

Abstract

WNT signaling is frequently deregulated in malignancy, particularly in colon cancer, and plays a key role in the generation and maintenance of cancer stem cells. We report the discovery and optimization of a 3,4,5-trisubstituted pyridine 9 using a high-throughput cell-based reporter assay of WNT pathway activity. We demonstrate a twisted conformation about the pyridine–piperidine bond of 9 by small-molecule X-ray crystallography. Medicinal chemistry optimization to maintain this twisted conformation, cognisant of physicochemical properties likely to maintain good cell permeability, led to 74 (CCT251545), a potent small-molecule inhibitor of WNT signaling with good oral pharmacokinetics. We demonstrate inhibition of WNT pathway activity in a solid human tumor xenograft model with evidence for tumor growth inhibition following oral dosing. This work provides a successful example of hypothesis-driven medicinal chemistry optimization from a singleton hit against a cell-based pathway assay without knowledge of the biochemical target.
Introduction
The WNT signaling network is a major regulator of mammalian development through control of cellular functions such as proliferation and differentiation.1 WNT signaling is frequently deregulated in malignancy,2 especially in colon cancer.3 Binding of WNT ligands, a family of secreted glycosylated proteins, to the Frizzled and LRP families of cell surface receptors initiates a series of signaling events via the cytoplasmic protein Dishevelled (DSH). Upon activation, DSH recruits Axin and destabilizes the destruction complex that normally acts to degrade the transcriptional cofactor β-catenin. In the absence of degradation, β-catenin accumulates and translocates to the nucleus, where it recruits coactivators and associates with the LEF/TCF family of DNA-binding proteins, thereby altering expression of a variety of genes, including cyclin D1 and c-MYC.4 In cells not subject to stimulation by WNT ligands, β-catenin degradation by the destruction complex limits β-catenin-mediated gene transcription. Key components of the β-catenin destruction complex include Axin, APC (the protein product of the adenomatous polyposis coli tumor suppressor gene), and glycogen synthase kinase-3-β (GSK3-β), which phosphorylates β-catenin and thereby renders β-catenin a substrate for ubiquitination and degradation by the 26S proteasome.5
Overexpression of WNT-regulated genes can cause transformation of mammalian epithelial cells.6 Clinically, 80% of colon cancers have defects in the APC gene, leading to high levels of β-catenin.7,8 A subset of colon cancers and melanomas harbor β-catenin mutations that prevent its phosphorylation and subsequent degradation.9 Many other cancers also show evidence of inappropriate WNT pathway activation, including raised levels of β-catenin.10−13 In addition, defective WNT signaling plays a key role in the generation and maintenance of cancer stem cells.14 Validation of a pivotal role for WNT signaling in cancer is therefore very strong, and compounds that dampen WNT pathway activity could provide useful molecularly targeted therapeutics for the treatment of cancer;6,15,16 indeed, the fully humanized monoclonal antibody OMP-18R5 that targets Frizzled receptors is efficacious in patient-derived mouse tumor models17 and is currently in a phase I clinical trial.18 Despite the importance of the WNT pathway, it is only recently that small-molecule inhibitors have been identified and progressed toward clinical trial.16 Compound 1 (ICG001) blocks the interaction between β-catenin and the transcriptional coactivating protein CREB binding protein (CBP), leading to a reduction of colon adenoma formation in mouse models.19 The β-catenin–CBP interaction was shown to promote stem/progenitor marker expression, while the related β-catenin–p300 interaction (not inhibited by 1) promoted expression of genes involved in proliferative responses such as c-Myc.20 PRI-724 (structure undisclosed), an inhibitor of the β-catenin–CBP interaction, has now been progressed to clinical trial.21 Compounds 2 (XAV939) and 3 (IWR-1) have been disclosed as inhibitors of WNT signaling via inhibition of the tankyrase activity required for degradation of Axin; 2 was discovered by target deconvolution from a cell-based pathway screen.22,234 (IWP-2), a small-molecule inhibitor of porcupine, the acyltransferase essential for secretion of functional WNT ligands, was also discovered by cell-based pathway screening (Figure 1).23 Subsequent to the identification of tankyrase and porcupine as validated molecular targets, many small-molecule inhibitors have been reported.24 Examplar tankyrase inhibitors include 5 (NVP-TNKS656),256 (G007-LK),26 and compound 7(27) from Amgen; the porcupine inhibitor 8 (LGK974)28 is currently in a phase I clinical trial.
Figure 1.

Small-molecule inhibitors of the WNT pathway: 1,192,223,224,23 5,256,267,27 and 8.28
Many WNT pathway mutations occur at, or upstream of, β-catenin. Therefore, if the pathway is blocked at or below this point, an inhibitor should be active against multiple tumors driven by a WNT-activating mutation. With this in mind, we set out to discover small-molecule inhibitors of WNT signaling and employed a cell-based pathway screening strategy to identify compounds that block WNT signaling at, or downstream of, β-catenin. While our work was in progress a number of reports appeared describing successful cell-based pathway screens against the WNT pathway22,23 and, for example, BMP signaling29 revealing hitherto undiscovered regulatory mechanisms, increasing our confidence in such an approach. We have previously reported our screening strategy30 and describe here the medicinal chemistry optimization of a 3,4,5-trisubstituted pyridine hit (9) to give potent and orally bioavailable small-molecule inhibitors of WNT signaling. We demonstrate a 350-fold potency enhancement in a cell-based assay of WNT pathway activity through hypothesis-driven medicinal chemistry design respectful of physicochemical properties likely to elicit good cell permeability.
