

Abstract
Hydrogen sulfide (H2S) is a signaling molecule which plays regulatory roles in many physiological and/or pathological processes. Therefore, regulation of H2S levels could have great potential therapeutic value. In this work, we report the design, synthesis, and evaluation of a class of N-mercapto (N-SH)-based H2S donors. Thirty-three donors were synthesized and tested. Our results indicated that controllable H2S release from these donors could be achieved upon structural modifications. Selected donors (NSHD-1, NSHD-2, and NSHD-6) were tested in cellular models of oxidative damage and showed significant cytoprotective effects. Moreover, NSHD-1 and NSHD-2 were also found to exhibit potent protective effects in a murine model of myocardial ischemia reperfusion (MI/R) injury.
Introduction
Hydrogen sulfide (H2S), which is known for its characteristic odor of rotten eggs, has been considered as a highly toxic air pollutant for centuries. However, this gaseous molecule has been recently recognized as a member of a gasotransmitter family along with nitric oxide (NO) and carbon monoxide (CO).1−7 Endogenous H2S formation has been attributed to at least three enzymes: cystathionine β-synthase (CBS), cystathionine γ-lyase (CSE), and 3-mercaptopyruvate sulfur-transferase (MPST).8−12 These enzymes convert cysteine or cysteine derivatives to H2S in different tissues and organs. A number of studies have revealed a variety of biological effects of H2S, which include vasodilation,13−15 anti-inflammation,16,17 anticancer,18,19 and cardioprotection.20−24 While the exact mechanisms of these actions are still under investigation, some interesting biochemical/catabolic reactions of H2S have been disclosed. For example, H2S reacts readily with methemoglobin to yield sulfhemoglobin.25 H2S inhibits cytochrome c oxidase, which accounts for its toxic action.26 H2S is an effective reducing agent and can react with reactive oxygen species such as hydrogen peroxide, superoxide, and peroxynitrite.27−29 It has been reported that H2S can cause protein S-sulfhydration (i.e., to form -S-SH),30−32 and this process is significant as it provides a possible route by which H2S can alter functions of a wide range of cellular proteins and enzymes.33−35 H2S can also interact with S-nitrosothiols to yield thionitrous acid (HSNO), the smallest S-nitrosothiol, which can freely diffuse through membranes to facilitate transnitrosation of proteins.36 These reactions may be responsible for the biological functions of H2S. It is also possible that additional important biological reactions of H2S will be discovered. All of these findings suggest that regulation of H2S levels may have great therapeutic value.
In the H2S field, H2S releasing agents (also known as H2S donors) are important research tools.37,38 Moreover, H2S donors could potentially be unique therapeutic agents, given the well-documented protective effects of H2S in many disease states.37,38 Among these donors, sulfide salts, including sodium sulfide (Na2S) and sodium hydrogen sulfide (NaHS), are most frequently used. These salts have the advantage of boosting H2S concentrations rapidly. However, they release H2S spontaneously in aqueous solution, making it hard to control the precise H2S concentrations. In addition, H2S concentrations in aqueous solution can rapidly decrease due to volatilization, thus significantly limiting the use of these two H2S precursors.39
Considering these drawbacks, synthetic H2S donors have received considerable attention, and a number of donors have been reported.40,41 For example, a widely used donor is GYY4137, a derivative of Lawesson’s reagent.42 GYY4137 has been demonstrated to release H2S both in vitro and in vivo. GYY4137 can induce H2S-related biological effects such as vasodilation and anti-inflammation.42−45 However, the mechanism of H2S release from GYY4137 is still unclear. The extremely slow release of H2S from GYY4137 could also be problematic for its applications.46 Dithiolthiones (DTTs) are another class of synthetic H2S donors.38 DTTs have been widely used to prepare nonsteroidal anti-inflammatory drug (NSAIDs)-H2S hybrids. The resultant HS-NSAIDs, compared to the parent NSAIDs, showed similar anti-inflammatory effects but significantly decreased gastric damage.47−49 Although DTTs have shown promising H2S-related biological activities, their H2S release is also uncontrollable, and their H2S release profile has not been explored yet.
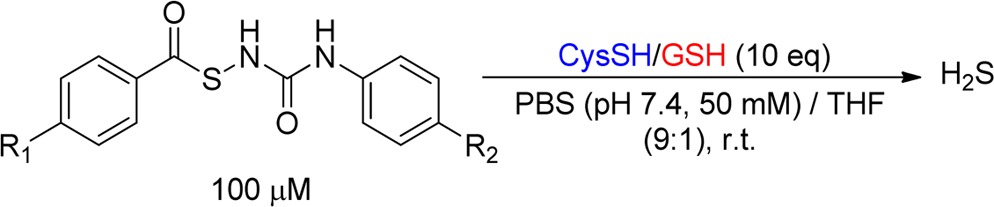
The problem with the majority of currently available donors is that the H2S generation from these compounds is uncontrollable. Commonly used donors such as sulfide salts and GYY4137 release H2S spontaneously upon hydrolysis, making it very difficult to obtain the precise physiological or pharmacological H2S levels needed. Modifications made between the time that a solution is prepared and the time that the biological effect is measured can dramatically affect research outcomes and sometimes can even get disparate results. From a therapeutic perspective and for applications in H2S-related biological research, ideal H2S donors should release H2S slowly in moderate amounts and/or in a highly controllable manner. To this end, our group introduced thiol-activated H2S donors in 2009.50 The idea is described in Scheme 1. It is known that S–N bonds are weak and can break under certain conditions.51−53 This property makes N-mercapto (N-SH) species, such as 1, useful templates for the design of H2S donors. As N-SH species are unstable and highly reactive, we expected that the protecting groups on −SH should enhance their stability. Moreover, the removal of protecting groups can be achieved by different strategies, making controllable H2S release possible. To prove this hypothesis, 12 N-(benzoylthio)benzamides were prepared as the first thiol-activated H2S donors. Some preliminary results regarding their H2S release profiles were reported.50 Recently, we expanded the scope of this type of donors and studied 33 substrates. Herein, we report the comprehensive results about these N-SH-based donors (NSHDs), including their synthesis, H2S release evaluation/mechanism, and structure–activity relationship (SAR). In addition, protective effects of selected donors on cellular oxidative damages and myocardial ischemia reperfusion (MI/R) injury are also reported.
Scheme 1. Design of NSHDs.

Results and Discussion
Synthesis of N-(Acylthio)amides as NSHDs
In the design of thiol-activated NSHDs, acyl groups were selected as the protecting groups on −SH. Both alkyl- and aryl-based acyl groups were used to prepare the donors. The synthesis is illustrated in Scheme 2. Readily available thiocarboxylic acids 2 were the starting materials. The treatment of 2 with hydroxylamine-O-sulfonic acid gave S-acylthiohydroxylamines 3 as the reactive intermediates, which were further reacted (without separation) with acid anhydrides to provide NSHDs 4. In total, 26 NSHDs were prepared using this route, and overall yields were 61–81%.
Scheme 2. Synthesis of NSHDs.

H2S Release of N-(Acylthio)amides
With these compounds in hand, we first tested their H2S release ability in aqueous buffers. N-(Acylthio)amides were found to be stable in buffers. Unlike hydrolysis-based donors, N-(acylthio)amides did not release H2S spontaneously. In addition, no H2S generation was observed in the presence of some potential cellular nucleophiles, such as serine and lysine. However, a time-dependent H2S release was detected when cysteine was added into the solutions, indicating that thiols are essential to trigger H2S release from these molecules.
