

Abstract
The role of the central melanocortin system in the regulation of energy metabolism has received much attention during the past decade since gene mutations of key components in melanocortin signaling cause monogenic forms of obesity in animals and humans. In the arcuate nucleus of the hypothalamus the prohormone proopiomelanocortin (POMC) is posttranslationally cleaved to produce α-melanocyte stimulating hormone (α-MSH), a peptide with anorexigenic effects upon activation of the melanocortin receptors (MCRs). α-MSH undergoes extensive post-translational processing and its in vivo activity is short lived due to rapid degradation. The enzymatic process that controls α-MSH inactivation is incompletely understood. Recent evidence suggests that prolyl carboxypeptidase (PRCP) is an enzyme responsible for α-MSH degradation. As for many key melanocortin peptides, gene mutation of PRCP causes a change in the metabolic phenotype of rodents. This review summarizes the current knowledge on the melanocortin system with particular focus on PRCP, a newly discovered component of the melanocortin system.
Keywords: Hypothalamus, arcuate nucleus, POMC processing, alpha-MSH, hormones, fuel, energy metabolism, Melanocortin receptors, signaling pathways, PRCP
Introduction
Obesity has profound consequences dramatically diminishing both the quality and length of life. Compelling evidence has been gathered to suggest that the underlying cause for fat accumulation in peripheral tissues arises, at least in part, from the central nervous system [18, 65, 73, 97, 109, 122, 188, 191, 203].
The major role played by the central nervous system was further supported by the discovery in 1994 and 1995 of the gene encoding the adipose signal, leptin, [24, 84, 169, 234], and its receptors [137, 208]. Leptin receptors are expressed predominantly in the ventromedial structure of the hypothalamus, the arcuate nucleus where melanocortin neurons are located [136, 168, 187]. The importance of the melanocortin system in the regulation of energy metabolism was revealed when to better understand the role of various melanocortin receptors in the determination of skin color [148, 149, 181], animals in which melanocortin receptor 4 (MC4R) was ablated became morbidly obese [69, 99]. The melanocortin system comprises of neurons expressing POMC and AgRP (the endogenous antagonist) and neurons expressing melanocortin receptors. Today, the melanocortin system is considered a major regulator of energy metabolism since alterations of many of its components have been shown to affect energy balance [39, 65, 73, 97, 98, 122, 187, 189, 191, 220].
1. Role of the hypothalamus in metabolism regulation
The critical role that the arcuate nucleus melanocortin system (the proopiomelanocortin (POMC) cells, the arcuate nucleus agouti-related protein (AgRP)- neuropeptide Y (NPY)-producing cells) [83], plays in mediating leptin’s effect on metabolism provided an exceptionally attractive and simple model of the central feeding center. In this model, activation of the POMC neurons by leptin [45, 62] triggers release of α-melanocyte stimulating hormone (α-MSH) from POMC axon terminals, which in turn, activates melanocortin receptors (MCRs) leading to suppressed food intake and increased energy expenditure. Simultaneously, leptin suppresses the activity of arcuate nucleus NPY/AgRP neurons [45, 62], which otherwise, through the release of AgRP, antagonize the effect of α-MSH on MCRs [162]. The NPY/AgRP system not only antagonizes anorexigenic melanocortin cells at their target sites where MCRs are located, but it also very robustly and directly inhibits POMC perikarya [94, 95]. Both NPY as well as the small inhibitory amino acid neurotransmitter, GABA [45], are involved in this inhibition, which occurs through basket-like synaptic innervations of POMC cells by NPY/AgRP terminals [46, 94]. This unidirectional [94] interaction between the NPY/AgRP and POMC perikarya is of critical significance, because it provides a tonic inhibition of the melanocortin cells whenever the NPY/AgRP neurons are active. Because there is no constitutive direct feedback mechanism from the POMC cells to disengage NPY/AgRP neurons, this simple anatomical constellation may provide the easiest explanation why the baseline blueprint of feeding circuits is more likely to promote feeding then satiety. While this bias towards positive energy balance is a necessity from an evolutionary perspective, it is also a likely contributor to the etiology of metabolic disorders, such as obesity.
Despite the important role of the NPY/AgRP neurons in the regulation of energy homeostasis by the melanocortin system, neither NPY nor AgRP single gene knockout mice [68] nor NPY/AgRP double knockout mice [174] exhibited the expected hypophagic phenotype. Because this could be due to compensatory mechanisms during development, two independent studies examined the role of NPY/AgRP and POMC neurons in mediating adult energy regulation [81, 130]. Both groups used the cell-targeted expression of diphteria toxin receptors that do not exist in mice, making these animals “immune” to diphtheria toxin-induced necrosis. When these mice, which have a normal development, were injected with two subsequent doses of diphtheria toxin, the targeted hypothalamic neurons rapidly died resulting in hypophagia in NPY neuron-ablated mice [81, 130] and hyperphagia in POMC neuron-ablated animals [81]. In addition, Luquet and collaborators [13]) found that ablation of NPY neurons during the early postnatal period did not result in an overt metabolic phenotype, arguing that functional effects of degenerated neurons in critical developmental periods may be overcome by reorganization of the circuits.
The PVN has emerged as an important nucleus of the hypothalamus for its role in the regulation of energy balance since a subpopulation of its neurons express MC4R, the target of arcuate projections [149]. The restoration of the MC4R gene selectively in the PVN of MC4R knockout mice by viral or genetic manipulation produced mice displaying lower body weight and reduced food intake compared to the MC4R knockout controls [6]. However, the restoration of the receptor was not sufficient to bring energy expenditure to the level of wild type controls. Thus, this suggests that melanocortin regulation of energy expenditure occurs elsewhere in the hypothalamus or through receptors other than MC4R [6]. In addition, MC4R neurons also express Single-minded 1 (SIM1), a transcription factor and one of only six genes implicated in human monogenic obesity [92]. Haploinsufficiency of SIM1 is associated with hyperphagic obesity and increased linear growth in both humans and mice [139]. Additionally, SIM1 heterozygous mice also show hyperphagia and obesity in response to a high-fat diet [93]. Thus, the phenotype of SIM1 haploinsufficiency is similar to that of agouti yellow (Ay), and MC4R knockout mice, both of which are defective in hypothalamic melanocortin signaling.
While the melanocortin system has emerged as key to the hypothalamic control of energy balance, other hypothalamic systems have also been closely tied to this regulatory mechanism. These include the lateral hypothalamic orexigenic neuronal populations producing either melanin concentrating hormone (MCH) [176] or orexin/hypocretin [49, 185], both of which, while in separate cell populations [19], appear to operate by the classic excitatory neurotransmitter, acetylcholine [33]. The lateral hypothalamus (LH) is considered a “feeding center” since lesions of this area induce hypophagia and weight loss [10, 140].
The discovery and functional characterization of two distinct neuronal populations in the LH that express orexigenic peptides, hypocretin/orexin and MCH, has supported the idea that the LH conveys important signals to promote feeding [11]. MCH and orexin/hypocretin expressing neurons are spatially distinct, but both regulate energy balance, among other functions [19, 61, 185, 205]. Leptin inhibits the expression of MCH and orexin/hypocretin in the LH, inhibits the activity of orexin neurons [22, 55, 193, 208, 223], and decreases excitatory inputs onto these neurons [99]. The mechanisms by which leptin regulates LH function are poorly understood. Projections from leptin responsive POMC- and AgRP-expressing neurons onto LH neurons have been identified. In addition, several neuroanatomical studies showed the presence of leptin receptors in LH neurons suggesting that direct leptin action on these neurons may regulate LH functions. Thus, leptin likely regulates LH functions by multiple mechanisms [63, 64].
Another important hypothalamic nucleus involved in the regulation of energy metabolism is the ventromedial nucleus (VMH). Its implication in the regulation of energy balance was shown when electrolytic lesions to this region in rats resulted in hyperphagia and obesity [17, 18, 89, 90]. Subsequent studies using pharmacological or chemical lesions have demonstrated that the VMH is a critical region that inhibits feeding and increases metabolism [5, 209]. The VMH was subsequently found to be one of the key regions in the hypothalamus where leptin receptors (LRb) are highly expressed [70, 137]. VMH neurons have been shown to be important in mediating leptin’s effect to control energy balance [111]. Animals with a selective deletion of the leptin receptor gene in neurons of the VMH expressing steroid factor-1 (SF-1), a transcription factor important for the development of the VMH [48, 166, 192], are obese and hyperphagic [50, 132].
VMH neurons have also been shown to play a role in the regulation of the activity of POMC neurons of the arcuate nucleus of the hypothalamus [201]. Using laser scanning photo-stimulation (LSPS) in combination with slice electrophysiology, it has been shown that VMH neurons project to POMC neurons and that these projections are mostly excitatory. Furthermore, food deprivation has been shown to decrease these excitatory inputs onto arcuate POMC cells [201].
The activity of the VMH neurons, on the other hand, is also modulated by projections from the arcuate nucleus. However, the projections from the arcuate to the VMH are less dense compared to those to other nuclei [233]. In addition, the VMH contains MC4R, as well as NPY Y1, Y2 and Y5 receptors [14, 85, 112, 125, 128, 219]. Studies have shown that infusions of NPY into the VMH increase feeding, and fasting is associated with elevated levels of NPY in this region [14, 109]. Furthermore, electrophysiological recordings have shown that VMH neuronal responses to α-MSH are decreased in fasted animals and in animals treated with AgRP prior to sacrifice compared to those of ad libitum fed animals [125].
Brain-derived neurotrophic factor (BDNF), that has been shown to affect the metabolic functions regulated by the VMH, is specifically and abundantly expressed in the VMH. Genetic manipulations of BDNF or its TrkB receptor resulted in obesity in both humans and mice [180, 222]. Interestingly, leptin increases BDNF transcripts [155, 156, 212], whereas fasting decreases them selectively in the VMH, suggesting that the BDNF is a regulatory component of leptin signaling. Because BDNF is known to affect synaptic morphology and function [180], synaptic plasticity of the VMH may be a part of the regulatory mechanism of energy regulation. Notably, the anorectic effects of BDNF are not directly mediated by the melanocortin system since BDNF reduces the body weight and food intake of mice that lack the MC4R [222]. This suggests that the VMH may influence POMC activity by other means, such as classic neurotransmitters and/or plasticity.
The redundant interaction of the melanocortin system with other hypothalamic and extrahypothalamic neuronal systems may occur so that alternative signaling modalities can be utilized if a pathway is blocked. Recent developments also suggest that the flexibility of the hypothalamic center of metabolism is not only due to redundant interconnectivity but is also a consequence of its recently uncovered soft wiring [98, 170].
2. Hypothalamic POMC
The importance of the role of POMC in the regulation of energy metabolism has been well documented by several studies in rodents and observations in humans in which POMC gene mutations are associated with an obese phenotype [28, 38, 118]. The POMC gene is expressed in the brain as well as in the periphery such as in the pituitary gland, immune system and skin [231]. Within the brain, POMC is expressed in the hypothalamus and in the nucleus tractus solitarius (NTS) of the brainstem [232]. The POMC gene encodes several peptides that are the products of a complex post-translational process. The bioactive peptides of the POMC gene include adrenocorticotrophin (ACTH), β-endorphin, α-, β- and γ-melanocyte stimulating hormones (MSH). Five melanocortin receptors (MC) have been cloned so far, MC1-5R. While MC1R, MC2R and MC5R have found to be implicated in the pigmentation in the skin, adrenal steroidogenesis and thermoregulation respectively [30, 37, 181], MC3R and MC4R have been shown to play a major role in the regulation of food intake and energy balance [39, 190]. Both MC3R and MC4R knockout mice, for example, are obese. However, while MC4R-deficient mice are hyperphagic [101], MC3R mice are not [21, 29]. The hypothalamic localization of these 2 receptor subtypes is also quite distinctive. Within the hypothalamus, MC3R is expressed in the POMC neurons of the arcuate nucleus, suggesting that MC3R plays an important role in the feedback mechanism of these neurons [45, 106]. Contrary to MC3Rs, MC4Rs are not expressed in the arcuate nucleus but in other areas of the hypothalamus that are important in the regulation of food intake including the PVN, the dorsomedial hypothalamus (DMH) and the LH [106, 127, 149].
The importance of the role of MC4R in the regulation of energy metabolism has been further established when mutations in the MC4R gene in humans were identified and associated with an obese phenotype [214, 230]. In fact, to date, mutations in MC4R are the most common monogenetic form of human obesity described [31].
The projections of the two components of the melanocortin system, POMC and NPY/AgRP neurons, grossly overlap both within and outside of the hypothalamus [19, 105, 138, 217], although they are rarely very long. The common targets of the NPY/AgRP and POMC projections are the hypothalamic parvicellular PVN [44], the lateral hypothalamic and perifornical areas, all of which express melanocortin receptors MC3/4R [149184]. The interplay between AgRP and α-MSH fibers on melanocortin receptor-expressing neurons in the PVN is suggested to be critical for metabolism regulation [44]. The signals that target the PVN can then be distributed in various directions as the PVN projects to brain stem nuclei, the spinal cord, cortex (via various routes), and thalamus, as well as to the portal vessels connecting the hypothalamus to the anterior pituitary.
Besides the PVN, other areas to which melanocortin neurons project include the medial preoptic area (MPA), the periacquedoctal gray (PAG), the nucleus of the solitary tract (NTS) and the parabrachial nucleus (PBN) [19].
An important target area of the mediobasal hypothalamic POMC neurons is the preganglionic sympathetic system in the thoracic segment of the spinal cord [61]. These POMC projections are unopposed by NPY/AgRP efferents [61] and are thought to play an important role in melanocortin regulation of the sympathetic nervous system [61]. Interestingly, POMC neurons that give rise to this spinal cord projection are not homogenously distributed within the mediobasal hypothalamus [61]. They are mainly located in the retrochiasmatic area of the hypothalamus and in an area lateral from the arcuate nucleus [61]. In fact, when analyzed closely, the appearance of POMC neurons is not homogenous throughout the mediobasal hypothalamus: within the arcuate nucleus, and specifically in its more medial aspects, POMC neurons are smaller compared to the laterally located, larger melanocortin cells. This enlarged soma size is consistent with neurons that have long projections, such as those POMC cells that project to the spinal cord.
Previous arguments have been made that because of the lack of blood brain barrier in the median eminence of the basal hypothalamus, POMC and NPY/AgRP neurons may be direct targets of circulating metabolic hormones [45], such as leptin ghrelin and insulin. However, the assertion that parts of the melanocortin system in close proximity to the median eminence (Fig. 1) would be outside of the blood brain barrier has never been proven. In a very recent study, the relationship between POMC and NPY/AgRP neurons and capillaries of the arcuate nucleus was analyzed [100]. When examined by electron microscopy, there was no example of either of these perikarya being directly associated with any phenestrated vessels or endothelial cells [100].
Figure 1.
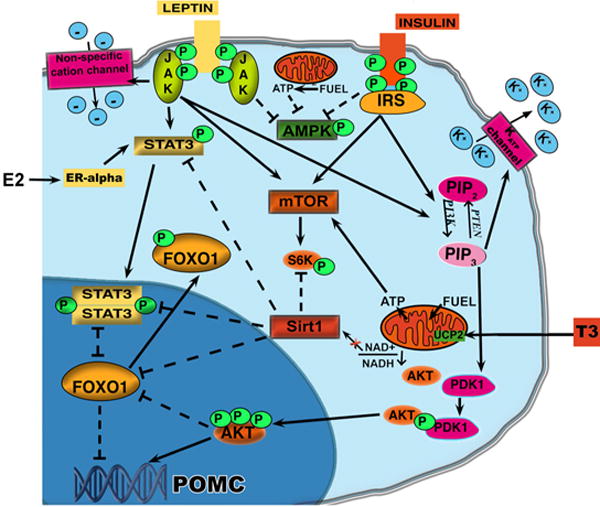
Schematic illustration showing the metabolic pathways activated by hormones and fuel availability in POMC neurons of the arcuate nucleus of the hypothalamus. Activation of PI3K in POMC neurons by either leptin or insulin receptors induces activation of AKT which leads to the phosphorylation of FoxO1 and thus its exclusion from the nucleus allowing the transcription of POMC. Concomitantly, PI3K activation also induces the opening of fewer potassium ATP channels thus inducing the leptin-dependent firing of the POMC neurons and leptin-dependent POMC transcription via Stat3. Stat3 has been also reported to be a target of estrogen signaling. However, the effect of estrogen on Stat3 is JAK independent. In satiety conditions when POMC neurons are activated, Sirt1 and NAD+ levels are decreased and Sirt1 activation of FoxO1 is also inhibited. Finally, the decrease in the ratio AMP/ATP will decrease AMPK while increase mTOR activity.