Chemistry
Our synthetic strategy to most desired 3,4,5-trisubstituted pyridines involved SNAr displacement of the 4-chloro substituent in either 3,4,5-trichloropyridine (10) or 3-bromo-4,5-dichloropyridine (11) using the appropriate N-boc-protected pyrrolidine 12, piperidine 13, or NH-piperidine 14 to give the 4-substituted analogues 9, 15–22, 39, 41, 43, 45, 47, 49, 51, 53, 57, and 84 according to our previously published methodology.31 Subsequent palladium-mediated cross-coupling of appropriate aryl boronic acids or esters gave the corresponding 3,4,5-trisubstituted pyridines 26, 28–38, 40, 42, 44, 46, 48, 50, 52, 54, and 65–81 (Scheme 1). Compounds 59, 60, and 62 described in Table 5 were prepared by sequential cross-coupling at the 3- and 5-positions of 8-(3-bromo-5-chloropyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (57). Compounds 63 and 64 were prepared directly from 8-(3-chloro-5-(4-morpholin-4-ylphenyl)pyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (44), while compound 56 was prepared by SNAr displacement of the 4-chloro substituent in 3-bromo-4-chloro-5-methylpyridine, followed by palladium-mediated cross-coupling at the 3-position. Preparations of noncommercial aryl boronic acids or esters are described in the Experimental Section and the Supporting Information.
Scheme 1. General Synthetic Route.

Table 5. Variation of the 3-Chloro Substituent.

MLM and HLM values indicate percentage parent remaining after 30 min of incubation.
Results and Discussion
To discover inhibitors of WNT signaling, we used an inducible luciferase reporter assay in human embryonic kidney cells (HEK293) that contained both estrogen receptor-DSH and TCF-luciferase-IRES-GFP constructs.30 GFP fluorescence was used to sort cells by flow cytometry that were highly induced by estradiol. Clone 7dF3 was selected for use in HTS format and gave a 40-fold induction of luciferase activity when DSH protein expression was induced using 10 μM estradiol. We screened 63 040 compounds at an assay concentration of 20 μM and selected compounds that reproducibly gave >50% inhibition in the 7dF3 luciferase reporter assay (n = 2) and did not inhibit luciferase enzymatic activity nor lead to >50% cell death at 20 μM.30 A control cell line incorporating a thymidine kinase promoter-driven renilla luciferase reporter construct was also utilized to deselect compounds that demonstrated potent and efficacious inhibition of non-WNT-mediated transcription.30
Hit compound 9 (IC50 = 1.76 μM) was the only exemplar of a 3,4,5-trisubstituted pyridine in our screening library at that time; however, despite being a singleton, it was among the most attractive chemicals, with a low molecular weight and acceptable physicochemical properties (Table 1 and Figure 2). Compound 9 blocked activation of the pathway by the GSK-3β inhibitor bromoindirubin-3′-oxime (BIO) (IC50 = 2.60 μM), indicative of action at or below GSK-3β-mediated phosphorylation of β-catenin. We then further characterized the locus of interaction for compound 9 using a cell-based assay cascade in which WNT signaling is activated at distinct levels by inducible expression of cDNA-encoded activating components of the canonical WNT pathway; a more robust development of our previously reported cell-based assay cascade.30 This cell-based assay cascade included activation of WNT signaling through expression of either ΔN-LRP6 (a constitutively activating component of the WNT receptor), Dvl-2 (a Disheveled homologue and activator of WNT signaling), Axin-GID (Axin-GSK3β Interaction Domain, a dominant negative form of Axin that binds to and inhibits GSK-3β), ΔN-β-catenin (a stabilized form of β-catenin resistant to degradation), or VP16-TCF (a TCF transcription factor that is constitutively active in the absence of β-catenin). Compound 9 demonstrated potency and efficacy in all these cell-based assays, similar to the potency observed in the 7dF3 primary assay and consistent with intracellular inhibition of WNT signaling at or below the TCF locale (IC50’s = 0.9–7.6 μM, Table 2).
Table 1. Modification of the Piperidine-4-carboxamide.

MLM and HLM values indicate percent turnover after 30 min of incubation.
Figure 2.

Attributes of hit compound 9.
Table 2. Assessment of Compounds 9 and 33 in Cell-Based Assays in Which WNT Signaling Is Activated at Distinct Levels by Inducible Expression of cDNAs Encoding Activating Components of the Canonical WNT Pathwaya.
| IC50 (μM) |
|||
|---|---|---|---|
| WNT pathway activation level | reporter | 9 | 33 |
| Dvl-ER (7dF3 primary assay) | TCF-luc | 1.76 | 0.037 |
| Tet-O-ΔN-LRP6 | TCF-luc | 0.91 | 0.020 |
| Tet-O-Dvl2 | TCF-luc | 1.20 | 0.020 |
| Tet-O-Axin-GID | TCF-luc | 1.50 | 0.024 |
| Tet-O-ΔN-β-catenin | TCF-luc | 2.73 | 0.070 |
| Tet-O-VP16-TCF4 | TCF-luc | 7.61 | 0.23 |
Activating components of the canonical WNT pathway: ΔN-LRP6 (a constitutively activating component of the WNT receptor), Dvl-2 (a Disheveled homologue and activator of WNT signaling), Axin-GID (Axin-GSK3β Interaction Domain, a dominant negative form of Axin that binds to and inhibits GSK-3β), ΔN-β-catenin (a stabilized form of β-catenin resistant to degradation), or VP16-TCF (a TCF transcription factor that is constitutively active in the absence of β-catenin). See the Supplementary Information for assay protocols.