In order to systematically compare the H2S generation capabilities of these donors, we studied the effects of donor concentrations, cysteine concentrations, and solvent systems, as well as reaction time/temperatures. Eventually, the optimized conditions were found to use 100 μM donors and 1000 μM cysteine in phosphate buffered saline (PBS) (pH 7.4) containing 10% tetrahydrofuran (THF). H2S release was monitored at room temperature for 2 h. The well-known methylene blue (MB) method was used to measure H2S generation. Using NSHD-1 as the example (Figure 1), a maximum of 56 μM of H2S (peak H2S concentration) at 50 min (peak time) was detected. As shown in Figure 1, higher concentrations of cysteine (i.e., 2 mM) did not significantly improve H2S release, while lower concentrations of cysteine (i.e 0.5 mM) dramatically decreased H2S production. In addition to cysteine, GSH’s ability to promote H2S release from NSHDs was also evaluated. Results showed that GSH (1 mM) can also successfully trigger H2S release at levels comparable to those of 1 mM cysteine.
Figure 1.
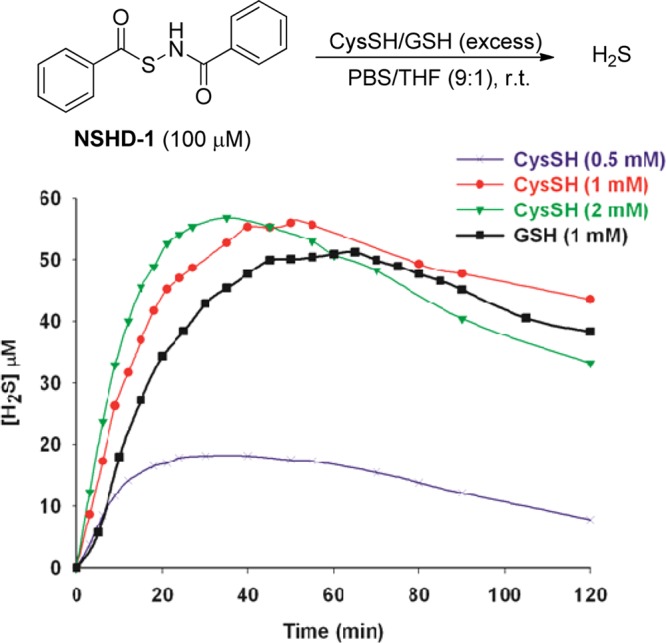
Cysteine and GSH-mediated H2S release from NSHD-1.
All NSHD substrates were then tested under the optimized conditions. Briefly, a solution of each donor (0.1 mM) and cysteine (or GSH, 1 mM) was prepared in a mixed solvent system PBS/THF (9:1, v/v). At different reaction times, 1.0 mL of reaction aliquots were taken and mixed with MB cocktails (0.5 mL). The MB reactions were carried out for 15 min, and absorbance at 670 nm was then measured. H2S concentrations were calculated based on a Na2S standard curve (please see the Experimental Procedures section for detailed procedures). Each donor was tested three times, and their average results are summarized in Table 1. The results showed that up to 68% of H2S can be detected within a 2-h experimental period, indicating that N-(acylthio)amides are potent H2S donors. As for SAR, electronic effects on H2S release were observed. Compared to the parent donor NSHD-1, donors with electron withdrawing groups (NSHD-2–NSHD-5) showed faster H2S release; donors with electron donating groups (NSHD-6–NSHD-12) exhibited slower H2S release. Steric effects were also observed as more sterically hindered substrates showed slower H2S release (NSHD-13–NSHD-15). In addition to the thioester moiety, SAR studies were also conducted on amide terminals (NSHD-20–NSHD-26). Results showed that all of these amide-modified donors exhibited similar H2S releasing abilities, suggesting that the amide end had little effects on the reactivity. In all GSH-treated groups, peak times were constantly 5–20 min slower than those in cysteine-treated groups. This is presumably due to the increased steric hindrance of GSH, therefore causing a slower reaction to release H2S. These findings demonstrated that structural modifications can regulate H2S release from NSHDs, therefore, achieving controllable H2S generation.
Table 1. H2S Release Profiles of N-(Acylthio)amides.
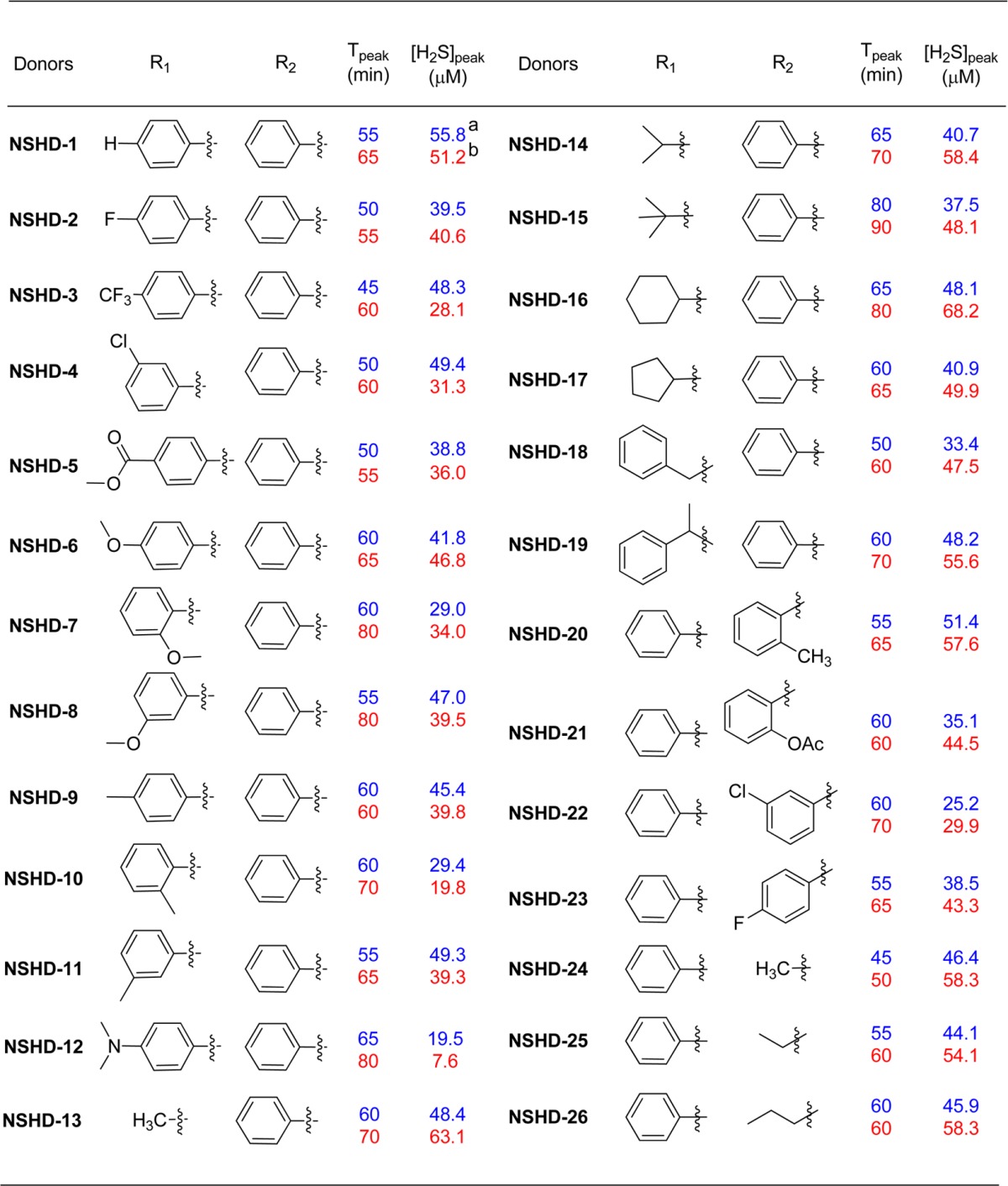
Data were obtained with cysteine.