3. Regulation of POMC in the Hypothalamus
3.1 Leptin
POMC is a regulator of appetite, energy expenditure and glucose metabolism [39]. Thus, its own regulation must be important for the control of energy homeostasis. The fat-derived hormone leptin is a major regulator of POMC. During fasting, low levels of leptin result in a reduction of POMC mRNA. In addition, leptin receptor-deficient rats (fa/fa) [117], leptin deficient (ob/ob) and leptin receptor (db/db) deficient mice [142] have reduced levels of POMC in the hypothalamus compared to their wild type controls, and leptin replacement to ob/ob mice induces an increase in POMC mRNA [8, 117, 142, 190].
AGRP is also directly regulated by leptin in the arcuate nucleus [143]. Both POMC and AGRP neurons express leptin receptor [32, 137] and leptin has been shown to directly increase action potentials in POMC neurons while decreasing the frequency of action potentials in AgRP neurons [45]. The signaling pathway activated by the binding of leptin to its receptors has been the object of many studies that have revealed a complex cascade of events (Fig. 1). Leptin binding to its receptors induces an alteration in the conformation of the receptors which enables the activation of Janus kinase/signal transducers and activators of transcription (STAT) (Fig. 1) as well as mitogen-activated protein kinase (MAPK) signal transduction pathways [213]. Stat 3 increases POMC expression by recruiting histone acetylases to the POMC promoter [114]. In addition, leptin increases the firing rates of POMC neurons via the activation of nonspecific cation channels [45] (Fig. 1). Leptin also induces the expression of suppressor of cytokine signaling (SOCS)-3 mRNA which is thought to act as inducible intracellular negative regulators of leptin signal transduction. In agreement with this, transfection data suggest that SOCS-3 is an inhibitor of leptin signaling [62]. Finally, the protein tyrosine phosphatase PTP1B has been shown to be an important regulator of leptin signaling in POMC neurons since its deletion in this specific neuronal population reduces diet-induced obesity weight gain and adiposity and increases energy expenditure [7]. On the other hand, specific deletion in POMC neurons of SHP2, a non-receptor tyrosine phosphatase, resulted in increased adiposity, weight gain and decreased energy expenditure [7].
In addition to these intracellular effects, leptin has been shown to induce a rearrangement of the synaptic input organization onto the melanocortin neurons [170]. A differential synaptic input organization has indeed been reported in leptin-deficient ob/ob mice compared to their wild type controls. Specifically, in ob/ob mice, a significantly lower number of synapses onto POMC neurons was reported [170]. Of those synapses, symmetrical putatively inhibitory synapses onto POMC cells were found to dominate over the asymmetrical excitatory synapses. The opposite was observed on the AgRP neurons [170]. Leptin treatment to ob/ob mice rapidly reversed the synaptic density to the levels of wild type mice [170]. In agreement with this, different postsynaptic currents onto POMC and AgRP neurons were also found between these two groups. Although synaptic plasticity may not directly trigger alterations in postsynaptic response, and does not alone explain changes in energy states, it suggests that, depending on peripheral metabolic need, the probability of either activation or inhibition of POMC and AgRP may be rapidly modified by synaptic rearrangements. The understanding of the mechanisms underlying the changes in synaptic plasticity in the face of metabolic states could provide important insights into the regulation of feeding, and potentially into the regulation of other complex behavior.
3.2 Insulin
In addition to leptin, other hormones such as insulin, glucocorticoids, estrogen and thyroid hormones are also regulators of POMC neurons [76, 108, 110, 144, 146, 142, 183] (Fig. 1).
Insulin receptors are expressed in POMC neurons and, like leptin, their activation induces the downstream activation of phosphatidylinositol-3 kinase (PI3K; Fig. 1). Indeed, central injections of a specific PI3K inhibitor block the anorexigenic effect of insulin [159]. However, recent data have challenged the notion that acute intracerebroventricularly injected insulin has an anorectic effect in rats [107].
In addition, a recent study mapping insulin and leptin responsive arcuate POMC neurons found that leptin and insulin are largely segregated in distinct subpopulations of the POMC neurons [218].
Insulin receptor (IR) is a membrane-bound tyrosine receptor. In the CNS, when insulin binds its receptor, it induced the autophosphorylation of IR and the recruitment of IRS proteins and subsequent activation of PI3K and MAPK cascades. Concomitantly, PI3K activation also induces the opening of fewer ATP-sensitive potassium channels (KATP) thus [196] enabling the increased firing of the POMC neurons [167] (Fig. 1).
PI3K activation generates phosphatidylinositol-3,4,5-triphosphate (PIP3), which binds phosphoinositide-dependent protein kinase 1 (PDK1) that consequently phosphorylates AKT protein kinase. Activated AKT then enters the nucleus to regulate neuropeptide expression through the phosphorylation and inactivation of the fork-head box-containing transcription factor of the O subfamily type 1 (FoxO1) [113] which is now excluded from the nucleus allowing POMC transcription (Figure 1) [74, 114, 226]. In support of these findings, mice with a deletion of FoxO1 specifically in POMC neurons have a leaner phenotype due to a significant decrease in food intake with unchanged energy expenditure and locomotor activity [171]. However, no effect on POMC mRNA levels was observed in these mice. On the other hand, FoxO1 inactivation in POMC neurons was found to act on POMC processing by affecting carboxypeptidase E (Cpe) mRNA and activity levels [171].
The central effects of insulin also include the regulation of glucose metabolism in the periphery [20]. By inactivating insulin receptors in neurons and glial cells, it has been found that mice are prone to diet-induced obesity, have mild insulin resistance and impaired hepatic glucose production [20]. Central injection of insulin reduces glucose production in the liver, whereas, blocking insulin signaling with an inhibitor of central KATP channels, diminishes the ability of insulin to suppress peripheral glucose output [160]. This regulation may involve electric transmission via the autonomic nervous system, since transection of the vagal nerve abolishes this effect [172]. Conditional ablation of insulin receptors in either NPY/AgRP or POMC neurons has shown that only AgRP neurons may be important regulators of hepatic glucose production [116]. In addition, targeted knock-ins of insulin receptors in either POMC or AgRP neurons in mice in which insulin receptors are significantly reduced [163] in liver, pancreatic beta cells and several brain areas including the arcuate nucleus (L1 mice) showed that while AgRP regulates hepatic glucose production, POMC neurons regulate energy expenditure and locomotor activity [126]. However, very recent data have shown that concomitant insulin and leptin action in POMC neurons is required to maintain glucose homeostasis and reproductive function [91]. Indeed, by knocking down both leptin and insulin receptors in POMC neurons (Pomc-Cre, Lepr(flox/flox) IR(flox/flox) mice), mice display systemic insulin resistance, which is distinct from the single deletion of either receptor. In addition, Pomc-Cre, Lepr(flox/flox) IR(flox/flox) female mice display elevated serum testosterone levels and ovarian abnormalities, resulting in reduced fertility [91]. Finally, recent evidence suggests that central insulin action also regulates fat metabolism [115]. However, the site of action within the brain is still unclear.
3.3 Estrogen
The known effect of estrogen on energy metabolism occurs by regulation of the melanocortin neurons [76–78]. Like leptin, estrogen has been shown to trigger robust synaptic input rearrangement on the POMC neurons of the arcuate nucleus [76] which leads to a corresponding change in the electric properties of these cells. Thus, estrogen regulation of energy homeostasis, like leptin, involves synaptic rearrangement onto the POMC neurons to induce an anti-obese neuronal response. Since both leptin and estrogen are also important regulators of reproduction, a similar mechanism underlying hypothalamic control of fertility may be hypothesized. Whether this involves a direct interaction between the two signals remains to be proved. However, this idea is supported by several data. For example, in females, chronic estrogen withdrawal, as in ovariectomy in rodents and postmenopause in humans, causes leptin resistance, while estrogen replacement prevents this phenotype [2]. Furthermore, estrogen increases leptin-induced STAT3 phosphorylation in the arcuate nucleus [for review see 77], indicating that estrogen may act on a similar target to that of leptin (Fig. 1). Interestingly, estrogen’s effect on energy homeostasis and synaptogenesis is independent of leptin and leptin receptors [59, 76, 80]. Thus, this suggests that interaction between estrogen and leptin signaling must bypass leptin receptors and directly target the downstream components.
STAT3 has been reported to be a target of estrogen signaling in cells [13, 194]. Distinct activation mechanisms by which estrogen activates STAT3 were observed. For example, STAT3 is phosphorylated and activated in response to estrogen in cells involving multiple intracellular signal pathways that include MAPK, Src-kinase and PI3K [13, 25] and/or direct physical association between estrogen receptors and STAT3 [34].
Peripheral administration of estrogen induced tyrosine phosphorylation of STAT3 in the hypothalamus in less than 30 minutes. This effect of estrogen on STAT3 tyrosine phosphorylation is JAK independent, since it is effective in leptin receptor knockout mice [76]. Finally, in neural STAT3 knockout (STAT3N-/-) mice, which exhibit an obese and infertile phenotype, highly resembling leptin deficiency [75], estrogen exhibited no effect on reducing body weight. These observations strongly suggest that STAT3 is downstream of estrogen signaling in neurons and is required for the anti-obesity effect of estrogen to occur.
The cross-talk between estrogen and STAT3 takes place, at least in part, through classic estrogen receptors (ERs), the ligand-dependent nuclear transcription factors. A potential role for the recently uncovered membrane estrogen receptors [175] in this matter may not be ruled out; however, it requires more study. Estrogen receptor-α (ERα) has been suggested to be the main estrogen receptor that mediates E2’s anti-obesity effect in both sexes [78, 182]. ERα is the dominant estrogen receptor in the arcuate nucleus of the hypothalamus where POMC neurons are located while levels of ERβ are negligible [165]. When ERα is knocked out, it causes obesity and infertility in both males and females [87, 88]. While ERα-null animals develop obesity, ERβ-null mice are lean. In addition, levels of circulating E2 in obese ERα-null animals are 10 times higher than those in their wild-type littermates [43], revealing estrogen resistance in these mice, and additional administration of E2 in these mice does not reduce either their food intake or their body weight [78]. Finally, estrogen administration to ERα-null mutants does not alter estrogen-induced synaptic plasticity onto the POMC perikarya [76]. Thus, it is reasonable to suggest that estradiol’s effect on feeding, metabolism, synaptic plasticity and fertility is mediated by the α-type estrogen receptor and potentially involves STAT3.
3.4 Thyroid Hormones
Thyroid hormone regulation of melanocortin neurons involves the activity of the thyroid hormone activating enzyme, 5′ deiodinase type II (DII) in glial cells of the hypothalamus. A differential expression of DII during fasting seems to contribute to the activation of NPY/AgRP and inhibition of α-MSH neurons. During negative energy balance such as fasting, DII mRNA and activity levels are elevated in the arcuate nucleus-median eminence region [51] and this elevation in DII production and activity seems to be regulated by the inverse shift in circulating levels of leptin and corticosterone [41]. Thus, the local hyperthyroidism [41, 42] caused by DII activation could then affect and alter neuropeptide expression in leptin-responsive arcuate neurons.
The signaling modality by which T3 affects arcuate neurons seems to involve a thyroid hormone regulated mitochondrial protein, uncoupling protein 2 (UCP2; Fig. 1) [42, 121]. UCP2, a protein belonging to a family of mitochondrial anion carrier, is located in the inner membrane of the mitochondria and its primary function is thought to leak hydrogen protons from the intermembrane space to the matrix of the mitochondria [16, 72]. Through this process, UCP2 may affect ATP and superoxide production, and decrease the entry of calcium to the mitochondrial matrix [157].
In the brain, UCP2 is expressed predominantly in neuronal populations of brain stem and hypothalamus involved in the central regulation of autonomic, endocrine, and metabolic processes in both rodents and primates. [53, 96, 179].
The involvement of UCP2 in central metabolic pathways is supported by the finding of an abundant expression of UCP2 in NPY/AgRP [42, 96] and POMC neurons [167]. Thus, thyroid hormone regulation of melanocortin neurons seems to act via a mitochondrial mechanism that by affecting ATP levels and free radical formation will then increase the activity of NPY/AgRP neurons and decrease the activity of POMC cells [3, 54].
3.5 Glucocorticoids
Glucocorticoids play a key function in metabolism regulation by regulating hypothalamic POMC. Many of the genetic models of obesity are characterized by elevated corticosterone levels. In addition, corticosterone withdrawal such as after adrenalectomy (ADX) reduces food intake, fat stores and body weight gain [for review see 47]. It has been shown that ADX induced an increase of POMC mRNA and Fos expression in α-MSH-immunoreactive neurons in rats [183]. In addition, ADX is able to reverse the obese phenotype and restore hypothalamic melanocortin tone in leptin-deficient ob/ob mice [133]. ADX has been shown to alter the sensitivity of the central melanocortin system to the effects of AgRP, the melanocortin antagonist [57]. This effect is restored by corticosterone replacement [57]. Glucocorticoids could also have a direct effect on POMC neurons since glucocorticoid receptors are expressed in these neurons and glucocorticoid response elements (GRE) have been identified in the POMC promoter [36, 58]. Recent data [82] suggest that the known orexigenic action of corticosteroids is mediated, at least in part, by synaptic changes and altered excitability of the melanocortin system.
3.6 Circulating nutrients
Nutritional signals have been known to regulate energy balance by hypothalamic neurons sensitive to the metabolic status [145]. Circulating nutrients such as glucose, amino acids and fatty acids, affect POMC neurons. Several metabolic fuel sensors have been shown to be activated in POMC neurons according to the metabolic state of the organism. A considerable amount of work has been devoted to decipher the mechanism by which “nutrient sensing” occurs in the regulation of whole body energy metabolism. A large amount of data has accumulated to suggest that specific mechanisms exist for the “sensing” of the levels of various circulating nutrients, such as carbohydrates (glucose), fatty- and amino acids, by the brain. These sensors include mammalian target of rapamycin (mTOR) kinase, Sirtuin1 (Sirt 1), AMP-activated protein kinase (AMPK) and glucokinase. In addition to PI3K cascade regulation, FoxO1 has been recently shown to be regulated by a NAD+ -dependent deacetylase, Sirt1 [23] (Fig. 1). Sirt1 is expressed in POMC neurons and its activity levels are increased by fasting [177]. Cakir and collaborators recently found that the fasting-induced decrease of FoxO1 acetylation and POMC expression are Sirt1-dependent [23]. Furthermore, they also showed that Sirt1 regulates S6 Kinase signaling which has been previously shown to be under the control of mTOR kinase activity [86, 220, 221]. mTOR kinase activity, which is inversely altered by changes in the AMP/ATP ratio, leads to the phosphorylation of its downstream targets including S6K1, S6 ribosomal protein (S6), and eukaryotic initiation factor 4E–binding protein (4E-BP1) [86, 220, 221]. The chronic activation of mTOR specifically in POMC neurons has been found to induce a defect in the development of POMC projections in a cell-autonomous manner as well as an increase in the size of the soma [146]. As mTOR signaling is critical for protein synthesis in general, it is reasonable to assume that alteration in mTOR activity will affect many neuronal parameters such as membrane potential and synaptic events. Although Sirt1 and mTOR seem to regulate POMC neurons, it remains to be established whether they act independently or whether one is upstream to the other. Furthermore, recent data have reported that in peripheral tissues such as muscle, Sirt 1 is regulated by AMPK [26] the activity of which changes inversely to that of mTOR and according to changes in the AMP/ATP ratio. As for Sirt 1, fasting induces an increase of AMPK by increasing its phosphorylation on Thr172 within its catalytic α subunit. The activation of AMPK in the hypothalamus has been shown to be involved in the fatty acid metabolic pathway [120]. Another sensing mechanism specific for glucose that was demonstrated first in pancreatic beta cells [211] and then in hypothalamic neurons [60, 229] is mediated by glucokinase. However, the mechanism by which glucokinase regulates glucose-sensing appears to be independent of ATP as shown by several studies [71, 228].
