon activation we observed an increase in tgRFPt intensity within the ROI and measured the average maximum ratio value to be 1.52 (Fig. 4A, B). Additionally, upon activation Myr-Venus-CRY2 formed large clusters at the membrane (Fig. 4A inset). To better understand CRY2 cluster formation and dissociation we performed the same ROI analysis on the Venus channel. During activation, we observed an increase in Venus fluorescence intensity within the ROI, which represents cluster formation. Interestingly, we observed a persistent increase in intensity for ~1 min after the light was turned off, suggesting that the clusters continue to form after the blue light is turned off (Fig. S6B).
In the experiments with Venus-CIB1N-CAAX anchored to the membrane, tgRFPt-CRY2PHR is diffuse throughout the cytoplasm before activation. Upon activation with blue light the tgRFPt intensity increases within the ROI and in some cases small tgRFPt-CRY2PHR clusters begin to form (Fig. 4A inset). Surprisingly, we measured the average maximum ratio value to be 4.98; significantly higher than when CRY2PHR is anchored to the membrane. However, we hypothesized that CRY2PHR oligomerization alone may be responsible for a portion of the increase in fluorescence measured at the ROI. The idea being that once oligomerized, diffusion of tgRFPt-CRY2PHR would slow while recruiting more monomers, increasing the signal. We tested this hypothesis by expressing and activating tgRFPt-CRY2PHR alone in cells and found that this was indeed the case. CRY2PHR alone had a maximum ratio value of 2.67 (Fig. S6C and D). We also hypothesized that expressing the photoactive half of the switch in the cytoplasm would provide less spatial control as compared to membrane anchored. Our reasoning was that once activated the CRY2PHR could more easily diffuse through the cytoplasm and bind to CIB1N outside of the ROI. Interestingly, the gradient of tgRFPt intensity peripheral to the activated ROI was similar to what we measured for the iLID switches where the photoactive domain was anchored to the membrane (Fig. S6A).
For all of the dimers, the dissociation rate constants in cells when the light is turned off were longer than the in vitro measured half-lives for the photoswitches. This may reflect that the in vitro experiments measured only the photocycle kinetics while in cells our measurement depends on photocycle kinetics as well as dissociation and diffusion. However, the measurements parallel the in vitro patterns and what has been previously observed. The LOV2-based switches are all similar at about 60 sec while the CRY2PHR/CIB1N switches are slower. Interestingly the CRY2PHR/CIB1N reversion half-lives are dependent on the orientation of the switch. The Myr-Venus-CRY2 has a faster half-life at 132 s while the tgRFPt-CIB1N-CAAX is significantly slower with a half-life of 242 s (Fig. 4C).
Except in the case of Venus-CIB1N-CAAX with tgRFPt-CRY2PHR the activation half-life seems to correlate with the dynamic range of the switch (larger dynamic range takes longer to reach equilibrium after activation) and not on the kinetics of the protein conformational change (Fig. 4B). This suggests that the rate-limiting step is diffusion. While we are unsure what causes the slower rate of Venus-CIB1N-CAAX with tgRFPt-CRY2PHR, we hypothesize that it is due to CRY2PHR oligomerization and is discussed further below.
Mitochondrial re-localization assay
A limitation of the membrane localization assay is that it is difficult to accurately determine the portion of tgRFPt labeled protein that is at the plasma membrane prior to activation due to the axial spatial resolution of the microscope (~600nm). In the relatively flat cultured fibroblasts we used for these experiments, the apical and basal membrane fluorescence values are captured but cannot be distinguished from the cytoplasmic fluorescence. By anchoring the Venus labeled half of the switch to the mitochondrial membrane we were able to more accurately determine the initial amount of tgRFPt labeled protein at the mitochondria relative to the cytoplasm and monitor its change during and after activation. Proteins were anchored to the mitochondrial membrane by fusion to TOM20 at the N-terminus or Mito anchor sequence from Listeria monocytogenes ActA protein 28 at the C-terminus. After co-expression of each half of the switch, whole cells were activated with 488 nm light and imaged. Using an automated ImageJ macro, we measured the ratio of mitochondrial to cytoplasmic fluorescence intensity throughout activation and reversion. For each switch this assay produces parameters describing the half-life of activation and reversion, a starting mito/cyto intensity (representative of dark state binding), a maximal mito/cyto intensity and the fold change in intensity (Table 1.). To establish a baseline for the recruitment assay cells were transfected with the mismatched pair, Venus-iLID Mito and tgRFPt-ePDZ. The cells were activated, and the data was analyzed as described above (Fig. 5A, B).
FIGURE 5. Targeted mitochondrial localization identifies differences in dark state binding dynamic range and kinetics.
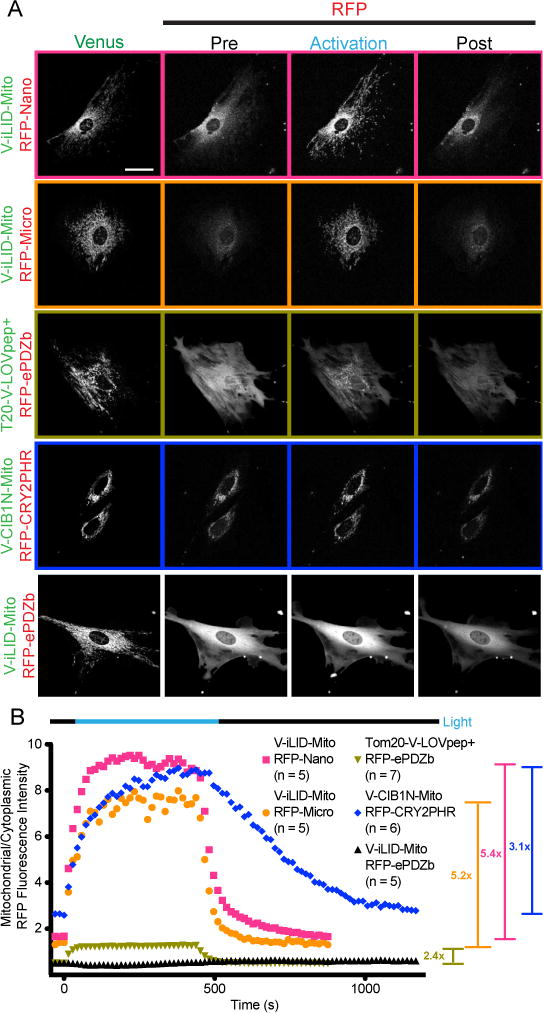
A) Representative images of the data analyzed in B. Cells transfected with each mitochondrial bound switch pair were visualized and activated by confocal microscopy. Venus labeled constructs are bound to the plasma membrane while tgRFPt labeled constructs are cytoplasmic. The entire field of view is activated. The activation and post activation images represent the final image of the specified time frame. (Bar = 50 μm) B) A ratio of mitochondrial to cytoplasmic tgRFPt fluorescence intensity throughout the experiments shown in A.
