

Abstract
Background
Mutations in parkin and PTEN-induced protein kinase (PINK1) represent the two most common causes of autosomal recessive parkinsonism. The possibility that heterozygous mutations in these genes also predispose to disease or lower the age of disease onset has been suggested, but currently there is insufficient data to verify this hypothesis conclusively.
Objective
To study the frequency and spectrum of parkin and PINK1 gene mutations and to investigate the role of heterozygous mutations as a risk factor for early-onset Parkinson’s disease (PD).
Methods
All exons and exon-intron boundaries of PINK1 and parkin were sequenced in 250 patients with early-onset PD and 276 normal controls. Gene dosage measurements were also performed, using high-density single-nucleotide polymorphism arrays.
Results
In total 41 variants were found, of which 8 have not been previously described (parkin: p.A38VfsX6, P.C166Y, P.Q171X, p.D243N, p.M458L; PINK1: p.P52L, P.T420T, P.A427E). 1.60% of patients were homozygous or compound heterozygous for pathogenic mutations. Heterozygosity for pathogenic parkin or PINK1 mutations was over-represented in patients compared with healthy controls (4.00% vs. 1.81%) but the difference was not significant (p = 0.13). The mean age at disease onset was significantly lower in patients with homozygous or compound heterozygous mutations than in patients with heterozygous mutations (mean difference 11 years, 95% Cl 1.4 to 20.6, p = 0.03). There was no significant difference in the mean age at disease onset in heterozygous patients compared with patients without a mutation in parkin or PINK1 (mean difference 2 years, 95% Cl −3.7 to 7.0, p = 0.54).
Conclusions
Our data support a trend towards a higher frequency of heterozygosity for pathogenic parkin or PINK1 mutations in patients compared with normal controls, but this effect was small and did not reach significance in our cohort of 250 cases and 276 controls.
Parkinson’s disease (PD) is a common neurodegenerative disorder characterised by bradykinesia, rigor, tremor and postural instability. Several lines of evidence suggest that genetic susceptibility plays an important role in the pathogenesis of PD.1 Mutations in parkin and PTEN-induced putative kinase 4 (PINK1) have been found to cause early-onset PD (disease onset before the age of 50 years) with autosomal recessive inheritance.2,3 Parkin, encoding the E3 ubiquitin ligase parkin, is one of the largest genes in the human genome, spanning approximately 1.4 Mb. It is located on chromosome 6q25.2-q27 in a highly unstable genomic region.4 Point mutations and exon rearrangements in parkin are the most common genetic causes of early-onset PD; approximately 18% of sporadic and 50% of all familial early-onset cases have been attributed to parkin mutations.5 PINK1, located on chromosome 1p36, has been implicated in PD based on linkage studies in families with autosomal recessive early-onset parkinsonism.2 In contrast to parkin, PINK1 is a much smaller gene, containing only eight exons that span about 18 kb. Homozygous or compound heterozygous loss of function mutations in PINK1 have been found to lead to mitochondrial dysfunction.6,7
There is growing evidence that heterozygosity for parkin or PINK1 mutations might act as a susceptibility factor for PD.8–10 There is an increased frequency of heterozygous mutations in patients with PD compared with healthy controls, and cross-sectional imaging studies have shown subclinical dopaminergic dysfunction in heterozygous parkin or PINK1 mutation carriers.9,11,12 In addition, an inverse relationship has been suggested between the number of pathogenic mutations and the age at disease onset.8,13 However, the literature is not consistent, as some studies report a similar frequency of heterozygous mutations in cases and in controls.14–16 Additional screening studies are therefore required to investigate the relevance of heterozygous pathogenic mutations to the pathogenesis of PD.
We undertook a comprehensive screening study of a large, publicly available cohort of patients with early-onset PD (defined as age at disease onset 50 years) and normal controls, to determine the frequencies and spectrum of mutations in parkin and PINK1 and to investigate the role of heterozygous mutations as a risk factor for PD. We identified several novel mutations of which some are likely to be pathogenic, and found a trend towards a higher frequency of heterozygosity for pathogenic parkin or PINK1 mutations in patients compared with normal controls.
METHODS
All participants gave written informed consent to participate in genetics research and the research protocol was approved by the institutional review board.
Subjects
We screened 250 early-onset patients with PD (mean age at disease onset 41 years, range 7–50) and 276 neurologically normal controls (mean age at sampling 39 years, range 15–53) for mutations in parkin and PINK1. These samples, precompiled in six plates (NDPT14, NDPT15, NDPT16, NDPT19, NDPT20 and NDPT21), are publicly available at the NINDS Neurogenetics Repository at the Coriell Institute for Medical Research, New Jersey, USA (http://ccr.coriell.org/ninds). Clinical characteristics of cases and controls are summarised in table 1.
Table 1.
Clinical description of cases and controls
| Characteristics | Patients | Controls |
|---|---|---|
| Subjects, n | 250 | 276 |
| Female/Male | 97/153 | 199/77 |
| Mean age at sampling, years | 54 | 39 |
| Mean age at onset (range), years | 41 (7 to 50) | MA |
| Ethnicity | ||
| Caucasian: Hispanic | 7 | 3 |
| Caucasian: not Hispanic | 156 | 273 |
| Caucasian: not specified | 87 | 0 |
| Positive family history* | 95 | 44 |
| No information on family history | 3 | 0 |
NA, not applicable.
Positive family history for one or more of: parkinsonism, tremor, dementia, restless legs syndrome, dystonia.