In addition to promising potency in the 7dF3 luciferase cell-based reporter assay (IC50 = 1.76 μM), compound 9 demonstrated moderate in vitro metabolic stability in human and mouse liver microsomes (Figure 2). However, we were concerned that 9 may be a nonspecific pharmacophore for ion channels by virtue of the 4-aminopyridine motif. A measured pKa of the pyridine nitrogen (pKa = 3.5)32 indicated that 9 is essentially nonbasic, in contrast to 4-aminopyridine (pKa = 9.2)33 and 4-(piperidin-1-yl)pyridine (measured pKa = 9.42).32 The inductive electron-withdrawing effect of two m-chloro substituents is likely to significantly reduce the pKa (pyridine pKa = 5.24; 3-chloropyridine pKa = 2.84).34,35 We hypothesized that a twisted conformation about the pyridine–piperidine bond, driven by the steric bulk of the flanking chlorine atoms, may also reduce the basicity of compound 9 by abrogating electron donation from the 4-amino substituent. Indeed, a twisted conformation was evident in the small molecule X-ray crystal structure of 9 with a torsion angle of 54.4° observed between the plane of the piperidine and pyridine rings [Figure 3 and Supplementary Figure S4, Supporting Information (SI)]. By contrast, 4-(piperidin-1-yl)pyridine adopts a planar conformation about the piperidine–pyridine bond in two reported small-molecule X-ray crystal structures.36 Thus, the 3,5-dichloro substitution pattern in compound 9 reduces the pKa of the pyridine nitrogen (inductive effect) and induces a twisted conformation about the piperidine–pyridine bond (steric effect), which may further reduce the pKa. The measured pKa of compound 9 is consistent with an observed lack of in vitro ion channel pharmacology (hERG IC50 > 10 μM) and absence of CYP450 enzyme inhibition apart from weak inhibition of CYP3A4 (IC50 = 7 μM, Supplementary Table S1, SI); these observations are in marked contrast to classical 4-aminopyridine-based ion channel modulators.37
Figure 3.
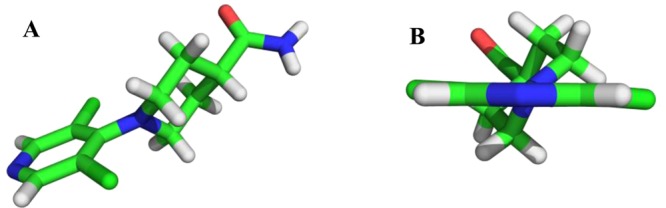
Small-molecule X-ray crystal structure of 9 (panel A) depicting a twisted conformation about the pyridine–piperidine bond with the plane of piperidine ring tilted 54.4° against the plane of the aromatic ring (panel B and Supplementary Figure S4, SI). Data has been deposited with the Cambridge Crystallographic Data Centre (deposition number CCDC 1020858).
In the absence of information about the precise biochemical target of compound 9, we followed a classical ligand-based design approach. We hypothesized, first, that the H-bond donor (HBD) and acceptor (HBA) properties of the primary amide moiety would be essential for activity; second, that the pyridine nitrogen would be an essential component of activity; and, third, that it would be important to maintain the observed twisted conformation about the pyridine–piperidine bond. We initially set out to test these three hypotheses, cognisant in our compound design of the cell-based primary assay whereby true SAR versus an intracellular biochemical target may be masked by poor cell penetration. Hit compound 9 (HBD count = 2; HBA count = 4) has excellent passive cell permeability (Caco-2: Papp = 36 × 10–6 cm s–1, no efflux) and compounds with equivalent or lower HBD/HBA count were prioritized to avoid masking of SAR trends against an intracellular target by poor cell permeability.
Methylation of the primary amide in compound 9 to give the secondary amide 15 or tertiary amide 16 led to 7- and 15-fold loss in activity, respectively, suggesting that at least one of the H-bond donors is required for activity. The greater loss of activity with more sterically demanding amides 17–19 suggested a lack of space adjacent to the amide moiety. Removal of the carboxamide completely (compound 20) or removal of the carbonyl moiety (compound 21) led to a reduction in potency compared with compound 9, consistent with the hypothesis that both H-bond donor and acceptor functions of the carboxamide are important components of the pharmacophore (Table 1).
To explore the extent to which the pyridine nitrogen is essential for activity, we prepared compound 22, where the pyridine nitrogen was replaced with a CH (Table 1, entry 9). A significant potency drop was observed, suggesting that the hydrogen bond acceptor capability of this nonbasic nitrogen is indeed crucial for activity.
We next turned our attention to the requirement for a twisted conformation about the pyridine–piperidine bond while the piperidine 4-carboxamide was kept constant. Replacement of chlorine atoms with protons at both the 3- and 5-positions on the pyridine ring led to loss of activity, while replacement of one chloride led to a 13-fold loss in potency (Table 3, entries 1 and 2). By contrast, replacement of both chlorine atoms with bulkier bromines led to a 5-fold potency increase (Table 3, entry 3). Pleasingly, replacement of one chlorine atom with a phenyl ring in compound 26 (Table 3, entry 4) resulted in a 20-fold increase in potency over compound 9; however, the combination of a 3-phenyl, 5-H (Table 3, entry 5) abrogated activity by 37-fold in comparison with 26 (Table 3, entry 4), consistent with a requirement for substitution at both the 3- and 5-positions of the pyridine ring.
Table 3. Modification of the Pyridine 3- and 5-Substituents.

MLM and HLM values indicate percent turnover after 30 min of incubation.