Again, the data for the iLID switch has been previously reported. However, the data was reanalyzed using an improved ImageJ macro, which was able to better differentiate the cytoplasm from background. In this assay iLID was fused to the Mito anchoring domain of ActA while Micro and Nano were cytoplasmic. We measured the average fold change for iLID-Nano and Micro to be 5.4 and 5.2 respectively (Fig. 5A, B). As expected, the initial relative mitochondrial fluorescence intensity for iLID-nano is higher than iLID-micro, paralleling the in vitro measured dark state affinity being tighter (Fig. 5B).
To test the TULIP switch we fused Venus labeled LOVpep+ to TOM20 at the N-terminus to preserve an accessible C-Terminal PDZ binding motif. Upon co-expression with tgRFPt-ePDZb and activation, the switch produces an average 2.4 fold change in relative mitochondrial tgRFPt fluorescence intensity (Fig. 5A, B). This fold change parallels the smaller in vitro measured dynamic range of binding in comparison to the iLID switches. TULIP also showed a lower starting mitochondrial tgRFPt intensity (0.59); again paralleling TULIP’s lower in vitro dark state affinity (Fig. 5B).
We tested the CRY2PHR/CIB1N switch in both orientations. We first tested TOM20-Venus-CRY2PHR with tgRFPt-CIB1N. Unfortunately, TOM20-Venus-CRY2PHR appeared to be toxic to the cells and therefore expression levels in the surviving cells were significantly lower than all other constructs to the extent that the laser power of the microscope had to be substantially increased to obtain a clear image. Additionally, the distribution of mitochondria within the surviving cells was abnormal. Upon activation the cells did not produce a measureable increase in mitochondrial tgRFPt intensity (Fig. S5B). We therefore reversed the orientation of the switch. By co-expressing and activating Venus-CIB1N-Mito and tgRFPt-CRY2PHR we measured an average 3.1 fold change in relative mitochondrial tgRFPt fluorescence intensity (Fig. 5A, B). The average initial value of mito/cyto tgRFPt was also high (2.88) compared to the other switches (Fig. 5B). This suggests a relatively tight dark state binding affinity.
Functional Comparison of the Switches
Light Controlled Transcription in Yeast
To examine if our findings from the in vitro binding assays and the localization studies correlate with outcomes in a functional assay, we used the light dimerization pairs to control transcription in yeast. The yeast two-hybrid approach has previously been used to demonstrate light dependent transcription for CRY2PHR with CIB1N as well as ePDZb and LOVpep. We used diploids generated from mating Y187 and Y2HGold strains to test for the activation of the lacZ, his3 and ade2 reporter genes (Fig. 6A) transformed with the split Gal4 transcription factor constructs (Fig. 6B). We observe an assortment of induced transcription levels dependent on the protein pair used and the reporter gene observed. We identified strong light dependent transcription using β-galactose expression as readout for iLID with Nano (19.5 fold) and iLID with Micro (9.4 fold) (Fig. 6C and D). We previously showed that iLID had an improved dynamic range when compared to its parental construct, oLID, by a multitude of measurements. However, we were curious how the two switches compared in their ability to control yeast transcription. As expected light dependent transcription was not detected for the oLID paired with either Nano or Micro (Fig. S7). In our hands there was also no detectable β-galactose expression for ePDZb paired with LOVpep or LOVpep+.
FIGURE 6. Yeast two hybrid transcription comparison.
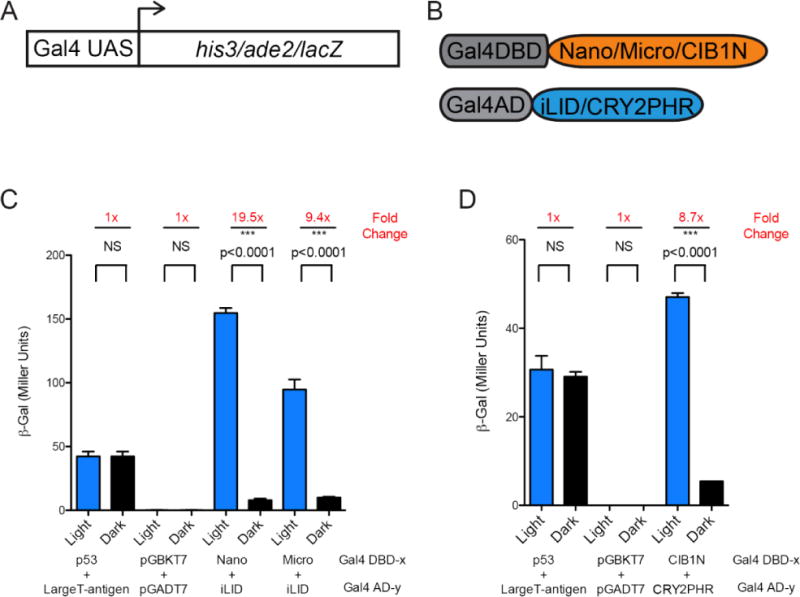
A) A schematic of the genome reporters. B) A schematic of the constructs tested. C) ß-galactose transcription induced with the iLID paired with Nano or the Micro (n = 9 each, mean reported ± SEM and statistical significance is calculated with unpaired two-tailed t-student’s test (p<0.0001)) and D) CIB1N with CRY2PHR (n = 3 each, mean reported ± SEM and statistical significance is calculated with unpaired two-tailed t-student’s test (p<0.0001)) (Blue Bars – growth under continuous blue light at 465nm, Black Bars – growth in the dark).