Sequencing
All exons and exon-intron boundaries of parkin (reference sequence: NM_004562.1) and PINK1 (reference sequence: NM_0032409.2) were amplified by PCR, in a reaction mix of 15 ng of genomic DNA, 10 nmol/μl forward primer, 10 nmol/μl reverse primer and 12 μl of a master mix (FastStart PCR Master; Roche, Indiana, USA). For exon 1 in parkin, 1 μl of 5% dimethylsulphoxide (American Bioanalytical, Massachusetts, USA) was added to the PCR reaction mix and PCR amplification for exon 1 in PINK1 was achieved by adding 12.5 nmol/μl 7-deaza-GTP (New England Biolabs, Massachusetts, USA) to the mix (primer sequences and fragment lengths are shown in supplementary table 1 online). For the PCR reaction, we used a 60 touch down 50 thermo-cycling program. PCR cycling conditions were as follows: initial denaturation step of 4 minutes at 94°C; 8 cycles of 30 seconds at 94°C, 30 seconds at 60°C and 30 seconds at 72°C; 20 cycles of 30 seconds at 94°C, 30 seconds at 60°C, decreasing by 0.5°C after each cycle, 30 seconds at 72°C; 16 cycles of 30 seconds at 94°C, 20 seconds at 50°C and 30 seconds at 72°C; and a final extension step of 5 min at 72°C.
After PCR product purification (AMPure; Agencourt Bioscience Corporation, Massachusetts, USA), we performed direct dye terminator sequencing (BigDye v3.1; Applied Biosystems, California, USA). The resulting reactions were cleaned with CleanSEQ (Agencourt Bioscience Corporation, Massachusetts, USA), processed on an automated analyser (3730 ×l DNA Analyzer; Applied Biosystems) and analysed with Sequencher V.4.1.4 (Gene Codes Corp., Michigan, USA).
For sample ND00548 from plate NDPT14, chromatography analysis showed a complex mutation in exon 2 of parkin. We therefore performed allele-specific sequencing in this sample. Exon 2 was amplified by PCR and then cloned (pCR8/GW/TOPO TA Cloning Kit; Invitrogen, Carlsbad, California, USA), in accordance with the manufacturer’s instructions. Transformed colonies were cultured overnight in LB medium containing 100 μg/ml spectinomycin and followed by isolation of plasmid DNA using PureLink Quick Plasmid Miniprep Kit (Invitrogen). DNA from 12 colonies was sequenced bidirectionally using direct sequencing as described above.
Gene dosage measurements
In all samples we performed genome-wide single-nucleotide polymorphism (SNP) genotyping using SNP chips (Infinium HumanHap 550K V.3; Illumina Inc., California, USA) in accordance with the manufacturer’s protocol. Raw genotypes were determined using the genotyping module of BeadStudio V.3.1.12 (Illumina Inc.). Gene dosage, inferred from signal intensity data and genotype calls at the parkin and PINK1 loci, was visualised using Genome Viewer V.3.1.4 within BeadStudio as previously described.17 A heterozygous duplication was called if the log R ratio (a metric for gene dosage) was increased by 50%, whereas a 50% decrease in the log R ratio in combination with absence of heterozygous SNP calls was considered a heterozygous deletion.
In samples with an exon rearrangement involving the coding sequence of parkin, we performed quantitative PCR (qPCR) assays (TaqMan; Applied Biosystems) to confirm each rearrangement. β-globin was used as an endogenous reference. Each reaction contained two fluorescent-label TaqMan MGB probes (Applied Biosystems), one VIC-labelled probe that specifically bound to the respective parkin exon sequence and one 6-FAM-labelled probe that was specific for the β-globin reference (primer and probe sequences are shown in supplementary tables 2 and 3 online). The qPCR reaction mix consisted of 25 ng genomic DNA, 10 μl of 2×TaqMan Universal PCR Master Mix (Applied Biosystems), 72 nmol/μl primers, 10 nmol/μl of the β-globin probe and 10 nmol/μl test probe. PCR cycling conditions were: 95°C for 10 minutes, 95°C for 15 seconds and 60°C for 1 minute (40 cycles). Each sample was replicated six times and for each exon, two control samples were measured. The dosage of each exon relative to β-globin and normalised to control DNA was determined using the 2−ΔΔCT method.
Bioinformatic analyses
For multiple sequence alignments, human PINK1 and parkin reference sequences were aligned with paralogues and orthologues from the NCBI conserved domain database using ClustalW2. First, the serine/threonine protein kinase catalytic domain (c109925) was identified in the PINK1 sequence by querying NCBI conserved domain database and then sequence-to-structure alignments were applied to the kinase domain of PINK1 to map the kinase functional subdomains to the PINK1 kinase region. Similar approaches were applied on the RING-finger domains of parkin.
Statistical analyses
For sequence analysis, we defined any alteration from the reference sequence as a variant or a mutation. Changes in exon copy number were referred to as exon rearrangements. Polymorphisms were defined as variants that occurred at a minor allele frequency of at least 1% in the control population. To ensure accuracy of the statistical analyses, we only called a mutation pathogenic if one of the following criteria applied: stop mutation, frameshift mutation, exon rearrangement or a missense mutation that has been reported to be pathogenic (details are presented in table 2).
Table 2.
Subjects with pathogenic mutations included in statistical analysis
| Sample ID | Mutation(s)* | Age at onset, years | Family history | |
|---|---|---|---|---|
| Pathogenic parkin mutations in cases | ||||
| ND00136 | Heterozygous for p.Q171X | Heterozygous for p.R275W | 38 | Present |
| ND00153 | Heterozygous deletion of exons 5–6 | 29 | Present | |
| ND00187† | Heterozygous duplication of exons 5–9 | 38 | Present | |
| ND00429† | Heterozygous duplication of exons 5–9 | 41 | Present | |
| ND00548 | Heterozygous for p.A38VfsX6 | Heterozygous for p.Q34RfsX5 | 36 | Absent |
| ND01119 | Heterozygous deletion exon 2 | 50 | Absent | |
| ND01122 | Heterozygous for p.R275W | 47 | Absent | |
| ND02639 | Homozygous for p.L112LfsX15 | 30 | Absent | |
| ND02798 | Heterozygous duplication of exon 6 | 32 | Present | |
| ND04581 | Heterozygous deletion exon 2 | Heterozygous for p.Q34RfsX5 | 22 | Present |
| ND05921 | Heterozygous deletion exon 4 | 49 | Present | |
| ND06330 | Heterozygous duplication exons 2–3 | 50 | Absent | |
| ND06635 | Heterozygous for p.N52MfsX29 | 42 | Present | |
| Pathogenic PINK1 mutations in cases | ||||
| ND08471 | Heterozygous for p.M318L | 47 | Absent | |
| Pathogenic parkin mutations in controls | ||||
| ND03967 | Heterozygous deletion exon 3 | NA | Present | |
| ND04990 | Heterozygous duplication exon 2 | NA | Absent | |
| ND08538 | Heterozygous deletion exons 2–4 | NA | Absent | |
| ND09912 | Heterozygous for p.R275W | NA | Absent | |
| ND10272 | Heterozygous for p.T415N | NA | Present | |
NA, not applicable.
