

Abstract
A sterically accessible tert-butyl-substituted dipyrrinato di-iron(II) complex [(tBuL)FeCl]2 possessing two bridging chloride atoms was synthesized from the previously reported solvento adduct. Upon treatment with aryl azides, the formation of high-spin FeIII species was confirmed by 57Fe Mössbauer spectroscopy. Crystallographic characterization revealed two possible oxidation products: (1) a terminal iron iminyl from aryl azides bearing ortho isopropyl substituents, (tBuL)FeCl(•NC6H3-2,6-iPr2); or (2) a bridging di-iron imido arising from reaction with 3,5-bis(trifluoromethyl)aryl azide, [(tBuL)FeCl]2(μ-NC6H3-3,5-(CF3)2). Similar to the previously reported (ArL)FeCl(•NC6H4-4-tBu), the monomeric iron imido is best described as a high-spin FeIII antiferromagnetically coupled to an iminyl radical, affording an S = 2 spin state as confirmed by SQUID magnetometry. The di-iron imido possesses an S = 0 ground state, arising from two high-spin FeIII centers weakly antiferromagnetically coupled through the bridging imido ligand. The terminal iron iminyl complex undergoes facile decomposition via intra- or intermolecular hydrogen-atom abstraction (HAA) from an imido aryl ortho isopropyl group, or from 1,4-cyclohexadiene, respectively. The bridging di-iron imido is a competent N-group transfer reagent to cyclic internal olefins as well as styrene. Although solid-state magnetometry indicates an antiferromagnetic interaction between the two iron centers (J = −108.7 cm−1) in [(tBuL)FeCl]2(μ-NC6H3-3,5-(CF3)2), we demonstrate that in solution the bridging imido can facilitate HAA as well as dissociate into a terminal iminyl species, which then can promote HAA. In situ monitoring reveals the di-iron bridging imido is a catalytically competent intermediate, one of several iron complexes observed in the amination of C–H bond substrates or styrene aziridination.

1. INTRODUCTION
Given the importance of C–N bond forming reactions in the pharmaceutical industry,1 the direct incorporation of nitrogen functionality into unreactive C–H bonds remains an important synthetic challenge. To this end, great emphasis is being placed on utilizing transition metal catalysts as a tool for promoting reactivity in a controlled fashion. Motivated by understanding the key attributes required to facilitate N-group transfer chemistry, isolation and characterization of reactive intermediates have been targeted with a variety of metal centers, ligand platforms, and nitrene sources.2–4 As a result, a range of possible metal-bound nitrene motifs has been identified, comprised of metal nitrenoid-type species via simple coordinate covalent interaction of the nitrene with the metal center (Figure 1a), open-shell metal iminyl electronic structures (Figure 1b), as well as closed-shell terminally bound or bridging imido complexes (Figure 1c and d). These electronic descriptors highlight differences in both the covalency of the metal–nitrene interaction as well as the valency of the nitrogen center, features that are thought to influence the activation and functionalization of C–H bonds.
Figure 1.

Possible metal-bound nitrene intermediates, varying the nuclearity and valency of the nitrogen center.
Late, first-row transition metals have been of interest for C–H functionalization chemistry given their inherent compressed ligand field allowing for tuning of reactivity. Manganese,5 iron,6–18 cobalt,7,19–27 and nickel28–37 terminal imido complexes have been isolated; however, few display C–H amination reactivity. Almost all reported species feature low or intermediate spin states, favoring strong metal–N multiple bonds and decreasing the propensity for C–H activation or functionalization. The few exceptions, metal imido complexes reported by Hillhouse,31 Theopold,21 and our group,11 all can access open-shell electronic configurations that enforce unpaired electron density along the M–N bond vector, affording compounds poised for transferring the N-functionality into C–H bonds.
Bridging metal imidos supported by multiple metal centers have also been described in the literature,38–41 although their reactivity toward functionalization of C–H bonds is relatively limited. Among these, the di-copper imidos reported by Warren42,43 exhibit productive C–H amination chemistry, a transformation that is believed to occur via dissociation into a reactive terminal copper imido species.43,44 Similar dinuclear motifs have been reported with iron,15,45–56 cobalt,57 and nickel,30,36 the latter of which have been shown to undergo two-electron-type chemistry by transferring the N-functionality to isocyanides or carbon monoxide.30 However, no other examples of C–H functionalization mediated by these bridging imido species have been previously discussed. Generally, the higher nuclearity bridging imido complexes are more robust, and, consequently, their role in C–H amination has not been widely explored. Questions regarding the ability for stepwise C–H activation at the dinuclear site, attenuation or enhancement of reactivity relative to terminal metal imidos, as well as factors that dictate the reactivity of these species remain interesting.
We have demonstrated that high-spin FeII complexes stabilized by weak-field dipyrrin ligands promote catalytic C–H bond amination both intra- and intermolecularly.58,59 The observed reactivity has been ascribed to the unique electronic structure of the isolated putative intermediate: a high-spin FeIII center antiferromagnetically coupled to an aryl-iminyl radical and supported by a sterically encumbered ligand.11 This species serves as a viable nitrene delivery reagent, where amination of toluene as well as aziridination of styrene at room temperature was demonstrated. While this electronic structure has been invoked for other metal imido complexes,21,29,42,60 no other isolated examples have been reported. To address whether the iron iminyl electronic configuration is enforced by the sterically protected dipyrrin system, we explored the influence of a less sterically hindered tert-butyl-substituted dipyrrin platform. Additionally, we sought to assess whether this ligand scaffold permits access to other modes of interaction between the nitrene and the metal center and establish the effect on N-group transfer chemistry in the context of C–H amination. Herein, we report investigations of both a terminal iron iminyl and a bridging iron imido species isolated with a more sterically accessible ligand. Our findings correlate the ability of these species to activate C–H bonds with the potential for accessing high-spin electronic configurations at the iron center. Studies conducted to understand the reactivity of the isolated bridging imido reveal the possibility for unprecedented direct C–H activation at the dinuclear site, as well as revealing its competency during catalysis.
2. RESULTS AND DISCUSSION
Following literature protocol, metalation of the lithiated tert-butyl-substituted dipyrromethene affords the solvated complex (tBuL)FeCl(OEt2) (1) consistent with published characterized data (tBuL = 1,9-di-tert-butyl-5-(2,6-dichlorophenyl)-dipyrromethene).59 Our previous work with isolated metal-nitrene intermediates has shown that these species display a high propensity for hydrogen-atom abstraction (HAA).11 As such, we sought to minimize access to weak C–H bonds by removing the bound diethyl ether molecule from the iron coordination sphere. Refluxing 1 in toluene, followed by heating under vacuum, furnishes a dimeric ferrous species [(tBuL)FeCl]2 (2) as a green microcrystalline powder (Scheme 1). Slow evaporation of a benzene solution of 2 at room temperature afforded crystals suitable for X-ray diffraction. The solid-state molecular structure of 2 (Figure 2a) displays two chloride atoms bridging the iron centers (Fe–Cl–Fe angle of 92.70(3)°) adopting a distorted trigonal monopyramidal geometry.
Scheme 1.