Data were obtained with GSH.
Synthesis and H2S Release of 1-(Benzoylthio)-3-phenylureas
To expand the scope of the donors, we next replaced the amide moiety with urea templates. A series of 1-mercapto-3-phenylurea-based H2S donors (6) were synthesized (Scheme 3). Briefly, readily available thiocarboxylic acids were treated with hydroxylamine-O-sulfonic acid to form S-acylthiohydroxylamines 3, which were then conjugated with phenyl isocyanate derivatives (5) to provide 1-(benzoylthio)-3-phenylureas (6) as the desired products.
Scheme 3. Chemical Synthesis of 1-Mercapto-3-phenylurea-Based Donors.

H2S release from these donors was tested using the same protocol described above, and the results are shown in Table 2. Interestingly, these 1-(benzoylthio)-3-phenylurea based donors showed lower and slower H2S release as compared to N-(acylthio)amides.
Table 2. H2S Release Profiles of 1-(Benzoylthio)-3-phenylurea Based Donorsa.
| CysSH |
GSH |
|||||
|---|---|---|---|---|---|---|
| donors | R1 | R2 | Tpeak (min) | [H2S]peak (μM) | Tpeak (min) | [H2S]peak (μM) |
| NSHD-27 | H | H | 95 | 21.5 | 110 | 29.9 |
| NSHD-28 | OCH3 | H | 105 | 14.1 | 120 | 26.4 |
| NSHD-29 | CH3 | H | 105 | 16.5 | 130 | 20.0 |
| NSHD-30 | F | H | 75 | 18.8 | 90 | 18.5 |
| NSHD-31 | Cl | H | 95 | 18.2 | 120 | 24.9 |
| NSHD-32 | H | CH3 | 90 | 21.8 | 130 | 27.4 |
| NSHD-33 | H | Cl | 110 | 10.7 | 120 | 20.9 |
Data were reported as the average value of three measurements.
H2S Release Mechanism
To understand the mechanism of H2S release from these N-mercapto-based donors, a model reaction using NSHD-1 and cysteine (10 equiv) was studied. Three products, N-benzoyl cysteine 8, benzamide 10, and cystine 12, were obtained. On the basis of these products, the H2S release mechanism was proposed as follows (Scheme 4): a reversible thiol exchange between NSHD-1 and cysteine initiates the reaction. The resultant S-benzoyl cysteine 7 should then undergo a native chemical ligation (NCL)-type acyl transfer to form a more stable N-benzoyl cysteine 8. Meanwhile, the free N-SH species, N-mercaptobenzamide 9, should react with cysteine to yield benzamide 10 and cysteine perthiol 11. The latter product can further react with cysteine to liberate H2S and form cystine 12.
Scheme 4. Proposed Mechanism of H2S Release from NSHDs.

Cytotoxicity of NSHDs
With these NSHDs in hand, we next explored their H2S-induced biological activities. Recent studies have suggested that H2S can protect the cardiovascular system against myocardial ischemia reperfusion (MI/R) injury. It has been demonstrated that H2S (using Na2S or NaHS), when applied at the time of reperfusion or as a preconditioning reagent, exhibits significant cardioprotection by different mechanisms, such as reducing oxidative stress, preserving mitochondrial function, decreasing myocardial inflammation, and improving angiogenesis.20−24 Among these, H2S’s effects as an antioxidant may play a prominent role. We envisioned that NSHDs should exhibit similar cardioprotective activities due to H2S release.
Before conducting experiments to investigate NSHD’s cardioprotective actions, we tested the cytotoxicity of three representative donors (NSHD-1, NSHD-2, and NSHD-6) in H9c2 cardiomyoblasts. These three donors were selected because of their varied H2S release abilities. The cells, originating from rat heart ventricular tissue, have been widely used as an in vitro model for cardiac muscle in virtue of their morphological features and biochemical properties.54 In our experiment, H9c2 cells were incubated with each donor at different concentrations (20–160 μM) for 1 h. Cells were then washed with PBS twice to remove extracellular donors, and cell viability was measured by the standard CCK-8 assay. As shown in Figure 2, these donors did not exhibit significant cytotoxicity at low concentrations (<40 μM). However, cytotoxicity was observed when higher concentrations (>80 μM) were applied. Considering that cysteine and GSH are rich in living systems, which are expected to react with NSHDs and trigger H2S release, we envisioned NSHD-induced cytotoxicity could be diminished or avoided when cysteine or GSH is presented. To prove this hypothesis, H9c2 cells were then incubated with each donor (160 μM) in the presence of cysteine (480 μM). As expected, NSHD’s cytotoxicity was completely suppressed, suggesting that NSHDs should be safe in cysteine-rich environments.
Figure 2.

Cytotoxicity of NSHDs in the absence or presence of cysteine. H9c2 cells were incubated with NSHD-1, NSHD-2, and NSHD-6 at varied concentrations (20–160 μM) for 1 h in the absence or presence of cysteine (480 μM). Excess NSHDs were then washed away with PBS. The CCK-8 assay was applied to measure cell viability. Data were expressed as the mean ± SEM. *P < 0.05, **P < 0.01 vs the control group.
Establishment of an in Vitro Model of MI/R Injury
Under the condition of MI/R injury, oxidative stress (OS) plays a central role.55 OS-induced cell injury can be triggered by reactive oxygen species (ROS), including superoxide (O2•–), hydroxyl radical (•OH), singlet oxygen (•O), and hydrogen peroxide (H2O2).56,57 Among these, H2O2 is the dominant form due to its stability.58 Therefore, H2O2-treated H9c2 cells are usually used as an in vitro model of MI/R injury.59,60 To establish the cellular model of MI/R injury, H9c2 cells were treated with increasing concentrations of H2O2 (100–800 μM) for 5 h. As shown in Figure 3, exposure of H9c2 cells to H2O2 led to a dose-dependent inhibition in cell viability (P < 0.01), indicating that H2O2 was able to induce the oxidative injury in cardiomyocytes. This can partly imitate in vivo I/R-triggered effects. Since the median lethal concentration of H2O2 in H9c2 cells was approximately 400 μM, this concentration was used for the following studies.
Figure 3.

Effects of H2O2 on cell viability. H9c2 cells were treated with varied concentrations of H2O2 for 5 h. The CCK-8 assay was performed to detect cell viability. Data were expressed as the mean ± SEM. **P < 0.01 vs the control group.
Effects of NSHDs on H2O2-Induced Damage in H9c2 Cells
We next tested the protective capabilities of NSHDs against H2O2-induced cellular damage. In these experiments, H9c2 cells were pretreated with NSHD-1, NSHD-2, or NSHD-6 at varied concentrations (40, 80, and 160 μM) in the presence of cysteine (480 μM) for 1 h. Cells were then washed with PBS to remove extracellular NSHDs and cysteine. The donor-treated cells were challenged by H2O2 (400 μM) for another 5 h, after which cell viability was analyzed. Results showed that NSHDs exhibited significant attenuation of H2O2-induced damage (Figure 4A, B, and C). In order to compare these new donors with known donors, we also tested the effects of GYY4137 and NaHS, two well-studied H2S donors, under the same conditions. Similar protective effects were observed with GYY4137 and NaHS (40, 80, and 160 μM) (Figure 4D and E). To further prove that the protective effects of NSHDs were due to H2S release, we studied the effects of cysteine only (480 μM) and byproducts of NSHDs (i.e., N-benzoyl cysteine (NBC) and benzamide (BZM), at 160 μM). These species did not exhibit protection against H2O2-induced damage (Figure 4F). Taken together, these results suggested that NSHDs can protect cardiomyocytes from oxidative injury and that their protective activities are likely due to H2S release.