Any of the aforementioned mechanisms, in turn, must trigger an orchestrated and predictable alteration of the activity of the POMC neuronal circuits that alter behavior as well as autonomic and endocrine functions so that adaption to the changing metabolic milieu can occur. Thus, when one considers fuel sensing, it is from the perspective of whole body responses to circulating carbohydrates, fatty acids or amino acids. The idea that there is a “sensor” of these fuels on neurons of the hypothalamus independent of fuel metabolism has never been proven to exist. In contrast, the utilization of specific fuels by hypothalamic neurons in support of the whole body metabolism of that class of nutrient as a way of “fuel sensing” is more in line with available data. For example, glucose is considered the main driver of POMC neuronal firing [154, 157, 158]. POMC neurons increase their firing when glucose levels rise and this, in turn, affects hepatic glucose homeostasis and insulin action [99, 154, 157, 158]. This is entirely consistent with the putative role of POMC cells as satiety signaling neurons, since at satiety, circulating glucose levels are elevated and POMC neurons exhibit increased firing [159]. In turn, the increase in glucose oxidation induces a rise of free radicals which seems to play an important role as a driving force of POMC neuronal firing [3]. Conversely, during negative energy balance when glucose levels are lower and circulating fatty acids increase, POMC neurons are silent while NPY/AgRP neurons show increased firing [3]. The intracellular mechanism of fatty acid oxidation occurs without an elevation of ROS levels [3].
4. POMC processing in the hypothalamus
The POMC-derived neuropeptide, α-MSH, is generated by extensive posttranslational processes that involve several enzymes such as the prohormone convertases 1 and 2 (PC1 and PC2), carboxypeptidase E (CPE) and peptidyl a-amidating monooxygenase (PAM). PC1 is responsible for the first step in which the 32 kDa preprotein is cleaved to form proACTH and β-lipotrophin (β-LPH) (Figure 2). While β-LPH will be further processed to form β-MSH and β-endorphin, ProACTH will be further cleaved by PC1 to generate ACTH 1-39. PC2 will then cleave ACTH 1-39 to generate ACTH1–17 [for review see 173]. The generation of mature α-MSH from ACTH1–17 is catalyzed by carboxypeptidase E (CPE), peptidyl α-amidating mono-oxygenase (PAM) and a n-acetyltransferase. First, carboxy-terminal basic amino acids are trimmed from ACTH1–17 by CPE (Fig. 2). The peptide is subsequently amidated by PAM to generate desacetyl α-MSH (Des α-MSH). This is converted to mature α-MSH by a n-acetyltransferase that has not yet been identified. CPE and PAM are also believed to have additional functions in peptide processing. CPE may play a role in targeting prohormones into secretory granules [36] while PAM may interact with components of the actin cytoskeleton and thus may influence the trafficking of secretory granules [35]. Thus, the generation of α-MSH in the hypothalamus requires a complex pathway of posttranslational modification, involving numerous enzymes. It is therefore possible that alterations in any step of the POMC posttranslational modifications may have effects on energy metabolism. Impaired POMC processing has indeed been reported to be responsible for mouse models of obesity [154] and human obesity [104]. For example, the Cpe fat mouse displays a recessive obesity syndrome which is associated with high circulating levels of prohormones [154] due to the inactivation of carboxypeptidase E. In humans, an obesity phenotype has been reported which is associated with mutations in the human prohormone convertase 1 gene [104] and increased levels of unprocessed POMC. Studies on the regulation of these enzymes have shown for example that leptin regulates PC2 in vivo [178]. In addition, PC1 and PC2 were found to be differentially regulated in the brain of Cpe fat mice [9] and in vitro data were provided suggesting that the biosynthesis and expression of PC1 and PC2 are regulated by leptin [158]. Furthermore, additional studies have reported that PC1 and PC2 are also regulated by other metabolic hormones such as glucocorticoids and thyroid hormones [56, 123, 124].
Figure 2.
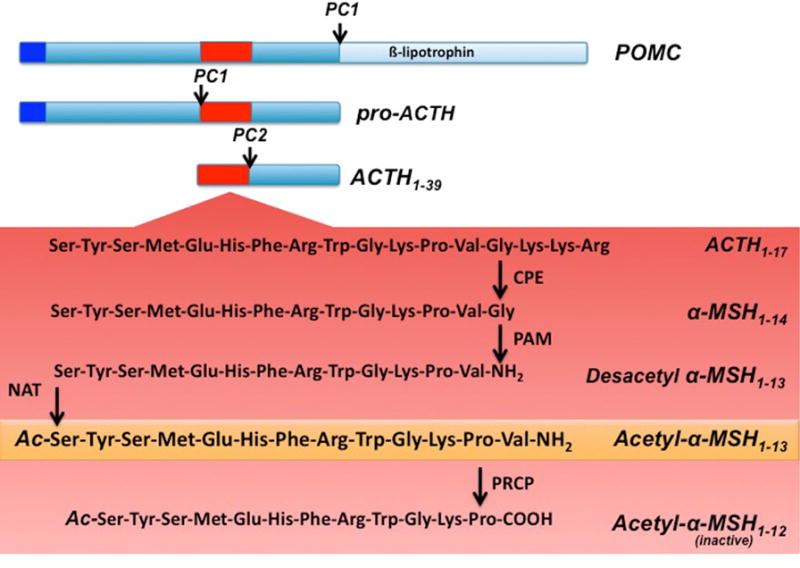
Schematic illustration of hypothalamic POMC processing. Prohormone convertase 1 (PC1) cleaves the 32 kDa preprotein into proACTH and β-lipotrophin . ProACTH is then further converted by PC1 to form ACTH1–39. Prohormone convertase 2 (PC2) will then cleave ACTH1–39, to form ACTH1–17. ACTH1–17 will be first processed by carboxypeptidase E (CPE) and then α-amidating mono-oxygenase (PAM) to form desacetyl α-MSH1–13. An n-actelyltransferase (NAT) will then convert desacetyl-α-MSH1–13 into the more active form acetyl-α-MSH1–13. Finally acetyl- α-MSH1–13 will be degraded by prolyl carboxypeptidase (PRCP) into the inactive product acetyl- α-MSH1–12.
Thus, impaired processing of POMC in the central nervous system by altering the melanocortin signaling, may ultimately result in obesity.
Despite the well described anorectic function of α-MSH [101, 186], peripheral administration of α-MSH has not been reported to reduce food intake. In fact, Des-α -MSH and Act-α -MSH have been shown to be potent anorexigenic peptides only when administered centrally. Additionally, Act-α-MSH seems to have a greater ability to decrease food intake than Des-α-MSH probably due to its greater stability. Nevertheless, these data strongly suggest that both Act-α-MSH and Des-α-MSH are rapidly degraded. Furthermore, the degradation of α-MSH is independent of the presence of MC4R, although it has been reported that MC4R undergoes an internalization process that is dependent on its binding to α-MSH [195].
5. Prolyl carboxypeptidase
New data have recently suggested that prolyl carboxypeptidase (PRCP) is the enzyme responsible for the degradation of α-MSH [216]. Evidence was provided that hypothalamic PRCP is in an anatomical position to determine the efficacy of released α-MSH, thereby controlling the output and thus the efficacy of the melanocortin system (Fig. 3). In support of this idea, mice lacking PRCP display a lean phenotype when exposed to either a regular diet or high fat diet. Furthermore, in support of its function, PRCP has been previously associated with metabolic syndrome in human males [135].
Figure 3.
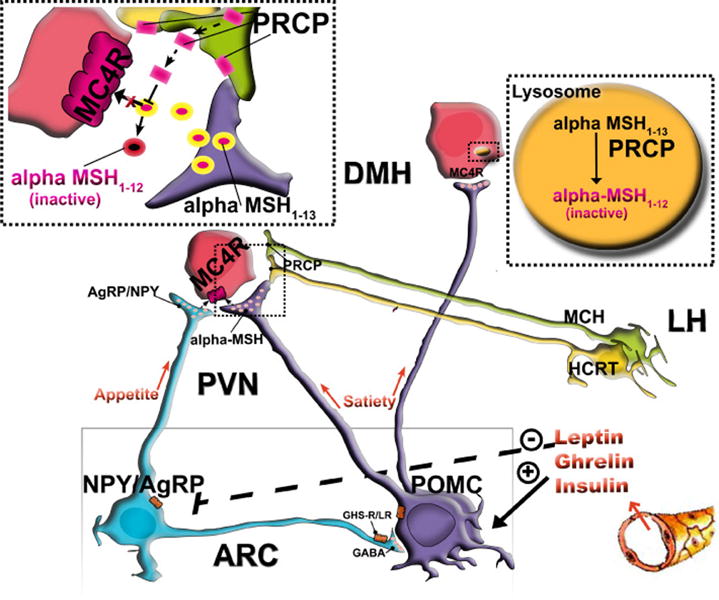
PRCP is in an anatomical position to determine the efficacy of released α-MSH, thus controlling the output of the melanocortin system. PRCP is mainly expressed in the lateral hypothalamus (LH) in hypocretin/orexin (Hcrt) and melanin-concentrating hormone (MCH) neurons and in the hypothalamic dorsomedial nucleus (DMH) where MC4R-expressing neurons are located. The Hcrt and MCH neurons project to various areas of the hypothalamus, such as the paraventricular nucleus, where α-MSH terminals strongly innervate MC4R-expressing neurons. When released from the Hcrt and/or MCH terminals, PRCP will degrade α-MSH extracellularly, thus increasing the antagonistic effect of AgRP. In the DMH, however, PRCP degrades α-MSH intracellularly, thereby enhancing the overall orexigenic tone of the system.
5.1 Enzymatic properties
Prolyl carboxypeptidase (PRCP) is an enzyme of the family of the carboxypeptidase (CPs) which contains a serine residue in the active center, essential for catalytic activity that reacts with organophosphorus inhibitors. PRCP cleaves only peptides with a penultimate proline residue.
PRCP was first discovered in 1968 during studies of bradykinin metabolism in kidney [225]. It was found that after removal of the C-terminal Arg of bradykinin, the Pro-Phe-OH bond, which is usually resistant to carboxypeptidases, was cleaved by a kidney extract [225]. Since angiotensin II has the same C-terminus, it was tested and found to be a substrate of the enzyme. The enzyme was initially named angiotensinase C but later it was shown that the enzyme cleaves a variety of Pro-X bonds, thus, it was renamed PRCP.
PRCP is a single chain protein of approximately 58 KDa [225, 226]. The cDNA for human PRCP was cloned from a human kidney library [206] and based on that, the molecular weight of the mature protein should be around 51 kDa, indicating the presence of about 12% carbohydrate by weight [206]. This is consistent with the existence of six potential N-linked glycosylation sites. The deduced protein sequence contains 496 amino acids and includes a 30 residue signal peptide and a 15 amino acid propeptide [206]. The C-terminal half of the sequence contains an interesting “serine repeat” in which Ser is repeated as the 26th residue 6 out of 9 times [206]. In the other three repeats, Ser is replaced by Thr, Gly or Glu. When the Ser residues are aligned to yield 26 residue repeats, the positions of some of the other residues also fall into a pattern. The significance of this repeat region is not clear, but it might be involved in the maintenance of a secondary or tertiary structural motif or in the formation of the homodimer. The gene encoding human PRCP is on chromosome 11, while in the mouse it is on chromosome 7.
PRCP is inhibited by compounds that react with the active site serine residue. However, this action is not true for all serine protease inhibitors, such as aprotinin, or by chelating agents or inhibitors that react specifically with the active site cysteine in sulfhydryl proteases. Particularly, PRCP is not inhibited by non-specific SH-reactive agents, but is relatively sensitive to the prolylendopeptidase inhibitor, Cbz-Pro-prolinal, because of its specificity for peptides with penultimate proline residues [161, 206].
5.2 Function
The functions of PRCP in regulating peptide actions in vivo have not been explored in detail. As a carboxypeptidase, PRCP catalyzes only the hydrolysis of the C terminal peptide bond, releasing one amino acid at a time. Although the cleavage of one amino acid from the C-terminus of a peptide or protein could appear to be of limited importance, it often results in profoundly altered physiological activity of such molecules [199, 216].
Specifically, PRCP cleaves peptides only if the penultimate residue is proline. Of the short synthetic substrates, Cbz-Pro-Ala is hydrolyzed fastest [161]. The enzyme does not cleave the Pro-Pro-COOH or (OH)-Pro-Pro-COOH bond [161]. Because the enzyme retains significant activity at neutral pH with naturally occurring substrates, such as angiotensin [102, 161], PRCP may also be active extracellularly.
The known substrates for PRCP are angiotensin II and III, prekallikrein [225, 226] and most recently α-MSH [216].
PRCP has an acidic pH optima (=5.0) when hydrolyzing short synthetic peptide substrates [102, 161, 206], but it has been found that it retains significant activity at neutral pH range. Indeed, at physiological pH, α-MSH has been shown to be a better substrate than the well studied angiotensin II [216]. The enzymatic degradation of α-MSH 1–13 by PRCP results in the formation of α-MSH 1–12. When centrally administrated, α-MSH 1–12, was found to be inactive in regulating food intake and also unable to induce increases in the frequency of action potentials of MC4R neurons of the hypothalamic paraventricular nucleus [216]. In addition, central and peripheral inhibition of PRCP enzymatic activity by inhibitors showed a significant reduction in food intake [216]. In addition, PRCP ablated (PRCPgt/gt) mice are more sensitive to peripheral administration of α-MSH compared to wild type controls and, PRCPgt/gt mice are leaner than controls on a regular and high-fat diet [216].
Besides α-MSH, PRCP could also regulate other peptides with a proline as the penultimate amino acid, the function of which is associated with body weight regulation. A few other hypothalamic peptides have a penultime proline and they include Neuropeptide-glutamic acid-isoleucine (NEI; P56942; EIGDEENSAKFPI), Apelin 13 (Q9R0R4; QRPRLSHKGPMPF) and ghrelin (Q9EQX0; GSSFLSPEHQKAQQRKESKKPPAKLQPR).
NEI, a peptide originating from the pro-MCH 131–164 region, has been found to be stronger than MCH in stimulating rat feeding behavior [134]. Thus, its effect on food intake would not be in line with the phenotype that was observed in the PRCP-ablated mouse. The same conclusion is true for ghrelin, the most potent circulating orexigenic hormone.
However, conflicting data have been reported on Apelin 13 [15, 164, 204, 207, 215]. Thus, future studies are needed to test whether Apelin 13 is a substrate for PRCP.
Finally, one cannot exclude the possibility that other peptides with a proline as the penultimate amino acid, the function of which is still unknown or not shown to be related to metabolism regulation, may be the cause of the PRCP-ablated mouse phenotype.
5.3 Localization
PRCP activity has been detected in a variety of cells and organs [102, 119, 198]. Within the cells, PRCP activity has been found in lysosomes and the cellular membrane [119, 131, 198–200]. PRCP has also been found to be released in response to stimulation and appears in the extracellular medium or biological fluids [141, 225]. In addition, lysosomal enzymes can associate with plasma membranes after exocytosis, bound to other transmembrane or membrane-associated proteins [200]. In addition, this serine carboxypeptidase can block part of the signal transduction stimulated by peptides. PRCP cleaves peptides after ligand-mediated receptor endocytosis [67, 150]. This usually involves fusion of the endosomes with lysosomes and results in peptide degradation and receptor recycling [226]. This process could be especially important in the metabolism of peptides that otherwise have a long-lasting effect.