Figure 2.

Solid-state molecular structures for (a) [(tBuL)FeCl]2 (2), (b) (tBuL)FeCl(•NC6H3-2,6-iPr2) (3), (c) (tBuL)FeCl(H2NC6H3-2-iPr2-6-C(CH2) (CH3)) (4), and (d) [(tBuL)FeCl]2(μ-NC6H3-3,5-(CF3)2) (6) with thermal ellipsoids at 50% probability level. Color scheme: Fe, orange; N, blue; Cl, aquamarine; F, green. Hydrogens, solvent molecules, and disordered isopropyl group in 3 are omitted for clarity.
57Fe Mössbauer analysis of 2 revealed the presence of a single iron environment with parameters (δ = 0.94 mm/s, |ΔEQ| = 1.96 mm/s) in line with other high-spin FeII dipyrrin complexes previously synthesized.11 Variable-temperature magnetic susceptibility measurements corroborate the S = 0 spin state assignment resulting from two weakly antiferromagnetically interacting FeII centers, with a χMT value of 6.40 cm3K/mol at 295 K and a coupling constant of −1.95 cm−1 (Figure S-2). The higher than expected magnetic moment (μeff = 7.15 μB at 295 K versus 6.93 μB) for two non-interacting high-spin ferrous centers is in line with the presence of large zero-field splitting as found upon fitting the reduced magnetization data collected at 1–7 T over the 1.8–10 K temperature range: g1 = g2 = 2.075, D = 13.48 cm−1, D2 = 13.16 cm−1, |E1/D1| = 0.33, and |E2/D2| = 0.33.
2.1. Canvassing Nitrene Capture Viability of 2
With 2 in hand, we began examining its efficacy for nitrene capture from aryl azides (Scheme 2). Addition of a thawing solution of 2 in benzene-d6 to a stoichiometric amount of 4-tert-butylphenyl azide immediately resulted in a new paramagnetically shifted 1H NMR spectrum. 57Fe Mössbauer analysis of the reaction mixture reveals a new iron-containing species with isomer shift and quadrupole splitting values supporting a high-spin FeIII formulation (δ = 0.30 mm/s, |ΔEQ| = 2.04 mm/s, Figure S-3a), as the parameters are in good agreement with those previously reported for the FeIII iminyl radical species (ArL)FeCl(•NC6H4-4-tBu) (δ = 0.28 mm/s, |ΔEQ| = 2.29 mm/s).11 This is not an isolated example; rather, rapid oxidation of 2 to afford trivalent iron complexes is observed with several aryl azides (4-methoxy-, 4-nitro-, 2,6-diisopropyl-, and 3,5-bis-(trifluoromethyl)phenyl azide) as evidenced via 57Fe Mössbauer analysis (Figure S-3). However, all of these reaction mixtures feature two distinct iron species, with contribution from a divalent iron species (Figure S-3, green trace). With the exception of 4-tert-butylphenyl azide and 3,5-bis-(trifluoromethyl)phenyl azide, which do not fully consume 2, we propose these ferrous species result upon fast decay of the FeIII intermediates, suggesting a reactive nature for the proposed ferric products. Despite our attempts to cleanly synthesize and isolate all of the products mentioned above, we were unsuccessful in confirming the identity of the species formed upon treatment of 2 with any of the para-substituted aryl azides. However, modified syntheses of the products formed in the presence of either 2,6-diisopropylphenyl azide and 3,5-bis(trifluoromethyl)phenyl azide were amenable for characterization.
Scheme 2.

2.2. Isolation and Characterization of Terminal Iminyl Complex
Stirring a solution of 2 with 2,6-diisopropylphenyl azide at −40 °C for 5 h (Scheme 3) cleanly affords a new product as confirmed by a single quadrupole doublet in the 57Fe Mössbauer spectrum of the reaction mixture (Figure 3a). Gratifyingly, layering a fluorobenzene solution of this product with pentane affords a green crystalline material, which was characterized by X-ray diffraction to unveil a terminally bound imido/iminyl moiety (tBuL)FeCl(•NC6H3-2,6-iPr2) (3) (Figure 2b).
Scheme 3.

Figure 3.