Only frameshift mutations, stop mutations, exon rearrangements that span the coding sequence and missense mutations that have been reported to be pathogenic are listed.
ND00187 and ND00429 are siblings.
Fisher’s exact tests on allelic association and tests for assessing departures from Hardy–Weinberg equilibrium were performed using PLINK (version 1.04; http://pngu.mgh.harvard.edu/~pur-cell/plink/).18 Statistical significance for differences in age at onset was tested using t tests after Kolmogorov–Smirnov tests showed a normal distribution of the data (SPSS V.12.0.1). For the age at onset analysis, p <0.05 was considered significant.
RESULTS
We found 41 sequence variants in a cohort of 250 patients with early-onset PD and 276 normal controls (fig 1, tables 3,4). No significant departures from Hardy–Weinberg equilibrium were found for any of these variants and none of the identified variants achieved significance for allelic association after Bonferroni correction for multiple testing (adjusted p <0.0012).
Figure 1.
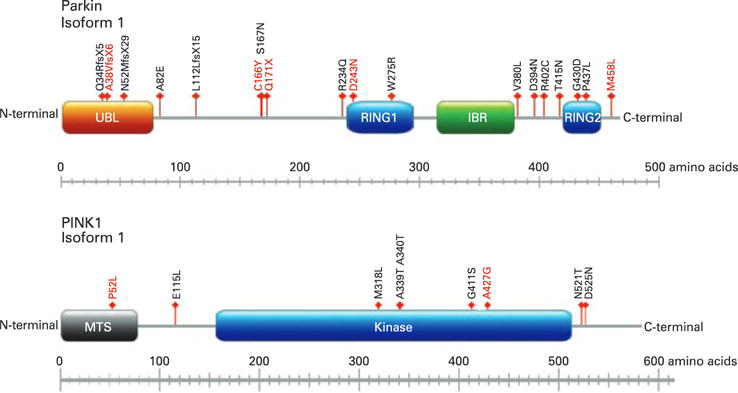
Missense, nonsense and frameshift mutations observed in this study. Arrows indicate the position of each respective mutation relative to the protein domains. Novel mutations are indicated by red font, whereas previously described mutations are black. UBL, ubiquitin-like domain; RING1, RING finger motif 1; IBR, in-between-RING domain; RING2, ring finger motif 2; MTS, mitochondrion transit sequence.
Table 3.
Variants in parkin in 250 PD cases and 276 controls
| Nucleotide | Location/rs number | Amino acid change | Mutation type | Genotypes in cases (A1A1/A1A2/A2A2) | Genotypes in controls (A1A1/A1A2/A2A2) | MAF cases/controls | Ref. |
|---|---|---|---|---|---|---|---|
| Novel mutations that are likely to be pathogenic | |||||||
| c.[214delC+216GR T] | Exon 2 | p.Ala38ValfsX6 | Frameshift | 0/1/249 | 0/0/276 | 0.002/0.000 | – |
| c.612CR T | Exon 4 | p.Gln171X | Stop | 0/1/249 | 0/0/276 | 0.002/0.000 | – |
| Novel mutations of unknown significance | |||||||
| C.598GR A | Exon 4 | p.Cys166Tyr | Missense | 0/1/249 | 0/0/276 | 0.002/0.000 | – |
| C.828GR A | Exon 6 | p.Asp243Asn | Missense | 0/0/250 | 0/1/275 | 0.000/0.002 | – |
| C.1473AR C | Exon 12 | p.Met458Leu | Missense | 0/1/249 | 0/0/276 | 0.002/0.000 | – |
| Previously described mutations that are likely to be pathogenic | |||||||
| c.202−203delAG | Exon 2 | p.Gln34ArgfsX5 | Frameshift | 0/2/248 | 0/0/276 | 0.004/0.000 | 25 |
| c.256delA | Exon 2 | p.Asn52MetfsX29 | Frameshift | 0/1/249 | 0/0/276 | 0.002/0.000 | 25 |
| c.437−477del | Exon 3 | p.Leu112LeufsX15 | Frameshift | 1/0/249 | 0/0/276 | 0.004/0.000 | 25 |
| c.924CR T | Exon 7/rs34424986 | p.Arg275Trp | Missense | 0/2/248 | 0/1/275 | 0.004/0.002 | 25,27 |
| c.1345CR A | Exon 11 | p.Thr415Asn | Missense | 0/0/250 | 0/1/275 | 0.000/0.002 | 25 |
| Previously described mutations with unknown significance | |||||||
| c.802GR A | Exon 6 | p.Arg234Gln | Missense | 0/1/249 | 0/0/276 | 0.002/0.000 | 28 |
| c.1305CR T | Exon 11 | p.Arg402Cys | Missense | 0/0/250 | 0/1/275 | 0.000/0.002 | 29 |
| c.1390GR A | Exon 12 | p.Gly430Asp | Missense | 0/1/249 | 0/0/276 | 0.002/0.000 | 5 |
| c.1444CR T | Exon 12 | p.Pro437Leu | Missense | 0/3/247 | 0/3/273 | 0.006/0.005 | 24 |
| Previously described mutations that are likely to be non-pathogenic | |||||||
| c.273−18A | Intron 2 | – | Intronic | 0/4/246 | 0/2/274 | 0.008/0.004 | 16 |
| c.34BCR A | Exon 3 | p.Ala82Glu | Missense | 0/0/250 | 0/2/274 | 0.000/0.004 | 30,31 |
| c.623CR T | Exon 4 | p.Leu174Leu | Silent | 0/1/249 | 0/2/274 | 0.002/0.004 | 15 |
| c.884TR C | Exon 7/rs9456711 | p.Leu261Leu | Silent | 0/2/248 | 0/0/276 | 0.004/0.000 | 28,32 |
| Previously described polymorphisms | |||||||
| c.272+25C | Intron 2/rs2075923 | – | Intronic | 13/83/154 | 10/95/171 | 0.218/0.208 | 25 |
| c.514−20T | Intron 3/rs4709583 | – | Intronic | 1/35/214 | 1/42/233 | 0.074/0.080 | 25 |
| c.601GR A | Exon 4/rs1801474 | p.Ser167Asn | Missense | 0/7/243 | 0/9/267 | 0.014/0.016 | 33 |
| c.973−35G | Intron 7/rs3765474 | – | Intronic | 52/126/72 | 52/138/86 | 0.460/0.438 | 25 |
| c.1034+48T | Intron 8/rs10945756 | – | Intronic | 16/87/147 | 17/98/161 | 0.238/0.239 | 16 |
| c.1239GR C | Exon 10/rs1801582 | p.Val380Leu | Missense | 6/78/166 | 5/89/182 | 0.180/0.179 | 25 |
| c.1281GR A | Exon 11/rs1801334 | p.Asp394Asn | Missense | 0/16/234 | 0/14/262 | 0.032/0.025 | 25 |
MAF, minor allele frequency.