Figure 4.

Effects of H2S donors (A–E) and NSHD byproducts (F) on H2O2-induced cellular damage. Prior to the 5-h H2O2 (400 μM) treatment, H9c2 cells were preconditioned with NSHD-1 (A), NSHD-2 (B), and NSHD-6 (C) in the presence of cysteine (480 μM) at the indicated concentrations for 1 h. GYY4137 (D) and NaHS (E) were used as positive controls under similar conditions. Effects of H2S releasing byproducts, such as NBC and BZM (160 μM), and cysteine alone (480 μM) were also investigated (F). The CCK-8 assay was performed to detect cell viability. Data were expressed as the mean ± SEM. **P < 0.01 vs the control group. †P < 0.05, ‡P < 0.01 vs the H2O2 treatment group.
Measurement of LDH and MMP Levels
In addition to the cell viability assay, several other methods were also used to validate NSHD’s protective effects in cells. Lactate dehydrogenase (LDH) is a stable cytoplasmic enzyme located in the cytoplasm under normal conditions. When the heart suffers from MI/R injury, the cell membrane of cardiomyocytes is damaged, leading to the increase of cell membrane permeability and LDH leakage from the cells into the blood.61 In the present study, by comparing the contents of LDH in cytoplasm and medium, the LDH release rate can be used to indicate the extent of cellular injury. As shown in Figure 5, preconditioning of H9c2 cells with cysteine (480 μM, 1 h) followed by the exposure to H2O2 (400 μM, 5 h) remarkably enhanced LDH release, indicating that H2O2 induced severe damage to cells and that cysteine (480 μM) did not provide protection. However, LDH release was significantly reduced when cells were pretreated with 160 μM NSHD donors in the presence of cysteine (480 μM), demonstrating that NSHDs can protect cells from oxidative damage.
Figure 5.

Effects of NSHDs on H2O2-induced LDH release. H9c2 cells were preconditioned with NSHD-1, NSHD-2, and NSHD-6 (160 μM) in the presence of cysteine (480 μM) for 1 h followed by treatment with H2O2 (400 μM) for 5 h. LDH in cells and medium was measured with a commercial kit. Data were expressed as the mean ± SEM. **P < 0.01 vs the control group. ‡P < 0.01 vs the H2O2 treatment group.
Mitochondria are the energy factory of cells and sensitive to noxious stimuli. Mitochondrial membrane potential (MMP) usually reflects whether mitochondria are healthy, which further indicates whether cells are suffering from noxious damage. Rh-123 staining is a useful tool to observe MMP. Figure 6A shows that under normal conditions H9c2 cells had bright green fluorescence. When cells were preconditioned with cysteine (480 μM) for 1 h and then treated with H2O2 (400 μM) for 5 h, a dramatic MMP loss was observed, evidenced by weak green fluorescence, suggesting that H2O2 induced severe damage to cells and that cysteine (480 μM) did not provide significant protection (Figure 6B). However, preconditioning with 160 μM of NSHDs greatly impeded this MMP loss (Figures 6C–F) by preserving mitochondrial functions. These results further confirmed that NSHDs exhibited potent cellular protection against oxidative injury.
Figure 6.

Effects of H2S donors on H2O2-induced MMP loss in H9c2 cells. (A–E) Rh123 staining followed by photofluorography to observe MMP in H9c2 cells. (A) Control group. (B) Cells were pretreated with cysteine (480 μM) for 1 h, then exposed to H2O2 (400 μM) for 5 h. Cells were preconditioned with 160 μM NSHD-1 (C), NSHD-2 (D), and NSHD-6 (E) in the presence of cysteine (480 μM) for 1 h prior to H2O2 treatment. (F) Quantitative analysis for Rh123 in panels A–E. Mean fluorescence intensities (MFI) were measured using IMAGEJ software. Data were expressed as the mean ± SEM. **P < 0.01 vs the control group. ‡P < 0.01 vs the H2O2 treated group.
Cardioprotective Effects of NSHD-1 and NSHD-2 in MI/R Injury
Finally, we tested the protective effects of two representative donors, i.e., NSHD-1 and NSHD-2, against myocardial ischemia/reperfusion (MI/R) injury in a murine model system. In these experiments, mice were subjected to 45 min of left ventricular ischemia followed by 24 h reperfusion. NSHD-1, NSHD-2, or vehicle was administered by direct intracardiac (i.c.) injection at the time of reperfusion at different doses. All animal groups displayed similar area-at-risk per left ventricle (AAR/LV), which means that surgery caused a similar risk. However, compared to vehicle treated mice, mice receiving NSHD-1 and NSHD-2 displayed a dose-dependent reduction in infarct size per area-at-risk (INF/AAR), assessed by 2,3,5-triphenyltetrazolium chloride (TTC) staining (Figure 7). A 100 μg/kg bolus of NSHD-1 maximally reduced INF/AAR by ∼35%. Similarly, 50 μg/kg and 100 μg/kg of NSHD-2 reduced INF/AAR by ∼50% (p < 0.01, p < 0.001, respectively). Moreover, circulating cardiac troponin I (TnI) levels, the marker for acute myocardial infarction, were significantly lowered with NSHD-2 treated animals (p < 0.05 for 50 ug/kg and p < 0.01 for 100 ug/kg) (Figure 8). These results strongly suggest that N-mercapto compounds are potent H2S donors and may have some pharmacological benefits in biological systems. In the present study, we injected these H2S donors directly into the left ventricular cavity at the time of reperfusion. In the clinical setting, we believe that these agents could be administered via a number of routes to patients suffering from an acute myocardial infarction. These compounds could be administered via intravenous bolus injection or infusion or via intracoronary injection during coronary catheterization to open the occluded coronary artery. In previous studies, we have administered H2S donors into the systemic circulation with excellent results.62
Figure 7.

Cardioprotective effects of NSHD-1 and NSHD-2 in myocardial ischemia-reperfusion injury. Doses of NSHD-1, NSHD-2, or vehicle were injected at the time of reperfusion. (Left) Myocardial area-at-risk (AAR) per left ventricle (AAR/LV) and infarct per area-at-risk (INF/AAR) were assessed in vehicle (n = 12), NSHD-1 (50 μg/kg) treated animals (n = 6), and NSHD-1 (100 μg/kg) treated animals (n = 12). AAR/LV was similar among all groups. INF/AAR was significantly smaller in the 100 μg/kg dose treated animals. (Right) Myocardial area-at-risk (AAR) per left ventricle (AAR/LV) and infarct per area-at-risk (INF/AAR) were assessed in the vehicle (n = 12), NSHD-2 (50 μg/kg) treated (n = 12), and NSHD-2 (100 μg/kg) treated animals (n = 17). AAR/LV was similar among all groups. INF/AAR was significantly smaller in animals treated with either dose.
Figure 8.