The increased potency of compound 26 was of particular interest, although in vitro microsomal stability was suboptimal. We set out to explore alternatives to the phenyl ring with greater stability to oxidative metabolism. Replacement with 4-substituted pyrazoles led to a loss of activity (Table 3, entries 6 and 7), although some activity loss exhibited by compound 28 (Table 3, entry 6) may result from the presence of an additional H-bond donor and consequent impaired cell penetration with respect to its matched pair (Table 3, entry 7). The thiophene analogue 30, while among the most potent replacements, exhibited unsurprising metabolic instability,38 and pyridine replacements had decreased potency (Table 3, entries 9 and 10). Substitution at the para-position of the phenyl ring proved more fruitful, with both electron-donating and -withdrawing substituents tolerated (entries 11–13); however, the strongly electron withdrawing sulfone was not tolerated (entry 14). Increased potency of the p-methoxy analogue 33 in the 7dF3 cell-based luciferase reporter assay (IC50 = 0.037 μM) compared to 9 (IC50 = 1.76 μM) was mirrored by increased potency and overall profile in cell-based assays incorporating different activating components of WNT signaling, suggesting that the locus of action on the WNT pathway had been maintained (Table 2).
Although compound 33 was among the most potent, its metabolic stability was poor; however, the p-morpholine derivative 38 (Table 3, entry 16) proved equipotent, with acceptable metabolic stability, while the corresponding meta-derivative 37 was 8-fold less potent (Table 3, entry 15). Despite acceptable metabolic stability, compound 38 demonstrated in vivo clearance greater than liver blood flow upon iv dosing in mouse pharmacokinetic studies; we suspected amidase-mediated cleavage of the primary amide, although this proved difficult to definitively confirm.
With compound 38 in hand, we were keen to further explore the piperidine carboxamide in order to understand the optimal geometry for the H-bond acceptor and donor motifs and to reduce the potential for putative amidase-mediated cleavage of the primary carboxamide. Replacement of the 4-carboxamide by nitrile, a highly directional H-bond acceptor, maintained activity in the reporter assay and reinforced the notion that an appropriately positioned H-bond acceptor motif was the dominant feature (Table 4, entry 1). Introduction of a geminal methyl group at the 4-position of the piperidine ring also maintained potency (Table 4, entry 2) and suggested that spirocyclic carboxamides might be tolerated. With this result in hand, a range of 6,5-spirocyclic ring scaffolds was explored and, pleasingly, resulted in compounds with good potency and metabolic stability. The 2,8-diazaspiro[4.5]decan-1-one 44 exhibited 25-fold improved potency over the isomeric spirocycle 46, consistent with a specific interaction from the amide motif (Table 4, entries 3 and 4). Methylation of the amide (entry 5) reduced potency 24-fold, indicating that, in this spirocyclic scaffold, both H-bond donor and acceptor motifs are important for activity. An isomeric spirocyclic ring system at the 3-position of the piperidine ring lost significant potency (entry 6), although potency could be restored with the corresponding 5,5-spirocyclic ring system incorporated as a racemate (entry 7). Improving the H-bond donor ability of the spirocyclic NH by incorporation of an additional carbonyl functionality to give the acidic imide 54 resulted in a very potent and metabolically stable cell-based inhibitor of WNT pathway activity; however, poor solubility (0.003 mg/mL in phosphate buffer at pH 7.4) was an unfavorable attribute and below our criteria for compound progression. Considering all the data together, compound 44 presented the most promising profile, and we were keen to explore the extent to which the chloro substituent could be varied (Table 5). Replacement with small alkyl, cycloalkyl, aryl, or heteroaryl groups (Table 5, entries 2–7), all of which were designed to maintain cell penetration, resulted in maintenance of potency for the prop-1-en-2-yl substituent in 59 (Table 5, entry 3) but a loss of potency for other exemplars, which we ascribed to a lack of toleration of steric bulk in this region of the putative binding site. In order to avoid potential metabolic liabilities with the presence of an alkene in compound 59, we henceforth adopted chloride as the preferred substituent.
Table 4. Optimization of the Piperidine-4-carboxamide.
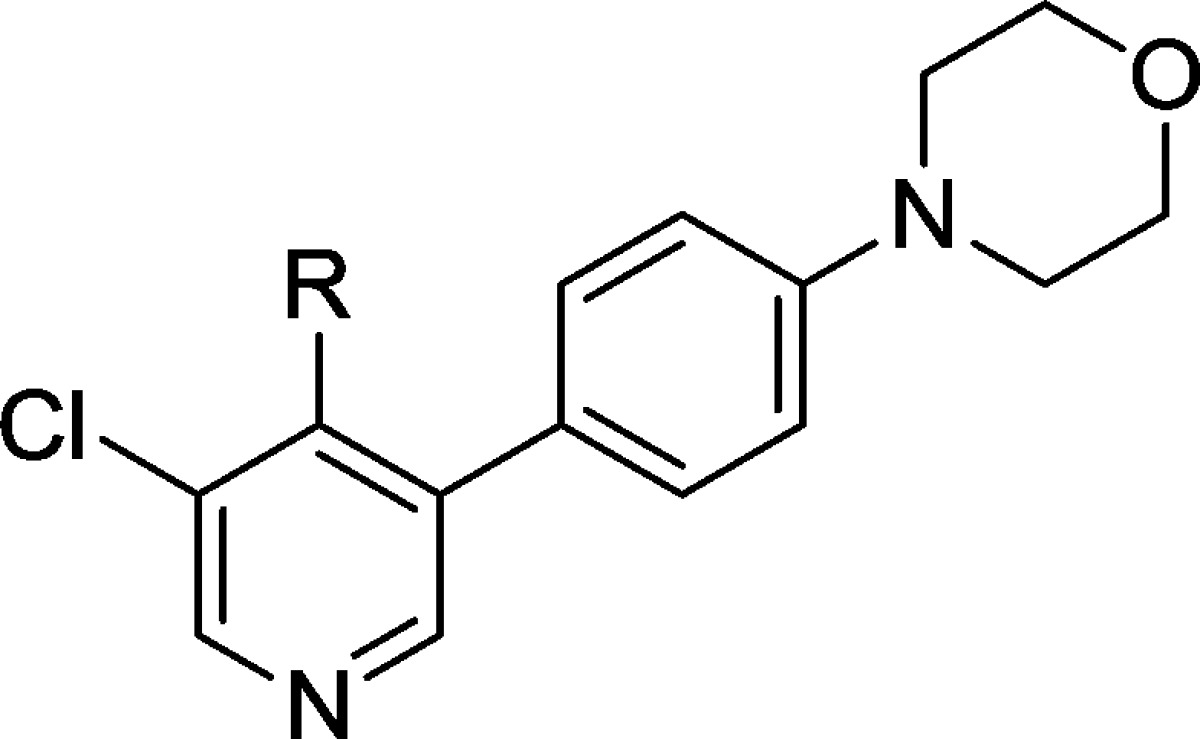

MLM and HLM values indicate percentage parent remaining after 30 min of incubation.