(a) Zero-field 57Fe Mössbauer spectrum of (tBuL)FeCl(•NC6H3-2,6-iPr2) (3) collected at 90 K: δ = 0.37 mm/s, |ΔEQ| = 2.17 mm/s. (b) Variable-temperature susceptibility data for of 3 collected at 1.0 T, with χMT = 2.76 cm3K/mol at 295 K; (inset) reduced magnetization collected at 7 fields (1–7 T) over the temperature range 1.8–10 K. Magnetization fit parameters obtained with PHI:62 g = 1.95, D = 2.91 cm−1, |E/D| = 0.21. (c) Mulliken spin density plot (α–β) calculated63 for 3, illustrating the antiferromagnetic coupling between the high-spin FeIII and the iminyl radical, J = −655 cm−1.
The solid-state molecular structure displays an Fe–Nim bond length of 1.768(4) Å, in excellent agreement with the metrics reported for (ArL)FeCl(•NC6H4-4-tBu) (1.768(2) Å)11 and significantly elongated with respect to previously isolated terminal iron imidos.6,9,10 Interestingly, the imido unit does not reside in the plane of the dipyrrin ligand, and it displays an almost linear Fe–Nim–CAr bond (178.7(4)° vs 156.43(17)° for (ArL)FeCl(•NC6H4-4-tBu)). The distinct iminyl moiety configuration around the iron center could result from the less sterically encumbered ligand platform; however, the influence of the ortho aryl substituents cannot be excluded.
Comparison of the C–C bond lengths within the aryl group (Cipso–Cortho 1.435(6), 1.439(6) Å; Cortho–Cmeta 1.374(6), 1.382(6); Cmeta–Cpara 1.385(6), 1.373(6) Å) with those of the respective benzene moiety in an isolated FeII aniline adduct (vide infra 4) (Cipso–Cortho 1.399(5), 1.402(5) Å; Cortho–Cmeta 1.396(5), 1.398(5) Å; Cmeta–Cpara 1.381(6), 1.384(6) Å) emphasizes the elongation of the Cipso–Cortho bonds, without any other significant distortions. Additionally, the double bond character of the N–Cipso bond (1.325(5) Å) is also indicative of the radical density not being fully localized on the nitrogen atom. Bimolecular reactivity to afford a C–C coupled product similar to that reported for a nickel 2,6-diisopropyl-imide bearing radical character on the imido linkage61 was not observed, suggesting a lack of electron density at the para-position. We have previously found that treatment of the corresponding (ArL)FeCl with phenyl azide furnishes [(ArL)-FeCl]2(μ-N(Ph)(C6H5)N) following C–N bond coupling of two (ArL)FeCl(•NPh) moieties.11 No such product was detected either; however, the additional imido ortho groups may be preventing such interaction. In conjunction with the 57Fe Mössbauer data, these results are suggestive of an FeIII iminyl radical electronic description.
To probe the electronic structure of iminyl 3, the magnetic behavior was investigated via SQUID magnetometry (Figure 3b). Variable-temperature (VT) direct-current (dc) susceptibility data collected over the temperature range 5–300 K provided a χMT of 2.76 cm3K/mol (4.70 μB) at 295 K, consistent with the spin-only value of 3.00 cm3K/mol (4.90 μB) for a quintet state. The persistence of the S = 2 molecular spin state at room temperature highlights the strength of the coupling interaction, which cannot be thermally overcome (208 cm−1 at 300 K). Magnetization measurements were collected under dc fields 1–7 T over the temperature range 1.8–10 K. The 7T isofield curve plateaus at 3.31 μB (at 1.8 K), significantly lower than the expected magnetic saturation of 4.0 μB for an S = 2 spin state, indicating the presence of magnetic anisotropy. The reduced magnetization data were fit using the program PHI62 to find an acceptable model for a quintet state with the following parameters: g = 1.95, D = 2.91 cm−1, and |E/D| = 0.21.
2.2.1. Theoretical Analysis for the Electronic Structure of 3
Further support for the proposed electronic structure of 3 came from DFT. An unrestricted, single-point calculation at the B3LYP level63 using the crystallographic coordinates determined for 3 converged to the broken-symmetry BS(5,1) solution, with an estimated exchange coupling constant of −655 cm−1 to corroborate the magnetometry. The calculated 57Fe Mössbauer parameters (δ = 0.39 mm/s, ΔEQ = −1.80 mm/s) were found to be in good agreement with the experimental data. The resulting corresponding orbitals reveal that the β-electron resides in an Fe–N π symmetric orbital, highlighting the attenuated bond order of the Fe–N bond, in line with the experimentally observed elongation. Furthermore, the Mulliken spin density plot (α–β) (Figure 3c) illustrates radical delocalization throughout the entire aryl ring. As such, we believe that the antiferromagnetically coupled FeIII iminyl radical formulation predicted is an appropriate description for 3, as implicated by all experimental results (57Fe Mössbauer spectroscopy, X-ray diffraction studies, and SQUID magnetometry).
2.2.2. Reactivity of the Iminyl Radical 3
Given the observed instability of 3 within minutes at room temperature, we hypothesized that the less sterically protected iron iminyl would be more reactive toward C–H bonds than the previously isolated iminyl (ArL)FeCl(•NC6H4-4-tBu). Indeed, within 12 h, a standing solution of 3 converts cleanly to an FeII aniline adduct 4, where one of the isopropyl groups has been dehydrogenated following two sequential HAA steps (Figure 2c, Scheme 4).
Scheme 4.

Furthermore, exposure of 2,6-diisopropylphenyl azide to a 10 mol % solution of 2 in toluene affords nearly quantitative conversion to the respective 2-isopropyl-6-(prop-1-en-2-yl)-aniline. Treatment of 3 with excess 1,4-cyclohexadiene generates the expected FeII (2,6-diisopropylaniline) (5) four-coordinate complex as confirmed by 1H NMR (Scheme 4). However, in the presence of a stronger C–H bond such as cyclohexene (BDE = 81 kcal/mol64), the facile intramolecular HAA pathway dominates and no C–H amination is detected.
Although 3 is not a viable N-group transfer reagent, its ability to perform two sequential HAA steps is of interest. Literature reports on metal imidos undergoing intramolecular HAA showcase either subsequent insertion of the nitrogen functionality into the supporting ligand,21,42 generation of a metallacycle,25 or further radical chemistry along the ligand scaffold.65 A recently reported FeIV bisimido complex was shown to undergo a similar dehydrogenation of the 2,6-diisopropylphenyl imido moiety to afford the corresponding iron amide [(IMes)Fe(HNC6H3-2,6-iPr2) (HNC6H3-2-iPr-6-(CH2) (CH3))].66 In the present example, nitrene insertion into the ortho benzylic C–H bond would afford a four-membered ring, which is thermodynamically disfavored, whereas formation of a new Fe–C bond following a rebound reaction with the benzylic radical is likely sterically prohibited. However, the posited FeIII amido complex resulting from HAA is competent to further activate C–H bonds.
2.3. Isolation of Di-iron Bridging Imido Complex
2.3.1. Synthesis and Characterization
Exposure of 2 to 5 equiv of 3,5-bis(trifluoromethyl)phenyl azide in hexanes at −40 °C for 6 h provided clean access to a new dark-green species (Scheme 5). X-ray diffraction studies on single crystals obtained from a 2:1 mixture of hexanes:benzene at −35 °C unveiled a di-iron complex bridged through an aryl imido unit [(tBuL)-FeCl]2(μ-NC6H3-3,5-(CF3)2) (6) (Figure 2d). The Fe–N–Fe linkage displays Fe–N bond lengths of 1.888(4) and 1.893(4) Å and an Fe–N–Fe angle of 132.1(2)°, parameters similar to those reported by West45 for a (μ-p-tolylimido) bis[(N,N′-ethane-1,2-diyl-bis(salicylaldiminato))iron(III)] (average Fe–N 1.88(1) Å, Fe–N–Fe 129.6(6)°), and slightly elongated as compared to those found by Borovik46 for bis[bis[(N′-tert-butylureaylato)-N-ethyl]-N-methylaminatoferrate(III)]-(l-xylylimido) (average Fe–N 1.870(2) Å, Fe–N–Fe 105.37(8)°) as a result of the additional bridging ligands present in the latter example.
Scheme 5.

The 57Fe Mössbauer spectrum of 6 (Figure 4a) displays one quadrupole doublet (with parameters δ = 0.33 mm/s, |ΔEQ| = 1.48 mm/s) indicative of a ferric oxidation state for the two metals. VT magnetic susceptibility measurements revealed an antiferromagnetic interaction of the two iron centers. A plot of χMT versus T displays a gradual decrease between 300 and 50 K as expected for an S = 0 ground state (Figure 4b). At 50 K, the χMT levels out at a value higher than zero due to the presence of an impurity in the sample. Assuming a high-spin FeIII impurity, we found a suitable fit for the susceptibility data using the program PHI62 (g = 1.95, 11.8% monomeric impurity) and extracted an exchange coupling constant67 (J) of −108.7 cm−1. Similar magnetic behavior has been described for the (μ-p-tolylimido) bis[(N,N′-ethane-1,2-diyl-bis(salicylaldiminato))-iron(III)], with an antiferromagnetic coupling interaction of −103 ± 0.5 cm−1.45 Formation of a bridging imido species could arise via the transient generation of a terminal iron iminyl analogous to 3, similar to the bimolecular coupling of two (ArL)FeCl(•NPh) moieties to afford [(ArL)FeCl]2(μ-N(Ph)-(C6H5)N).11 In the present example, however, intermolecular reactivity is not manifest at the para position as a result of the increased steric protection provided by the meta trifluoromethyl groups. Instead, the exposed nitrogen center on the imido unit undergoes further reaction with 0.5 equiv of 2 to afford a closed-shell bridging imido. Evidence for the intermediacy of an iron iminyl species can be gathered from 19F NMR spectra of reactions of 2 with varying amounts of 3,5-bis(trifluoromethyl)phenyl azide conducted in benzene-d6. Under stoichiometric conditions (1 equiv of azide per dimer) or small excess of azide, 6 is found to be the major complex observed in the 19F NMR immediately upon mixing the thawing solutions of 2 and azide (19F NMR δ −67.03 ppm, Figure S-19). If 2 is exposed to a large quantity of azide, a separate species, with a resonance at −29.5 ppm in the 19F NMR, is present in appreciable quantities (Figure S-19), although the 57Fe Mössbauer spectrum still reveals 6 as the dominant product (Figure S-20). In the absence of crystallographic confirmation and on the basis of these observations, we have tentatively assigned the signal at −29.5 ppm as the monomeric FeIII iminyl (tBuL)FeCl(•NC6H3-3,5-(CF3)2).
Figure 4.