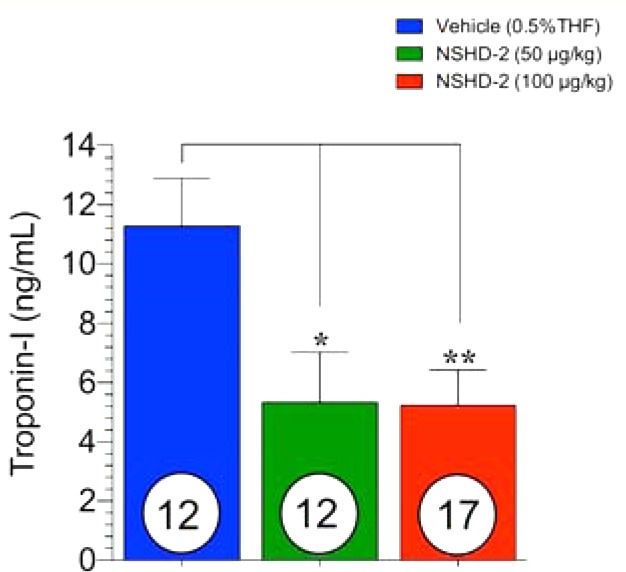
Blood was collected at 4 h of reperfusion, and circulating cardiac troponin I levels were measured. Troponin I level was significantly reduced with either 50 μg/kg or 100 μg/kg of NSHD-2 treatment.
Conclusions
In summary, we have developed a series of H2S donors based on a N-mercapto template. Their H2S release is triggered by cellular thiols such as cysteine or GSH. H2S release from these donors can be regulated by structural modifications. Moreover, we demonstrated that these donors are safe in cysteine-rich environments. Their protective effects on cellular oxidative injury and myocardial ischemia reperfusion injury were also observed. Taken together, our studies demonstrate that N-mercapto species are potent H2S donors and that they possess potential therapeutic benefits.
Experimental Procedures
Chemical Synthesis
All solvents were of reagent grade. THF was freshly distilled from sodium/benzophenone under argon. Reactions were magnetically stirred and monitored by thin layer chromatography (TLC) with 0.25 mm precoated silica gel plates. Flash chromatography was performed with silica gel 60 (particle size 0.040–0.062 mm). Yields refer to chromatographically and spectroscopically pure compounds, unless otherwise stated. Proton and carbon-13 NMR spectra were recorded on a 300 MHz spectrometer. Chemical shifts are reported relative to chloroform (δ7.26) for 1H NMR and chloroform (δ77.0) for 13C NMR. The purity of all of the compounds was at least 95%, determined by LC/MS. Compounds were also characterized by TLC, melting points, and NMR spectroscopy.
Synthesis of (N-Acylthio)amides (NSHD-1–NSHD-26)
General procedure: thiocarboxylic acids (1 mmol) were mixed with KOH (5 mmol) in water (10 mL). To this mixture was added hydroxylamine-O-sulfonic acid (3 mmol). The reaction mixture was stirred at room temperature for 5 min and then extracted with 15 mL of CH2Cl2 three times. Organic layers were then combined, dried (by MgSO4), and concentrated to give the crude product S-acylthiohydroxylamines 3. Without further purification, 3 was dissolved in CH2Cl2 (5 mL) containing acid anhydride (2 mmol). The reaction solution was stirred at room temperature for 12 h. Crude products were precipitated out and collected. Purification by column chromatography or recrystallization gave the final products. The characterization data of each donor is provided in the Supporting Information.
Synthesis of 1-(Benzoylthio)-3-phenylureas (NSHD-27–NSHD-33)
Each freshly prepared S-acylthiohydroxylamine 3 (1 mmol, see procedure described above) was mixed with the corresponding phenyl isocyanate derivative 5 (2 mmol) in CH2Cl2 (2 mL). The mixture was stirred at room temperature for 12 h. The crude products were precipitated out and collected. Recrystallization in CH2Cl2/hexanes gave the final products. The characterization data of each donor is provided in the Supporting Information.
H2S Measurement
H2S generation was initiated by adding 100 μL of a donor’s stock solution (30 mM in THF) into a 30 mL PBS (pH 7.4, 50 mM)/THF (9:1) solution containing cysteine or GSH (1.0 mM). Then, 1.0 mL of reaction aliquots were periodically taken and transferred to UV cuvettes containing MB cocktail (100 μL of zinc acetate (1% w/v), 200 μL of N,N-dimethyl-1,4-phenylenediamine sulfate (20 mM in 7.2 M HCl), and 200 μL of ferric chloride (30 mM in 1.2 M HCl)). The MB reaction was carried out for 15 min, and the absorbance (670 nm) of the resultant solution was determined using an UV–vis spectrometer (Thermo Evolution 300). The H2S concentration of each sample was calculated against a calibration curve, which was obtained with a series of Na2S solutions.
Product Analysis
NSHD-1 (129 mg 0.5 mmol) in 2 mL of MeOH was added to a stirred solution of l-cysteine (605 mg, 5 mmol) in 50 mL of PBS buffer (pH 7.4, 20 mM). After 1 h, 0.1 mL of solution was taken via syringe equipped with a 45 μM syringe filter, diluted with 0.4 mL of MeOH, and analyzed by HPLC (Thermo Scientific Surveyor, Hypersil gold column, 4.6 × 100 mm, water/CH3CN 80/20 2 min; 60/40 4 min; 20/80 2 min; 80/20 2 min, flow rate 1 mL/min). Compounds 8, 10, and 12 were identified and quantified as compared to standard compounds. These compounds were also isolated from the reaction mixture and characterized. The yields were 80%, 96%, and 95%, respectively.
Cell Culture
H9c2 cells were purchased from the American Type Culture Collection (Manassas, VA, USA). The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) high glucose medium supplemented with 10% fetal bovine serum (FBS) at 37 °C under 5% CO2.
Cell Viability Assay
The cell counting kit (CCK)-8 (Dojindo Laboratory, Kumamoto, Japan) was applied to measure the cell viability of H9c2 cells cultured in 96-well plates. After the indicated cell treatment, 100 μL of CCK-8 solution at 1:10 dilution was added to each well, and cells were incubated for a further 3 h at 37 °C. Absorbance was measured at 450 nm with a microplate reader (Tecan Infinite F200, Switzerland). The mean optical density (OD) of 4 wells in each group was used to calculate cell viability as follows (experiments were performed in triplicate):
Protective Activities of NSHDs on H2O2-Induced Cellular Damage
H9c2 cells were treated with NSHD-1, NSHD-2, and NSHD-6 at varied concentrations (40–160 μM) for 1 h in the presence of cysteine (480 μM). Excess donors and cysteine were washed away with PBS (pH 7.4). Cells were then incubated with H2O2 (400 μM) for an additional 5 h. The control group was set up by incubating H9c2 cells with H2O2 (400 μM) only. Cell viability was measured using the CCK-8 assay. GYY4137 and NaHS were used as positive controls, and experiments were set up in a similar manner in the absence of cysteine. Cysteine (480 μM), NBC (160 μM), and BZM (160 μM) were incubated with cells as well to rule out their effects on cell viability.
LDH Measurement
The rate of LDH release was determined with a commercial LDH kit supplied by Thermo Fisher Scientific Inc. (Pittsburgh PA, US). Briefly, H9c2 cells were inoculated in a 96-well plate and grew to about 70% confluence. After the indicated treatments, 50 μL of supernatant per well was carefully removed and transferred into corresponding wells for the determination of extracellular LDH levels. Then, 100 μL of DMEM with 2% Triton X-100 was added to the adherent cells to lyse the cells. Fifty microliters of cell lysate was transferred to a 96-well plate to determine intracellular LDH levels. The same volume of prepared reaction mixture was added to the supernatant or homogenate, separately, and reacted for 10 min at room temperature by gentle shaking. Stopping solution was added to stop the reaction. Absorbance was measured at 490 and 600 nm with a microplate reader (Tecan Infinite F200, Switzerland). LDH release rate was calculated as follows (experiments were performed 4 times):
MMP Measurement
Mitochondrial membrane potential (MMP) was observed using a fluorescent dye, rhodamine 123 (Rh123), a cell-permeable cationic dye that preferentially enters the mitochondria based on the highly negative MMP. Depolarization of MMP usually leads to a decreased intake of Rh123 and an intracellular weak fluorescence. After the indicated treatments, 10 mg/L Rh123 was added to cell cultures for 15 min at 37 °C, and fluorescence was measured over the entire field of vision using a fluorescent microscope (Advanced Microscopy Group, Seattle, US). The mean fluorescent intensity (MFI) of Rh123 from four random fields was analyzed using IMAGEJ 1.41o software.