PRCP has been found to be widely distributed, and its mRNA expression is highest in human placenta, lung and liver [206]. PRCP is also present in white blood cells and fibroblasts, and is highly expressed in endothelial cells [119, 200]. In mice, like humans, PRCP is expressed in peripheral tissues such as kidney, liver, heart and spleen. Centrally, PRCP is expressed in the hypothalamus and brainstem. Within the hypothalamus, higher expression of PRCP was detected in the perifornical-lateral hypothalamic region and dorsomedial nucleus [216]. This anatomical data suggest that hypothalamic PRCP is in an ideal position to determine the efficacy of released α-MSH, thereby controlling the output of the melanocortin system. PRCP is mainly expressed in the DMH where MC4R-expressing neurons are localized and in the lateral hypothalamic hypocretin/orexin [Hcrt] and melanin-concentrating hormone (MCH) neurons. A relatively small number of MC4-R-expressing neurons have been found in the LH, including the perifornical area [127]. However, Hcrt and MCH neurons project to various areas of the hypothalamus, including the PVN, where α-MSH terminals strongly innervate MC4R-expressing neurons.
Thus, PRCP could be released in the PVN from the Hcrt and MCH axon terminals into the synaptic space where it would then degrade α-MSH extracellularly (Fig. 3). However, high expression of MC4-R-containing neurons has been shown in the hypothalamic DMH [127] where abundant expression of PRCP was detected and where arcuate α -MSH–containing fibers project [197]. Thus, in the DMH, PRCP could be involved in the intracellular peptide degradation and receptor recycling (Figure 3). In conclusion, PRCP may act in different hypothalamic sites to modulate melanocortin efficacy. Once released from the Hcrt and/or MCH terminals in the PVN, PRCP will degrade α -MSH 1–13 extracellularly (Fig. 3) thus increasing the antagonist effect of AgRP, while in the DMH, PRCP will degrade α-MSH intracellularly thus shortening the effect of α -MSH on MC4R and enhancing the overall orexigenic tone of the melanocortin system (Fig. 3).
5.4 A potential role for PRCP in leptin resistance
A postulated cause for the development of obesity is leptin resistance. As fat stores increase during the development of obesity, circulating leptin levels are continuously rising. However, these elevated leptin levels or administration of exogenous leptin to obese subjects does not lead to decreased body weight. This discrepancy between leptin levels and the metabolic phenotype is attributed to leptin resistance [152]. A primary site of leptin resistance is suggested to be the POMC neurons [152]. Indeed, intracellular signaling cascades triggered by leptin receptor activation were found to be impaired in diet-induced obese animals [12, 151]. Specifically, activation of the transcription factor STAT3 by leptin is diminished in these animals [12, 151], an event that is attributed to the inhibitory action of SOCS3 on STAT3 phosphorylation [12]. Additionally, ex vivo studies revealed that leptin induced secretion of α-MSH1–13 is reduced from hypothalamic slices taken from diet-induced obese mice [66]. An alternative explanation for the development of leptin resistance was the decreased ability of leptin from the circulation to reach hypothalamic neurons [27]). Despite of all these elegant and logical data, several parameters of POMC neurons in diet induced obesity show alterations completely consistent with elevated leptin levels. For example, the wiring of the melanocortin system and transcript levels of POMC and AgRP/NPY of obese animals in response to a hypercaloric environment reflects an adequate and possibly maximal responsiveness of these cells to leptin [100]. The synaptic organization triggered in these animals shows similar trends of changes to those that were described previously for highly leptin sensitive ob/ob mice in response to leptin administration [170]. Although reactive gliosis was also detected in response to high fat diet specifically associated with arcuate nucleus capillaries [100], this gliosis does not appear to block the effect of leptin on the synaptic input organization of the POMC neurons and POMC, NPY and AgRP transcript levels, and hence, may not be the cause of leptin resistance as proposed earlier [27]. On the other hand, it is conceivable that assays using hypothalamic slices to measure α -MSH release [66] may be skewed by gliosis, an event that could be a structural obstacle for the release of measurable α -MSH into the medium, thereby limiting the usefulness of these assays for the study of α -MSH release kinetics. Thus, if neurobiological attributes of leptin action are present in the hypothalamus of diet-induced obese animals, it is a reasonable suggestion that the degradation of released α -MSH1–13 may interfere with the ability of MC4R activation in POMC target sites. As described above, PRCP location and function in the hypothalamus makes this carboxypeptidase, although previously unsuspected, a player in the emergence of systemic leptin resistance as it relates to the regulation of food intake, energy expenditure and glucose homeostasis.
Concluding remarks
Many hormonal and nutritional signals have been shown to directly affect hypothalamic melanocortin signaling by either activating or inhibiting transcriptional, translational and post-translational POMC processes. In addition, many of the signaling pathways activated by these circulating signals have been found to act via common denominators. Most recently, a new component of the melanocortin signaling, PRCP, has been revealed.
Although it was discovered over 40 years ago, limited studies on PRCP function and regulation have been performed. Some functions are related to its specificity to cleave after proline residues, restricting its substrates to fewer peptides. In the hypothalamus, α-MSH has been recently shown to be a substrate for PRCP [216]. Thus, due to its involvement in α-MSH degradation, it is possible that PRCP is an important component of the melanocortin signaling downstream of the melanocortin neurons thereby determining the efficacy of the melanocortin signaling. Thus, an increase in the activity of PRCP as a result of a decrease in α-MSH levels in the hypothalamus, may promote metabolic dysfunctions, including obesity and type 2 diabetes. Therefore, future studies on the role, function and regulation of PRCP under different metabolic conditions are needed and may reveal it to be a potential drug target for such metabolic disorders.
The central melanocortin system plays a critical role in the regulation of metabolism.
α-melanocyte stimulating hormone (α-MSH) is the most potent anorectic peptide.
Many circulating signals regulate α-MSH production and activity.
The enzymatic inactivation of α-MSH plays an important role in metabolism
Acknowledgments
This work was supported by NIDDK/NIH (Grant no. R01 DK084065).
The author thanks Giuseppe D’Agostino, Claudia Horvath-Diano and Marya Shanabrough for their assistance in the preparation of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure statement
The author has nothing to disclose.
References
- 1.Ainscow EK, et al. Dynamic imaging of free cytosolic ATP concentration during fuel sensing by rat hypothalamic neurones: evidence for ATP-independent control of ATP-sensitive K(+) channels. J Physiol. 2002;544:429–45. doi: 10.1113/jphysiol.2002.022434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ainslie DA, Morris MJ, Wittert G, Turnbull H, Proietto J, Thorburn AW. Estrogen deficiency causes central leptin insensitivity and increased hypothalamic neuropeptide Y. Int J Obes Relat Metab Disord. 2001;25:1680–8. doi: 10.1038/sj.ijo.0801806. [DOI] [PubMed] [Google Scholar]
- 3.Andrews ZB, Liu ZW, Walllingford N, Erion DM, Borok E, Friedman JM, Tschöp MH, Shanabrough M, Cline G, Shulman GI, Coppola A, Gao XB, Horvath TL, Diano S. Uncoupling protein-2 mediates ghrelin’s action on NPY/AgRP neurons. Nature. 2008;454:846–51. doi: 10.1038/nature07181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bady I, Marty N, Dallaporta M, Emery M, Gyger J, Tarussio D, Foretz M, Thorens B. Evidence from glut2-null mice that glucose is a critical physiological regulator of feeding. Diabetes. 2006;55:988–95. doi: 10.2337/diabetes.55.04.06.db05-1386. [DOI] [PubMed] [Google Scholar]
- 5.Bagnasco M, Dube MG, Kalra PS, Kalra SP. Evidence for the existence of distinct central appetite, energy expenditure, and ghrelin stimulation pathways as revealed by hypothalamic site-specific leptin gene therapy. Endocrinology. 2002;143:4409–4421. doi: 10.1210/en.2002-220505. [DOI] [PubMed] [Google Scholar]
- 6.Balthasar N, Dalgaard LT, Lee CE, Yu J, Funahashi H, Williams T, Ferreira M, Tang V, McGovern RA, Kenny CD, Christiansen LM, Edelstein E, Choi B, Boss O, Aschkenasi C, Zhang CY, Mountjoy K, Kishi T, Elmquist JK, Lowell BB. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell. 2005;123:493–505. doi: 10.1016/j.cell.2005.08.035. [DOI] [PubMed] [Google Scholar]
- 7.Banno R, Zimmer D, De Jonghe BC, Atienza M, Rak K, Yang W, Bence KK. PTP1B and SHP2 in POMC neurons reciprocally regulate energy balance in mice. J Clin Invest. 2010;120:720–34. doi: 10.1172/JCI39620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bergendahl M, Wiemann JN, Clifton DK, Huhtaniemi I, Steiner RA. Short-term starvation decreases POMC mRNA but does not alter GnRH mRNA in the brain of adult male rats. Neuroendocrinology. 1992;56:913–20. doi: 10.1159/000126324. [DOI] [PubMed] [Google Scholar]
- 9.Berman Y, Mzhavia N, Polonskaia A, Devi LA. Impaired prohormone convertases in Cpe(fat)/Cpe(fat) mice. J Biol Chem. 2001;276:1466–73. doi: 10.1074/jbc.M008499200. [DOI] [PubMed] [Google Scholar]
- 10.Bernardis LL, Bellinger LL. The lateral hypothalamic area revisited: ingestive behavior. Neurosci Biobehav Rev. 1996;20:189–287. doi: 10.1016/0149-7634(95)00015-1. [DOI] [PubMed] [Google Scholar]
- 11.Beuckmann CT, Yanagisawa M. Orexins: from neuropeptides to energy homeostasis and sleep/wake regulation. J Mol Med. 2002;80:329–342. doi: 10.1007/s00109-002-0322-x. [DOI] [PubMed] [Google Scholar]
- 12.Bjorbak C, Lavery HJ, Bates SH, Olson RK, Davis SM, Flier JS, Myers MG., Jr SOCS3 mediates feedback inhibition of the leptin receptor via Tyr985. J Biol Chem. 2000;275:40649–57. doi: 10.1074/jbc.M007577200. [DOI] [PubMed] [Google Scholar]
- 13.Björnström L, Sjöberg M. Signal transducers and activators of transcription as downstream targets of nongenomic estrogen receptor actions. Mol Endocrinol. 2002;16:2202–14. doi: 10.1210/me.2002-0072. [DOI] [PubMed] [Google Scholar]
- 14.Bouali SM, Fournier A, St-Pierre S, Jolicoeur FB. Effects of NPY and NPY2-36 on body temperature and food intake following administration into hypothalamic nuclei. Brain Res Bull. 1995;36:131–135. doi: 10.1016/0361-9230(94)00177-3. [DOI] [PubMed] [Google Scholar]
- 15.Boucher J, Masri B, Daviaud D, Gesta S, Guigné C, Mazzucotelli A, Castan-Laurell I, Tack I, Knibiehler B, Carpéné C, Audigier Y, Saulnier-Blache JS, Valet P. Apelin, a newly identified adipokine up-regulated by insulin and obesity. Endocrinology. 2005;146:1764–71. doi: 10.1210/en.2004-1427. [DOI] [PubMed] [Google Scholar]
- 16.Bouillaud F, Ricquier D, Thibault J, Weissenbach J. Molecular approach to thermogenesis in brown adipose tissue: cDNA cloning of the mitochondrial uncoupling protein. Proc Natl Acad Sci U S A. 1985;82:445–8. doi: 10.1073/pnas.82.2.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brobeck JR, Tepperman J, Long CNH. Experimental hypothalamic hyperphagia in the albino rat. Yale Journal of Biology and Medicine. 1943;15:831–853. [PMC free article] [PubMed] [Google Scholar]
- 18.Brobeck JR. Mechanism of the development of obesity in animals with hypothalamic lesions. Physiol Rev. 1946;26:541–559. doi: 10.1152/physrev.1946.26.4.541. [DOI] [PubMed] [Google Scholar]
- 19.Broberger C, De Lecea L, Sutcliffe JG, Hokfelt T. Hypocretin/orexin- and melanin-concentrating hormone-expressing cells form distinct populations in the rodent lateral hypothalamus: relationship to the neuropeptide Y and agouti gene-related protein systems. J Comp Neurol. 1998;402:460–74. [PubMed] [Google Scholar]
- 20.Brüning JC, Gautam D, Burks DJ, Gillette J, Schubert M, Orban PC, Klein R, Krone W, Müller-Wieland D, Kahn CR. Role of brain insulin receptor in control of body weight and reproduction. Science. 2000;289:2122–5. doi: 10.1126/science.289.5487.2122. [DOI] [PubMed] [Google Scholar]
- 21.Butler AA, Kesterson RA, Khong K, Cullen MJ, Pelleymounter MA, Dekoning J, Baetscher M, Cone RD. A unique metabolic syndrome causes obesity in the melanocortin-3 receptor-deficient mouse. Endocrinology. 2000;141:3518–3521. doi: 10.1210/endo.141.9.7791. [DOI] [PubMed] [Google Scholar]
- 22.Cai XJ, Widdowson PS, Harrold J, Wilson S, Buckingham RE, Arch JR, Tadayyon M, Clapham JC, Wilding J, Williams G. Hypothalamic orexin expression: modulation by blood glucose and feeding. Diabetes. 1999;48:2132–2137. doi: 10.2337/diabetes.48.11.2132. [DOI] [PubMed] [Google Scholar]
- 23.Cakir I, Perello M, Lansari O, Messier NJ, Vaslet CA, Nillni EA. Hypothalamic Sirt1 regulates food intake in a rodent model system. PLoS One. 2009;4:e8322. doi: 10.1371/journal.pone.0008322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Campfield LA, Smith FJ, Guisez Y, Devos R, Burn P. Recombinant mouse OB protein: evidence for a peripheral signal linking adiposity and central neural networks. Science. 1995;269:546–549. doi: 10.1126/science.7624778. [DOI] [PubMed] [Google Scholar]