(a) Zero-field 57Fe Mössbauer spectrum for [(tBuL)FeCl]2(μ-NC6H3-3,5-(CF3)2) (6) collected at 90 K: δ = 0.33 mm/s, |ΔEQ| = 1.48 mm/s. (b) Variable-temperature susceptibility data for 6: χMT vs T collected at 1.0 T. Fit parameters obtained with PHI:62 g = 1.95, J = −108.7 cm−1, 11.8% monomeric impurity displayed in green. (c) Mulliken spin density plot (α–β) calculated63 for 6, illustrating the antiferromagnetic coupling between the two high-spin FeIII centers, Jcalc = −120 cm−1.
2.3.2. Electronic Structure of Bridging Imido 6
Electronic investigations of 6 via an unrestricted single-point DFT calculation63 using the crystallographic coordinates determined for 6 corroborate the presence of the antiferromagnetic coupling between the two iron centers with an estimated exchange coupling constant of −120 cm−1 (Figure 4c), in very good agreement with the experimentally observed value. Furthermore, analysis of the broken symmetry corresponding orbitals reveals both σ (Fe dz2, Npz, overlap integral of 0.32)-and π (Fe dxz, Npx, overlap integral of 0.35)-mediated exchange pathways, reflecting the strength of the coupling interaction (Figure S-8). Calculated 57Fe Mössbauer parameters (Fe1, δ = 0.34 mm/s, ΔEQ = +1.35 mm/s; Fe2, δ = 0.35 mm/s, ΔEQ = −1.22 mm/s) correlate well with experimental data.
Given the large metal–metal distance (3.4549(10) Å), a direct Fe–Fe coupling mechanism is unlikely.68 As suggested by the DFT calculations, the antiferromagnetic interaction is instead mediated via a superexchange pathway through the imido moiety. The moderate magnitude of the exchange coupling constant reveals the diminished degree of orbital overlap between the metal d orbitals and the corresponding p orbitals of the bridging unit relative to what would be expected for a perfectly linear Fe–N–Fe linkage, as predicted by the Goodenough–Kanamori rules.69–71 Indeed, strong antiferromagnetic interactions have been inferred for the recently reported bridging nitrido di-iron porphyrin complexes displaying nearly linear Fe–N–Fe linkages (estimated exchange coupling constant values exceeded −250 cm−1).72 Similar correlations between the Fe–X–Fe angle and the degree of coupling have been previously described for a series of bridging oxo or sulfido complexes, which were shown to display larger exchange coupling constant values with increasing Fe–X–Fe angles (X = O or S).73
As a result of the direct effect of the bridging angle on the extent of coupling, geometrical changes that may occur in solution could significantly alter the magnetic behavior of these complexes. In the case of 6, a 1H NMR silent spectrum is detected at room temperature consistent with a paramagnetic high-spin ferric system rather than a diamagnetic species that would be expected upon antiferromagnetic coupling of the two iron centers. Unfortunately, solution magnetic susceptibility measurements were not feasible due to rapid decay of solubilized 6 above −40 °C.
Considering the electronic structure of 6, the strong antiferromagnetic interaction between the two high-spin ferric centers detected via solid-state magnetometry gives rise to a spin ladder comprised of five possible dimer states with a total spin ranging between 0 and 5, states that can be used to describe the electronic configuration of the system as affected by thermal energy.74–78 The 2J energy difference of −217.4 cm−1 between the ground state singlet and excited state triplet is comparable to the thermal energy at room temperature (208 cm−1), suggesting that in the solid state at room temperature, the S = 1 state is significantly populated, but complete uncoupling of the two metals is not achieved. In solution, however, molecular motions can disrupt the geometry of the Fe–N–Fe linkage leading to diminished orbital overlap, thus decreasing the magnitude of the antiferromagnetic interaction. As a result, thermal energy can overcome the exchange coupling to afford a dimeric system consisting of two non-interacting high-spin FeIII ions. These, in turn, possess unpaired electron density along the Fe–N bond vector, a feature that we believe is essential for C–H activation reactivity. Therefore, the electronic structure analysis suggests the potential for 6 to undergo direct HAA, reactivity that contrasts the chemistry of most synthetic di-iron bridging oxo complexes, which are rather unreactive toward C–H bonds unless they can unveil a terminal oxo unit.79
2.3.3. Reactivity of the Bridging Imido
The limited reports on C–H functionalization mediated by metal imido intermediates prompted us to probe the viability of the isolated bridging imido as a reactive nitrene source. In the presence of excess 1,4-cyclohexadiene, consumption of 6 is observed within 30 min at room temperature to form several new compounds, including the FeII aniline adduct (tBuL)FeIICl(H2NC6H3-3,5-(CF3)2) (7) as confirmed by both 1H and 19F NMR (Scheme 6, Figure S-21). If instead 6 is exposed to a substrate possessing a single hydrogen atom source such as 2,4,6-tri-tert-butylphenol (BDE = 83 kcal/mol64), one major species can be detected via 19F NMR at −148.1 ppm (Figure S-22, inset). This complex can be independently prepared via a three-step synthesis (Scheme 6): direct metathesis of 2 with lithium 3,5-bis(trifluoromethyl)anilide affords the corresponding FeII anilide derivative 8 (Figure 5a); further treatment of 8 with tetrabutylammonium chloride and ferrocinium hexafluorophosphate affords the FeIII chloride anilide complex 9 as evidenced by the solid-state molecular structure shown in Figure 5b. 9 is also slowly generated upon addition of 2,4,6-tri-tert-butylphenoxyl radical to 7, suggesting that the BDEs of the N–H bond for the FeII aniline adduct 7 and 2,4,6-tri-tert-butylphenol are comparable.64
Scheme 6.

Figure 5.

