

Abstract
The biosynthesis of nitriles is known to occur in specialized pathways involving multiple enzymes; however, in bacterial and archeal biosyntheses of 7-deazpurines, a single enzyme, ToyM, catalyzes the conversion of the carboxylic acid containing 7-carboxy-7-deazaguanine (CDG) to its corresponding nitrile, 7-cyano-7-deazaguanine (preQ0). The mechanism of this unusual direct transformation is shown to entail adenylation of CDG activating it to form the newly discovered amide intermediate, 7-amido-7-dezaguanine (ADG), which is subsequently dehydrated to form the nitrile consuming a second equivalent of ATP. The authentic amide intermediate is shown to be chemically and kinetically competent. The ability of ToyM to activate two substrates, acid and amide, accounts for this unprecedented one-enzyme catalysis of nitrile synthesis, and the differential rates of these two half reactions suggest this catalytic ability is derived from an amide synthetase that gained this new function.
Keywords: biosynthesis, bioorganic chemistry, enzymes, antibiotics, evolution
Graphical Abstract
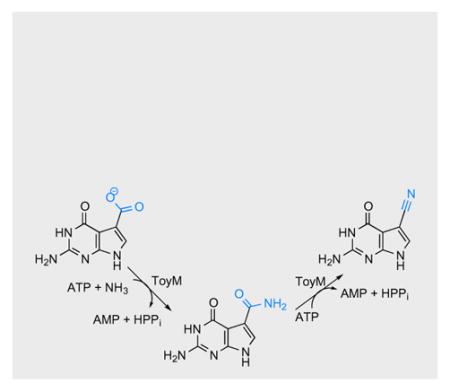
Nitrile containing products are widespread in nature in secondary metabolites such as cyanoglucosides and alkanenitriles.[1] Though these are increasingly being recognized as important next generation antibiotics and cytotoxic agents with pharmaceutical potential,[2,3] the biosynthesis of these compounds is very poorly understood. Only one biosynthetic route utilizing at least three separate enzymes has previously been described with mechanistic detail involving aldoxime formation[4] and a subsequent dehydration[5,6] (Figure 1). The discovery of the 7-deazapurine biosynthetic operon of Streptomyces rimosus led to the first enzyme capable of acting on a carboxylic acid to form a nitrile[7] (Figure 1). This nitrile synthetase, ToyM, was shown to be ATP dependent and to use ammonia as the source of the nitrile nitrogen.[8] Homologs of ToyM are widely distributed as this nitrilation is essential in the biosynthesis of the modified nucleosides archaeosine and the ubiquitous queuosine, as well as many pyrrolopyrimidine nucleoside antibiotics including toyocamycin and sangivamycin.[9]
Figure 1.
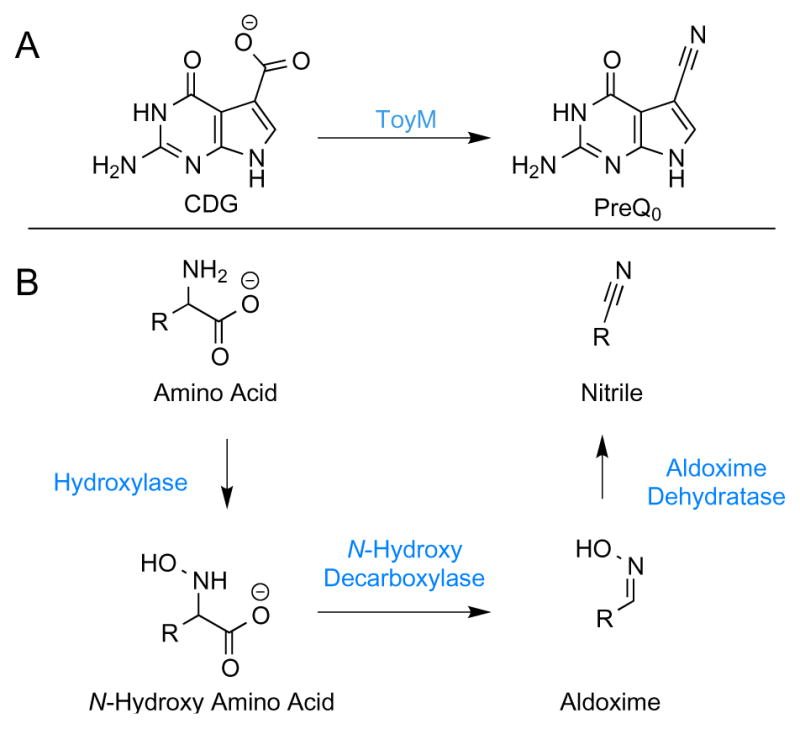
Two biosynthetic paradigms to nitrile secondary moieties in nature.