In addition to monitoring β-galactose expression, we also tested for light-dependent survival on histidine and histidine/adenine dropout plates. Interestingly, iLID when paired with Nano or Micro conveyed growth in the light and dark for single and double dropout plates. In contrast, yeast expressing the LOVpep did not survive in the dark, but there was growth on histidine dropout plates in the light. On the other hand, ePDZb paired with LOVpep+ exhibited no detectable transcription for any reporter, which is consistent with the weaker affinities observed for this pair (Fig. S8). These results are consistent with the survival assays being more sensitive to low levels of expression. iLID Nano and Micro have stronger binding affinities in the dark than LOVpep and LOVpep+ to ePDZb, and in this context this “leakiness” is sufficient to allow growth even when the switch is in the inactive/dark state.
In previous studies CRY2-DBD paired with CIB1 or CIB1N has been shown to activate transcription in yeast 12,23, although the overall levels of transcription with these constructs were lower when compared to results with full-length CRY2. To date the inversed orientation has not been reported to our knowledge. When we paired CIB1N-DBD with CRY2PHR-AD we observed strong light dependent expression of lacZ achieving about 9 fold difference, similar to when combining Micro with iLID, but overall lower levels for both light and dark levels. In contrast, when testing CRY2PHR-DBD with CIB1N-AD we saw no significant transcriptional activation of lacZ but only of his3 reporter genes, which as expected was in a light-dependent manner.
Manipulation of lamellipodial protrusion
To test each pair’s ability to functionally manipulate a mammalian cell we targeted the Rho GTPase family. The Rho family of small GTPases is known to regulate the dynamics of the actin cytoskeleton and therefore the cells shape. Canonically, activation of the membrane bound Rac family member produces highly branched actin, leading to dynamic lamellipodial protrusions 29. The inactive/active state of GTPases is determined by the state of the bound nucleotide (GDP/GTP respectively). Guanine nucleotide exchange factors (GEFs) activate GTPase by aiding in the exchange of GDP for GTP 30,31. Using iLID we have previously shown that by localizing the catalytic DH/PH domain of a Rac GEF (Tiam) to a portion of the plasma membrane we can induce lamellipodial protrusions in that region 14. We therefore used this approach as a functional test of the CRY2PHR/CIB1N or TULIP switches. To this end, we fused the Tiam DH/PH domain to each of the tgRFPt labeled halves of the switch. The Tiam constructs were then co-expressed with the appropriate membrane bound switch half. Cells were imaged and activated in a similar manner to the previous membrane localization experiments, though here the activation ROIs were located at the edge of the cell. For each cell, the maximal protrusion distance at the ROI was then quantified by kymography 28. In order to,control for the background flux of a cell membrane under light stimulation, tgRFPt-Nano without the DH/PH domain was recruited to the edge of the cell. In the time frame of activation, minor changes in membrane position were measured by kymography and the average displacement was negligible (Fig. 7A, B). iLID-Nano and Micro produced on average a maximal protrusion distance of 12.0 and 14.5 μm respectively (Fig. 7A, B). The TULIP switch caused an average protrusion distance of 1.5 μm, significantly less than the iLID switches (Fig. 7A, B). The CRY2PHR/CIB1N switches were again tested in both orientations (CRY2PHR or CIB1N anchored at the membrane) and produced an average protrusion distance of 4.2 and 2.2 μm respectively (Fig. 7A, B). It is interesting that the scenario with CIB1N anchored in the membrane produces such a small effect in this assay as this configuration showed more robust co-localization of CRY2PHR and CIB1N with light-activation. It may be that homo-oligomerized TIAM DH/PH-CRY2PHR has reduced activity. In fact, CRY2 oligomerization has previously been used to inhibt GEFs and GTPase activity 32.
FIGURE 7. Targeting Tiam DH/PH domains to the plasma membrane with each switch causes varying degrees of protrusion.
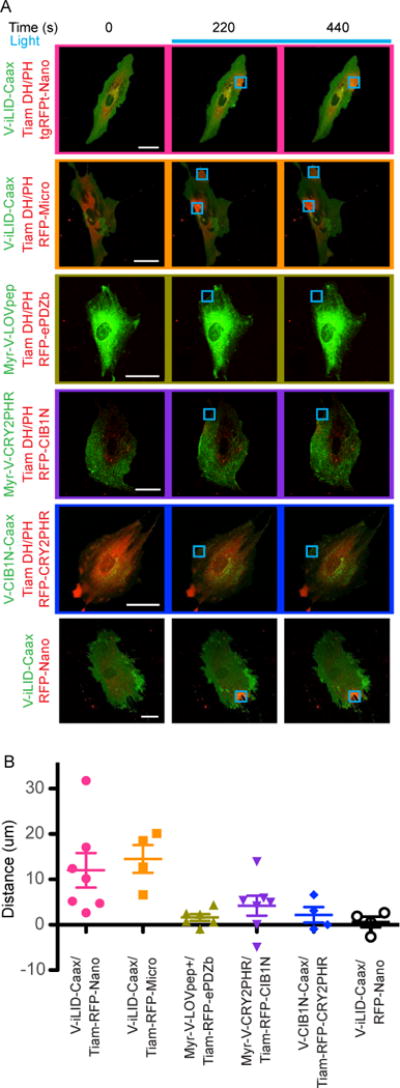
A) Representative images of the data analyzed in B. Cells transfected with each membrane bound Tiam DH/PH switch pair were visualized and activated by confocal microscopy. Venus labeled constructs are bound to the plasma membrane while tgRFPt labeled constructs are cytoplasmic. The activated ROI is is represented by the blue square. (Bar = 50 μm) B) Protrusion distances for each cell were measured by kymography.