Solid-state molecular structures for (a) [(tBuL)Fe(NHAr′)]2 (8), (b) (tBuL)FeCl(NHAr′) (9), and (c) (tBuL)Fe(NHAr′)2 (11) with thermal ellipsoids at 50% probability level. Color scheme: Fe, orange; N, blue; Cl, aquamarine; F, green. Hydrogens, solvent molecules, and disordered trifluoromethyl groups in 11 are omitted for clarity; Ar′ = 3,5-bis(trifluoromethyl)phenyl. (d) Mulliken spin density plot calculated63 for 9, illustrating 7.72% spin density on the nitrogen atom.
We propose two possible pathways for the observed HAA reactivity of 6 (Scheme 7). Analysis of the 19F NMR spectrum of a solution of 6 in benzene-d6 upon standing at room temperature for 3 min reveals the emergence of the proposed monomeric iron iminyl species (signal at −29.5 ppm, Figure 6). This observation suggests 6 may undergo dissociation to the iron iminyl (Scheme 7), which is competent for HAA as showcased via the two isolated monomeric iron iminyl radical complexes.11
Scheme 7.

Figure 6.

19F NMR spectra of a benzene-d6 solution of 6 upon standing at room temperature over time. Proposed observed species (δ in ppm): red (δ −29.5) (tBuL)FeCl(•NC6H3-3,5-(CF3)2), dark blue (δ −67.03) [(tBuL)FeCl]2(μ-NC6H3-3,5-(CF3)2) 6, green (δ −77.34) [(tBuL)FeCl]2(μ-HNC6H3-3,5-(CF3)2), purple (δ −128.0) (tBuL)Fe-(HNC6H3-3,5-(CF3)2)2 11, light blue (δ −148.1) (tBuL)FeCl-(HNC6H3-3,5-(CF3)2) 9.
A closer look at the decay of 6 in solution over time unveils the rapid formation of an additional fluorine containing species prior to generation of the iron(III) anilide 9 (Figure 6). A frozen solution EPR spectrum displays a rhombic signal consistent with an S = ½ spin state in addition to signals in line with high-spin FeIII centers (Figure S-23). To justify the doublet spin state, we propose formation of a mixed valent di-iron bridging amido complex (corresponding to the fluorine resonance at −77.34 ppm) that could be accessed via direct HAA from 6 (Scheme 7). An antiferromagnetic coupling of high-spin ferric and ferrous centers would result in a S = ½ molecular spin state. Following formation of a di-iron amido compound, dissociation to furnish 9 should be facile as the Fe–N bonds are weakened; and, thus, the coupling interaction is expected to decrease as well.80
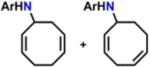
In the presence of C–H substrates, we were pleased to find that 6 serves as a viable N-group transfer reagent, mediating C–H amination of allylic (cyclohexene, cyclooctene, cyclooctadiene) or benzylic (toluene) C–H bonds as well as aziridination of styrene (Scheme 8). Interestingly, a 5:3 mixture of products can be detected upon activation of cyclooctadiene (GC–MS). The two compounds differ by a C=C bond transposition, providing indirect evidence for the generation of an allylic radical during the C–H activation step. Consequently, we propose that reaction of 6 with C–H substrates proceeds via initial HAA, furnishing the iron(III) amide 9, and subsequent radical recombination to afford the functionalized products.
Scheme 8.

To test the viability of the proposed stepwise mechanism, the isolated Fe amide 9 proved suitable for investigation of the radical recombination step. The frozen solution EPR of 9 (Figure S-14) displays features in line with a high-spin ferric formulation, an electronic description further corroborated by 57Fe Mössbauer spectroscopy (Figure S-13). Examination of complex 9 by DFT63 unveils 7.72% spin density on the nitrogen atom (Figure 5d), suggesting the possible involvement of 9 in radical-type chemistry. Indeed, treatment of 9 with triphenylmethyl radical results in rapid formation of 3,5-bis(trifluoromethyl)-N-tritylaniline and regeneration of 2 (Scheme 9, Figure S-30). The ability of 9 to capture the carbon-based radical species and afford the functionalized amine product highlights the role of this species in the C–H amination process mediated by 6. Interestingly, heating a benzene-d6 solution containing 9 in the presence of 1,4-cyclohexadiene at 65 °C results in slow conversion to the aniline adduct (tBuL)FeIICl(H2NC6H3-3,5-(CF3)2) (7) (Figure S-29), emphasizing the importance of the unpaired electron density along the Fe–N π-vector to engender reactivity toward C–H bonds.
Scheme 9.

2.3.4. Catalytic N-Group Transfer Chemistry
Motivated by our previously reported catalytic intermolecular C–H amination protocol,59 we sought to address the viability to transfer the N-functionality provided by 3,5-bis(trifluoromethyl)phenyl azide into C–H bonds. Toward this end, exposing a 10 mol % solution of 2 in cyclohexene, cyclooctene, or cyclooctadiene to 3,5-bis(trifluoromethyl)phenyl azide (Scheme 10) produces the corresponding aminated products in modest to good yields (Table 1). Reaction with the stronger C–H bond of toluene results in formation of the corresponding benzyl amine in 21% yield as determined by 1H NMR. Additionally, detection of 3,5-bis(trifluoromethyl)aniline, diazene, the overoxidized imine product, as well as ligand functionalization via either 1H NMR or mass spectrometry accounts for the lower yield observed for C–H amination. If the reaction is conducted in styrene instead, the expected aziridine is obtained in 92% yield as quantified via 19F NMR.
Scheme 10.

Table 1.
Catalytic Reactivity of 2 and 3,5-Bis(trifluoromethyl)phenyl Azided
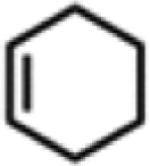
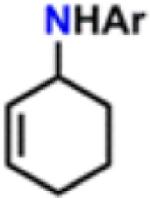
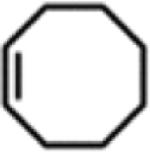
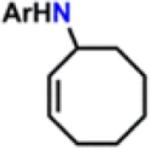
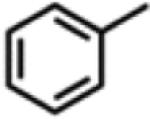
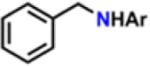
Isolated yields.
1H NMR yield using ferrocene as internal standard.
19F NMR yield using 1,2-difluorobenzene as internal standard.
All reactions were conducted neat in substrate.
2.3.5. In Situ Monitoring during Catalysis
Prompted by these results and the reactivity of the isolated bridging imido complex, we envisioned that 6 can function as a catalytically competent intermediate. To elucidate the catalytic viability of any of the species associated with the bridging imido complex, a time-course analysis for the reaction of 2 with excess 3,5-bis(trifluoromethyl)phenyl azide in cyclohexene was conducted (Scheme 11). The 19F NMR spectrum collected immediately upon warming the reaction to room temperature features resonances consistent with formation of the postulated monomeric iron iminyl and bridging amido complexes as well as the isolated bridging imido 6 (Figure 7b), albeit in insignificant amounts relative to the quantity of azide or corresponding cyclohexyl amine product (Figure 7a and Figure S-26). These signals, however, do not persist at room temperature as would be expected for catalytically relevant intermediates (Figure 7b).
Scheme 11.