- 25.Canesi L, Ciacci C, Betti M, Lorusso LC, Marchi B, Burattini S, Falcieri E, Gallo G. Rapid effects of 17beta-estradiol on cell signaling and function of Mytilus hemocytes. Gen Comp Endocrinol. 2004;136:58–71. doi: 10.1016/j.ygcen.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 26.Cantó C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–60. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caro JF, Kolaczynski JW, Nyce MR, Ohannesian JP, Opentanova I, Goldman WH, Lynn RB, Zhang PL, Sinha MK, Considine RV. Decreased cerebrospinal-fluid/serum leptin rationin obesity: a possible mechanism for leptin resistance. Lancet. 1996;348:159–161. doi: 10.1016/s0140-6736(96)03173-x. [DOI] [PubMed] [Google Scholar]
- 28.Challis BG, Coll AP, Yeo GS, Pinnock SB, Dickson SL, Thresher RR, Dixon J, Zahn D, Rochford JJ, White A, Oliver RL, Millington G, Aparicio SA, Colledge WH, Russ AP, Carlton MB, O’Rahilly S. Mice lacking pro-opiomelanocortin are sensitive to high-fat feeding but respond normally to the acute anorectic effects of peptide-YY(3-36) Proc Natl Acad Sci U S A. 2004;101:4695–700. doi: 10.1073/pnas.0306931101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen AS, Marsh DJ, Trumbauer ME, Frazier EG, Guan XM, Yu H, Rosenblum CI, Vongs A, Feng Y, Cao L, et al. Inactivation of the mouse melanocortin-3 receptor results in increased fat mass and reduced lean body mass. Nature Genetics. 2000;26:97–102. doi: 10.1038/79254. [DOI] [PubMed] [Google Scholar]
- 30.Chen W, Kelly MA, Opitz-Araya X, Thomas RE, Low MJ, Cone RD. Exocrine gland dysfunction in MC5-R-deficient mice: evidence for coordinated regulation of exocrine function by melanocortin peptides. Cell. 1997;91:789–798. doi: 10.1016/s0092-8674(00)80467-5. [DOI] [PubMed] [Google Scholar]
- 31.Chiesi M, Huppertz C, Hofbauer KG. Pharmacotherapy of obesity: targets and perspectives. Trends in Pharmacological Sciences. 2001;22:247–254. doi: 10.1016/s0165-6147(00)01664-3. [DOI] [PubMed] [Google Scholar]
- 32.Cheung CC, Clifton DK, Steiner RA. Proopiomelanocortin neurons are direct targets for leptin in the hypothalamus. Endocrinology. 1997;138:4489–4492. doi: 10.1210/endo.138.10.5570. [DOI] [PubMed] [Google Scholar]
- 33.Chou TC, Rotman SR, Saper CB. Lateral hypothalamic acetylcholinesterase-immunoreactive neurons co-express either orexin or melanin concentrating hormone. Neurosci Lett. 2004;370:123–6. doi: 10.1016/j.neulet.2004.08.070. [DOI] [PubMed] [Google Scholar]
- 34.Ciana P, Ghisletti S, Mussi P, Eberini I, Vegeto E, Maggi A. Estrogen receptor alpha, a molecular switch converting transforming growth factor-alpha-mediated proliferation into differentiation in neuroblastoma cells. J Biol Chem. 2003;278:31737–44. doi: 10.1074/jbc.M301525200. [DOI] [PubMed] [Google Scholar]
- 35.Ciccotosto GD, Schiller MR, Eipper BA, Mains RE. Induction of integral membrane PAM expression in AtT-20 cells alters the storage and trafficking of POMC and PC1. J Cell Biol. 1999;144:459–71. doi: 10.1083/jcb.144.3.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cintra A, Bortolotti F. Presence of strong glucocorticoid receptor immunoreactivity within hypothalamic and hypophyseal cells containing pro-opiomelanocortic peptides. Brain Res. 1992;577:127–33. doi: 10.1016/0006-8993(92)90545-k. [DOI] [PubMed] [Google Scholar]
- 37.Clark AJ, McLoughlin L, Grossman A. Familial glucocorticoid deficiency associated with point mutation in the adrenocorticotropin receptor. Lancet. 1993;341:461–462. doi: 10.1016/0140-6736(93)90208-x. [DOI] [PubMed] [Google Scholar]
- 38.Coll AP, Farooqi IS, Challis BG, Yeo GS, O’Rahilly S. Proopiomelanocortin and energy balance: insights from human and murine genetics. J Clin Endocrinol Metab. 2004;89:2557–62. doi: 10.1210/jc.2004-0428. [DOI] [PubMed] [Google Scholar]
- 39.Cone RD. Anatomy abd regulation of the central melanocortin system. Nature Neuroscience. 2005;8:571–578. doi: 10.1038/nn1455. [DOI] [PubMed] [Google Scholar]
- 40.Cool DR, Normant E, Shen F, Chen HC, Pannell L, Zhang Y, Loh YP. Carboxypeptidase E is a regulated secretory pathway sorting receptor: genetic obliteration leads to endocrine disorders in Cpe(fat) mice. Cell. 1997;88:73–83. doi: 10.1016/s0092-8674(00)81860-7. [DOI] [PubMed] [Google Scholar]
- 41.Coppola A, Meli R, Diano S. Inverse shift in circulating corticosterone and leptin levels elevates hypothalamic deiodinase type 2 in fasted rats. Endocrinology. 2005;146:2827–33. doi: 10.1210/en.2004-1361. [DOI] [PubMed] [Google Scholar]
- 42.Coppola A, Liu ZW, Andrews ZB, Paradis E, Roy MC, Friedman JM, Ricquier D, Richard D, Horvath TL, Gao XB, Diano S. A central thermogenic-like mechanism in feeding regulation: an interplay between arcuate nucleus T3 and UCP2. Cell Metab. 2007;5:21–33. doi: 10.1016/j.cmet.2006.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Couse JF, Curtis SW, Washburn TF, Lindzey J, Golding TS, Lubahn DB, Smithies O, Korach KS. Analysis of transcription and estrogen insensitivity in the female mouse after targeted disruption of the estrogen receptor gene. Mol Endocrinol. 1995;9:1441–54. doi: 10.1210/mend.9.11.8584021. [DOI] [PubMed] [Google Scholar]
- 44.Cowley MA, Pronchuk N, Fan W, Dinulescu DM, Colmers WF, Cone RD. Integration of NPY, AGRP, and melanocortin signals in the hypothalamic paraventricular nucleus: evidence of a cellular basis for the adipostat. Neuron. 1999;24:155–63. doi: 10.1016/s0896-6273(00)80829-6. [DOI] [PubMed] [Google Scholar]
- 45.Cowley MA, Smart JL, Rubinstein M, Cerdar MG, Diano S, Horvath TL, Cone RD, Low MJ. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480–484. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- 46.Csiffary A, Gorcs TJ, Palkovits M. Neuropeptide Y innervation of ACTH-immunoreactive neurons in the arcuate nucleus of rats: a correlated light and electron microscopic double immunolabeling study. Brain Res. 1990;506:215–22. doi: 10.1016/0006-8993(90)91253-d. [DOI] [PubMed] [Google Scholar]
- 47.Dallman MF, la Fleur SE, Pecoraro NC, Gomez F, Houshyar H, Akana SF. Minireview: glucocorticoids–food intake, abdominal obesity, and wealthy nations in 2004. Endocrinology. 2004;145:2633–8. doi: 10.1210/en.2004-0037. [DOI] [PubMed] [Google Scholar]
- 48.Davis AM, Seney ML, Stallings NR, Zhao L, Parker KL, Tobet SA. Loss of steroidogenic factor 1 alters cellular topography in the mouse ventromedial nucleus of the hypothalamus. J Neurobiol. 2004;60:424–36. doi: 10.1002/neu.20030. [DOI] [PubMed] [Google Scholar]
- 49.De Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, Fukuhara C, Battenberg EL, Gautvik VT, Bartlett FS, II, Frankel WN, van den Pol AN, Bloom FE, Gautvik KM, Sutcliff JG. The hypocretins: hypothalamic-specific peptide with neuroexicitatory activity. Proc Natl Acad Sci USA. 1998;95:322–327. doi: 10.1073/pnas.95.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dhillon H, Zigman JM, Ye C, Lee CE, McGovern RA, Tang V, Kenny CD, Christiansen LM, White RD, Edelstein EA, Coppari R, Balthasar N, Cowley MA, Chua S, Jr, Elmquist JK, Lowell BB. Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron. 2006;49:191–203. doi: 10.1016/j.neuron.2005.12.021. [DOI] [PubMed] [Google Scholar]
- 51.Diano S, Naftolin F, Goglia F, Horvath TL. Fasting-induced increase in type II iodothyronine deiodinase activity and messenger ribonucleic acid levels is not reversed by thyroxine in the rat hypothalamus. Endocrinology. 1998;139:2879–84. doi: 10.1210/endo.139.6.6062. [DOI] [PubMed] [Google Scholar]
- 52.Diano S, Naftolin F, Goglia F, Csernus V, Horvath TL. Evidence for a monosynaptic pathway between the arcuate nucleus expressing glial type II iodothyronine 5′ deiodinase mRNA and the median eminence-projective TRH cells of the rat paraventricular nucleus. J Neuroendocrinology. 1998;10:731–742. doi: 10.1046/j.1365-2826.1998.00204.x. [DOI] [PubMed] [Google Scholar]
- 53.Diano S, Urbanski HF, Horvath B, Bechmann I, Kagiya A, Nemeth G, Naftolin F, Warden CH, Horvath TH. Mitochondrial uncoupling protein 2 (UCP2) in the nonhuman primate brain and pituitary. Endocrinology. 2000;141:4226–38. doi: 10.1210/endo.141.11.7740. [DOI] [PubMed] [Google Scholar]
- 54.Diano S, Matthews RT, Patrylo P, Yang L, Beal MF, Barnstable CJ, Horvath TL. Uncoupling protein 2 prevents neuronal death including that occurring during seizure: a mechanism for preconditioning. Endocrinology. 2003;144:5014–21. doi: 10.1210/en.2003-0667. [DOI] [PubMed] [Google Scholar]
- 55.Diano S, Horvath B, Urbanski HF, Sotonyi P, Horvath TL. Fasting activates the nonhuman primate hypocretin (orexin) system and its postsynaptic targets. Endocrinology. 2003;144:3774–3778. doi: 10.1210/en.2003-0274. [DOI] [PubMed] [Google Scholar]
- 56.Dong W, Seidel B, Marcinkiewicz M, Chrétien M, Seidah NG, Day R. Cellular localization of the prohormone convertases in the hypothalamic paraventricular and supraoptic nuclei: selective regulation of PC1 in corticotrophin-releasing hormone parvocellular neurons mediated by glucocorticoids. J Neurosci. 1997;17:563–75. doi: 10.1523/JNEUROSCI.17-02-00563.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Drazen DL, Wortman MD, Schwartz MW, Clegg DJ, van Dijk G, Woods SC, Seeley RJ. Adrenalectomy alters the sensitivity of the central nervous system melanocortin system. Diabetes. 2003;52:2928–34. doi: 10.2337/diabetes.52.12.2928. [DOI] [PubMed] [Google Scholar]
- 58.Drouin J, Maira M, Philips A. Novel mechanism of action for Nur77 and antagonism by glucocorticoids: a convergent mechanism for CRH activation and glucocorticoid repression of POMC gene transcription. J Steroid Biochem Mol Biol. 1998;65:59–63. doi: 10.1016/s0960-0760(97)00180-5. [DOI] [PubMed] [Google Scholar]
- 59.Dubuc PU. Effects of estrogen on food intake, body weight, and temperature of male and female obese mice. Proc Soc Exp Biol Med. 1985;180:468–73. doi: 10.3181/00379727-180-42204. [DOI] [PubMed] [Google Scholar]
- 60.Dunn-Meynell AA, et al. Glucokinase is the likely mediator of glucosensing in both glucose-excited and glucose-inhibited central neurons. Diabetes. 2002;51:2056–65. doi: 10.2337/diabetes.51.7.2056. [DOI] [PubMed] [Google Scholar]
- 61.Elias CF, Saper CB, Maratos-Flier E, Tritos NA, Lee C, Kelly J, Tatro JB, Hoffman GE, Ollmann MM, Barsh GS, et al. Chemically defined projections linking the mediobasal hypothalamus and the lateral hypothalamic area. J Comparative Neurology. 1998;402:442–459. [PubMed] [Google Scholar]
- 62.Elias CF, Aschkenasi C, Lee C, Kelly J, Ahima RS, Bjorbaek C, Flier JS, Saper CB, Elmquist JK. Leptin differentially regulates NPY and POMC neurons projecting to the lateral hypothalamic area. Neuron. 1999;23:775–86. doi: 10.1016/s0896-6273(01)80035-0. [DOI] [PubMed] [Google Scholar]
- 63.Elias CF, Kelly JF, Lee CE, Ahima RS, Drucker DJ, Saper CB, Elmquist JK. Chemical characterization of leptin-activated neurons in the rat brain. J Comp Neurol. 2000;423:261–281. [PubMed] [Google Scholar]
- 64.Elmquist JK, Bjorbaek C, Ahima RS, Flier JS, Saper CB. Distributions of leptin receptor mRNA isoforms in the rat brain. J Comp Neurol. 1998;395:535–547. [PubMed] [Google Scholar]
- 65.Elmquist JK, Elias CF, Saper CB. From lesions to leptin: hypothalamic control of food intake and body weight. Neuron. 1999;22:221–232. doi: 10.1016/s0896-6273(00)81084-3. [DOI] [PubMed] [Google Scholar]
- 66.Enriori PJ, Evans AE, Sinnayah P, Jobst EE, Tonelli-Lemos L, Billes SK, Glavas MM, Grayson BE, Perello M, Nillni EA, Grove KL, Cowley MA. Diet-induced obesity causes severe but reversible leptin resistance in arcuate melanocortin neurons. Cell Metab. 2007;5:181–94. doi: 10.1016/j.cmet.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 67.Erdos EG, Wagner BA, Harbury CB, Painter RG, Skidgel RA, Fa XG. Down-regulation and inactivation of neutral endopeptidase 24.11 (enkephalinase) in human neutrophils. J Biol Chem. 1989;264:14519–14523. [PubMed] [Google Scholar]
- 68.Erickson JC, Clegg KE, Palmiter RD. Sensitivity to leptin and susceptibility to seizures of mice lacking neuropeptide Y. Nature. 1996;381:415–21. doi: 10.1038/381415a0. [DOI] [PubMed] [Google Scholar]
- 69.Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature. 1997;385:165–168. doi: 10.1038/385165a0. [DOI] [PubMed] [Google Scholar]
- 70.Fei H, Okano HJ, Li C, Lee GH, Zhao C, Darnell R, Friedman JM. Anatomic localization of alternatively spliced leptin receptors (Ob-R) in mouse brain and other tissues. Proc Natl Acad Sci U S A. 1997;94:7001–5. doi: 10.1073/pnas.94.13.7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fioramonti X, et al. A new ATP-sensitive K+ channel-independent mechanism is involved in glucose-excited neurons of mouse arcuate nucleus. Diabetes. 2004;53:2767–75. doi: 10.2337/diabetes.53.11.2767. [DOI] [PubMed] [Google Scholar]
- 72.Fleury C, Sanchis D. The mitochondrial uncoupling protein-2: current status. Int J Biochem Cell Biol. 1999;31:1261–78. doi: 10.1016/s1357-2725(99)00049-7. [DOI] [PubMed] [Google Scholar]
- 73.Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature. 1998;395:763–770. doi: 10.1038/27376. [DOI] [PubMed] [Google Scholar]