Correlating in vitro binding measurements with in vivo activities
The protein switches tested here cover a wide range of dark and lit state affinities; each with different dynamic ranges. For the LOV2 based switches, we found a correlation between in vitro affinities and behavior in living cells. The iLID switches had the largest fold-change in binding affinity upon light stimulation and were the most effective at localizing protein to the plasma membrane, inducing cellular protrusions via localization of Tiam DH/PH, and controlling β-galactose expression in yeast. However, the iLID switches also had higher dark state affinities than the TULIP switch, which was evident in the mitochondrial localization assay where more dark-state localization was observed for both iLID pairs than for the TULIP switch. Also, the iLID pairs exhibited more dark-state activity in the yeast two-hybrid survival assays.
The results for CRY2 and CIB1 present a more complicated story. First, we did not observe a light dependent change in CIB1N binding with full-length CRY2 purified from insect cells. This may be due to an unrecognized issue with the purification of CRY2, such as a missing cofactor or post-translational modification. However, the CRY2/CIB1 results also provide evidence for another hypothesis; which is that changes in the oligomerization state of CRY2 are what lead to co-localization with CIB1 in cells, rather than an intrinsic change in affinity for CIB1. Evidence for this hypothesis comes from the sensitivity of the CRY2/CIB1 system to different fusion strategies. In cases where CRY2 homo-oligomerization could lead to multivalent interactions with CIB1, we observed more robust light-dependent changes. In contrast, when multivalent interactions were not created, only small changes were observed with light stimulation. For example, when CIB1N is fused to the plasma membrane, there is stronger recruitment of CRY2PHR to the membrane with light stimulation. In this scenario, multivalent interactions are possible between light-induced CRY2PHR oligomers and CIB1N, which is already co-localized via membrane anchoring (Fig. S9A). In the reverse scenario where CRY2 is localized to the membrane, cytoplasmic CIB1N is monomeric and there is no way to generate a multivalent interaction with CRY2PHR oligomers. With this setup, we only observed weak recruitment of CIB1N to CRY2PHR (Fig. S9B). An alternative explanation for the observed behavior is that the large clusters that form when membrane anchored CRY2PHR is light-activated may preclude robust CIB1N binding.
Changes in valency that accompany CRY2 oligomerization may also help explain the yeast two-hybrid results with CRY2/CIB1. We observed more robust light-dependent changes in transcription when CIB1N was fused to the DNA binding domain. The Gal4 DNA binding domain forms a dimer when bound to DNA, and therefore CIB1N–DBD is presented as a dimer to CRY2PHR-AD. This may allow for a multivalent interaction when CRY2PHR oligomerizes and therefore enhance the affinity between CRY2PHR-AD and CIB1N-DBD. The same multivalent interaction would not be created with the CRY2PHR-DBD/CIB1N-AD pair. When taken together, our results and results from previous studies 19,20,32 indicate that CRY2 homo-oligomerization is likely to play a significant role in the activity of the switch, and this can be used to enhance light-dependent signaling if multivalent interactions can be created.
Correlating in vitro and in vivo kinetics of activation and reversion
The activation and reversion kinetics of the switches become important when planning experiments as they determine how often you must expose the proteins to blue light in order to maintain dimerization. In the context of a cell this may be important in avoiding phototoxicity or regulating fast signaling processes. In turn, this needs to be balanced with the rate at which the switch needs to be fully off in the context of the experiment. The CRY2 switch reverted to dark state with the slowest kinetics of all switches tested. The quicker kinetics of iLID and the LOVpep switches give more precise temporal resolution, allowing for less lag time between light removal and dissociation. One important point to note is the photocycle of AsLOV2 switches can be tuned with some previously discovered mutations 33,34, so these switches can be altered to fit a variety of contexts. These mutations haven’t been tested in the heterodimerization context, but they may have little impact on dynamic range as they don’t directly interact with the Jα helix. The in cell rate of binding, except in one CRY2PHR/CIB1N orientation, seems to be limited by diffusion. The Venus-CIB1N-CAAX / tgRFPt-CRY2PHR produced a maximal plasma membrane recruitment level similar to the iLID switches. However the rate of activation was significantly slower. This is surprising, as the other switches recruitment half-life were proportional to their dynamic range. This suggests that in addition to CRY2PHR – CIB1N binding, an additional process is driving the increase in tgRFPt fluorescence intensity, such as CRY2 oligomerization. The rates of dissociation maintain the same rank order as the in vitro measurements but are longer in cells. This is most likely due to rates of diffusion out of the measured ROIs. Interestingly, in the membrane localization assay, the CRY2PHR/CIB1N reversion rates are orientation dependent and may also be explained by CRY2PHR oligomerization.
Practical considerations
Each switch contains other characteristics that we found to influence experimental design. While both components of iLID can be tagged on either the N- or C-terminus, we have found that both CRY2 and LOVpep C-terminal fusions inhibit binding to their partners. These stipulations have hindered particular applications in the past 10. When using the CRY2/CIB1 pair, orientation specific effects must be considered. Fusions of CRY2 with a protein of interest can have activating or inhibiting effects, which can be used advantageously if designed to do so. The time scale of experiment and physical light activation should also play a role in switch selection. We noticed that for multi-day experiments, weak dark state affinity was crucial as even ~50 μM binding was enough to elicit activation in the yeast growth assays (3 days). However, for shorter timescale responses like GTPase activation or transcription, tighter affinity pairs created a quicker functional output. On these shorter timescales, dark state activity had less of an effect.
While blue light induced dimers provide a powerful set of tools for use in cellular optogenetics there are some technical challenges that are worth considering. In our hands high intensity blue light is cytotoxic. For this reason it is imperative to optimize the light conditions used for your experiment. We have found that for confocal microscopy, that exposure to 1% of our 25mW Argon laser every 10s is enough to fully activate each of the switches tested without causing cytotoxic effects within the time periods presented here. Furthermore, imaging multiple fluorescent proteins without spectral overlap with the photoactive domain proved challenging. Here we have imaged Venus and tgRFPt but have relied upon a suboptimal GFP filter set for imaging of Venus which leads to a high signal to noise ratio for that channel as we are not collecting the entirety of light emitted from the fluorophore. While this setup works we recommend labeling your protein or signal of interest with tgRFPt. Alternatively, filter sets compatible with photoactivation and desired fluorophores can be custom ordered.