To benchmark the chemical series, we conducted mouse in vivo pharmacokinetic studies on compound 44, which revealed improved in vivo clearance compared to compound 38; however, clearance remained unacceptably high with corresponding low oral bioavailability (Table 6). Despite an unfavorable pharmacokinetic profile, other properties of compound 44 were attractive: it was inactive when tested at 10 μM across a panel of 55 receptors, ion channels, and enzymes and demonstrated no significant activity at 1 μM across a broad panel of 209 nonmutated kinases (see Supplementary Tables S2 and S3, SI); in addition, 44 did not strongly inhibit the known intracellular WNT pathway target tankyrase (TNKS1 IC50 = 19.0 ± 0 μM, TNKS2 IC50 = 16.0 ± 1.41 μM). Permeability, as assessed by Caco-2 flux, was high (Papp = 37 × 10–6 cm s–1) with no efflux, while kinetic and thermodynamic aqueous solubilities were acceptable (164 μM and 0.017 mg/mL in phosphate buffer at pH 7.4, respectively) and no plasma instability was observed. We concluded that the poor oral bioavailability was largely the result of high first pass clearance and adopted a more accurate measure of intrinsic metabolic clearance (Clint) across three species to better inform our selections for in vivo pharmacokinetic studies. In these in vitro metabolism studies, compound 44 showed high intrinsic clearance in the mouse consistent with our in vivo observations (Table 7).
Table 6. Mouse Pharmacokinetics of Compound 44.
| dose (mg/kg) | route | Cl (mL/min/kg) | Vss (L/kg) | Tmax (h) | AUC (h ng/mL) | bioavailability (%) | T1/2 (h) |
|---|---|---|---|---|---|---|---|
| 0.2 | iv | 65 | 1.44 | 0.10 | 50.8 | NA | 0.31 |
| 0.5 | po | 0.25 | 7.4 | 6 |
Table 7. In Vitro Metabolic Stability of Key Spirocyclic Compounds.
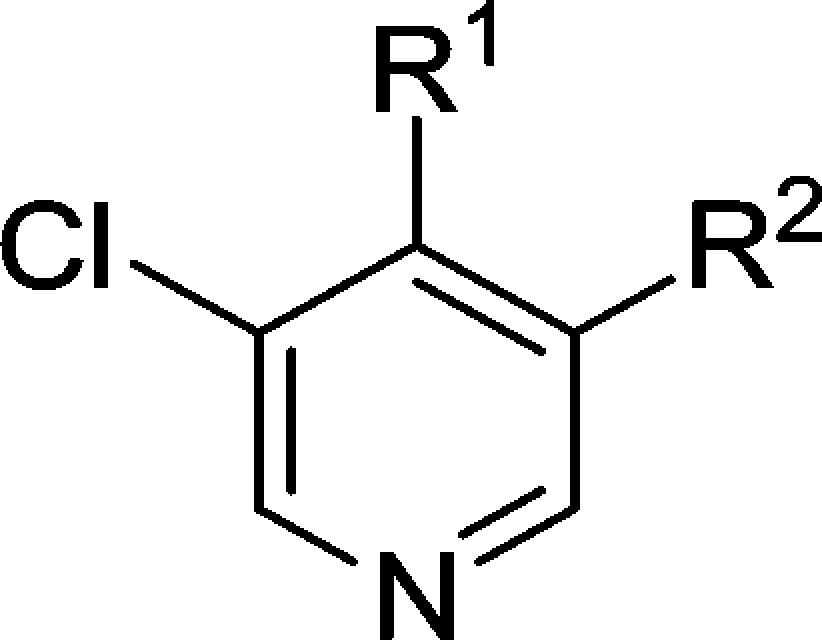

MLM and HLM figures indicate percent turnover after 30 min of incubation.
Compound 44 also exhibited increased inhibition of CYP enzymes (CYP2C9, IC50 = 2.9 μM; CYP3A4, IC50 = 2.1 μM) in comparison with the original hit 9 and generated a glutathione adduct in an in vitro reactive metabolite assay (data not shown); we were therefore keen to replace the morpholine moiety that has been implicated in reactive metabolite formation.39 A diverse set of compounds was prepared bearing saturated rings or aromatic heterocycles as morpholine replacements (Table 8, entries 1–4 and 5–10, respectively) as well as 6,6- and 6,5-fused ring systems as isosteres for the 4-morpholinophenyl (Table 8, entries 11–13). Removal of either morpholine heteroatom reduced potency versus the parent compound 44 (Table 8, entries 1 and 2). Incorporation of an unsubstituted piperazine proved detrimental, whereas the N-methylpiperazine 68 restored potency with some improvement in metabolic stability; however, an unfavorable Caco-2 permeability profile (efflux ratio >500) precluded further progression of this compound.