ToyM bears evolutionary similarity to glutamine dependent asparagine synthetases and the lysidine forming enzyme, TilS, all of which employ ATP in the adenylation of substrate molecules poising them for condensation with amine nitrogens.[10,11] The crystal structure of ToyM, solved before its enzymatic function was known, showed the presence of a structural zinc divalent cation and revealed that it adopts a PPi loop structure characteristic of ATP utilizing enzymes.[12] This suggests the mechanistic strategy employed by ToyM may involve adenylation. The simplest mechanism for ToyM (Figure 2) would predict adenylation to activate CDG, followed by addition of ammonia to generate 7-amido-7-deazaguanine (ADG). Activation of ADG by a second equivalent of ATP would precede collapse to form the nitrile product. Such a mechanism would be a rare example of a single enzyme activating two functional groups, both the acid as well and the resultant amide intermediate, and present a new paradigm for biosynthesis of nitriles from carboxylic acids.
Figure 2.
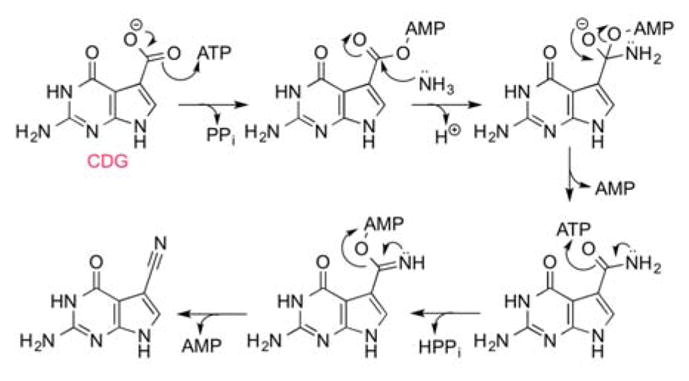
Proposed mechanism of ToyM
To examine the nitrilation catalyzed by ToyM the recombinant protein was produced from E. coli expression host. Protein was quantified by amino acid analysis, and metals analysis confirmed the presence of 1.1 ± 0.2 equivalents of Zn2+ per monomer. To test the mechanistic proposal in Figure 2, we incubated ToyM with CDG, ATP, and magnesium, and monitored the reaction by LC-MS (Supporting Figure S1). We found that magnesium supplementation significantly increased product formation (Supporting Figure S2 A), and it was routinely included in the assays. The HPLC traces showed that preQ0 was produced as previously shown, as well as an intermediate that increased as CDG was consumed and decreased as preQ0 formed (Figure 3A). This intermediate exhibited a UV-visible spectrum highly similar to that of CDG (Supporting Figure S3) and a mass consistent with 7-amido-7-deazaguanine (ADG) ([M+H]+ m/z of 194.0670; theoretical 194.0678). As with preQ0, added magnesium was required to observe maximal quantities of the intermediate (Supporting Figure S2 B).
Figure 3.
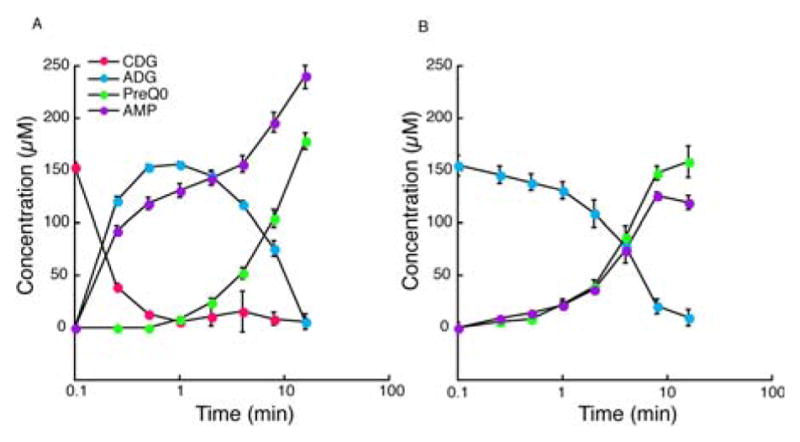
A) Reaction of ToyM with CDG, ammonium chloride, and ATP. B) Reaction of ToyM with ADG and ATP.
The identity of this intermediate was confirmed by comparison with the UV-visible spectra and the mass spectra of authentic ADG, which was synthesized enzymatically from preQ0 using ToyJKL, a nitrile hydratase from the same pathway as ToyM.[13] To determine if ADG is a bona fide intermediate, it was incubated with ToyM and ATP. In these experiments (Figure 3B), preQ0 was formed at the expense of ADG with nearly identical t1/2 values (~ 4 min). In addition, AMP also formed with a profile that nearly mirrored the formation of preQ0, suggesting that adenylation of ADG is the likely mode of activation. Therefore, the formation of preQ0 from CDG appears to occur in two ATP-dependent half reactions, the first of which leads to formation of ADG, which is converted to preQ0 in the second step.