In conclusion, through rigorous benchmarking we have determined in vitro, in vivo and functional characteristics of three sets of blue light inducible dimers. This information can be used to guide future efforts aimed at cellular optogenetics.
Methods
Cloning
All clones are available via Addgene. iLID, LOVpep, and WT AsLOV2 were all cloned into pQE-80L BamHI and HindIII sites for E. coli expression with an N-terminal 6x His tag. The respective binding partners, SspB Nano & Micro, and ePDZb were cloned into a modified pQE-80L vector (BamH1/HindIII sites) with an N-terminal 6xHis-MBP-TEV tag. Both full length AtCRY2 as well as the PHR domain alone was cloned into the SalI site of the pFastBac HT A vector for insect cell expression. Recombinant bacmid DNA was made in DH10Bac E. coli cells and virus amplified in Sf9 insect cells. CIB1N was cloned into the BamH1 and HindIII sites of pQE-80L for expression in E. coli with a 6xHis-tag. All mammalian constructs were cloned into the pLL7.0 lentiviral vectors. Expression is therefore driven by a CMV promoter. The constructs were assembled by PCR based overlap extension, enzyme restriction, and ligation or through Gibson assembly.
Expression and Purification
Bacterial expression was performed as follows: BL21(DE3) cells were transformed through heat shock with each of the expression vectors. For each construct, 1.5L of LB media was inoculated and grown at 37°C to OD 0.6 and induced with 333mM IPTG. iLID, LOVpep, AsLOV2, SspB nano & micro, and ePDZb were expressed at 18°C for 16 hours. CIB1N was expressed at 25°C for 6 hours. After expression, cells were spun down at 3500 rpm for 10 minutes and pellets were frozen until purification. Insect cell expression was performed as follows: SF9 cells were inoculated with baculovirus at an MOI of 10 and expressed at 27°C for 48 hours according to 24. After 48 hours, cells were spun down at 2000 rpm, washed with cold PBS buffer and frozen at -80°C until purification. Bacterial cell pellets of LOV based switches and their binding partners were resuspended in phosphate lysis buffer (50 mM sodium phosphate pH 7.5, 500 mM NaCl, 20 mM Imidazole, 100 μM PMSF) and sonicated. Cell lysates were spun down for 30 minutes at 20,000 rpm. Cell supernatants were filtered with a 5 μm filter, run over HisTrap HP columns (GE) and eluted with elution buffer (50 mM sodium phosphate pH 7.5, 500 mM NaCl, 500 mM Imidazole, 100 μM PMSF). Proteins expressed as 6xHis-MBP fusions were dialyzed overnight in PBS with TEV protease and re-run over HisTrap columns to separate the protein of interest from His-MBP. Finally, all proteins were passed over at Superdex 75 column (GE) as a final clean up and buffer exchange to PBS (10 mM dibasic sodium phosphate, 1.8 mM monobasic potassium phosphate, 137 mM NaCl, 2.7 mM KCl, pH 7.4) for characterization. AtCRY2 and CIB1N purification was similar to the above protocol except Tris buffers were used instead of phosphates buffers as previously published 24. Insect cells were lysed by sonication without detergents to prevent contamination. The final size exclusion buffer for both AtCRY2 and CIB1N was 50 mM Tris-HCl pH 7.4, 250 nM NaCl, 5 mM BME. We also expressed AtCRY2PHR domain (domain necessary for dimerization) of AtCRY2 alone, but poor yields precluded in vitro experiments with this variant. Most of the dimer systems expressed highly and we had little handling or solubility issues with them. The notable exception was AtCRY2, which precipitated at concentrations above 20 μM.
Fluorescent Probe Generation
To measure direct binding between AtCRY2-CIB1N, CIB1N was labeled with 5(6)-TAMRA (Anaspec) at a single cysteine residue; position 103. Purified proteins were buffer exchanged on PD-10 desalting columns (GE) into 20mM Tris-HCl pH 7.5, 150mM NaCl, 1mM TCEP. Ten-fold excess dye was added to the prep and was left on a rotator at 4°C overnight. Labeled proteins were then passed through another PD-10 column to remove free dye. Absorbance at 555 nm (ε=65,000 M−1 cm−1) was used to quantify dye concentration and BCA assay (Thermo Scientific) was used to quantify protein concentration. Competitive binding assays were used to measure binding for the iLID and LOVpep systems. The sequence for the LOVpep competitor peptide was 5(6)TAMRA-EEIDKAVDTWV and the sequence for the iLID competitor peptide was 5(6)TAMRA-QIEEAANDENY.
Fluorescent Polarization Binding Assay
Fluorescence polarization measurements were recorded using a Jobin Yvon Horiba FluoroMax3 fluorescence spectrometer. All binding assays except CRY2/CIB1N were performed in PBS buffer in either a 1 cm or 1 mm quartz cuvette at 25 °C. CRY2/CIB1N binding was performed in a Tris (20mM Tris-HCl, pH 7.4, 200 mM NaCl, 5 mM BME) buffer due to solubility issues. Polarization of TAMRA was measured with excitation at 555 nm and emission at 584 nm. For AtCRY2-CIB1N binding, the concentration of TAMRA-CIB1N started at 200 nM and AtCRY2 was titrated in. At each titration point, the sample chamber was illuminated with 6.0 mW cm-2 blue light using a collimated blue led array. A lit state time point was taken immediately after removal of the blue light and another 5 minutes later for AsLOV2 binding and 10 minutes later for AtCRY2 binding. Initial affinities of the iLID and LOVpep competitor peptides were measured through direct binding titrations. Starting peptide concentrations were 25 nM for the iLID peptide and 250 nM for the LOVpep peptide. For iLID nano competitive binding assays, 25 nM peptide and 40 nM SspB nano were incubated with enough competitor to bind approximately 60% of peptide prior to titration. Competitive binding titrations were illuminated with blue light as in the direct binding assays and dark state measurements were taken after 5 minutes of darkness.