Table 8. Optimization of the Pyridine 5-Substituent.
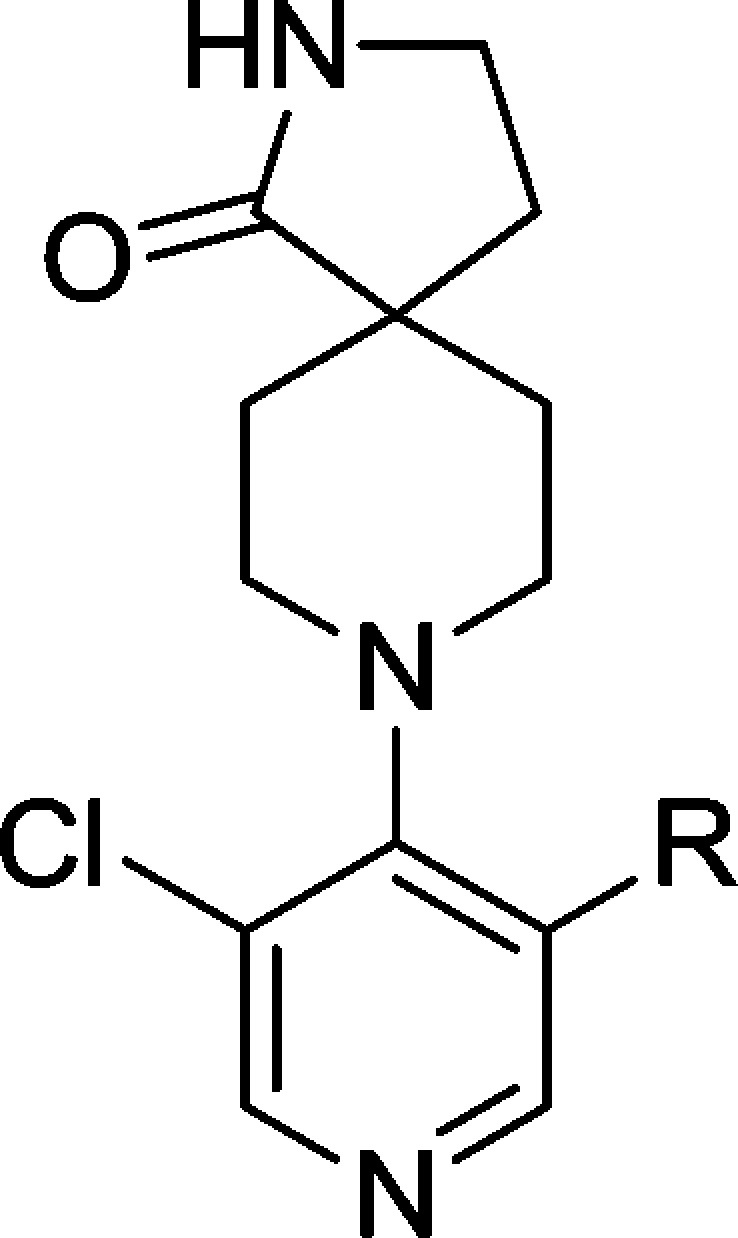

Pleasingly, 5- or 6-membered heterocyclic replacements for the morpholine provided compounds with good potency and equivalent or improved metabolic stability. The electron-withdrawing oxadiazole 70 proved the least potent modification in the set, consistent with the poor activity of the electron-withdrawing sulfone 36 (Table 3, entry 14). The N-methylpyrazole 74 proved more potent than its desmethyl matched pair 73, possibly due to poorer cell penetration of the latter compound, while the regioisomeric pyrazole 71 also displayed lower potency. Replacement of the 4-morpholinophenyl moiety with fused 6,6- or 5,6-heterocycles (Table 8, entries 11–13) retained potency but at the expense of higher metabolic clearance compared to compound 74, which provided the optimal balance of potency and metabolic stability in this subseries.
We were keen to explore the combination of the preferred 1-methyl-4-phenyl-1H-pyrazole with selected spirocyclic carboxamides from Table 4. In comparison with compound 74, we noted that the corresponding 5,5-spirocyclic carboxamide 78 was significantly less metabolically stable as a racemate (Table 9). The most potent enantiomer 80 (absolute stereochemistry not assigned) displayed the highest metabolic stability of the pair; however, this was still inferior to the parent molecule 74 and halted further investigation of this compound. Combining a spirocyclic imide with our favored 1-methyl-4-phenyl-1H-pyrazole substituent gave the potent and stable inhibitor 81; however, in common with other imides prepared in this series, aqueous solubility proved to be very poor (<0.001 mg/mL in phosphate buffer at pH 7.4), which precluded further progression of this compound. 8-(3-Chloro-5-(4-(1-methyl-1H-pyrazol-4-yl)phenyl)pyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (CCT251545, 74) was therefore selected for progression to in vivo studies.
Table 9. Combination of Optimal Pyridine 4- and 5-Substituents.