Figure 7.

19F NMR spectra for the time-course of the catalytic reaction of 3,5-bis(trifluoromethyl)phenyl azide with 20 mol % of 2 in cyclohexene. (a) Spectrum after 10 min, showing the relative amount of azide and the corresponding cyclohexylamine product (signals at −62.97 ppm) to the iron complexes. (b) Decay of iron complexes over time (increased intensity to show the boxed region in top spectrum). Proposed observed species (δ in ppm): red (δ −29.04) (tBuL)FeCl-(•NC6H3-3,5-(CF3)2), dark blue (δ −66.46) [(tBuL)FeCl]2(μ-NC6H3-3,5-(CF3)2) 6, green (δ −77.10) [(tBuL)FeCl]2(μ-HNC6H3-3,5-(CF3)2), purple (δ −128.0) (tBuL)Fe(HNC6H3-3,5-(CF3)2)2 11, light blue (δ −144.1) (tBuL)FeCl(HNC6H3-3,5-(CF3)2) 9.
On the basis of these findings, we can formulate a mechanism for the catalytic C–H amination transformation mediated by 2: treatment of 2 with the aryl azide furnishes a terminal FeIII iminyl radical species, which can either undergo HAA through interaction with the C–H substrate or couple to 2 giving rise to 6. As demonstrated above, the bridging imido also manifests HAA ability to afford the FeIII anilide complex 9. Finally, generation of 9 via either pathway is succeeded by radical recombination with the carboradical to release the amine product and regenerate 2 (Scheme 12).
Scheme 12.

2.3.6. Observation of Other Reactive Intermediates
The in situ 19F NMR monitoring of the stoichiometric (Figures 6 and S-25) and catalytic (Figures 7 and S-26) reactions of 6 reveals several unaccounted for 19F signals, suggesting a more complex reactivity profile of 6. In an effort to understand the implications of these alternate reactivity paths arising from 6 toward the C–H amination process, investigations to identify these unknown species were undertaken.
We hypothesized that, given the high-spin nature of the iron complexes, ligand exchange should be a facile process such that disproportionation of 6 into two trivalent iron species could occur yielding (tBuL)FeIIICl2 (10) and a terminally bound imido (tBuL)FeIII(NC6H3-3,5-(CF3)2). Attempts to synthesize the latter compound via treatment of 9 with an additional equivalent of lithium 3,5-bis(trifluoromethyl)anilide that could act as a base were unsuccessful. Instead, a new ferric bis-anilide (tBuL)FeIII(NHC6H3-3,5-(CF3)2)2 (11) was isolated and confirmed crystallographically (Figure 5c). Preparation of 11 could also be achieved directly via metathesis of (tBuL)FeCl2 (10) with 2 equiv of lithium 3,5-bis(trifluoromethyl)anilide (Scheme 13). 19F NMR analysis of a solution of 11 reveals a single resonance at −128.0 ppm, a signal that is consistent with one of the 19F species detected following decomposition of 6 in solution (Figure 6) or during the reaction of 2 with 3,5-bis(trifluoromethyl)phenyl azide (Figure S-25).
Scheme 13.

Complex 11 could arise from the corresponding ferrous anilide species (8) either upon reaction with the 3,5-bis(trifluoromethyl)phenyl azide and subsequent HAA or via ligand exchange with 9 (Scheme 14). Indeed, treatment of 8 with aryl azide rapidly furnishes 11 in solution as judged via both 19F NMR and frozen solution EPR (Figure S-28), whereas mixing solutions of 8 and 9 results in formation of both 11 and 2 (Figure S-27). Unlike the mono-anilide 9, 11 does not undergo HAA in the presence of 1,4-cyclohexadiene at 65 °C. Though, treatment of 11 with triphenylmethyl radical furnishes the corresponding trityl aniline product and the FeII anilide complex 8 (Figure S-31). Despite the reduced oxidative power to effect HAA, the ability of 11 to undergo radical recombination suggests that 11 could be a competent intermediate in the C–H functionalization process.
Scheme 14.