Genotype distribution of the major allele (A2) and minor allele (A1) is described for each variant.
Table 4.
Variants in PINK1 in 250 and 276 controls
| Nucleotide | Location/rs number | Amino acid change | Mutation type | Genotypes in cases A1A1/A1A2/A2A2) | Genotypes in controls (A1A1/A1A2/A2A2) | MAF cases/controls | Ref. |
|---|---|---|---|---|---|---|---|
| Novel mutation that is likely to be pathogenic | |||||||
| c.1374CR A | Exon 7 | p.Ala427Glu | Missense | 0/2/248 | 0/0/276 | 0.004/0.000 | – |
| Novel mutation of unknown significance | |||||||
| c.249CR T | Exon 1 | p.Pro52Leu | Missense | 0/1/249 | 0/0/276 | 0.002/0.000 | – |
| Novel mutation that is likely to be non-pathogenic | |||||||
| c.1354GR A | Exon 7 | p.Thr420Thr | Silent | 0/1/249 | 0/0/276 | 0.002/0.000 | – |
| Previously described mutation that is likely to be pathogenic | |||||||
| c.1046AR T | Exon 4 | p.Met318Leu | Missense | 0/1/249 | 0/0/276 | 0.002/0.000 | 34,35 |
| Previously described mutations with unknown significance | |||||||
| c.1109GR A | Exon 5/rs55831733 | p.Ala339Thr | Missense | 0/2/248 | 0/0/276 | 0.004/0.000 | 34 |
| c.1325GR A | Exon 6/rs45478900 | p.Gly411Ser | Missense | 0/0/250 | 0/1/275 | 0.000/0.002 | 36 |
| c.1667GRA | Exon 8 | p.Asp525Asn | Missense | 0/1/249 | 0/0/276 | 0.002/0.000 | 37 |
| c.1822AR G | Exon 8 | p.Ser576Ser | Silent | 0/1/249 | 0/0/276 | 0.002/0.000 | 34 |
| Previously described mutations that are likely to be non-pathogenic | |||||||
| c.1030GR A | Exon 4/rs56200357 | p.Arg312Arg | Silent | 0/1/249 | 0/0/276 | 0.002/0.000 | 2 |
| c.1267TR C | Exon 6/rs45499398 | p.Asp391Asp | Silent | 0/1/249 | 0/0/276 | 0.002/0.000 | 34,38 |
| Previously described polymorphisms | |||||||
| c.283CR T | Exon 1 | p.Leu63Leu | Silent | 9/81/160 | 5/78/193 | 0.198/0.159 | 34 |
| c.438AR T | Exon 1 | p.Gln115Leu | Missense | 0/17/233 | 0/32/244 | 0.034/0.058 | 18 |
| c.482−7A | Intron 1/rs2298298 | – | Intronic | 2/48/200 | 3/63/210 | 0.104/0.125 | 34 |
| c.1054−5G | Intron 4/rs3131713 | – | Intronic | 2/51/197 | 2/63/211 | 0.110/0.121 | 34 |
| c.111 MR A | Exon 5/rs3738136 | p.Ala340Thr | Missense | 1/33/216 | 0/18/258 | 0.070/0.033 | 34 |
| c.1656AR C | Exon 8/rs1043424 | p.Asn521Thr | Missense | 15/110/125 | 21/110/145 | 0.280/0.275 | 34 |
Genotype distribution of the major allele (A2) and minor allele (A1) is described for each variant.
MAF, minor allele frequency.
Parkin analysis
The following variants were seen in parkin: 4 frameshift mutations, 13 missense mutations, one stop mutation, 5 intronic variants and 2 silent mutations (table 3). Five of these variants have not been previously described (p.A38VfsX6, p.C166Y, p.Q171X, p.D243N, p.M458L; chromatograms of all novel variants are shown in supplementary figs 1 and 2 online). In addition, we identified four patients (1.6%) and two controls (0.7%) with a heterozygous exon deletion, and four cases (1.6%) and one control (0.4%) with a heterozygous exon duplication (table 2, supplementary figures 4–6 online). Nine patients (3.6%) and five controls (1.8%) were heterozygous for pathogenic parkin mutations, three patients (1.2%) were compound heterozygotes as they carried two different pathogenic mutations, and one patient (0.4%) was homozygous for a pathogenic mutation.