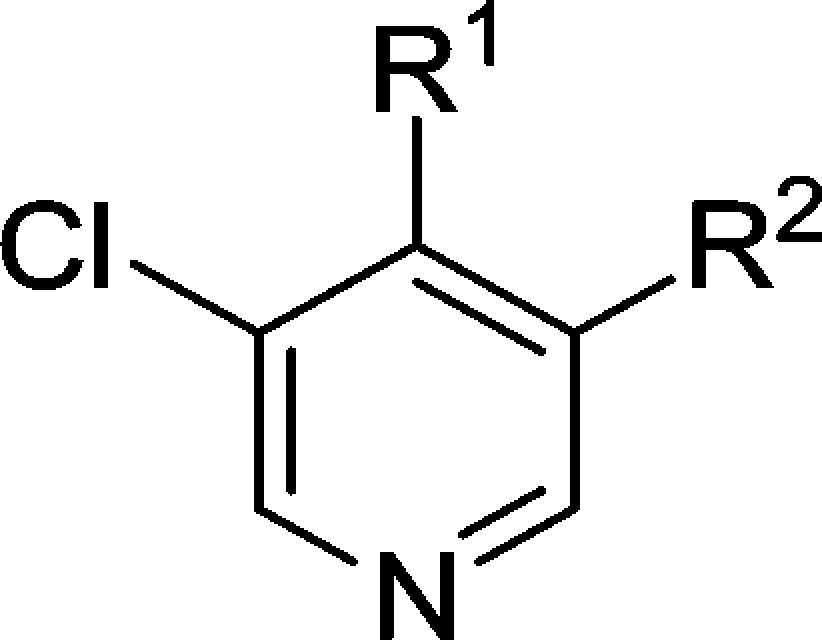

Absolute stereochemistry undefined.
In vivo pharmacokinetic studies on compound 74 revealed moderate clearance in both mouse and rat with moderate to high oral bioavailability; the volume of distribution in both species was low, consistent with the pharmacokinetic profile of a neutral compound (Table 10). Compound 74 displayed high flux (Papp = 24 × 10–6 cm s–1), acceptable efflux (ER = 2.5), and minimal hERG inhibition (IC50 > 10 μM); aqueous kinetic and thermodynamic solubilities were also sufficient for further progression (94 μM and 0.006 mg/mL, respectively, in pH 7.4 phosphate buffer). Compound 74 demonstrated minimal activity when tested across a panel of 55 receptors, ion channels, and enzymes at 1 μM (Table S4, SI); weak inhibition of CYPs 2C8, 2C9, and 3A4 was observed (see Supplementary Table S1, SI); however, this was not considered sufficient to preclude further investigation. Testing versus a panel of 291 kinases at 1 μM revealed weak inhibition of GSK3α and -β as the only hits (IC50 = 0.462 and 0.690 μM, respectively; see Supplementary Table S5, SI), insufficiently potent to account for the observed effects on cell-based WNT pathway activity; moreover, inhibition of GSK3β may be expected to activate rather than inhibit the WNT signaling. Compound 74 demonstrated weak inhibition of tankyrase enzymes (TNKS1 IC50 > 10 μM, TNKS2 IC50 = 15.0 ± 0 μM).
Table 10. Pharmacokinetics of Compound 74.
| species | dose (mg/kg) | route | Cl (mL/min/kg) | Vss (L/kg) | Tmax (h) | AUC (h ng/mL) | bioavailability (%) | t1/2 (h) |
|---|---|---|---|---|---|---|---|---|
| mouse | 0.2 | iv | 31 | 1.1 | 0.10 | 106.8 | NA | 0.55 |
| mouse | 0.5 | po | NA | NA | 0.25 | 143.0 | 54 | NA |
| rat | 0.2 | iv | 26 | 1.8 | 0.1 | 129.6 | NA | 0.97 |
| rat | 0.5 | po | NA | NA | 0.25 | 287.5 | 88 | NA |
To confirm activity on the WNT pathway, a number of studies were undertaken with compound 74. RT-PCR of 7dF3 cells treated with 74 demonstrated a 12-fold inhibition of endogenous cMYC transcription (IC50 = 2.3 nM, Supplementary Figure S1, SI), a commonly studied WNT target gene.40 Compound 74 was also tested in human colorectal cancer cell lines LS174T (β-catenin mutant) and SW480 (APC mutant) that have constitutively activated WNT signaling or PA-1 human teratocarcinoma cells that are WNT ligand dependent; 74 potently inhibited reporter-based readouts of basal WNT pathway activity in LS174T and SW480 cells in the absence of WNT ligand or other stimulants (IC50 = 0.023 ± 0.011 μM and 0.190 ± 0.030 μM, respectively) and demonstrated potent inhibition of WNT3a ligand-dependent reporter readout in PA-1 cells (IC50 = 0.007 μM). No activity was observed in RKO-F1522 cells bearing a firefly luciferase reporter downstream of the EF1-α promoter, a commonly used, ubiquitously active, house-keeping promoter that has high activity in most mammalian cells, irrespective of their WNT dependence (IC50 > 10 μM). We developed an APC mutant human colorectal cancer cell line (COLO205-F1756 clone 4) engineered to express a modified luciferase-based WNT pathway reporter. Compound 74 demonstrated potent inhibition of WNT pathway activity in this clone (IC50 = 0.035 ± 0.003 μM, n = 8); in addition, we were able to demonstrate that compound 74 did not affect endogenous levels of TCF1 or TCF4 at concentrations that reduce luciferase expression in this cell line (Supplementary Figure S2, SI). Taken together, these data confirm that suppression of the WNT pathway activity by 74 is not unique to a specific cell line, is independent of exogenous WNT pathway stimulation, does not result from destabilization of TCF1 or TCF4, and is not due to interference with the luciferase-based reporter readout.
Extension of WNT pathway inhibition to the whole animal setting was achieved using the APC mutant human colorectal cancer cell line COLO205-F1756 clone 4. Mice bearing established COLO205-F1756 clone 4 tumor xenografts were treated with compound 74 (70 mg/kg po bid for 9 days), at which point WNT reporter activity was measured by whole body bioluminescent imaging and therapeutic response by excised tumor weights. Significant inhibition of WNT signaling was observed, and treated tumors were smaller than controls: flux normalized to tumor weight was reduced by 80% (P = 0.0003) and tumor weights by 37.5% (P = 0.0379, Mann–Whitney U test; Figure 4A,B). Plasma and tumor levels of compound 74 were determined 2 and 6 h after the last dose, and at these time points, total plasma and tumor levels significantly exceeded those required for inhibition of luciferase reporter activity in a cell-based assay. Taken together, these data demonstrate inhibition of WNT pathway activity in a human colorectal cancer xenograft model and some indication of tumor growth inhibition following oral dosing at exposures consistent with inhibition of WNT pathway activity.