Multi-Angle Light Scattering
SEC-MALS experiments were performed on a Wyatt DAWN HELEOS II light scattering instrument interfaced to an Agilent FPLC System with a Superdex S200 column, Wyatt T-rEX refractometer and Wyatt dynamic light scattering module. CRY2 samples were prepared at 15 μM (in 50 mM Tris-HCl pH 7.4, 250 nM NaCl, 5 mM BME) and run through the S200 either in the presence of blue light (2 mW cm−2, blue led array) or in darkness.
Dynamic Light Scattering
CRY2 oligomerization was measured in a DynaPro Dynamic Light Scattering Plate Reader at room temperature. CRY2 at 15 μM was illuminated with blue light (6.0 mW cm-2 blue light, collimated blue led array) for 1 minute and placed in the instrument. Measurements were taken every 5 seconds for 20 minutes.
Absorption Recovery after Activation
Excited state recovery times were measured using a Cary 50 UV-Visible Spectrophotometer. Samples were irradiated with blue light (6.0 mW cm−2 blue light, collimated blue led array) and absorbance at 450 nm was recorded until recovery.
Yeast Plasmids Generation
Clontech pGBKT7 vector was modified to substitute the 2μ origin with CEN4 origin of replication by restriction digest with SacI and XmaI introduced from primers of a vector PCR and CEN4 from pNIA-CEN-MBP 25 yielding pGBKT7-CEN. CRY2PHR (1-498) was cloned with NdeI and NotI, ePDZb with NdeI and BamHI and finally, SspB Nano and Micro were cloned with EcoRI and BamHI into the newly generated pGBKT7-CEN plasmid. Additionally, CIB1N (1-170) was cloned in the original pGBKT7 vector using NdeI and BamHI. CIB1N was cloned in pGADT7 with NdeI and BamHI, LOV-pep and variant were cloned as well as oLID and iLID were cloned with EcoRI and BamHI. Finally, CRY2PHR was cloned in pGADT7 via NdeI and NotI restriction digest as well. All plasmids were sequence verified using Eurofins DNA sequencing service.
Yeast Transformation and Mating
The resultant plasmids were transformed via high efficiency lithium acetate transformation 26 in Y187 for pGADT7-derived plasmids and Y2HGold for pGBKT7-derived plasmids. After about 72 hours, single colonies for each were isolated and inoculate 0.5 mL YPD culture overnight in order to mate them and generate the respective diploids. The next day, the mated yeast were pelleted at 3000 rpm for 5 minutes and plated on douple dropout plates (SC-Leucine/-Tryptophane).
β-Galactose Assay
β-Galactose assay were performed as follows: Freshly mated yeast colonies were grown for about 36h at 30°C in 5 ml SC-Leu/-Trp. Cell density was measured at OD600 and 2.5 mL cultures were diluted to OD600 = 0.2 in duplicates – one for a light and another for a dark condition (falcon tubes were wrapped in aluminium foil). Cultures were grown at 30°C in a shaking incubator (250 rpm) for 3 hours in the dark and then for another 4 hours under blue light (465 nm) at 500 μW/cm2 via LED strip light wrapped around the tube rack. The resulting cultures were pelleted in triplicates and β-Galactose assay using CPRG for a substrate was performed according Clontech yeast handling protocols.
Yeast Growth Assays
Survival assays were performed as follows: Fresh colonies were grown for about 36 h at 30°C in 5 ml SC-Leu/-Trp. Cell density was measured at OD600 and cultures diluted in 200 μl of OD600 = 1, followed by eight 5-fold serial dilutions. Then, 2 μl of each of the dilutions were pipetted and spotted using a multichannel pipette (Gilson) onto respective dropout plates. The dark condition plates were wrapped in aluminum foil and placed in the same incubator as the lit condition at 30°C. Continuous blue light (465 nm) at 500 μW/cm2 was provided with LED strip lights attached at the incubator. Yeast plates were imaged after 70 hours incubation, the resulting images were cropped and arranged using Adobe Photoshop.
Mammalian Cell Culture and Transfection
Mouse IA32 fibroblasts were cultured in DMEM supplemented with 10% (vol/vol) FBS (HyClone), 100 U/ml penicillin, 100 μg/mL streptomycin, and 292 μg/mL L-glutamine. Cells were cultured at a constant 37 °C and 5% (vol/vol) CO2. Cells were transiently transfected in 6 well cell culture dishes using 1 μg total DNA at 1:1 ratio and NanoJuice (EMD Millipore) transfection reagent as recommended by manufacturer.
Mammalian Cell localization/GEF Microscopy
Experiments were performed according to the methods found in Guntas et al. Briefly, cells were co-transfected with two vectors containing the sequences encoding each component of the switch in equal parts. 24 hr later tranfected cells were trypsonized and transferred to 3.5 cm MatTek glass bottom dishes coated with a 10 ug/ml solution of fibronectin. 24 – 48 hr later cells were imaged and photo-activated with an Olympus FV1000 confocal microscope equipped with a 1.30 N.A. 40× oil immersion objective. The Fluoview software Time Controller was used to produce a timeline of image acquisition and photo-activation using the same standard parameters found in Guntas et al. These standard settings were meant to keep protein expressions levels similar between the switches as only cells whose fluorescence fell within the dynamic range that these settings could capture were imaged. Activation parameters were also kept constant between samples. In short laser power was set at 1% for the 488nm line. For whole cell activation the entire field of view was activated in a 512×512 pixel grid with a 2us/pixel dwell time and repeated 5×. before the next image was acquired. For ROI activation a 60×60 pixel grid was activated with a 8us/pixel dwell time and repeated 10× before the next image was taken.