Cardioprotective Effects in MI/R Injury
Animals
Male C57BL/6J mice, 10–12 weeks of age (Jackson Laboratories, Bar Harbor, ME) were used in the present study. All animals were housed in a temperature-controlled animal facility with a 12-h light/dark cycle, with water and rodent chow provided ad libitum. All animals received humane care in compliance with the Principles of Laboratory Animal Care formulated by the National Society of Medical Research and the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health. All animal procedures were approved by the Louisiana State University Health Science Center Institutional Animal Care and Use Committee.
Myocardial Ischemia/Reperfusion (MI/R) Protocol
Surgical occlusion of the left coronary artery (LCA), infarct size determination, and troponin I (TnI) measurements were performed as previously described.63 C57BL6/J male mice (10–12 weeks of age) were anesthetized using pentobarbital (50 mg/kg) and ketamine (60 mg/kg) and maintained under anesthesia throughout the procedure with additional administration of pentobarbital (50 mg/kg). Mice were intubated and mechanically ventilated using a Hugo Sachs type 845 minivent. A hair removal agent was used to remove the hair on the chest of mice. The area was then cleaned using betadine and alcohol wipes alternating 3 times. An incision was made proximally left of the midline of the chest, exposing the second and third ribs. The chest was opened by cauterizing through the second and third rib. The LCA was visualized, and using a 7–0 silk suture, a 3–5 mm piece of PE-10 tubing was secured over the LCA, occluding the artery. Mice were subjected to 45 min of myocardial injury, and at the time of reperfusion, an injection of vehicle (0.5% THF), NSHD-1 (50 μg/kg and 100 μg/kg), or NSHD-2 (50 μg/kg) was given i.c. The chest was closed in layers, closing the muscle layer first, then the skin. Mice were removed from ventilation once pedal reflex returned and it was determined that respiration was spontaneous. Mice were placed in a recovery cage and supplied with 100% O2. Reperfusion was allowed for 24 h, and blood samples for TnI measurements were taken at 4 h after time of reperfusion. At 24 h of reperfusion, the LV area at risk (AAR) and infarct size were determined by Evans blue and 2,3,5-tetrazolium chloride stainin, as previously described.63
Drug Preparation
On the day of experimentation, test compounds (NSHD-1 or NSHD-2) were diluted in 0.2 mL of 100% THF solution. For in vivo experiments, the test compounds were further diluted in sterile saline to final concentrations of 0.05 or 0.1 mg/mL. Then, various volumes were administered based on the animals’ weights to obtain the correct dosage. The resulting concentration of THF in each dosage was 0.5% v/v. The vehicle consisted of a solution of 0.5% v/v THF in sterile saline.
Cardiac Troponin-I Assay
Blood samples were collected via a tail vein at 4 h of reperfusion. Cardiac troponin I level was measured in serum using the Life Diagnostic highsensitivity mouse cardiac troponin I ELISA kit (Mouse Cardiac Tn-I ELISA Kit; Life Diagnostics, West Chester, PA) following the manufacturer’s instructions.
Acknowledgments
We would like to thank Orleiquis Guerra Mulgado for technical support. This work is supported by an American Chemical Society-Teva USA Scholar Grant to M.X., National Institutes of Health (R01HL116571 to M.X. and D.J.L.), and Natural Science Foundation of China (81200606) to Y.C.
Glossary
Abbreviations Used
- H2S
hydrogen sulfide
- NO
nitric oxide
- CO
carbon monoxide
- CBS
cystathionine β-synthase
- CSE
cystathionine γ-lyase
- MPST
3-mercaptopyruvate sulfur-transferase
- S-SH
S-sulfhydration
- HSNO
thionitrous acid
- Na2S
sodium sulfide
- NaHS
sodium hydrogen sulfide
- DTTs
dithiolthiones
- NSAIDs
nonsteroidal anti-inflammatory drugs
- N-SH
N-mercapto
- NSHDs
N-SH-based donors
- SAR
structure–activity relationship
- MI/R
myocardial ischemia reperfusion
- MB
methylene blue
- GSH
reduced glutathione
- PBS
phosphate buffered saline
- NCL
native chemical ligation
- CCK-8
cell counting kit-8
- OS
oxidative stress
- ROS
reactive oxygen species
- O2•–
superoxide
- •OH
hydroxyl radical
- •O
singlet oxygen
- H2O2
hydrogen peroxide
- NBC
N-benzoyl cysteine
- BZM
benzamide
- LDH
lactate dehydrogenase
- MMP
mitochondrial membrane potential
- i.c.
intracardiac
- AAR/LV
area-at-risk per left ventricle
- INF/AAR
infarct size per area-at-risk
- TTC
2,3,5-triphenyltetrazolium chloride
- TnI
troponin I
- AAR
area-at-risk
- THF
tetrahydrofuran
- TLC
thin layer chromatography
- DMEM
Dulbecco’s modified Eagle’s medium
- FBS
fetal bovine serum
- OD
optical density
- MFI
mean fluorescent intensity
- LCA
left coronary artery
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.5b01033.
Detailed synthetic procedures, characteristic data, and experimental procedures (PDF)
Author Contributions
⊥ Z.Y. and Y.C. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Li L.; Moore P. K. H2S and cell signaling. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 169–187. 10.1146/annurev-pharmtox-010510-100505. [DOI] [PubMed] [Google Scholar]
- Fukuto J. M.; Carrington S. J.; Tantillo D. J.; Harrison J. G.; Ignarro L. J.; Freeman B. A.; Chen A.; Wink D. A. Small molecule signaling agents: the integrated chemistry and biochemistry of nitrogen oxides, oxides of carbon, dioxygen, hydrogen sulfide, and their derived species. Chem. Res. Toxicol. 2012, 25, 769–793. 10.1021/tx2005234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandiver M. S.; Snyder S. H. Hydrogen sulfide: a gasotransmitter of clinical relevance. J. Mol. Med. 2012, 90, 255–263. 10.1007/s00109-012-0873-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol. Rev. 2012, 92, 791–896. 10.1152/physrev.00017.2011. [DOI] [PubMed] [Google Scholar]
- Olson K. R.; Donald J. A.; Dombkowski R. A.; Perry S. F. Evolutionary and comparative aspects of nitric oxide, carbon monoxide and hydrogen sulfide. Respir. Physiol. Neurobiol. 2012, 184, 117–129. 10.1016/j.resp.2012.04.004. [DOI] [PubMed] [Google Scholar]