PINK1 analysis
We found the following variants for PINK: nine missense mutations, five silent mutations and two intronic variants (table 4). Of these, three mutations have not been previously reported (p.P52L, p.T420T, p.A427G; chromatograms of all novel variants are shown in supplementary fig 3 online). None of the studied subjects had a PINK1 gene duplication or deletion. One patient (0.4%) was heterozygous for a pathogenic mutation in PINK1, whereas none of the controls carried a heterozygous mutation. No subject had pathogenic PINK1 mutations.
Combined analysis
In all, 14 patients (5.6%) and 5 controls (1.8%) had 1 pathogenic mutations in parkin or PINK1, and this difference was significant (p = 0.02). Of the 14 patients with 1 pathogenic mutation, 8 (57.1%) had a family history of parkinsonism, compared with 87 of 250 (34.8%) patients without a pathogenic mutation (missing family history data: 3 patients without a pathogenic mutation (1.2%)). Of 5 controls with a pathogenic mutation, 2 (40.0%) had a positive family history for PD, compared with only 7 of 271 (2.6%) controls without a pathogenic mutation.
Bioinformatic analysis of sequence conservation
To determine the extent of sequence conservation at newly discovered mutated sites, we aligned the human PINK1 and parkin sequences with human domain paralogues and with orthologues from various species. Based on this analysis, at least three variants were probably pathogenic: p.A38VfsX6 and p.Q171X lead to an early termination of the coding sequence, and p.A427E is located in close proximity to the highly conserved serine-threonine kinase activation loop, and it is likely that mutations at this site will interfere with substrate binding to this crucial domain (fig 2).
Figure 2.
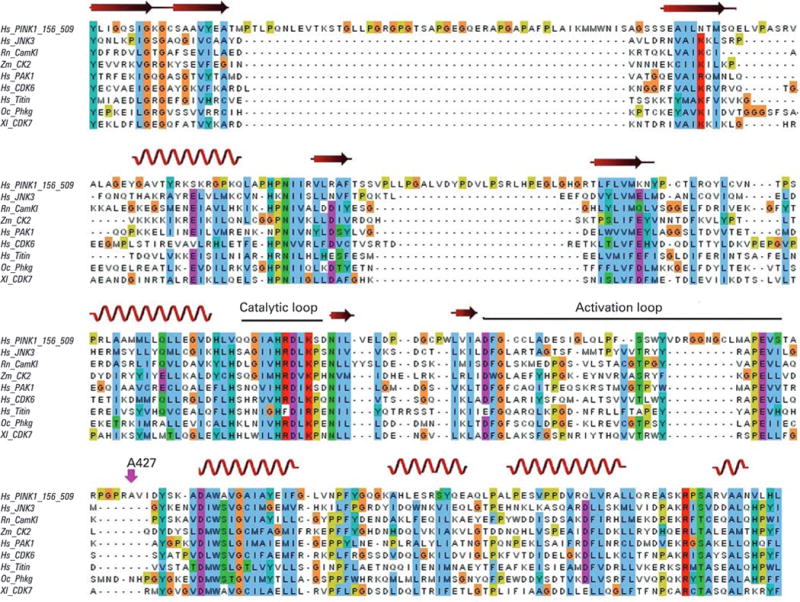
Alignment of PINK1 kinase domain with conserved serine-threonine protein kinases. Sequence alignment between the PINK1 kinase domain and several serine-threonine protein kinase members in the NCBI conserved domain database. The red swirl ribbons and arrowed ribbons stand for the predicted conserved α-helix and β-strand in three dimensional structures, respectively. The mutation p.Ala427Glu lies close to the highly conserved APE triple that is the C-terminal anchor of the activation loop. It is likely that mutations in or close to this crucial domain interfere with substrate recognition.
DISCUSSION
We report the results of a comprehensive screening study for parkin and PINK1 mutations in 250 patients with early-onset PD and 276 normal controls. We identified 41 sequence variants: 8 novel and 33 previously described. Conservation across various species strongly suggests that at least three of the novel mutations are likely to be pathogenic (p.A38VfsX6, p.A171X, p.A427E). In addition to sequence variants, we also found eight cases and three controls with a heterozygous exon rearrangement (table 2).
Overall, patients with pathogenic changes in parkin or PINK1 accounted for 5.6% of all cases in this study. This frequency is lower than previous reports have suggested.5,19 Possible explanations for this discrepancy could be a population-specific variability in allele frequencies, different case selection criteria, different age at onset distributions in the studied cohorts, and different criteria for determining which variant is considered pathogenic.
Increasing evidence indicates a role of heterozygous pathogenic mutations as a susceptibility factor for PD. Positron emission tomography imaging studies have reported a subclinical dopaminergic dysfunction in heterozygous parkin or PINK1 mutation carriers.11,12,20 This observation is supported by transcranial ultrasound studies that reported hyperechogenicity of the substantia nigra, which was shown to depend on mutation status.11,21,22 Furthermore, subclinical sensory abnormalities have been described in patients with heterozygous PINK1 mutations.23 The frequency of heterozygous pathogenic mutations has been reported to be significantly increased in patients with parkinsonism compared with normal controls.9 However, several studies have provided contradictory data.14–16 The lack of cohesion between these studies may have arisen due to the differences in how variants were defined as pathogenic, which determined if they were included in the statistical analysis. For our study, we used conservative criteria, and only included mutations in the analysis that were frameshift or stop mutations, exon rearrangements or mutations that have been reported as probably pathogenic (table 2). Based on these strict criteria, we found an increased frequency of patients with a heterozygous pathogenic mutation compared with controls (4.0% vs. 1.8%), but the difference was not significant (p = 0.13). None of the patients was heterozygous for pathogenic mutations in both genes. Therefore, tests for possible additive effects were not feasible.