Image analysis and quantification
All images were analyzed using FIJI software. Spot localization was quantified according to Guntas et al. Briefly the tgRFPt fluorescence intensity was measured within the activated ROI and an initial intensity and size matched area outside the activated ROI. A ratio of fluorescence intensity inside : outside the ROI was analyzed throughout time. The values that correspond to the period of activation were fit to the equation Y= 1 + Ymax*(1-exp(−K*X)). The values that correspond to the period of reversion were normalized to the maximum values and fit to the equation Y=(Y0 − Plateau)*exp(−K*X) + Plateau. Whole cell activation was quantified with an improved version of the method described in Guntas et al. Mitochondrial ROIs (Mito) are determined by automated thresholding of the tgRFPt channel. Cytoplasmic ROIs (Cyto) were determined by first creating a 5 pixel buffer outside of the Mito and selecting a ROI 10 pixels outside of that. The cytoplasmic ROIs were further refined by removing a small subset of pixels that are representative of the background. The values described in the paper are the average tgRFPt fluorescence intensities from the algorithmically determined ROIs expressed as (Mito-Cyto)/Cyto. Cytoplasmic values were first subtracted from mitochondrial values to remove any fluosecence signal contributed by the cytoplasm above and below the mitochondria. Curves were fit to the values during the activation and reversion periods using the equations Y=S+Ymax*(1−exp(−K*X)) & Y=(Y0 − Plateau)*exp(−K*X) + Plateau respectively. The fold change was determined by (S+Ymax)/S. All curve fittings were performed using Prism (GraphPad) software. The protrusion distance reported in the Tiam DH/PH localization experiments was measured by kymography. A line one pixel thick was drawn through each of the activated ROIs. The image values through time along that line were concatenated to form a new image. This image was then used to determine the initial and maximal position of the membrane within the time of activation to determine the maximum protrusion distance.
Supplementary Material
Acknowledgments
We thank Dr. Ashutosh Tripathy for help with SEC-MALS. We would also like to thank Dr. Chandra Tucker and Dr. Devin Strickland for providing DNA for the TULIP and Cryptochrome constructs. This work was supported by NIH grants RO1GM093208 and R01DA036887 to B.K as well as R01GM111557 to J.B.
Abbreviations
- LOV
Light Oxygen Voltage
- AsLOV2
Avena sativa phot1 LOV2 domain
- MTHF
510-methyltetrahydrofolate
- PHR
Photolyase homology region
- CCT
Cryptochrome carboxyl-terminus
- GTPase
Guanosine triphosphate phosphotase
- GEF
Guanine Nucleotide Exchange Factor
- TAMRA
Carboxytetramethylrhodamine
- iLID
Improved light inducible dimer
- TULIPs
Tunablelight-controlled interacting protein tags for cell biology
- ePDZb
Engineered PDZ domain version b
- CPRG
Red-β-D-galactopyranoside
- LOVpep+
LOVpep with T406-7AI532A mutations
- CRY2
Cryptochrome 2
- CIB1N
N-Terminus of CIB1
- PCB
phycocyanobilin
- ROI
Region of Interest
Footnotes
Supporting Information.
CRY2-CIB1N binding data measured by fluorescence polarization with additional cofactors, CRY2-CIB1N binding measured by gel shift assay, CRY2 oligomerization state in the presence of additional cofactor, CRY2-CIB1N co-elution data, localization with CRY2PHR-CIB1N and LOVpep-ePDZb constructs with orientation dependent effects, the effects of CRY2PHR oligomerization in mammalian cells, oLID yeast two hybrid, yeast survival assays, cartoon model of how CRY2 oligomerization affects localization in cells.
Author Contribution
R.H. and S.Z. contributed equally. R.H. designed, performed and analyzed all in vitro experiments, and contributed to writing and editing of the manuscript. S.Z. designed, performed and analyzed all mammalian cell experiments, and contributed to writing and editing of the manuscript. H.Y. designed, performed and analyzed all yeast cell experiments, and contributed to writing and editing of the manuscript. J.B. designed experiments, provided equipment and edited the manuscript. B.K. designed experiments, provided equipment and contributed to writing and editing of the manuscript.
References
- 1.Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K. Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci. 2005;8:1263–8. doi: 10.1038/nn1525. [DOI] [PubMed] [Google Scholar]
- 2.Nagel G, Szellas T, Huhn W, Kateriya S, Adeishvili N, Berthold P, Ollig D, Hegemann P, Bamberg E. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc Natl Acad Sci U S A. 2003;100:13940–5. doi: 10.1073/pnas.1936192100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shimizu-Sato S, Huq E, Tepperman JM, Quail PH. A light-switchable gene promoter system. Nat Biotechnol. 2002;20:1041–4. doi: 10.1038/nbt734. [DOI] [PubMed] [Google Scholar]
- 4.Levskaya A, Weiner OD, Lim WA, Voigt CA. Spatiotemporal control of cell signalling using a light-switchable protein interaction. Nature. 2009;461:997–1001. doi: 10.1038/nature08446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu YI, Frey D, Lungu OI, Jaehrig A, Schlichting I, Kuhlman B, Hahn KM. A genetically encoded photoactivatable Rac controls the motility of living cells. Nature. 2009;461:104–108. doi: 10.1038/nature08241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yazawa M, Sadaghiani AM, Hsueh B, Dolmetsch RE. Induction of protein-protein interactions in live cells using light. Nat Biotechnol. 2009;27:941–945. doi: 10.1038/nbt.1569. [DOI] [PubMed] [Google Scholar]
- 7.Renicke C, Schuster D, Usherenko S, Essen LO, Taxis C. A LOV2 domain-based optogenetic tool to control protein degradation and cellular function. Chem Biol. 2013;20:619–26. doi: 10.1016/j.chembiol.2013.03.005. [DOI] [PubMed] [Google Scholar]