Direct evidence of the nature of the activated intermediate was obtained by combining ToyM, CDG, and ATP in the absence of ammonia. This reaction was filtered to remove enzyme and analyzed with ion pairing LC-MS in the positive ion mode. In these experiments, we detect formation of an intermediate which elutes at ~25 min. Examination of the MS suggested that the peak has m/z of 524.1030, which is consistent with adenylated CDG (theoretical m/z 524.1044) (Figure 4). The appearance of this new peak is dependent on the presence of ToyM, CDG, and ATP, but it is abolished in the presence of ammonia (Figure 4A).
Figure 4.
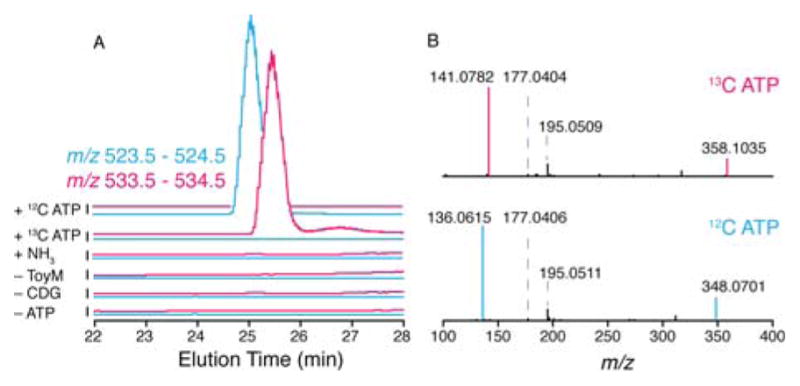
A) Extracted ion traces from ToyM reactions with CDG and ATP in the absence of ammonium ions showing the adenylated intermediate forms only when all three are present. B) MSMS analysis of the adenylated intermediate showing fragments of CDG and AMP.
The identity of adenylated CDG was confirmed using U-13C10-labeled ATP. The resulting covalent adduct exhibited the expected mass shift from m/z of 524.1030 to 534.1376 (theoretical m/z 534.1379), consistent with incorporation of ten 13C from the labeled ATP. MS/MS analysis of the intermediate further confirmed its identity by revealing fragments consistent with expulsion of [CDG+H]+ (m/z 195.0511) and a ketene-like fragment of [CDG]+ (m/z 177.0406). We also observed fragments due to [adenosine+H]+ (m/z 348.0701) and [adenine+H]+ (m/z 136.0615), both of which shift when13C-lableled ATP is used. With the labeled substrate, the [13C10-adenosine+H]+ is observed at m/z 358.1035 and the [13C5-adenine+H]+at m/z 141.0782 (Figure 4B and Supporting Figure S4). We note that presence of magnesium abolished the buildup of the adenylated intermediate, presumably as a result of the magnesium cat alyzing the hydrolysis of the adenylated intermediate in solution. Therefore, these assays were all carried out in the absence of added divalent cation.
To further examine whether both the acid and amide substrates are activated in a similar manner, ToyM was incubated with ATP, CDG and ammonia, or ADG for 8.5 hours after which these reaction mixtures were tested with a coupled enzyme pyrophosphate detection assay.[14] Reactions with both substrates showed the characteristic signal for the presence of pyrophosphate in at least the same stoichiometry as that of the preQ0 product (Figure 5). Taken together with the formation of AMP with both substrates, this demonstrates conclusively that ToyM is capable of catalyzing the same adenylation reaction on two substrates, CDG and ADG, to eventually form the nitrile, preQ0.
Figure 5.
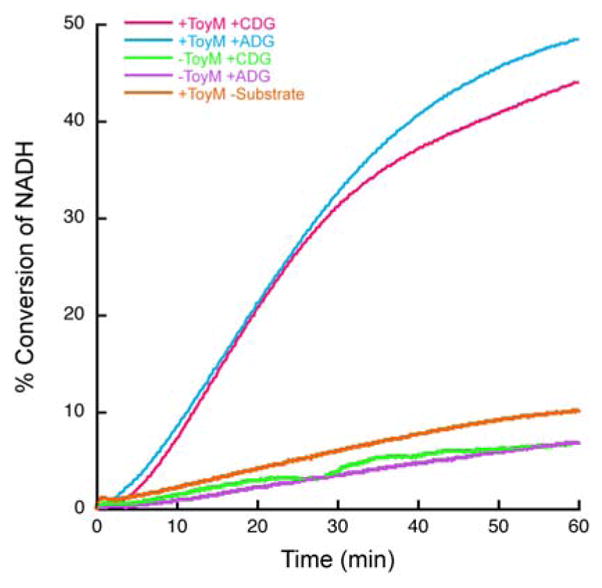
Pyrophosphate dependent consumption of NADH through a coupled enzyme assay showing with both substrates, CDG and ADG, ToyM produces pyrophosphate whereas with no substrate or no enzyme, only background consumption of NADH is observed.