- 74.Fukuda M, Jones JE, Olson D, Hill J, Lee CE, Gautron L, Choi M, Zigman JM, Lowell BB, Elmquist JK. Monitoring FoxO1 localization in chemically identified neurons. J Neurosci. 2008;28:13640–8. doi: 10.1523/JNEUROSCI.4023-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gao Q, Wolfgang MJ, Neschen S, Morino K, Horvath TL, Shulman GI, Fu XY. Disruption of neural signal transducer and activator of transcription 3 causes obesity, diabetes, infertility, and thermal dysregulation. Proc Natl Acad Sci U S A. 2004;101:4661–6. doi: 10.1073/pnas.0303992101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gao Q, Mezei G, Nie Y, Rao Y, Choi CS, Bechmann I, Leranth C, Toran-Allerand D, Priest CA, Roberts JL, Gao XB, Mobbs C, Shulman GI, Diano S, Horvath TL. Anorectic estrogen mimics leptin’s effect on the rewiring of melanocortin cells and Stat3 signaling in obese animals. Nat Med. 2007;13:89–94. doi: 10.1038/nm1525. [DOI] [PubMed] [Google Scholar]
- 77.Gao Q, Horvath TL. Cross-talk between estrogen and leptin signaling in the hypothalamus. Am J Physiol Endocrinol Metab. 2008;294:E817–26. doi: 10.1152/ajpendo.00733.2007. [DOI] [PubMed] [Google Scholar]
- 78.Geary N, Asarian L, Korach KS, Pfaff DW, Ogawa S. Deficits in E2-dependent control of feeding, weight gain, and cholecystokinin satiation in ER-alpha null mice. Endocrinology. 2001;142:4751–7. doi: 10.1210/endo.142.11.8504. [DOI] [PubMed] [Google Scholar]
- 79.Gilbert M, Magnan C, Turban S, Andre J, Guerre-Millo M. Leptin receptor-deficient obese zucker rats reduce their food intake in response to a systemic supply of calories from glucose. Diabetes. 2003;52:277–282. doi: 10.2337/diabetes.52.2.277. [DOI] [PubMed] [Google Scholar]
- 80.Grasa MM, Vilà R, Esteve M, Cabot C, Fernandez-López JA, Remesar X, Alemany M. Oleoyl-estrone lowers the body weight of both ob/ob and db/db mice. Horm Metab Res. 2000;32:246–50. doi: 10.1055/s-2007-978629. [DOI] [PubMed] [Google Scholar]
- 81.Gropp E, Shanabrough M, Borok E, Xu AW, Janoschek R, Buch T, Plum L, Balthasar N, Hampel B, Waisman A, Barsh GS, Horvath TL, Brüning JC. Agouti-related peptide-expressing neurons are mandatory for feeding. Nat Neurosci. 2005;8:1289–91. doi: 10.1038/nn1548. [DOI] [PubMed] [Google Scholar]
- 82.Gyengesi E, Liu ZW, D’Agostino G, Gan G, Horvath TL, Gao XB, Diano S. Corticosterone regulates synaptic input organization of POMC and NPY/AgRP neurons in adult mice. Endocrinology in press. doi: 10.1210/en.2010-0681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hahn TM, Breininger JF, Baskin DG, Schwartz MW. Coexpression of Agrp and NPY in fasting-activated hypothalamic neurons. Nat Neurosci. 1998;1:271–272. doi: 10.1038/1082. [DOI] [PubMed] [Google Scholar]
- 84.Halaas JL, et al. Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 1995;269:543–546. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- 85.Harrold JA, Widdowson PS, Williams G. Altered energy balance causes selective changes in melanocortin-4(MC4-R), but not melanocortin-3 (MC3-R), receptors in specific hypothalamic regions: further evidence that activation of MC4-R is a physiological inhibitor of feeding. Diabetes. 1999;48:267–271. doi: 10.2337/diabetes.48.2.267. [DOI] [PubMed] [Google Scholar]
- 86.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–45. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 87.Heine PA, Taylor JA, Iwamoto GA, Lubahn DB, Cooke PS. Increased adipose tissue in male and female estrogen receptor-alpha knockout mice. Proc Natl Acad Sci U S A. 2000;97:12729–34. doi: 10.1073/pnas.97.23.12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hess RA, Bunick D, Lee KH, Bahr J, Taylor JA, Korach KS, Lubahn DB. A role for oestrogens in the male reproductive system. Nature. 1997;390:509–12. doi: 10.1038/37352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hetherington AW, Ranson SW. Hypothalamic lesions and adipocity in the rat. Anatomical Record. 1940;78:149. [Google Scholar]
- 90.Hetherington AW, Ranson SW. The relation of various hypothalamic lesions to adiposity in the rat. Journal of Comparative Neurology. 1942;76:475–499. [Google Scholar]
- 91.Hill JW, Elias CF, Fukuda M, Williams KW, Berglund ED, Holland WL, Cho YR, Chuang JC, Xu Y, Choi M, Lauzon D, Lee CE, Coppari R, Richardson JA, Zigman JM, Chua S, Scherer PE, Lowell BB, Brüning JC, Elmquist JK. Direct insulin and leptin action on pro-opiomelanocortin neurons is required for normal glucose homeostasis and fertility. Cell Metab. 2010;11:286–97. doi: 10.1016/j.cmet.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Holder JL, Jr, Butte NF, Zinn AR. Profound obesity associated with a balanced translocation that disrupts the SIM1 gene. Hum Mol Genet. 2000;9:101–108. doi: 10.1093/hmg/9.1.101. [DOI] [PubMed] [Google Scholar]
- 93.Holder JL, Jr, Zhang L, Kublaoui BM, DiLeone RJ, Oz OK, Bair CH, Lee YH, Zinn AR. Sim1 gene dosage modulates the homeostatic feeding response to increased dietary fat in mice. Am J Physiol Endocrinol Metab. 2004;287:E105–E113. doi: 10.1152/ajpendo.00446.2003. [DOI] [PubMed] [Google Scholar]
- 94.Horvath TL, Naftolin F, Kalra SP, Leranth C. Neuropeptide-Y innervation of beta-endorphin-containing cells in the rat mediobasal hypothalamus: a light and electron microscopic double Immunostaining analysis. Endocrinology. 1992;131:2461–2467. doi: 10.1210/endo.131.5.1425443. [DOI] [PubMed] [Google Scholar]
- 95.Horvath TL, Garcia-Segura LM, Naftolin F. Control of gonadotropin feedback: the possible role of estrogen-induced hypothalamic synaptic plasticity. Gynecol Endocrinol. 1997;11:139–143. doi: 10.3109/09513599709152525. [DOI] [PubMed] [Google Scholar]
- 96.Horvath TL, Warden CH, Hajos M, Lombardi A, Goglia F, Diano S. Brain uncoupling protein 2: uncoupled neuronal mitochondria predict thermal synapses in homeostatic centers. J Neurosci. 1999;19:10417–27. doi: 10.1523/JNEUROSCI.19-23-10417.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Horvath TL, Diano S, Tschop M. Brain Circuits Regulating Energy Homeostasis. The Neuroscientist. 2004;10:235–246. doi: 10.1177/1073858403262151. [DOI] [PubMed] [Google Scholar]
- 98.Horvath TL, Diano S. The floating blueprint of hypothalamic feeding circuits. Nat Rev Neurosci. 2004;5:662–7. doi: 10.1038/nrn1479. [DOI] [PubMed] [Google Scholar]
- 99.Horvath TL, Gao XB. Input organization and plasticity of hypocretin neurons: possible clues to obesity’s association with insomnia. Cell Metab. 2005;1:279–286. doi: 10.1016/j.cmet.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 100.Horvath TL, Sarman B, García-Cáceres C, Enriori PJ, Shanabrough M, Borok E, Argente J, Chowen JA, Perez-Tilve D, Pfluger PT, Brönneke HS, Levin BE, Diano S, Cowley MA, Tschöp MH. Synaptic Input Organization of the Melanocortin System Predicts Diet-Induced Hypothalamic Reactive Gliosis and Obesity. PNAS. doi: 10.1073/pnas.1004282107. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, Lee F. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 102.Jackman HL, et al. A peptidase in human platelets that deamidates tachykinins: Probable identity with the Iysosomal “protective protein. J Biol Chem. 1990;265:265–11272. [PubMed] [Google Scholar]
- 103.Jackman HL, et al. Plasma membrane-bound and Iysosomal peptidases in human alveolar macrophages. Am J Respir Cell Mol Biol. 1995;13:196–204. doi: 10.1165/ajrcmb.13.2.7626287. [DOI] [PubMed] [Google Scholar]
- 104.Jackson RS, Creemers JW, Ohagi S, Raffin-Sanson ML, Sanders L, Montague CT, Hutton JC, O’Rahilly S. Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nat Genet. 1997;16:303–6. doi: 10.1038/ng0797-303. [DOI] [PubMed] [Google Scholar]
- 105.Jacobowitz DM, O’Donohue TL. alpha-Melanocyte stimulating hormone: immunohistochemical identification and mapping in neurons of rat brain. Proc Natl Acad Sci U S A. 1978;75:6300–4. doi: 10.1073/pnas.75.12.6300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jegou S, Boutelet I, Vaudry H. Melanocortin-3 receptor mRNA expression in pro-opiomelanocortin neurones of the rat arcuate nucleus. Journal of Neuroendocrinology. 2000;12:501–505. doi: 10.1046/j.1365-2826.2000.00477.x. [DOI] [PubMed] [Google Scholar]
- 107.Jessen L, Clegg DJ, Bouman SD. Evaluation of the lack of anorectic effect of intracerebroventricular insulin in rats. Am J Physiol Regul Integr Comp Physiol. 2010;298:R43–50. doi: 10.1152/ajpregu.90736.2008. [DOI] [PubMed] [Google Scholar]
- 108.Jo YH, Su Y, Gutierrez-Juarez R, Chua S., Jr Oleic acid directly regulates POMC neuron excitability in the hypothalamus. J Neurophysiol. 2009;101:2305–16. doi: 10.1152/jn.91294.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kalra SP, Dube MG, Pu S, Xu B, Horvath TL, Kalra PS. Interacting appetite-regulating pathways in the hypothalamic regulation of body weight. Endocr Rev. 1999;20:68–100. doi: 10.1210/edrv.20.1.0357. [DOI] [PubMed] [Google Scholar]
- 110.Kim EM, Grace MK, Welch CC, Billington CJ, Levine AS. STZ-induced diabetes decreases and insulin normalizes POMC mRNA in arcuate nucleus and pituitary in rats. American Journal of Physics. 1999;276:R1320–R1326. doi: 10.1152/ajpregu.1999.276.5.R1320. [DOI] [PubMed] [Google Scholar]
- 111.King BM. The rise, fall, and resurrection of the ventromedial hypothalamus in the regulation of feeding behavior and body weight. Physiol Behav. 2006;87:221–44. doi: 10.1016/j.physbeh.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 112.Kishi T, Aschkenasi CJ, Lee CE, Mountjoy KG, Saper CB, Elmquist JK. Expression of melanocortin 4 receptor mRNA in the central nervous system of the rat. J Comp Neurol. 2003;457:213–235. doi: 10.1002/cne.10454. [DOI] [PubMed] [Google Scholar]
- 113.Kitamura T, Nakae J, Kitamura Y, Kido Y, Biggs WH, 3rd, Wright CV, White MF, Arden KC, Accili D. The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic beta cell growth. J Clin Invest. 2002;110:1839–47. doi: 10.1172/JCI200216857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kitamura T, Feng Y, Kitamura YI, Chua SC, Jr, Xu AW, Barsh GS, Rossetti L, Accili D. Forkhead protein FoxO1 mediates Agrp-dependent effects of leptin on food intake. Nat Med. 2006;12:534–40. doi: 10.1038/nm1392. [DOI] [PubMed] [Google Scholar]
- 115.Koch L, Wunderlich FT, Seibler J, Könner AC, Hampel B, Irlenbusch S, Brabant G, Kahn CR, Schwenk F, Brüning JC. Central insulin action regulates peripheral glucose and fat metabolism in mice. J Clin Invest. 2008;118:2132–47. doi: 10.1172/JCI31073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Könner AC, Janoschek R, Plum L, Jordan SD, Rother E, Ma X, Xu C, Enriori P, Hampel B, Barsh GS, Kahn CR, Cowley MA, Ashcroft FM, Brüning JC. Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab. 2007;5:438–49. doi: 10.1016/j.cmet.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 117.Korner J, Chua SC, Williams JA, Leibel RL, Wardlaw SL. Regulation of hypothalamic proopiomelanocortin by leptin in lean and obese rats. Neuroendocrinology. 1999;70:377–83. doi: 10.1159/000054499. [DOI] [PubMed] [Google Scholar]
- 118.Krude H, Biebermann H, Luck W, Horn R, Brabant G, Gruters A. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat Genet. 1998;19:155–7. doi: 10.1038/509. [DOI] [PubMed] [Google Scholar]
- 119.Kumamoto K, Stewart TA, Johnson AR, Erdos EG. Prolylcarboxypeptidase (angiotensinase C) in human cultured cells. J Clin Invest. 1981;67:210–215. doi: 10.1172/JCI110015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lage R, Dieguez C, Vidal-Puig A, Lopez M. AMPK: a metabolic gauge regulating whole-body energy homeostasis. Trends in Molecular Medicine. 2008;14:539–549. doi: 10.1016/j.molmed.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 121.Lanni A, De Felice M, Lombardi A, Moreno M, Fleury C, Ricquier D, Goglia F. Induction of UCP2 mRNA by thyroid hormones in rat heart. FEBS Lett. 1997;418:171–4. doi: 10.1016/s0014-5793(97)01375-6. [DOI] [PubMed] [Google Scholar]
- 122.Leibel RL, Chung WK, Chua SC. The molecular genetics of rodent single gene obesities. J Biol Chem. 1997;272:31937–31940. doi: 10.1074/jbc.272.51.31937. [DOI] [PubMed] [Google Scholar]
- 123.Li QL, Jansen E, Brent GA, Friedman TC. Regulation of prohormone convertase 1 (PC1) by thyroid hormone. Am J Physiol Endocrinol Metab. 2001;280:E160–70. doi: 10.1152/ajpendo.2001.280.1.E160. [DOI] [PubMed] [Google Scholar]
- 124.Li QL, Jansen E, Brent GA, Naqvi S, Wilber JF, Friedman TC. Interactions between the prohormone convertase 2 promoter and the thyroid hormone receptor. Endocrinology. 2000;141:3256–66. doi: 10.1210/endo.141.9.7674. [DOI] [PubMed] [Google Scholar]
- 125.Li YZ, Davidowa H. Food deprivation decreases responsiveness of ventromedial hypothalamic neurons to melanocortins. J Neurosci Res. 2004;77:596–602. doi: 10.1002/jnr.20192. [DOI] [PubMed] [Google Scholar]
- 126.Lin HV, Plum L, Ono H, Gutiérrez-Juárez R, Shanabrough M, Borok E, Horvath TL, Rossetti L, Accili D. Divergent regulation of energy expenditure and hepatic glucose production by insulin receptor in agouti-related protein and POMC neurons. Diabetes. 2010;59:337–46. doi: 10.2337/db09-1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Liu H, Kishi T, Roseberry AG, Cai X, Lee CE, Montez JM, Friedman JM, Elmquist JK. Transgenic mice expressing green fluorescent protein under the control of the melanocortin-4 receptor promoter. J Neurosci. 2003;23:7143–54. doi: 10.1523/JNEUROSCI.23-18-07143.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lopez-Valpuesta FJ, Nyce JW, Griffin-Biggs TA, Ice JC, Myers RD. Antisense to NPY-Y1 demonstrates that Y1 receptors in the hypothalamus underlie NPY hypothermia and feeding in rats. Proc Biol Sci. 1996;263:881–886. doi: 10.1098/rspb.1996.0130. [DOI] [PubMed] [Google Scholar]
- 129.Ludwig DS, Mountjoy KG, Tatro JB, Gillette JA, Frederich RC, Flier JS, Maratos- Flier E. Melanin-concentrating hormone: A functional melanocortin antagonist in the hypothalamus. Am J Physiol. 1998;37:E627–E633. doi: 10.1152/ajpendo.1998.274.4.E627. [DOI] [PubMed] [Google Scholar]