Age at onset analysis found a significantly lower mean age at disease onset in patients who were homozygous or compound heterozygous for pathogenic parkin mutations (mean (SD) 32 (7) years) than in patients with a heterozygous pathogenic mutation (43 (8) years) (mean difference 11 years, 95% CI 1.4 to 20.6, p = 0.03). In contrast to previous reports that have shown a significant decrease in age at onset in heterozygous carriers compared with patients without a heterozygous mutation,8,24 our study did not reveal a significant difference (43 (8) years vs. 41 (9) years; mean difference 2 years, 95% CI −3.7 to 7.0, p = 0.54).
A possible limitation of this study is the moderate resolution of genome-wide SNP chips for the identification of exon rearrangements. On average, the Illumina 550K chip contains about 0.35 SNPs per kb at the parkin locus and 0.39 SNPs per kb at the PINK1 locus. Small duplications or deletions could therefore have been missed, and our finding of 11 exon rearrangements in the entire series could be an underestimation.
In conclusion, we present the results of a comprehensive analysis of variation in the parkinsonism genes parkin and PINK1. Our data found that there was a trend towards a higher frequency of heterozygosity for pathogenic parkin or PINK1 mutations in patients compared with controls.
Acknowledgments
We thank the participants of this study and the submitters for depositing samples at the Coriell Institute for Medical Research (http://www.coriell.org/). This research was supported entirely by the Intramural Research Program of the NIH, National Institute on Aging, National Institutes of Health, Department of Health and Human Services, USA (Z-number: Z01 AG000957-05).
Footnotes
Competing interests: None.
References
- 1.Scholz S, Singleton A. Susceptibility genes in movement disorders. Mov Disord. 2008;23:927–34. doi: 10.1002/mds.21983. quiz 1064. [DOI] [PubMed] [Google Scholar]
- 2.Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, Gonzalez-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158–60. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 3.Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–8. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 4.McAvoy S, Zhu Y, Perez DS, James CD, Smith DI. Disabled-1 is a large common fragile site gene, inactivated in multiple cancers. Genes Chromosomes Cancer. 2008;47:165–74. doi: 10.1002/gcc.20519. [DOI] [PubMed] [Google Scholar]
- 5.Lucking CB, Durr A, Bonifati V, Vaughan J, De Michele G, Gasser T, Harhangi BS, Meco G, Denefle P, Wood NW, Agid Y, Brice A. Association between early-onset Parkinson’s disease and mutations in the parkin gene. N Engl J Med. 2000;342:1560–7. doi: 10.1056/NEJM200005253422103. [DOI] [PubMed] [Google Scholar]
- 6.Gautier CA, Kitada T, Shen J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc Natl Acad Sci U S A. 2008;105:11364–9. doi: 10.1073/pnas.0802076105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wood-Kaczmar A, Gandhi S, Yao Z, Abramov AS, Miljan EA, Keen G, Stanyer L, Hargreaves I, Klupsch K, Deas E, Downward J, Mansfield L, Jat P, Taylor J, Heales S, Duchen MR, Latchman D, Tabrizi SJ, Wood NW. PINK1 is necessary for long term survival and mitochondrial function in human dopaminergic neurons. PLoS ONE. 2008;3:e2455. doi: 10.1371/journal.pone.0002455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun M, Latourelle JC, Wooten GF, Lew MF, Klein C, Shill HA, Golbe LI, Mark MH, Racette BA, Perlmutter JS, Persian A, Guttman M, Nicholson G, Xu G, Wilk JB, Saint-Hilaire MH, DeStefano AL, Prakash R, Williamson S, Suchowersky O, Labelle N, Growdon JH, Singer C, Watts RL, Goldwurm S, Pezzoli G, Baker KB, Pramstaller PP, Burn DJ, Chinnery PF, Sherman S, Vieregge P, Litvan I, Gillis T, MacDonald ME, Myers RH, Gusella JF. Influence of heterozygosity for parkin mutation on onset age in familial Parkinson disease: the GenePD study. Arch Neurol. 2006;63:826–32. doi: 10.1001/archneur.63.6.826. [DOI] [PubMed] [Google Scholar]
- 9.Klein C, Lohmann-Hedrich K, Rogaeva E, Schlossmacher MG, Lang AE. Deciphering the role of heterozygous mutations in genes associated with parkinsonism. Lancet Neurol. 2007;6:652–62. doi: 10.1016/S1474-4422(07)70174-6. [DOI] [PubMed] [Google Scholar]
- 10.Abou-Sleiman PM, Muqit MM, McDonald NQ, Yang YX, Gandhi S, Healy DG, Harvey K, Harvey RJ, Deas E, Bhatia K, Quinn N, Lees A, Latchman DS, Wood NW. A heterozygous effect for PINK1 mutations in Parkinson’s disease? Ann Neurol. 2006;60:414–19. doi: 10.1002/ana.20960. [DOI] [PubMed] [Google Scholar]
- 11.Binkofski F, Reetz K, Gaser C, Hilker R, Hagenah J, Hedrich K, van Eimeren T, Thiel A, Buchel C, Pramstaller PP, Siebner HR, Klein C. Morphometric fingerprint of asymptomatic Parkin and PINK1 mutation carriers in the basal ganglia. Neurology. 2007;69:842–50. doi: 10.1212/01.wnl.0000267844.72421.6c. [DOI] [PubMed] [Google Scholar]