Enzyme promiscuity has been integral in the formation of the immense biosynthetic repertoire found in nature.[15,16] According to this model, the efficiency of enzymes for their ancestral, primary, reaction is likely higher than that of the reactions for which they gain abilities to catalyze over time.[17] ToyM bears tantalizing clues to its evolution in that the first half reaction, the amidation, proceeds more rapidly than the dehydration to the nitrile in the second half reaction. It is thus likely that this unprecedented and simplified nitrile synthesis is an elegant example of finding new chemical capability through the promiscuity of an amide synthetase enzyme to achieve what requires at least three enzymes in other known synthetic pathways.
In conclusion, ToyM is an amide synthetase as well as a nitrile synthetase in its capability of accepting both the acid and amide forms of CDG allowing for sequential amidation and dehydration to the nitrile. This provides a new paradigm through which to study the origin of the many nitrile containing secondary metabolites whose biosynthetic provenance is currently unknown, and it gives molecular detail to the synthesis of an important intermediate in many pathways, the nitrile-containing preQ0.
Supplementary Material
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.Fleming FF. Nat Prod Rep. 1999;16:597–606. [Google Scholar]
- 2.Schulze JC, Bray WM, Loganzo F, Lam M, Szal T, Villalobos A, Koehn FE, Linington RG. J Nat Prod. 2014;77:2570–2574. doi: 10.1021/np500727g. [DOI] [PubMed] [Google Scholar]
- 3.Lane AL, Nam S, Fukuda T, Yamanaka K, Kaufmman CA, Jensen PR, Fenical W, Moore BS. J Am Chem Soc. 2013;135:4171–4174. doi: 10.1021/ja311065v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oldfield MF, Bennet RN, Kiddle G, Wallsgrove RM, Botting NP. Plant Physiol Biochem. 1999;37:99–108. [Google Scholar]
- 5.Kato Y, Tsuda T, Asano Y. Biochem Biophys Acta. 2007;1774:856–865. doi: 10.1016/j.bbapap.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 6.Oinuma KI, Hashimoto Y, Konishi K, Masahiko G, Noguchi T, Higashibata H, Kobayashi M. J Biol Chem. 2003;278:29600–29608. doi: 10.1074/jbc.M211832200. [DOI] [PubMed] [Google Scholar]
- 7.McCarty RM, Bandarian V. Chem Biol. 2008;15:790–798. doi: 10.1016/j.chembiol.2008.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCarty RM, Bandarian V. Biochemistry. 2009;48:3847–3852. doi: 10.1021/bi900400e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCarty RM, Bandarian V. Bioorg Chem. 2012;43:15–25. doi: 10.1016/j.bioorg.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fresquet V, Thoden JB, Holden HM, Raushel FM. Bioorg Chem. 2004;32:63–75. doi: 10.1016/j.bioorg.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 11.Suzuki T, Miyauchi K. FEBS Lett. 2010;584:272–277. doi: 10.1016/j.febslet.2009.11.085. [DOI] [PubMed] [Google Scholar]
- 12.Cicmil N, Huang RH. Proteins. 2008;72:1084–1088. doi: 10.1002/prot.22098. [DOI] [PubMed] [Google Scholar]
- 13.Nelp MT, Astashkin AV, Breci LA, Bandarian V. Biochemistry. 2014;53:3990–3994. doi: 10.1021/bi500260j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Brien WE. Analytical Biochemistry. 1976;43:423–3994. doi: 10.1016/0003-2697(76)90337-7. [DOI] [PubMed] [Google Scholar]
- 15.Schimdt MZ, Mundorff EC, Dojka M, Bermudez E, Ness JE, Govindarajan S, Babbit PC, Minshull J, Gerlt JA. Biochemistry. 2003;28:8387–8393. doi: 10.1021/bi034769a. [DOI] [PubMed] [Google Scholar]
- 16.O’Brien PJ, Herschlag D. Chem Biol. 1999;6:R91–R105. doi: 10.1016/S1074-5521(99)80033-7. [DOI] [PubMed] [Google Scholar]
- 17.Copley SD. Curr Opin Chem Biol. 2003;7:265–272. doi: 10.1016/s1367-5931(03)00032-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.