- Kolluru G. K.; Shen X.; Bir S. C.; Kevil C. G. Hydrogen sulfide chemical biology: Pathophysiological roles and detection. Nitric Oxide 2013, 35, 5–20. 10.1016/j.niox.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono K.; Akaike T.; Sawa T.; Kumagai Y.; Wink D.; Tantillo D. J.; Hobbs A. J.; Nagy P.; Xian M.; Lin J.; Fukuto J. M. The redox chemistry and chemical biology of H2S, hydropersulfides, and derived species: implications to their possible biological activity and utility. Free Radical Biol. Med. 2014, 77, 82–94. 10.1016/j.freeradbiomed.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabil O.; Banerjee R. Enzymology of H2S biogenesis, decay and signaling. Antioxid. Redox Signaling 2014, 20, 770–782. 10.1089/ars.2013.5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King B. S. Potential biological chemistry of hydrogen sulfide (H2S) with the nitrogen oxides. Free Radical Biol. Med. 2013, 55, 1–7. 10.1016/j.freeradbiomed.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C. Roles of hydrogen sulfide in the pathogenesis of diabetes mellitus and its complications. Antioxid. Redox Signaling 2012, 17, 68–80. 10.1089/ars.2011.4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H. Production and physiological effects of hydrogen sulfide. Antioxid. Redox Signaling 2014, 20, 783–793. 10.1089/ars.2013.5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolluru G. K.; Shen X.; Kevil C. G. A tale of two gases: NO and H2S, foes or friends for life?. Redox Biol. 2013, 1, 313–318. 10.1016/j.redox.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhn C.; Dubrovska G.; Huang Y.; Gollasch M. Hydrogen sulfide: potent regulator of vascular tone and stimulator of angiogenesis. Int. J. Biomed. Sci. 2012, 8, 81–86. [PMC free article] [PubMed] [Google Scholar]
- Jackson-Weaver O.; Osmond J. M.; Riddle M. A.; Naik J. S.; Gonzalez Bosc L. V.; Walker B. R.; Kanagy N. L. Hydrogen sulfide dilates rat mesenteric arteries by activating endothelial large-conductance Ca2+-activated K+ channels and smooth muscle Ca2+ sparks. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1446–H1454. 10.1152/ajpheart.00506.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariyaratnam P.; Loubani M.; Morice A. H. Hydrogen sulphide vasodilates human pulmonary arteries: a possible role in pulmonary hypertension. Microvasc. Res. 2013, 90, 135–137. 10.1016/j.mvr.2013.09.002. [DOI] [PubMed] [Google Scholar]
- Wallace L.; Vong L.; Mcknight W.; Dicay M.; Martin G. R. Endogenous and exogenous hydrogen sulfide promotes resolution of colitis in rats. Gastroenterology 2009, 137, 569–578. 10.1053/j.gastro.2009.04.012. [DOI] [PubMed] [Google Scholar]
- Wallace J. L. Hydrogen sulfide-releasing anti-inflammatory drugs. Trends Pharmacol. Sci. 2007, 28, 501–505. 10.1016/j.tips.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Kashfi K. Anti-cancer activity of new designer hydrogen sulfide-donating hybrids. Antioxid. Redox Signaling 2014, 20, 831–846. 10.1089/ars.2013.5308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaium M. A.; Liu Y.; Zhu Q.; Liu C.; Duan J.; Tan B.; Zhu Y. Z. H2S donor, S-propargyl-cysteine, increases CSE in SGC-7901 and cancer-induced mice: evidence for a novel anti-cancer effect of endogenous H2S?. PLoS One 2011, 6, e20525. 10.1371/journal.pone.0020525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefer D. J. A new gaseous signaling molecule emerges: cardioprotective role of hydrogen sulfide. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 17907–17908. 10.1073/pnas.0709010104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polhemus D. J.; Lefer D. J. Emergence of hydrogen sulfide as an endogenous gaseous signaling molecule in cardiovascular disease. Circ. Res. 2014, 114, 730–737. 10.1161/CIRCRESAHA.114.300505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Predmore B. L.; Lefer D. J.; Gojon G. Hydrogen sulfide in biochemistry and medicine. Antioxid. Redox Signaling 2012, 17, 119–140. 10.1089/ars.2012.4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King A. L.; Lefer D. J. Cytoprotective actions of hydrogen sulfide in ischemia-reperfusion injury. Exp. Physiol. 2011, 96, 840–846. 10.1113/expphysiol.2011.059725. [DOI] [PubMed] [Google Scholar]
- Calvert J. W.; Coetzee W. A.; Lefer D. J. Novel insight into hydrogen sulfide-mediated cytoprotection. Antioxid. Redox Signaling 2010, 12, 1203–1217. 10.1089/ars.2009.2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haouzi P.; Bell H.; Philmon M. Hydrogen sulfide oxidation and the arterial chemoreflex: effect of methemoglobin. Respir. Physiol. Neurobiol. 2011, 177, 273–283. 10.1016/j.resp.2011.04.025. [DOI] [PubMed] [Google Scholar]
- Dorman D. C.; Moulin F. J.; McManus B. E.; Mahle K. C.; James R. A.; Struve M. Cytochrome oxidase inhibition induced by acute hydrogen sulfide inhalation: correlation with tissue sulfide concentrations in the rat brain, liver, lung, and nasal epithelium. Toxicol. Sci. 2002, 65, 18–25. 10.1093/toxsci/65.1.18. [DOI] [PubMed] [Google Scholar]
- Filipovic M. R.; Miljkovic J.; Allgauer A.; Chaurio R.; Shubina T.; Herrmann M.; Ivanovic-Burmazovic I. Biochemical insight into physiological effects of H2S: reaction with peroxynitrite and formation of a new nitric oxide donor, sulfinyl nitrite. Biochem. J. 2012, 441, 609–621. 10.1042/BJ20111389. [DOI] [PubMed] [Google Scholar]
- Jones C. M.; Lawrence A.; Wardman P.; Burkitt M. J. Electron paramagnetic resonance spin trapping investigation into the kinetics of glutathione oxidation by the superoxide radical: re-evaluation of the rate constant. Free Radical Biol. Med. 2002, 32, 982–990. 10.1016/S0891-5849(02)00791-8. [DOI] [PubMed] [Google Scholar]
- Carballal S.; Trujillo M.; Cuevasanta E.; Bartesaghi S.; Möller M. N.; Folkes L. K.; Garcia-Bereguian M. A.; Gutierrez-Merino C.; Wardmann P.; Denicola A.; Radi R.; Alvarez B. Reactivity of hydrogen sulfide with peroxynitrite and other oxidants of biological interest. Free Radical Biol. Med. 2011, 50, 196–205. 10.1016/j.freeradbiomed.2010.10.705. [DOI] [PubMed] [Google Scholar]
- Paulsen C. E.; Carroll K. S. Cysteine-mediated redox signaling: chemistry, biology, and tools for discovery. Chem. Rev. 2013, 113, 4633–4679. 10.1021/cr300163e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J.; Carroll K. S. Persulfide reactivity in the detection of protein s-sulfhydration. ACS Chem. Biol. 2013, 8, 1110–1116. 10.1021/cb4001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D.; Macinkovic I.; Devarie-Baez N. O.; Pan J.; Park C. M.; Carroll K. S.; Filipovic M. R.; Xian M. Detection of protein S-sulfhydration by a tag-switch technique. Angew. Chem., Int. Ed. 2014, 53, 575–581. 10.1002/anie.201305876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadalla M. M.; Snyder S. H. Hydrogen sulfide as a gasotransmitter. J. Neurochem. 2010, 113, 14–26. 10.1111/j.1471-4159.2010.06580.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan N.; Fu C.; Pappin D. J.; Tonks N. K. H2S-Induced sulfhydration of the phosphatase PTP1B and its role in the endoplasmic reticulum stress response. Sci. Signaling 2011, 4, ra86. 10.1126/scisignal.2002329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul B. D.; Snyder S. H. H2S signalling through protein sulfhydration and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 499–507. 10.1038/nrm3391. [DOI] [PubMed] [Google Scholar]