- 12.Khan NL, Scherfler C, Graham E, Bhatia KP, Quinn N, Lees AJ, Brooks DJ, Wood NW, Piccini P. Dopaminergic dysfunction in unrelated, asymptomatic carriers of a single parkin mutation. Neurology. 2005;64:134–6. doi: 10.1212/01.WNL.0000148725.48740.6D. [DOI] [PubMed] [Google Scholar]
- 13.Kumazawa R, Tomiyama H, Li Y, Imamichi Y, Funayama M, Yoshino H, Yokochi F, Fukusako T, Takehisa Y, Kashihara K, Kondo T, Elibol B, Bostantjopoulou S, Toda T, Takahashi H, Yoshii F, Mizuno Y, Hattori N. Mutation analysis of the PINK1 gene in 391 patients with Parkinson disease. Arch Neurol. 2008;65:802–8. doi: 10.1001/archneur.65.6.802. [DOI] [PubMed] [Google Scholar]
- 14.Lincoln SJ, Maraganore DM, Lesnick TG, Bounds R, de Andrade M, Bower JH, Hardy JA, Farrer MJ. Parkin variants in North American Parkinson’s disease: cases and controls. Mov Disord. 2003;18:1306–11. doi: 10.1002/mds.10601. [DOI] [PubMed] [Google Scholar]
- 15.Marongiu R, Ferraris A, Ialongo T, Michiorri S, Soleti F, Ferrari F, Elia AE, Ghezzi D, Albanese A, Altavista MC, Antonini A, Barone P, Brusa L, Cortelli P, Martinelli P, Pellecchia MT, Pezzoli G, Scaglione C, Stanzione P, Tinazzi M, Zecchinelli A, Zeviani M, Cassetta E, Garavaglia B, Dallapiccola B, Bentivoglio AR, Valente EM. PINK1 heterozygous rare variants: prevalence, significance and phenotypic spectrum. Hum Mutat. 2008;29:565. doi: 10.1002/humu.20719. [DOI] [PubMed] [Google Scholar]
- 16.Kay DM, Moran D, Moses L, Poorkaj P, Zabetian CP, Nutt J, Factor SA, Yu CE, Montimurro JS, Keefe RG, Schellenberg GD, Payami H. Heterozygous parkin point mutations are as common in control subjects as in Parkinson’s patients. Ann Neurol. 2007;61:47–54. doi: 10.1002/ana.21039. [DOI] [PubMed] [Google Scholar]
- 17.Simon-Sanchez J, Scholz S, Matarin Mdel M, Fung HC, Hernandez D, Gibbs JR, Britton A, Hardy J, Singleton A. Genomewide SNP assay reveals mutations underlying Parkinson disease. Hum Mutat. 2008;29:315–22. doi: 10.1002/humu.20626. [DOI] [PubMed] [Google Scholar]
- 18.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klein C, Djarmati A, Hedrich K, Schafer N, Scaglione C, Marchese R, Kock N, Schule B, Hiller A, Lohnau T, Winkler S, Wiegers K, Hering R, Bauer P, Riess O, Abbruzzese G, Martinelli P, Pramstaller PP. PINK1, Parkin, and DJ-1 mutations in Italian patients with early-onset parkinsonism. Eur J Hum Genet. 2005;13:1086–93. doi: 10.1038/sj.ejhg.5201455. [DOI] [PubMed] [Google Scholar]
- 20.Hilker R, Klein C, Hedrich K, Ozelius LJ, Vieregge P, Herholz K, Pramstaller PP, Heiss WD. The striatal dopaminergic deficit is dependent on the number of mutant alleles in a family with mutations in the parkin gene: evidence for enzymatic parkin function in humans. Neurosci Lett. 2002;323:50–4. doi: 10.1016/s0304-3940(01)02529-0. [DOI] [PubMed] [Google Scholar]
- 21.Hagenah JM, Konig IR, Becker B, Hilker R, Kasten M, Hedrich K, Pramstaller PP, Klein C, Seidel G. Substantia nigra hyperechogenicity correlates with clinical status and number of Parkin mutated alleles. J Neurol. 2007;254:1407–13. doi: 10.1007/s00415-007-0567-y. [DOI] [PubMed] [Google Scholar]
- 22.Hagenah JM, Becker B, Bruggemann N, Djarmati A, Lohmann K, Sprenger A, Klein C, Seidel G. Transcranial sonography findings in a large family with homozygous and heterozygous PINK1 mutations. J Neurol Neurosurg Psychiatry. 2008;79:1071–4. doi: 10.1136/jnnp.2007.142174. [DOI] [PubMed] [Google Scholar]
- 23.Fiorio M, Valente EM, Gambarin M, Bentivoglio AR, Ialongo T, Albanese A, Barone P, Pellecchia MT, Brancati F, Moretto G, Fiaschi A, Tinazzi M. Subclinical sensory abnormalities in unaffected PINK1 heterozygotes. J Neurol. 2008;255:1372–7. doi: 10.1007/s00415-008-0923-6. [DOI] [PubMed] [Google Scholar]
- 24.Foroud T, Uniacke SK, Liu L, Pankratz N, Rudolph A, Halter C, Shults C, Marder K, Conneally PM, Nichols WC. Heterozygosity for a mutation in the parkin gene leads to later onset Parkinson disease. Neurology. 2003;60:796–801. doi: 10.1212/01.wnl.0000049470.00180.07. [DOI] [PubMed] [Google Scholar]
- 25.Abbas N, Lucking CB, Ricard S, Durr A, Bonifati V, De Michele G, Bouley S, Vaughan JR, Gasser T, Marconi R, Broussolle E, Brefel-Courbon C, Harhangi BS, Oostra BA, Fabrizio E, Bohme GA, Pradier L, Wood NW, Filla A, Meco G, Denefle P, Agid Y, Brice A. A wide variety of mutations in the parkin gene are responsible for autosomal recessive parkinsonism in Europe. French Parkinson’s Disease Genetics Study Group and the European Consortium on Genetic Susceptibility in Parkinson’s Disease. Hum Mol Genet. 1999;8:567–74. doi: 10.1093/hmg/8.4.567. [DOI] [PubMed] [Google Scholar]