- 130.Luquet S, Perez FA, Hnasko TS, Palmiter RD. NPY/AgRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science. 2005;310:683–5. doi: 10.1126/science.1115524. [DOI] [PubMed] [Google Scholar]
- 131.Madar ZS, Rahimy E, Mahdi F, Schmaier AH. Overexpression of prolylcarboxypeptidase enhances plasma prekallikrein activation on Chinese hamster ovary cells. Am J Physiol Heart Circ Physiol. 2005;289:H2697–703. doi: 10.1152/ajpheart.00715.2005. [DOI] [PubMed] [Google Scholar]
- 132.Majdic G, Young M, Gomez-Sanchez E, Anderson P, Szczepaniak LS, Dobbins RL, McGarry JD, Parker KL. Knockout mice lacking steroidogenic factor 1 are a novel genetic model of hypothalamic obesity. Endocrinology. 2002;143:607–14. doi: 10.1210/endo.143.2.8652. [DOI] [PubMed] [Google Scholar]
- 133.Makimura H, Mizuno TM, Roberts J, Silverstein J, Beasley J, Mobbs CV. Adrenalectomy reverses obese phenotype and restores hypothalamic melanocortin tone in leptin-deficient ob/ob mice. Diabetes. 2000;49:1917–23. doi: 10.2337/diabetes.49.11.1917. [DOI] [PubMed] [Google Scholar]
- 134.Maulon-Feraille L, Della Zuana O, Suply T, Rovere-Jovene C, Audinot V, Levens N, Boutin JA, Duhault J, Nahon JL. Appetite-boosting property of pro-melanin-concentrating hormone(131–165) (neuropeptide-glutamic acid-isoleucine) is associated with proteolytic resistance. J Pharmacol Exp Ther. 2002;302:776–773. doi: 10.1124/jpet.302.2.766. [DOI] [PubMed] [Google Scholar]
- 135.McCarthy JJ, Meyer J, Moliterno DJ, Newby LK, Rogers WJ, Topol EJ. Evidence for substrantial effect modification by gender in a large scale genetic association study of the metabolic syndrome among coronary heart disease patients. Hum Genet. 2003;114:87–98. doi: 10.1007/s00439-003-1026-1. [DOI] [PubMed] [Google Scholar]
- 136.Mercer JG, Hoggard N, Williams LM, Lawrence CB, Hannah LT, Trayhurn P. Localization of leptin receptor mRNA and the long splice variant (O-Rb) in mouse hypothalamus and adjacent brain regions by in situ hybridization. FEBS Lett. 1996;387:113–116. doi: 10.1016/0014-5793(96)00473-5. [DOI] [PubMed] [Google Scholar]
- 137.Mercer JG, Hoggard N, Williams LM, Lawrence CB, Hannah LT, Trayhurn P. Coexpression of leptin receptor and neproneuropeptide Y mRNA in arcuate nucleus of mouse hypothalamus. J Neuroendocrinol. 1996;8:733–735. doi: 10.1046/j.1365-2826.1996.05161.x. [DOI] [PubMed] [Google Scholar]
- 138.Mezey E, Kiss JZ, Mueller GP, Eskay R, O’Donohue TL, Palkovits M. Distribution of the pro-opiomelanocortin derived peptides, adrenocorticotrope hormone, alpha-melanocyte-stimulating hormone and beta-endorphin (ACTH, alpha-MSH, beta-END) in the rat hypothalamus. Brain Res. 1985;328:341–347. doi: 10.1016/0006-8993(85)91046-7. [DOI] [PubMed] [Google Scholar]
- 139.Michaud JL, Boucher F, Melnyk A, Gauthier F, Goshu E, Levy E, Mitchell GA, Himms-Hagen J, Fan CM. Sim1 haploinsufficiency causes hyperphagia, obesity and reduction of the paraventricular nucleus of the hypothalamus. Hum Mol Genet. 2001;10:1465–1473. doi: 10.1093/hmg/10.14.1465. [DOI] [PubMed] [Google Scholar]
- 140.Mieda M, Yanagisawa M. Sleep, feeding, and neuropeptides: roles of orexins and orexin receptors. Curr Opin Neurobiol. 2002;12:339–345. doi: 10.1016/s0959-4388(02)00331-8. [DOI] [PubMed] [Google Scholar]
- 141.Miller JJ, Changaris DG, Levy RS. Angiorensin carboxypeptidase activity in urine from normal subjects and patients with kidney damage. Life Sci. 1991;48:1529–1535. doi: 10.1016/0024-3205(91)90277-i. [DOI] [PubMed] [Google Scholar]
- 142.Mizuno TK, Kleopoulos SP, Bergen HT, Roberts JL, Priest CA, Mobbs CV. Hypothalamic pro-opiomelanocortin mRNA is reduced by fasting and in ob/ob and db/db mice, but is stimulated by leptin. Diabetes. 1998;47:294–7. doi: 10.2337/diab.47.2.294. [DOI] [PubMed] [Google Scholar]
- 143.Mizuno TM, Mobbs CV. Hypothalamic agouti-related protein messenger ribonucleic acid is inhibited by leptin and stimulated by fasting. Endocrinology. 1991;140:814–817. doi: 10.1210/endo.140.2.6491. [DOI] [PubMed] [Google Scholar]
- 144.Mizuno TM, Makimura H, Silverstein J, Roberts JL, Lopingco T, Mobbs CV. Fasting regulates hypothalamic neuropeptide Y, agouti related peptide, and proopiomelanocortin in diabetic mice independent of changes in leptin or insulin. Endocrinology. 1999:4551–4557. doi: 10.1210/endo.140.10.6966. [DOI] [PubMed] [Google Scholar]
- 145.Mobbs CV, Mizuno T, Isoda F, Mastaitis J, Yang XJ. Impaired glucose signaling as a cause of obesity and the metabolic syndrome: The glucoadipostatic hypothesis. Physiol Behav. 2005;85:3–23. doi: 10.1016/j.physbeh.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 146.Mori H, Inoki K, Münzberg H, Opland D, Faouzi M, Villanueva EC, Ikenoue T, Kwiatkowski D, MacDougald OA, Myers MG, Jr, Guan KL. Critical role for hypothalamic mTOR activity in energy balance. Cell Metab. 2009;9:362–74. doi: 10.1016/j.cmet.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Mounien L, Marty N, Tarussio D, Metref S, Genoux D, Preitner F, Foretz M, Thorens B. Glut2-dependent glucose-sensing controls thermoregulation by enhancing the leptin sensitivity of NPY and POMC neurons. FASEB J. 2010;24:1747–58. doi: 10.1096/fj.09-144923. [DOI] [PubMed] [Google Scholar]
- 148.Mountjoy KG, Robbins LS, Mortrud MT, Cone RD. The cloning of a family of genes that encode the melanocortin receptors. Science. 1992;257:1248–51. doi: 10.1126/science.1325670. [DOI] [PubMed] [Google Scholar]
- 149.Mountjoy KG, Mortrud MT, Low MJ, Simerly RB, Cone RD. Localization of the melanocortin-4 receptor (MC4-R) in neuroendocrine and autonomic control circuits in the brain. Mol Endocrinol. 1994;8:1298–1308. doi: 10.1210/mend.8.10.7854347. [DOI] [PubMed] [Google Scholar]
- 150.Munoz CM, Leeb-Lundberg LMF. Receptormediated internalization of bradykinin. DDT, MF-2 smooth muscle cells process internalized bradykinin via multiple degradative pathways. J Biol Chem. 1992;267:303–309. [PubMed] [Google Scholar]
- 151.Münzberg H, Flier JS, Bjørbaek C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology. 2004;145:4880–9. doi: 10.1210/en.2004-0726. [DOI] [PubMed] [Google Scholar]
- 152.Münzberg H, Myers MG., Jr Molecular and anatomical determinants of central leptin resistance. Nat Neurosci. 2005;8:566–70. doi: 10.1038/nn1454. [DOI] [PubMed] [Google Scholar]
- 153.Mystkowski P, Schwartz MW. Gonadal steroids and energy homeostasis in the leptin era. Nutrition. 2000;16:937–46. doi: 10.1016/s0899-9007(00)00458-5. [DOI] [PubMed] [Google Scholar]
- 154.Naggert JK, Fricker LD, Varlamoy O, Nishina PM, Rouille Y, Steiner DF, Carroll RJ, Paigen BJ, Leiter EH. Hyperproinsulinaemia in obese fat/fat mice associated with a carboxypeptidase E mutation which reduces enzyme activity. Nat Genet. 1995;10:135–42. doi: 10.1038/ng0695-135. [DOI] [PubMed] [Google Scholar]
- 155.Nakagawa T, Ono-Kishino M, Sugaru E, Yamanaka M, Taiji M, Noguchi H. Brain-derived neurotrophic factor (BDNF) regulates glucose and energy metabolism in diabetic mice. Diabetes Metab Res Rev. 2002;18:185–191. doi: 10.1002/dmrr.290. [DOI] [PubMed] [Google Scholar]
- 156.Nakagawa T, Ogawa Y, Ebihara K, Yamanaka M, Tsuchida A, Taiji M, Noguchi H, Nakao K. Anti-obesity and anti-diabetic effects of brain-derived neurotrophic factor in rodent models of leptin resistance. Int J Obes Relat Metab Disord. 2003;27:557–565. doi: 10.1038/sj.ijo.0802265. [DOI] [PubMed] [Google Scholar]
- 157.Nègre-Salvayre A, Hirtz C, Carrera G, Cazenave R, Troly M, Salvayre R, Pénicaud L, Casteilla L. A role for uncoupling protein-2 as a regulator of mitochondrial hydrogen peroxide generation. FASEB J. 1997;11:809–15. [PubMed] [Google Scholar]
- 158.Nillni EA, Aird F, Seidah NG, Todd RB, Koenig JI. PreproTRH(178–199) and two novel peptides (pFQ7 and pSE14) derived from its processing, which are produced in the paraventricular nucleus of the rat hypothalamus, are regulated during suckling. Endocrinology. 2001;142:896–906. doi: 10.1210/endo.142.2.7954. [DOI] [PubMed] [Google Scholar]
- 159.Niswender KD, Morton GJ, Stearns WH, Rhodes CJ, Myers MG, Jr, Schwartz MW. Intracellular signalling. Key enzyme in leptin-induced anorexia. Nature. 2001;413:794–5. doi: 10.1038/35101657. [DOI] [PubMed] [Google Scholar]
- 160.Obici S, Zhang BB, Karkanias G, Rossetti L. Hypothalamic insulin signaling is required for inhibition of glucose production. Nat Med. 2002;8:1376–82. doi: 10.1038/nm1202-798. [DOI] [PubMed] [Google Scholar]
- 161.Odya CE, Marinkovic DV, Hammon KJ, Stewart TA, Erdos EG. Purification and properties of prolylcarboxypeptidase (Angiotensinase C) from human kidney. J Biol Chem. 1978;253:5927–5931. [PubMed] [Google Scholar]
- 162.Ollmann MM, Wilson BD, Yang YK, Kerns JA, Chen Y, Gantz I, Barsh GS. Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science. 1997;278:135–8. doi: 10.1126/science.278.5335.135. [DOI] [PubMed] [Google Scholar]
- 163.Okamoto H, Nakae J, Kitamura T, Park BC, Dragatsis I, Accili D. Transgenic rescue of insulin receptor-deficient mice. J Clin Invest. 2004;114:214–23. doi: 10.1172/JCI21645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.O’Shea M, Hansen MJ, Tatemoto K, Morris AJ. Inhibitory effect of Apelin 12 on nocturnal food intake in rats. Nutr Neurosci. 2003;6:163–167. doi: 10.1080/1028415031000111273. [DOI] [PubMed] [Google Scholar]
- 165.Osterlund M, Kuiper GG, Gustafsson JA, Hurd YL. Differential distribution and regulation of estrogen receptor-alpha and -beta mRNA within the female rat brain. Brain Res Mol Brain Res. 1998;54:175–80. doi: 10.1016/s0169-328x(97)00351-3. [DOI] [PubMed] [Google Scholar]
- 166.Parker KL, Rice DA, Lala DS, Ikeda Y, Luo X, Wong M, Bakke M, Zhao L, Frigeri C, Hanley NA, Stallings N, Schimmer BP. Steroidogenic factor 1: an essential mediator of endocrine development. Recent Prog Horm Res. 2002;57:19–36. doi: 10.1210/rp.57.1.19. [DOI] [PubMed] [Google Scholar]
- 167.Parton LE, Ye CP, Coppari R, Enriori PJ, Choi B, Zhang CY, Xu C, Vianna CR, Balthasar N, Lee CE, Elmquist JK, Cowley MA, Lowell BB. Glucose sensing by POMC neurons regulates glucose homeostasis and is impaired in obesity. Nature. 2007;449:228–32. doi: 10.1038/nature06098. [DOI] [PubMed] [Google Scholar]
- 168.Pelletier G, Dube D. Electron microscopic immunohistochemical localization of alpha-msh in the rat brain. Am J Anat. 1977;150:201–5. doi: 10.1002/aja.1001500115. [DOI] [PubMed] [Google Scholar]
- 169.Pelleymounter MA, Culler MJ, Baker MB, Hecht R, Winters D, Boone T, Collins F. Effects of the obese gene product on body weight regulation in ob/ob mice. Science. 1995;269:540–543. doi: 10.1126/science.7624776. [DOI] [PubMed] [Google Scholar]
- 170.Pinto S, Roseberry AG, Liu H, Diano S, Shanabrough M, Cai X, Friedman JM, Horvath TL. Rapid rewiring of arcuate nucleus feeding circuits by leptin. Science. 2004;304:110–115. doi: 10.1126/science.1089459. [DOI] [PubMed] [Google Scholar]
- 171.Plum L, Lin HV, Dutia R, Tanaka J, Aizawa KS, Matsumoto M, Kim NX, Cawley AJ, Paik JH, Loh YP, DePinho RA, Wardlaw SL, Accili D. The obesity susceptibility gene Cpe links FoxO1 signaling in hypothalamic pro-opiomelanocortin neurons with regulation of food intake. Nat Med. 2009;15:1195–201. doi: 10.1038/nm.2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Pocai A, Obici S, Schwartz GJ, Rossetti L. A brain-liver circuit regulates glucose homeostasis. Cell Metab. 2005;1:53–61. doi: 10.1016/j.cmet.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 173.Pritchard LE, Turnbull AV, White A. Pro-opiomelanocortin processing in the hypothalamus:impact on melanocortin signaling and obesity. J Endocrinol. 2002;172:411–421. doi: 10.1677/joe.0.1720411. [DOI] [PubMed] [Google Scholar]
- 174.Qian S, Chen H, Weingarth D, Trumbauer ME, Novi DE, Guan X, Yu H, Shen Z, Feng Y, Frazier E, Chen A, Camacho RE, Shearman LP, Gopal-Truter S, MacNeil DJ, Van der Ploeg LH, Marsh DJ. Neither agouti-related protein nor neuropeptide Y is critically required for the regulation of energy homeostasis in mice. Mol Cell Biol. 2002;22:5027–35. doi: 10.1128/MCB.22.14.5027-5035.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Qiu J, Bosch MA, Tobias SC, Krust A, Graham SM, Murphy SJ, Korach KS, Chambon P, Scanlan TS, Rønnekleiv OK, Kelly MJ. A G-protein-coupled estrogen receptor is involved in hypothalamic control of energy homeostasis. J Neurosci. 2006;26:5649–55. doi: 10.1523/JNEUROSCI.0327-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Qu D, Ludwig DS, Gammeltoft S, Piper M, Pelleymouter MA, Culler MJ, Mathes WF, Przypek R, Kanarek R, Maratos-Flier E. A role for melanin-concentrating hormone in the central regulation of feeding behavior. Nature. 1996;380:243–247. doi: 10.1038/380243a0. [DOI] [PubMed] [Google Scholar]
- 177.Ramadori G, Lee CE, Bookout AL, Lee S, Williams KW, Anderson J, Elmquist JK, Coppari R. Brain SIRT1: anatomical distribution and regulation by energy availability. J Neurosci. 2008;28:9989–96. doi: 10.1523/JNEUROSCI.3257-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Renz M, Tomlinson E, Hultgren B, Levin N, Gu Q, Shimkets RA, Lewin DA, Stewart TA. Quantitative expression analysis of genes regulated by both obesity and leptin reveals a regulatory loop between leptin and pituitary-derived ACTH. J Biol Chem. 2000;275:10429–36. doi: 10.1074/jbc.275.14.10429. [DOI] [PubMed] [Google Scholar]
- 179.Richard D, Rivest R, Huang Q, Bouillaud F, Sanchis D, Champigny O, Ricquier D. Distribution of the uncoupling protein 2 mRNA in the mouse brain. J Comp Neurol. 1998;397:549–60. [PubMed] [Google Scholar]
- 180.Rios M, Fan G, Fekete C, Kelly J, Bates B, Kuehn R, Lechan RM, Jaenisch R. Conditional deletion of brain-derived neurotrophic factor in the postnatal brain leads to obesity and hyperactivity. Mol Endocrinol. 2001;15:1748–1757. doi: 10.1210/mend.15.10.0706. [DOI] [PubMed] [Google Scholar]