- 8.Rao MV, Chu PH, Hahn KM, Zaidel-Bar R. An optogenetic tool for the activation of endogenous diaphanous-related formins induces thickening of stress fibers without an increase in contractility. Cytoskeleton (Hoboken) 2013;70:394–407. doi: 10.1002/cm.21115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu Y, Hyun YM, Lim K, Lee H, Cummings RJ, Gerber SA, Bae S, Cho TY, Lord EM, Kim M. Optogenetic control of chemokine receptor signal and T-cell migration. Proc Natl Acad Sci U S A. 2014;111:6371–6. doi: 10.1073/pnas.1319296111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Bergeijk P, Adrian M, Hoogenraad CC, Kapitein LC. Optogenetic control of organelle transport and positioning. Nature. 2015;518:111–4. doi: 10.1038/nature14128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Konermann S, Brigham MD, Trevino AE, Hsu PD, Heidenreich M, Cong L, Platt RJ, Scott DA, Church GM, Zhang F. Optical control of mammalian endogenous transcription and epigenetic states. Nature. 2013;500:472–6. doi: 10.1038/nature12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kennedy MMJ, Hughes RMRM, Peteya LA, Schwartz JW, Ehlers MD, Tucker CL. Rapid blue-light-mediated induction of protein interactions in living cells. Nat Methods. 2010;7:973–975. doi: 10.1038/nmeth.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Strickland D, Lin Y, Wagner E, Hope CM, Zayner J, Antoniou C, Sosnick TR, Weiss EL, Glotzer M. TULIPs: tunable, light-controlled interacting protein tags for cell biology. Nat Methods. 2012;9:379–84. doi: 10.1038/nmeth.1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guntas G, Hallett Ra, Zimmerman SP, Williams T, Yumerefendi H, Bear JE, Kuhlman B. Engineering an improved light-induced dimer (iLID) for controlling the localization and activity of signaling proteins. Proc Natl Acad Sci. 2015;112:112–117. doi: 10.1073/pnas.1417910112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Más P, Devlin PF, Panda S, Kay SA. Functional interaction of phytochrome B and cryptochrome 2. Nature. 2000;408:207–11. doi: 10.1038/35041583. [DOI] [PubMed] [Google Scholar]
- 16.Liu H, Gomez G, Lin S, Lin S, Lin C. Optogenetic control of transcription in zebrafish. PLoS One. 2012;7:e50738. doi: 10.1371/journal.pone.0050738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hughes RM, Bolger S, Tapadia H, Tucker CL. Light-mediated control of DNA transcription in yeast. Methods. 2012;58:385–91. doi: 10.1016/j.ymeth.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Polstein LR, Gersbach CA. A light-inducible CRISPR-Cas9 system for control of endogenous gene activation. Nat Chem Biol. 2015;11:198–200. doi: 10.1038/nchembio.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bugaj LJ, Choksi AT, Mesuda CK, Kane RS, Schaffer DV. Optogenetic protein clustering and signaling activation in mammalian cells. Nat Methods. 2013;10:249–52. doi: 10.1038/nmeth.2360. [DOI] [PubMed] [Google Scholar]
- 20.Taslimi A, Vrana JD, Chen D, Borinskaya S, Mayer BJ, Kennedy MJ, Tucker CL. An optimized optogenetic clustering tool for probing protein interaction and function. Nat Commun. 2014;5:4925. doi: 10.1038/ncomms5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harper SM, Neil LC, Gardner KH. Structural basis of a phototropin light switch. Science. 2003;301:1541–4. doi: 10.1126/science.1086810. [DOI] [PubMed] [Google Scholar]
- 22.Lungu OI, Hallett Ra, Choi EJ, Aiken MJ, Hahn KM, Kuhlman B. Designing Photoswitchable Peptides Using the AsLOV2 Domain. Chem Biol. 2012;19:507–517. doi: 10.1016/j.chembiol.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pathak GP, Strickland D, Vrana JD, Tucker CL. Benchmarking of optical dimerizer systems. ACS Synth Biol. 2014;3:832–8. doi: 10.1021/sb500291r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ozgür S, Sancar A. Analysis of autophosphorylating kinase activities of Arabidopsis and human cryptochromes. Biochemistry. 2006;45:13369–74. doi: 10.1021/bi061556n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marshall KS, Zhang Z, Curran J, Derbyshire S, Mymryk JS. An improved genetic system for detection and analysis of protein nuclear import signals. BMC Mol Biol. 2007;8:6. doi: 10.1186/1471-2199-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gietz RD, Schiestl RH. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc. 2007;2:31–4. doi: 10.1038/nprot.2007.13. [DOI] [PubMed] [Google Scholar]
- 27.Liu H, Yu X, Li K, Klejnot J, Yang H, Lisiero D, Lin C. Photoexcited CRY2 interacts with CIB1 to regulate transcription and floral initiation in Arabidopsis. Science. 2008;322:1535–9. doi: 10.1126/science.1163927. [DOI] [PubMed] [Google Scholar]
- 28.Bear JE, Loureiro JJ, Libova I, Fässler R, Wehland J, Gertler FB. Negative regulation of fibroblast motility by Ena/VASP proteins. Cell. 2000;101:717–28. doi: 10.1016/s0092-8674(00)80884-3. [DOI] [PubMed] [Google Scholar]
- 29.Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9:690–701. doi: 10.1038/nrm2476. [DOI] [PubMed] [Google Scholar]
- 30.Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- 31.Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 2005;6:167–80. doi: 10.1038/nrm1587. [DOI] [PubMed] [Google Scholar]
- 32.Lee S, Park H, Kyung T, Kim NY, Kim S, Kim J, Do Heo W. Reversible protein inactivation by optogenetic trapping in cells. Nat Methods. 2014;11:633–636. doi: 10.1038/nmeth.2940. [DOI] [PubMed] [Google Scholar]
- 33.Salomon M, Christie JM, Knieb E, Lempert U, Briggs WR. Photochemical and mutational analysis of the FMN-binding domains of the plant blue light receptor, phototropin. Biochemistry. 2000;39:9401–9410. doi: 10.1021/bi000585+. [DOI] [PubMed] [Google Scholar]
- 34.Zayner JP, Sosnick TR. Factors That Control the Chemistry of the LOV Domain Photocycle. PLoS One. 2014;9:e87074. doi: 10.1371/journal.pone.0087074. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.