3. CONCLUSIONS
In this report, we have showcased the isolation of two iron complexes that possess either a terminal iminyl or a bridging imido. Characterization of these species suggests that the antiferromagnetically coupled high-spin FeIII iminyl radical formulation previously described11 is a prominent electronic feature not dictated or affected by the steric demands of the dipyrrin framework. Both of these complexes demonstrate high reactivity toward C–H bonds: the terminal aryl iminyl bearing ortho-isopropyl substituents was shown to undergo two sequential HAA steps intramolecularly, whereas the di-iron bridging 3,5-bis(trifluoromethyl)-imido was found to successfully deliver the nitrene functionality into allylic and benzylic C–H bonds. Given the access to a maximally high-spin electronic configuration at the metal center, the iron-imido multiple bond is significantly attenuated, resulting in enhanced reactivity in comparison to most isolated metal imidos. As evidenced by several of the species presented herein, we propose that the high-spin nature of these metal complexes that places unpaired electron density along the Fe–N bond vector is essential for reactivity toward C–H bonds.
Furthermore, we have established the ability of the di-iron imide to perform C–H amination, and our investigations suggest that both dissociation to a terminal iron iminyl as well as direct C–H activation at the dinuclear imido site are viable pathways to accomplish this transformation. We propose that, despite the antiferromagnetic interaction manifested in the solid state, two independent high-spin FeIII centers can be unveiled in solution, permitting reactivity. Additionally, isolation of several reactive metal complexes arising from the bridging imido 6 not only emphasizes the complexity of our system, it also highlights the diverse reactivity pathways accessed by 6. We believe that the reactivity showcased by the bridging imido as presented in this report may have implications toward the highly regio- and chemoselective intermolecular allylic C–H amination process mediated by an adamantyl-substituted dipyrrin scaffold,59 revealing another possible role of how the bridging motif may contribute to the C–H functionalization chemistry.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the NSF (CHE-0955885) and NIH (GM-115815), and from Harvard University.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b12582.
General experimental considerations and procedures, multinuclear NMR data, 57Fe Mössbauer and EPR spectra, magnetometry data, solid-state molecular structures, computational details, and crystallographic CIF files and crystallography data (PDF)
X-ray data (CIF)
REFERENCES
- 1.Roughley SD, Jordan AM. J. Med. Chem. 2011;54:3451. doi: 10.1021/jm200187y. [DOI] [PubMed] [Google Scholar]
- 2.Gephart RT, Warren TH. Organometallics. 2012;31:7728. [Google Scholar]
- 3.Roizen JL, Harvey ME, Du Bois J. Acc. Chem. Res. 2012;45:911. doi: 10.1021/ar200318q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang L, Deng L. Chin. Sci. Bull. 2012;57:2352. [Google Scholar]
- 5.Eikey RA, Khan SI, Abu-Omar MM. Angew. Chem., Int. Ed. 2002;41:3591. doi: 10.1002/1521-3773(20021004)41:19<3591::AID-ANIE3591>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 6.Bart SC, Lobkovsky E, Bill E, Chirik PJ. J. Am. Chem. Soc. 2006;128:5302. doi: 10.1021/ja057165y. [DOI] [PubMed] [Google Scholar]
- 7.Betley TA, Peters JC. J. Am. Chem. Soc. 2003;125:10782. doi: 10.1021/ja036687f. [DOI] [PubMed] [Google Scholar]
- 8.Brown SD, Betley TA, Peters JC. J. Am. Chem. Soc. 2003;125:322. doi: 10.1021/ja028448i. [DOI] [PubMed] [Google Scholar]
- 9.Brown SD, Peters JC. J. Am. Chem. Soc. 2005;127:1913. doi: 10.1021/ja0453073. [DOI] [PubMed] [Google Scholar]
- 10.Cowley RE, DeYonker NJ, Eckert NA, Cundari TR, DeBeer S, Bill E, Ottenwaelder X, Flaschenriem C, Holland PL. Inorg. Chem. 2010;49:6172. doi: 10.1021/ic100846b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.King ER, Hennessy ET, Betley TA. J. Am. Chem. Soc. 2011;133:4917. doi: 10.1021/ja110066j. [DOI] [PubMed] [Google Scholar]
- 12.Lu CC, Saouma CT, Day MW, Peters JC. J. Am. Chem. Soc. 2007;129:4. doi: 10.1021/ja065524z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scepaniak JJ, Young JA, Bontchev RP, Smith JM. Angew. Chem., Int. Ed. 2009;48:3158. doi: 10.1002/anie.200900381. [DOI] [PubMed] [Google Scholar]
- 14.Thomas CM, Mankad NP, Peters JC. J. Am. Chem. Soc. 2006;128:4956. doi: 10.1021/ja0604358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Verma AK, Nazif TN, Achim C, Lee SC. J. Am. Chem. Soc. 2000;122:11013. [Google Scholar]
- 16.Nieto I, Ding F, Bontchev RP, Wang H, Smith JM. J. Am. Chem. Soc. 2008;130:2716. doi: 10.1021/ja0776834. [DOI] [PubMed] [Google Scholar]
- 17.Ni C, Fettinger JC, Long GJ, Brynda M, Power PP. Chem. Commun. 2008:6045. doi: 10.1039/b810941a. [DOI] [PubMed] [Google Scholar]
- 18.Moret ME, Peters JC. Angew. Chem., Int. Ed. 2011;50:2063. doi: 10.1002/anie.201006918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jenkins DM, Betley TA, Peters JC. J. Am. Chem. Soc. 2002;124:11238. doi: 10.1021/ja026852b. [DOI] [PubMed] [Google Scholar]
- 20.Hu X, Meyer K. J. Am. Chem. Soc. 2004;126:16322. doi: 10.1021/ja044271b. [DOI] [PubMed] [Google Scholar]
- 21.Shay DT, Yap GPA, Zakharov LN, Rheingold AL, Theopold KH. Angew. Chem., Int. Ed. 2005;44:1508. doi: 10.1002/anie.200462529. [DOI] [PubMed] [Google Scholar]
- 22.Mehn MP, Brown SD, Jenkins DM, Peters JC, Que L., Jr. Inorg. Chem. 2006;45:7417. doi: 10.1021/ic060670r. [DOI] [PubMed] [Google Scholar]
- 23.Cowley RE, Bontchev RP, Sorrell J, Sarracino O, Feng Y, Wang H, Smith JM. J. Am. Chem. Soc. 2007;129:2424. doi: 10.1021/ja066899n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jones C, Schulten C, Rose RP, Stasch A, Aldridge S, Woodul WD, Murray KS, Moubaraki B, Brynda M, La Macchia G, Gagliardi L. Angew. Chem., Int. Ed. 2009;48:7406. doi: 10.1002/anie.200900780. [DOI] [PubMed] [Google Scholar]
- 25.King ER, Sazama GT, Betley TA. J. Am. Chem. Soc. 2012;134:17858. doi: 10.1021/ja307699u. [DOI] [PubMed] [Google Scholar]
- 26.Zhang L, Liu Y, Deng L. J. Am. Chem. Soc. 2014;136:15525. doi: 10.1021/ja509731z. [DOI] [PubMed] [Google Scholar]