- 181.Robbins LS, Nadeau JH, Johnson KR, Kelly MA, Roselli-Rehfuss L, Baack E, Mountjoy KG, Cone RD. Pigmentation phenotypes of variant extension locus alleles result from point mutations that alter MSH receptor function. Cell. 1993;72:827–834. doi: 10.1016/0092-8674(93)90572-8. [DOI] [PubMed] [Google Scholar]
- 182.Roesch DM. Effects of selective estrogen receptor agonists on food intake and body weight gain in rats. Physiol Behav. 2006;87:39–44. doi: 10.1016/j.physbeh.2005.08.035. [DOI] [PubMed] [Google Scholar]
- 183.Rorato R, Castro M, Borges BC, Benedetti M, Germano CM, Antunes-Rodrigues J, Elias LL. Adrenalectomy enhances endotoxemia-induced hypophagia: higher activation of corticotrophin-releasing-factor and proopiomelanocortin hypothalamic neurons. Horm Behav. 2008;54:134–42. doi: 10.1016/j.yhbeh.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 184.Sahm UG, Qarawi MA, Olivier GW, Ahmed AR, Branch SK, Moss SH, Pouton CW. The melanocortin (MC3) receptor from rat hypothalamus: photoaffinity labelling and binding of alanine-substituted alpha-MSH analogues. FEBS Lett. 1994;350:29–32. doi: 10.1016/0014-5793(94)00725-x. [DOI] [PubMed] [Google Scholar]
- 185.Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, Williams SC, Richardson JA, Kozlowski GP, Wilson S, Arch JR, Buckingham RE, Haynes AC, Carr SA, Annan RS, McNulty DE, Liu WS, Terrett JA, Elshourbagy NA, Bergsma DJ, Yanagisawa M. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92:573–585. doi: 10.1016/s0092-8674(00)80949-6. [DOI] [PubMed] [Google Scholar]
- 186.Savontaus E, Breen TL, Kim A, Yang LM, Chua SC, Wardlaw SL. Metabolic effects of transgenic melanocyte-stimulating hormone overexpression in lean and obese mice. Endocrinology. 2004;145:3881–91. doi: 10.1210/en.2004-0263. [DOI] [PubMed] [Google Scholar]
- 187.Schwartz MW, Seeley RJ, Campfield LA, Burn P, Baskin DG. Identification of leptin action in rat hypothalamus. J Clin Invest. 1996;98:1101–1106. doi: 10.1172/JCI118891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Schwartz MW, Seeley RJ, Woods SC, Weigle DS, Campfield LA, Burn P, Baskin DG. Leptin increases hypothalamic pro-opiomelanocortin mRNA expression in the rostral arcuate nucleus. Diabetes. 1997;46:2119–23. doi: 10.2337/diab.46.12.2119. [DOI] [PubMed] [Google Scholar]
- 189.Schwartz MW, Woods SC, Jr, Porte D, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404:661–671. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- 190.Seeley RJ, Yagaloff KA, Fisher SL, Burn P, Thiele TE, van Dijk G, Baskin DG, Schwartz MW. Melanocortin receptors in leptin effects. Nature. 1997;390:349. doi: 10.1038/37016. [DOI] [PubMed] [Google Scholar]
- 191.Seeley RJ, Woods SC. Monitoring of stored and available fuel by the CNS: implications for obesity. Nat Rev Neurosci. 2003;4:901–909. doi: 10.1038/nrn1245. [DOI] [PubMed] [Google Scholar]
- 192.Segal JP, Stallings NR, Lee CE, Zhao L, Socci N, Viale A, Harris TM, Soares MB, Childs G, Elmquist JK, Parker KL, Friedman JM. Use of laser-capture microdissection for the identification of marker genes for the ventromedial hypothalamic nucleus. J Neurosci. 2005;25:4181–8. doi: 10.1523/JNEUROSCI.0158-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Segal-Lieberman G, Bradley RL, Kokkotou E, Carlson M, Trombly DJ, Wang X, Bates S, Myers MG, Flier JS, Maratos-Flier E. Melanin-concentrating hormone is a critical mediator of the leptin-deficient phenotype. Proc Natl Acad Sci USA. 2003;100:10085–10090. doi: 10.1073/pnas.1633636100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Sekine Y, Yamamoto T, Yumioka T, Imoto S, Kojima H, Matsuda T. Crosstalk between endocrine-disrupting chemicals and cytokine signaling through estrogen receptors. Biochem Biophys Res Commun. 2004;315:692–8. doi: 10.1016/j.bbrc.2004.01.109. [DOI] [PubMed] [Google Scholar]
- 195.Shinyama H, Masuzaki H, Fang H, Flier JS. Regulation of melanocortin-4 receptor signaling: agonist-mediated desensitization and internalization. Endocrinology. 2003;144:1301–14. doi: 10.1210/en.2002-220931. [DOI] [PubMed] [Google Scholar]
- 196.Shyng SL, Nichols CG. Membrane phospholipid control of nucleotide sensitivity of KATP channels. Science. 1998;282:1138–41. doi: 10.1126/science.282.5391.1138. [DOI] [PubMed] [Google Scholar]
- 197.Singru PS, Sánchez E, Fekete C, Lechan RM. Importance of melanocortin signaling in refeeding-induced neuronal activation and satiety. Endocrinology. 2007;148:648–656. doi: 10.1210/en.2006-1233. [DOI] [PubMed] [Google Scholar]
- 198.Skidgel RA, Wickstrom F, Kumamoto K, Erdos EG. Rapid radioassay for prolylcarboxypeptidase (angiotensinase C) Anal Biochem. 1981;118:113–119. doi: 10.1016/0003-2697(81)90165-2. [DOI] [PubMed] [Google Scholar]
- 199.Skidge RA. Basic carboxypepcidases:regulators of peptide hormone activity. Trends Pharmacol Sci. 1988;9:299–304. doi: 10.1016/0165-6147(88)90015-6. [DOI] [PubMed] [Google Scholar]
- 200.Skidgel RA, Jackman HL, Erdos EG. Metabolism of substance P and bradykinin by human neutrophils. Biochem Pharmacol. 1991;41:1335–1344. doi: 10.1016/0006-2952(91)90106-f. [DOI] [PubMed] [Google Scholar]
- 201.Sterson SM, Shepherd GM, Friedman JM. Topographic mapping of VMH –>arcuate nucleus microcircuits and their reorganization by fasting. Nat Neurosci. 2005;8:1356–63. doi: 10.1038/nn1550. [DOI] [PubMed] [Google Scholar]
- 202.Stolarczyk E, Guissard C, Michau A, Even PC, Grosfeld A, Serradas P, Lorsignol A, Pénicaud L, Brot-Laroche E, Leturque A, Le Gall M. Detection of extracellular glucose by GLUT2 contributes to hypothalamic control of food intake. Am J Physiol Endocrinol Metab 2. 2010;98:E1078–87. doi: 10.1152/ajpendo.00737.2009. [DOI] [PubMed] [Google Scholar]
- 203.Strominger JL, Brobeck JR. A mechanism of regulation of food intake. Yale J Biol Med. 1953;25:383–390. [PMC free article] [PubMed] [Google Scholar]
- 204.Sunter D, Hewson AK, Dickson SL. Intracerebroventricular injection of apelin-13 reduces food intake in the rat. Neurosci Lett. 2003;353:1–4. doi: 10.1016/s0304-3940(03)00351-3. [DOI] [PubMed] [Google Scholar]
- 205.Swanson LW, Sanchez-Watts G, Watts AG. Comparison of melanin-concentrating hormone and hypocretin/orexin mRNA expression patterns in a new parceling scheme of the lateral hypothalamic zone. Neurosci Lett. 2005;387:80–84. doi: 10.1016/j.neulet.2005.06.066. [DOI] [PubMed] [Google Scholar]
- 206.Tan F, Morris PW, Skidgel RA, Erdos EG. Sequencing and cloning of human prolylcarboxypeptidase (angiotensinase C). Similarity to both serine carboxypeptidase and prolylendopeptidase families. J Biol Chem. 1993;268:16631–8. [PubMed] [Google Scholar]
- 207.Taheri S, Murphy K, Cohen M, Sujkovic E, Kennedy A, Dhillo W, Dakin C, Sajedi A, Ghatei M, Bloom S. The effects of centrally administered apelin-13 on food intake, water intake and pituitary hormone release in rats. Biochem Biophys Res Commun. 2002;291:1208–12. doi: 10.1006/bbrc.2002.6575. [DOI] [PubMed] [Google Scholar]
- 208.Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, Richards GJ, Campfield LA, Clark FT, Deeds J. Identification and expression cloning of a leptin receptor, OB-R. Cell. 1995;83:1263–71. doi: 10.1016/0092-8674(95)90151-5. [DOI] [PubMed] [Google Scholar]
- 209.Tejwani GA, Richard CW. Effect of electrolytic and chemical ventromedial hypothalamic lesions on food intake, body weight, analgesia and the CNS opioid peptides in rats and mice. NIDA Res Monogr. 1986;75:497–500. [PubMed] [Google Scholar]
- 210.Tritos NA, Mastaitis JW, Kokkotou E, Maratos-Flier E. Characterization of melanin concentrating hormone and preproorexin expression in the murine hypothalamus. Brain Res. 2001;895:160–166. doi: 10.1016/s0006-8993(01)02066-2. [DOI] [PubMed] [Google Scholar]
- 211.Trus MD, et al. Regulation of glucose metabolism in pancreatic islets. Diabetes. 1981;30:911–22. doi: 10.2337/diab.30.11.911. [DOI] [PubMed] [Google Scholar]
- 212.Tsuchida A, Nonomura T, Nakagawa T, Itakura Y, Ono-Kishino M, Yamanaka M, Sugaru E, Taiji M, Noguchi H. Brain-derived neurotrophic factor ameliorates lipid metabolism in diabetic mice. Diabetes Obes Metab. 2002;4:262–269. doi: 10.1046/j.1463-1326.2002.00206.x. [DOI] [PubMed] [Google Scholar]
- 213.Vaisse C, Halaas JL, Horvath CM, Darnell JE, Stoffel M, Friedman JM. Leptin activation of Stat3 in the hypothalamus of wild-type and ob/ob mice but not db/db mice. Nat Genet. 1996;14:95–7. doi: 10.1038/ng0996-95. [DOI] [PubMed] [Google Scholar]
- 214.Vaisse C, Clement K, Guy-Grand B, Froguel P. A frameshift mutation in human MC4R is associated with a dominant form of obesity. Nature Genetics. 1998;20:113–114. doi: 10.1038/2407. [DOI] [PubMed] [Google Scholar]
- 215.Valle A, Hoggard N, Adams AC, Roca P, Speakman JR. Chronic central administration of apelin-13 over 10 days increases food intake, body weight, locomotor activity and body temperature in C57BL/6 mice. J Neuroendocrinol. 2008;20:79–84. doi: 10.1111/j.1365-2826.2007.01617.x. [DOI] [PubMed] [Google Scholar]
- 216.Wallingford N, Perroud B, Gao Q, Coppola A, Gyengesi E, Liu ZW, Gao XB, Diament A, Haus KA, Shariat-Madar Z, Mahdi F, Wardlaw SL, Schmaier AH, Warden CH, Diano S. Prolylcarboxypeptidase regulates food intake by inactivating alpha-MSH in rodents. J Clin Invest. 2009;119:2291–303. doi: 10.1172/JCI37209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Watson SJ, Richard CW, Barchas JD. Adrenocorticotropin in rat brain: immunocytochemical localization in cells and axons. Science. 1978;200:1180–2. doi: 10.1126/science.206967. [DOI] [PubMed] [Google Scholar]
- 218.Williams KW, Margatho LO, Lee CE, Choi M, Lee S, Scott MM, Elias CF, Elmquist JK. Segregation of acute leptin and insulin effects in distinct populations of arcuate proopiomelanocortin neurons. J Neurosci. 2010;30:2472–9. doi: 10.1523/JNEUROSCI.3118-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Wisialowski T, Parker R, Preston E, Sainsbury A, Kraegen E, Herzog H, Cooney G. Adrenalectomy reduces neuropeptide Y-induced insulin release and NPY receptor expression in the rat ventromedial hypothalamus. J Clin Invest. 2000;105:1253–1259. doi: 10.1172/JCI8695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Woods SC, Seeley RJ, Cota D. Regulation of food intake through hypothalamic signaling networks involving mTOR. Annu Rev Nutr. 2008;28:295–311. doi: 10.1146/annurev.nutr.28.061807.155505. [DOI] [PubMed] [Google Scholar]
- 221.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–84. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 222.Xu B, Goulding EH, Zang K, Cepoi D, Cone RD, Jones KR, Tecott LH, Reichardt LF. Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat Neurosci. 2003;6:736–742. doi: 10.1038/nn1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Yamanaka A, Beuckmann CT, Willie JT, Hara J, Tsujino N, Mieda M, Tominaga M, Yagami K, Sugiyama F, Goto K, et al. Hypothalamic orexin neurons regulate arousal according to energy balance in mice. Neuron. 2003;38:701–713. doi: 10.1016/s0896-6273(03)00331-3. [DOI] [PubMed] [Google Scholar]
- 224.Yamashiro DJ, Maxfield FR. Regulation of endocytic process by pH. Trends Pharmacol Sci. 1988;9:190–194. doi: 10.1016/0165-6147(88)90078-8. [DOI] [PubMed] [Google Scholar]
- 225.Yang HYT, Erdos EG, Chiang TS. New enzymatic route for the inactivation of angiotensin. Nature. 1968;218:1224–1226. doi: 10.1038/2181224a0. [DOI] [PubMed] [Google Scholar]
- 226.Yang HYT, Erdos EG, Chiang TS, Jenssen TA, Rodgers JG. Characteristics of an enzyme that inactivates angiotensin II (angiotensinase C) Biochem Pharmacol. 1970;19:1201–1211. [Google Scholar]
- 227.Yang G, Lim CY, Li C, Xiao X, Radda GK, Li C, Cao X, Han W. FoxO1 inhibits leptin regulation of pro-opiomelanocortin promoter activity by blocking STAT3 interaction with specificity protein 1. J Biol Chem. 2009;284:3719–27. doi: 10.1074/jbc.M804965200. [DOI] [PubMed] [Google Scholar]
- 228.Yang XJ, Mastaitis J, Mizuno T, Mobbs CV. Glucokinase regulates reproductive function, glucocorticoid secretion, food intake, and hypothalamic gene expression. Endocrinology. 2007;148:1928–32. doi: 10.1210/en.2006-1312. [DOI] [PubMed] [Google Scholar]
- 229.Yang XJ, et al. Hypothalamic glucose sensor: similarities to and differences from pancreatic beta-cell mechanisms. Diabetes. 1999;48:1763–72. doi: 10.2337/diabetes.48.9.1763. [DOI] [PubMed] [Google Scholar]
- 230.Yeo GS, Farooqi S, Aminian S, Halsall DJ, Stanhope RJ, O’Rahilly S. A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nature Genetics. 1998;20:111–112. doi: 10.1038/2404. [DOI] [PubMed] [Google Scholar]
- 231.Yeo GS, Farooqi IS, Challis BG, Jackson RS, O’Rahilly S. The role of melanocortin signalling in the control of body weight: evidence from human and murine genetic models. Quarterly Journal of Medicine. 2000;93:7–14. doi: 10.1093/qjmed/93.1.7. [DOI] [PubMed] [Google Scholar]
- 232.Young JI, Otero V, Cerdan MG, Falzone TL, Chan EC, Low MJ, Rubinstein M. Authentic cell-specific and developmentally regulated expression of pro-opiomelanocortin genomic fragments in hypothalamic and hindbrain neurons of transgenic mice. Journal of Neurosciences. 1998;18:6631–6640. doi: 10.1523/JNEUROSCI.18-17-06631.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Záborszky L, Makara GB. Intrahypothalamic connections: an electron microscopic study in the rat. Exp Brain Res. 1979;34:201–15. doi: 10.1007/BF00235668. [DOI] [PubMed] [Google Scholar]
- 234.Zhang Y, et al. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]