- Filipovic M. R.; Miljkovic J. L.; Nauser T.; Royzen M.; Klos K.; Shubina T.; Koppenol W. H.; Lippard S. J.; Ivanović-Burmazović I. Chemical characterization of the smallest S-nitrosothiol, HSNO; cellular cross-talk of H2S and S-nitrosothiols. J. Am. Chem. Soc. 2012, 134, 12016–12027. 10.1021/ja3009693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Biggs T. D.; Xian M. Hydrogen sulfide (H2S) releasing agents: chemistry and biological applications. Chem. Commun. 2014, 50, 11788–11805. 10.1039/C4CC00968A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caliendo G.; Cirino G.; Santagada V.; Wallace J. L. Synthesis and biological effects of hydrogen sulfide (H2S): development of H2S-releasing drugs as pharmaceuticals. J. Med. Chem. 2010, 53, 6275–6286. 10.1021/jm901638j. [DOI] [PubMed] [Google Scholar]
- DeLeon E. R.; Stoy G. F.; Olson K. R. Passive loss of hydrogen sulfide in biological experiments. Anal. Biochem. 2012, 421, 203–207. 10.1016/j.ab.2011.10.016. [DOI] [PubMed] [Google Scholar]
- Kashfi K.; Olson K. R. Biology and therapeutic potential of hydrogen sulfide and hydrogen sulfide-releasing chimeras. Biochem. Pharmacol. 2013, 85, 689–703. 10.1016/j.bcp.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W.; Cheng Y. Z.; Zhu Y. Z. Hydrogen sulfide and translational medicine. Acta Pharmacol. Sin. 2013, 34, 1284–1291. 10.1038/aps.2013.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Whiteman M.; Guan Y. Y.; Neo K. L.; Cheng Y.; Lee S. W.; Zhao Y.; Baskar R.; Tan C. H.; Moore P. K. Characterization of a novel, water-soluble hydrogen sulfide-releasing molecule (GYY4137): new insights into the biology of hydrogen sulfide. Circulation 2008, 117, 2351–2360. 10.1161/CIRCULATIONAHA.107.753467. [DOI] [PubMed] [Google Scholar]
- Whiteman M.; Li L.; Rose P.; Tan C. H.; Parkinson D. B.; Moore P. K. The effect of hydrogen sulfide donors on lipopolysaccharide-induced formation of inflammatory mediators in macrophages. Antioxid. Redox Signaling 2010, 12, 1147–1154. 10.1089/ars.2009.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Fox B.; Keeble J.; Salto-Tellez M.; Winyard P. G.; Wood M. E.; Moore P. K.; Whiteman M. The complex effects of the slow-releasing hydrogen sulfide donor GYY4137 in a model of acute joint inflammation and in human cartilage cells. J. Cell Mol. Med. 2013, 17, 365–376. 10.1111/jcmm.12016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Salto-Tellez M.; Tan C. H.; Whiteman M.; Moore P. K. GYY4137, a novel hydrogen sulfide-releasing molecule, protects against endotoxic shock in the rat. Free Radical Biol. Med. 2009, 47, 103–113. 10.1016/j.freeradbiomed.2009.04.014. [DOI] [PubMed] [Google Scholar]
- Lee Z. W.; Zhou J.; Chen C. S.; Zhao Y.; Tan C. H.; Li L.; Moore P. K.; Deng L. W. The slow-releasing hydrogen sulfide donor, GYY4137, exhibits novel anti-cancer effects in vitro and in vivo. PLoS One 2011, 6, e21077. 10.1371/journal.pone.0021077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gargallo C. J.; Lanas A. Is NSAIDs-related gastrointestinal damage preventable?. J. Dig. Dis. 2013, 14, 55–61. 10.1111/1751-2980.12019. [DOI] [PubMed] [Google Scholar]
- Sparetore A.; Santus G.; Giustarini D.; Rossi R.; Del Soldato P. Therapeutic potential of new hydrogen sulfide-releasing hybrids. Expert Rev. Clin. Pharmacol. 2011, 4, 109–121. 10.1586/ecp.10.122. [DOI] [PubMed] [Google Scholar]
- Chan M. V.; Wallace J. L. Hydrogen sulfide-based therapeutics and gastrointestinal diseases: translating physiology to treatments. Am. J. Physiol. 2013, 305, G467–G473. 10.1152/ajpgi.00169.2013. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Wang H.; Xian M. Cysteine-activated hydrogen sulfide donors. J. Am. Chem. Soc. 2011, 133, 15–17. 10.1021/ja1085723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Xian M. Fast reductive ligation of S-nitrosothiols. Angew. Chem., Int. Ed. 2008, 47, 6598–6601. 10.1002/anie.200801654. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Wang H.; Xian M. An unexpected bis-ligation of S-nitrosothiols. J. Am. Chem. Soc. 2009, 131, 3854–3855. 10.1021/ja900370y. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Li S.; Zhang D.; Wang H.; Whorton A. R.; Xian M. Reductive ligation mediated one-step disulfide formation of S-nitrosothiols. Org. Lett. 2010, 12, 4208–4211. 10.1021/ol101863s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branco A. F.; Pereira S. L.; Moreira A. C.; Holy J.; Sardao V. A.; Oliveira P. J. Isoproterenol cytotoxicity is dependent on the differentiation state of the cardiomyoblast H9c2 cell line. Cardiovasc. Toxicol. 2011, 11, 191–203. 10.1007/s12012-011-9111-5. [DOI] [PubMed] [Google Scholar]
- Dhalla N. S.; Golfman L.; Takeda S.; Takeda N.; Nagano M. Evidence for the role of oxidative stress in acute ischemic heart disease: a brief review. Can. J. Cardiol. 1999, 15, 587–593. [PubMed] [Google Scholar]
- Takano H.; Zou Y.; Hasegawa H.; Akazawa H.; Nagai T.; Komuro I. Oxidative stress-induced signal transduction pathways in cardiac myocytes: involvement of ROS in heart diseases. Antioxid. Redox Signaling 2003, 5, 789–794. 10.1089/152308603770380098. [DOI] [PubMed] [Google Scholar]
- D’Autreaux B.; Toledano M. B. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- Rhee S. G.; Chang T. S.; Bae Y. S.; Lee S. R.; Kang S. W. Cellular regulation by hydrogen peroxide. J. Am. Soc. Nephrol. 2003, 14, S211–S215. [DOI] [PubMed] [Google Scholar]
- Chou H. C.; Chen Y. W.; Lee T. R.; Wu F. S.; Chan H. T.; Lyu P. C.; Timms J. F.; Chan H. L. Proteomics study of oxidative stress and Src kinase inhibition in H9C2 cardiomyocytes: a cell model of heart ischemia-reperfusion injury and treatment. Free Radical Biol. Med. 2010, 49, 96–108. 10.1016/j.freeradbiomed.2010.04.001. [DOI] [PubMed] [Google Scholar]
- Szabo G.; Veres G.; Radovits T.; Gero D.; Modis K.; Miesel-Groschel C.; Horkay F.; Karck M.; Szabo C. Cardioprotective effects of hydrogen sulfide. Nitric Oxide 2011, 25, 201–210. 10.1016/j.niox.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy L. J.; Goeddel D. V.; Wong G. H. Tumor necrosis factor-alpha pretreatment is protective in a rat model of myocardial ischemia-reperfusion injury. Biochem. Biophys. Res. Commun. 1992, 184, 1056–1059. 10.1016/0006-291X(92)90698-K. [DOI] [PubMed] [Google Scholar]
- Calvert J. W.; Jha S.; Gundewar S.; Elrod J. W.; Ramachandran A.; Pattillo C. B.; Kevil C. G.; Lefer D. J. Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ. Res. 2009, 105, 365–374. 10.1161/CIRCRESAHA.109.199919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvert J. W.; Elston M.; Nicholson C. K.; Gundewar S.; Jha S.; Elrod J. W.; Ramachandran A.; Lefer D. J. Genetic and pharmacologic hydrogen sulfide therapy attenuates ischemia-induced heart failure in mice. Circulation 2010, 122, 11–19. 10.1161/CIRCULATIONAHA.109.920991. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.