- 27.Du J, Wang L, Xie M, Deng L. Angew. Chem., Int. Ed. 2015;54:12640. doi: 10.1002/anie.201505937. [DOI] [PubMed] [Google Scholar]
- 28.Iluc VM, Miller AJM, Anderson JS, Monreal MJ, Mehn MP, Hillhouse GL. J. Am. Chem. Soc. 2011;133:13055. doi: 10.1021/ja2024993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kogut E, Wiencko HL, Zhang L, Cordeau DE, Warren TH. J. Am. Chem. Soc. 2005;127:11248. doi: 10.1021/ja0533186. [DOI] [PubMed] [Google Scholar]
- 30.Laskowski CA, Hillhouse GL. Organometallics. 2009;28:6114. [Google Scholar]
- 31.Laskowski CA, Miller AJM, Hillhouse GL, Cundari TR. J. Am. Chem. Soc. 2011;133:771. doi: 10.1021/ja1101213. [DOI] [PubMed] [Google Scholar]
- 32.Mindiola DJ, Hillhouse GL. J. Am. Chem. Soc. 2001;123:4623. doi: 10.1021/ja010358a. [DOI] [PubMed] [Google Scholar]
- 33.Mindiola DJ, Hillhouse GL. J. Am. Chem. Soc. 2002;124:9976. doi: 10.1021/ja0269183. [DOI] [PubMed] [Google Scholar]
- 34.Waterman R, Hillhouse GL. J. Am. Chem. Soc. 2003;125:13350. doi: 10.1021/ja0381914. [DOI] [PubMed] [Google Scholar]
- 35.Waterman R, Hillhouse GL. J. Am. Chem. Soc. 2008;130:12628. doi: 10.1021/ja805530z. [DOI] [PubMed] [Google Scholar]
- 36.Wiese S, Aguila MJB, Kogut E, Warren TH. Organometallics. 2013;32:2300. [Google Scholar]
- 37.Wiese S, McAfee JL, Pahls DR, McMullin CL, Cundari TR, Warren TH. J. Am. Chem. Soc. 2012;134:10114. doi: 10.1021/ja302149k. [DOI] [PubMed] [Google Scholar]
- 38.Decker A, Fenske D, Maczek K. Angew. Chem., Int. Ed. Engl. 1996;35:2863. [Google Scholar]
- 39.Kamijyo N, Watanabe T. Bull. Chem. Soc. Jpn. 1974;47:373. [Google Scholar]
- 40.Nugent WA, Haymore BL. Coord. Chem. Rev. 1980;31:123. [Google Scholar]
- 41.Powers TM, Betley TA. J. Am. Chem. Soc. 2013;135:12289. doi: 10.1021/ja405057n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Badiei YM, Dinescu A, Dai X, Palomino RM, Heinemann FW, Cundari TR, Warren TH. Angew. Chem., Int. Ed. 2008;47:9961. doi: 10.1002/anie.200804304. [DOI] [PubMed] [Google Scholar]
- 43.Badiei YM, Krishnaswamy A, Melzer MM, Warren TH. J. Am. Chem. Soc. 2006;128:15056. doi: 10.1021/ja065299l. [DOI] [PubMed] [Google Scholar]
- 44.Aguila MJB, Badiei YM, Warren TH. J. Am. Chem. Soc. 2013;135:9399. doi: 10.1021/ja400879m. [DOI] [PubMed] [Google Scholar]
- 45.Nichols PJ, Fallon GD, Murray KS, West BO. Inorg. Chem. 1988;27:2795. [Google Scholar]
- 46.Zart MK, Powell D, Borovik AS. Inorg. Chim. Acta. 2007;360:2397. doi: 10.1016/j.ica.2006.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brown SD, Mehn MP, Peters JC. J. Am. Chem. Soc. 2005;127:13146. doi: 10.1021/ja0544509. [DOI] [PubMed] [Google Scholar]
- 48.Chen X-D, Duncan JS, Verma AK, Lee SC. J. Am. Chem. Soc. 2010;132:15884. doi: 10.1021/ja106478k. [DOI] [PubMed] [Google Scholar]
- 49.Duncan JS, Nazif TM, Verma AK, Lee SC. Inorg. Chem. 2003;42:1211. doi: 10.1021/ic025825j. [DOI] [PubMed] [Google Scholar]
- 50.Duncan JS, Zdilla MJ, Lee SC. Inorg. Chem. 2007;46:1071. doi: 10.1021/ic061133+. [DOI] [PubMed] [Google Scholar]
- 51.Li Y, Wong W-T. Coord. Chem. Rev. 2003;243:191. [Google Scholar]
- 52.Link H, Decker A, Fenske DZ. Anorg. Allg. Chem. 2000;626:1567. [Google Scholar]
- 53.Ohki Y, Takikawa Y, Hatanaka T, Tatsumi K. Organometallics. 2006;25:3111. [Google Scholar]
- 54.Takemoto S, Ogura S-I, Yo H, Hosokoshi Y, Kamikawa K, Matsuzaka H. Inorg. Chem. 2006;45:4871. doi: 10.1021/ic060744z. [DOI] [PubMed] [Google Scholar]
- 55.Zdilla MJ, Verma AK, Lee SC. Inorg. Chem. 2011;50:1551. doi: 10.1021/ic1021627. [DOI] [PubMed] [Google Scholar]
- 56.Zhang Q, Xiang L, Deng L. Organometallics. 2012;31:4537. [Google Scholar]
- 57.Dai X, Kapoor P, Warren TH. J. Am. Chem. Soc. 2004;126:4798. doi: 10.1021/ja036308i. [DOI] [PubMed] [Google Scholar]
- 58.Hennessy ET, Betley TA. Science. 2013;340:591. doi: 10.1126/science.1233701. [DOI] [PubMed] [Google Scholar]
- 59.Hennessy ET, Liu RY, Iovan DA, Duncan RA, Betley TA. Chem. Sci. 2014;5:1526. [Google Scholar]
- 60.Lu CC, De Beer George S, Weyhermüller T, Bill E, Bothe E, Wieghardt K. Angew. Chem., Int. Ed. 2008;47:6384. doi: 10.1002/anie.200800669. [DOI] [PubMed] [Google Scholar]
- 61.Bai G, Stephan D. Angew. Chem., Int. Ed. 2007;46:1856. doi: 10.1002/anie.200604547. [DOI] [PubMed] [Google Scholar]
- 62.Chilton NF, Anderson RP, Turner LD, Soncini A, Murray KS. J. Comput. Chem. 2013;34:1164. doi: 10.1002/jcc.23234. [DOI] [PubMed] [Google Scholar]
- 63.Neese F. ORCA - An ab initio, Density Functional and Semi-empirical Electronic Structure Package, Version 2.9-00. Universitat Bonn; Bonn, Germany: 2009. [Google Scholar]
- 64.Luo Y-R. Handbook of Bond Dissociation Energies in Organic Compounds. CRC Press LLC; Boca Raton, FL: 2003. [Google Scholar]
- 65.Cowley RE, Eckert NA, Vaddadi S, Figg TM, Cundari TR, Holland PL. J. Am. Chem. Soc. 2011;133:9796. doi: 10.1021/ja2005303. [DOI] [PubMed] [Google Scholar]
- 66.Wang L, Hu L, Zhang H, Chen H, Deng L. J. Am. Chem. Soc. 2015;137:14196. doi: 10.1021/jacs.5b09579. [DOI] [PubMed] [Google Scholar]
- 67.The Heisenberg, Dirac, and Van Vleck spin Hamiltonian H = −2JSFe1•SFe2 (ref 77) was used to describe the exchange interaction between the two iron centers.
- 68.Hay PJ, Thibeault JC, Hoffmann RJ. Am. Chem. Soc. 1975;97:4884. [Google Scholar]
- 69.Goodenough JB. Phys. Rev. 1955;100:564. [Google Scholar]
- 70.Goodenough JB. J. Phys. Chem. Solids. 1958:6. [Google Scholar]
- 71.Kanamori JJ. Phys. Chem. Solids. 1959;10:87. [Google Scholar]
- 72.Kudrik EV, Afanasiev P, Alvarez LX, Dubourdeaux P, Clémancey M, Latour JM, Blondin G, Bouchu D, Albrieux F, Nefedov SE, Sorokin AB. Nat. Chem. 2012;4:1024. doi: 10.1038/nchem.1471. [DOI] [PubMed] [Google Scholar]
- 73.Mukherjee RN, Stack TDP, Holm RH. J. Am. Chem. Soc. 1988;110:1850. [Google Scholar]
- 74.Heisenberg W. Eur. Phys. J. A. 1926;38:411. [Google Scholar]
- 75.Heisenberg W. Eur. Phys. J. A. 1928;49:619. [Google Scholar]
- 76.Dirac PAM. Proc. R. Soc. London, Ser. A. 1929;123:714. [Google Scholar]
- 77.Van Vleck JH. The Theory of Electric and Magnetic Susceptibilities. Oxford University Press; Oxford: 1932. [Google Scholar]
- 78.Murray KS. Coord. Chem. Rev. 1974;12:1. [Google Scholar]
- 79.Kurtz DM. Chem. Rev. 1990;90:585. [Google Scholar]
- 80.Theisen RM, Shearer J, Kaminsky W, Kovacs JA. Inorg. Chem. 2004;43:7682. doi: 10.1021/ic0491884. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.