- 26.Hedrich K, Marder K, Harris J, Kann M, Lynch T, Meija-Santana H, Pramstaller PP, Schwinger E, Bressman SB, Fahn S, Klein C. Evaluation of 50 probands with early-onset Parkinson’s disease for Parkin mutations. Neurology. 2002;58:1239–46. doi: 10.1212/wnl.58.8.1239. [DOI] [PubMed] [Google Scholar]
- 27.Wang C, Lu R, Ouyang X, Ho MW, Chia W, Yu F, Lim KL. Drosophila overexpressing parkin R275W mutant exhibits dopaminergic neuron degeneration and mitochondrial abnormalities. J Neurosci. 2007;27:8563–70. doi: 10.1523/JNEUROSCI.0218-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hertz JM, Ostergaard K, Juncker I, Pedersen S, Romstad A, Moller LB, Guttler F, Dupont E. Low frequency of Parkin, Tyrosine Hydroxylase, and GTP Cyclohydrolase I gene mutations in a Danish population of early-onset Parkinson’s Disease. Eur J Neurol. 2006;13:385–90. doi: 10.1111/j.1468-1331.2006.01249.x. [DOI] [PubMed] [Google Scholar]
- 29.Bartoli-Avella AM, Giroud-Benitez JL, Akyol A, Barbosa E, Schaap O, van der Linde HC, Martignoni E, Lopiano L, Lamberti P, Fincati E, Antonini A, Stocchi F, Montagna P, Squitieri F, Marini P, Abbruzzese G, Fabbrini G, Marconi R, Dalla Libera A, Trianni G, Guidi M, De Gaetano A, Boff Maegawa G, De Leo A, Gallai V, de Rosa G, Vanacore N, Meco G, van Duijn CM, Oostra BA, Heutink P, Bonifati V. Novel parkin mutations detected in patients with early-onset Parkinson’s disease. Mov Disord. 2005;20:424–31. doi: 10.1002/mds.20343. [DOI] [PubMed] [Google Scholar]
- 30.Hedrich K, Kann M, Lanthaler AJ, Dalski A, Eskelson C, Landt O, Schwinger E, Vieregge P, Lang AE, Breakefield XO, Ozelius LJ, Pramstaller PP, Klein C. The importance of gene dosage studies: mutational analysis of the parkin gene in early-onset parkinsonism. Hum Mol Genet. 2001;10:1649–56. doi: 10.1093/hmg/10.16.1649. [DOI] [PubMed] [Google Scholar]
- 31.Oliveira SA, Scott WK, Martin ER, Nance MA, Watts RL, Hubble JP, Koller WC, Pahwa R, Stern MB, Hiner BC, Ondo WG, Allen FH, Jr, Scott BL, Goetz CG, Small GW, Mastaglia F, Stajich JM, Zhang F, Booze MW, Winn MP, Middleton LT, Haines JL, Pericak-Vance MA, Vance JM. Parkin mutations and susceptibility alleles in late-onset Parkinson’s disease. Ann Neurol. 2003;53:624–9. doi: 10.1002/ana.10524. [DOI] [PubMed] [Google Scholar]
- 32.Clark LN, Afridi S, Karlins E, Wang Y, Mejia-Santana H, Harris J, Louis ED, Cote LJ, Andrews H, Fahn S, Waters C, Ford B, Frucht S, Ottman R, Marder K. Case-control study of the parkin gene in early-onset Parkinson disease. Arch Neurol. 2006;63:548–52. doi: 10.1001/archneur.63.4.548. [DOI] [PubMed] [Google Scholar]
- 33.Satoh J, Kuroda Y. Association of codon 167 Ser/Asn heterozygosity in the parkin gene with sporadic Parkinson’s disease. Neuroreport. 1999;10:2735–9. doi: 10.1097/00001756-199909090-00008. [DOI] [PubMed] [Google Scholar]
- 34.Rogaeva E, Johnson J, Lang AE, Gulick C, Gwinn-Hardy K, Kawarai T, Sato C, Morgan A, Werner J, Nussbaum R, Petit A, Okun MS, McInerney A, Mandel R, Groen JL, Fernandez HH, Postuma R, Foote KD, Salehi-Rad S, Liang Y, Reimsnider S, Tandon A, Hardy J, St George-Hyslop P, Singleton AB. Analysis of the PINK1 gene in a large cohort of cases with Parkinson disease. Arch Neurol. 2004;61:1898–904. doi: 10.1001/archneur.61.12.1898. [DOI] [PubMed] [Google Scholar]
- 35.Djarmati A, Hedrich K, Svetel M, Lohnau T, Schwinger E, Romac S, Pramstaller PP, Kostic V, Klein C. Heterozygous PINK1 mutations: a susceptibility factor for Parkinson disease? Mov Disord. 2006;21:1526–30. doi: 10.1002/mds.20977. [DOI] [PubMed] [Google Scholar]
- 36.Mellick GD, Siebert GA, Funayama M, Buchanan DD, Li Y, Imamichi Y, Yoshino H, Silburn PA, Hattori N. Screening PARK genes for mutations in early-onset Parkinson’s disease patients from Queensland, Australia. Parkinsonism Relat Disord. 2009;15:105–9. doi: 10.1016/j.parkreldis.2007.11.016. [DOI] [PubMed] [Google Scholar]
- 37.Valente EM, Salvi S, Ialongo T, Marongiu R, Elia AE, Caputo V, Romito L, Albanese A, Dallapiccola B, Bentivoglio AR. PINK1 mutations are associated with sporadic early-onset parkinsonism. Ann Neurol. 2004;56:336–41. doi: 10.1002/ana.20256. [DOI] [PubMed] [Google Scholar]
- 38.Bonifati V, Rohe CF, Breedveld GJ, Fabrizio E, De Mari M, Tassorelli C, Tavella A, Marconi R, Nicholl DJ, Chien HF, Fincati E, Abbruzzese G, Marini P, De Gaetano A, Horstink MW, Maat-Kievit JA, Sampaio C, Antonini A, Stocchi F, Montagna P, Toni V, Guidi M, Dalla Libera A, Tinazzi M, De Pandis F, Fabbrini G, Goldwurm S, de Klein A, Barbosa E, Lopiano L, Martignoni E, Lamberti P, Vanacore N, Meco G, Oostra BA. Early-onset parkinsonism associated with PINK1 mutations: frequency, genotypes, and phenotypes. Neurology. 2005;65:87–95. doi: 10.1212/01.wnl.0000167546.39375.82. [DOI] [PubMed] [Google Scholar]