Figure 4.

(A) Inhibition of WNT-driven luciferase reporter activity. (B) Reduced tumor growth with compound 74 in COLO205 human colon cancer xenografts after oral dosing (70 mg/kg bid). (C) Plasma and tumor total concentrations of 74 at 2 and 6 h after the last dose.
Conclusions
We have described the optimization of a small-molecule inhibitor of the WNT pathway identified as a singleton from an HTS of a diverse 63 040 compound library versus a cell-based luciferase reporter assay. Application of classical hypothesis-driven, ligand-based SAR enabled the discovery of highly potent cell-based WNT pathway inhibitors, with compound 74 demonstrating good oral pharmacokinetics in mouse and rat. With knowledge from our cell-based assay cascade, we hypothesized that this series of compounds acts at an intracellular target. Therefore, our medicinal chemistry optimization necessitated design criteria likely to maintain cell permeability to avoid masking true WNT-pathway SAR, and with this in mind, attention to H-bond donor and acceptor count was critical. The SAR is consistent with the requirement for a distinct twisted conformation about the pyridine–piperidine bond, as observed in the small-molecule X-ray crystal structure of the original hit compound 9, and also is consistent with the lower pKa of the pyridine nitrogen and the lack of significant ion channel pharmacology across exemplars of the chemical series. Enhanced potency was achieved through maintenance of this twisted conformation and replacement of one of the chlorine atoms with a para-substituted aryl ring. Methylpyrazole was found to be the optimal para-substituent in terms of potency, metabolic stability, and membrane permeability. Combination of the favored methylpyrazole substituent with a 6,5-spirocyclic carboxamide, in place of the original piperidine carboxamide in hit compound 9, led to 74, a potent small-molecule inhibitor of the WNT pathway with good oral pharmacokinetics and in vivo activity against the WNT pathway. Compound 74 also demonstrated a lack of transporter-mediated efflux and a lack of activity across broad in vitro panels of enzyme, receptor, and ion channel targets. Further details of the in vivo pharmacological profile of this chemical tool, including studies to determine its molecular target, will be reported in due course.
The canonical WNT pathway is of significant interest in oncology, and the paucity of druggable targets on the pathway has hampered drug discovery efforts. Pathway-based screening approaches are becoming increasingly widespread as greater appreciation of the benefits of this strategy become apparent.22,23,29 In particular, hits from such screens, by necessity, have good cell permeability, and components of protein complexes are more likely to be in a relevant endogenous conformation, which may reveal new druggable binding sites or alternative targetable conformations of known sites; moreover, key signal-determining pathway nodes are more likely to be discovered as a result of cell-based readouts at the transcriptional level. Here we describe a successful example of hypothesis-driven medicinal chemistry optimization from a singleton hit against a cell-based assay readout for the WNT pathway in the absence of knowledge of the biochemical target.
Experimental Section
Chemistry
Commercially available starting materials, reagents, and dry solvents were used as supplied. Flash column chromatography was performed using Merck silica gel 60 (0.025–0.04 mm). Column chromatography was also performed on a Biotage SP1 purification system using Thomson or Biotage Flash silica cartridges. Preparative TLC was performed on Merck plates. Ion exchange chromatography was performed using acidic Isolute Flash SCX-II columns or basic Isolute Flash NH2 columns. Preparative HPLC was conducted using a Phenomenex Luna column (5 μm, 250 × 21.2 mm, C18, Phenomenex, Torrance, CA) using a Gilson GX-281 Liquid Handler system combined with a Gilson 322 HPLC pump (Gilson, Middleton, WI), over a 15 min gradient elution from 10:90 to 100:0 MeOH:water (both modified with 0.1% formic acid) at a flow rate of 20 mL/min or over a 15 min gradient elution from 40:60 to 100:0 MeOH:water (both modified with 0.1% formic acid) at a flow rate of 20 mL/min. UV–vis spectra were acquired at 254 nm on a Gilson 156 UV–vis detector (Gilson, Middleton, WI). Collection was triggered by UV signal and collected using a Gilson GX-281 Liquid Handler system (Gilson, Middleton, WI). Raw data was processed using Gilson Trilution Software. 1H NMR spectra were recorded on a Bruker Avance-500 or Bruker Avance-400. Samples were prepared as solutions in a deuterated solvent and referenced to the appropriate internal nondeuterated solvent peak. 13C NMR spectra were recorded at 126 MHz using an internal deuterium lock. The following internal references were used: CDCl3 (δC 77.0), CD3OD (δC 49.0), and DMSO-d6 (δC 39.5). Unobserved resonances for quaternary carbon atoms are denoted by “Cq not observed”. LC–MS and HRMS analyses were performed on an Agilent 1200 series HPLC and diode array detector coupled to a 6210 time-of-flight mass spectrometer with dual multimode atmospheric pressure CI/ESI source or coupled to an Agilent 6520 quadrupole-time-of-flight mass spectrometer with dual multimode atmospheric pressure CI/ESI source. LC–MS analysis was also performed on a Waters Alliance 2795 separations module and Waters 2487 dual wavelength absorbance detector coupled to a Waters/Micromass LCt time-of-flight mass spectrometer with ESI source. Analytical separation was carried out according to the following methods. For method A, analytical separation was carried out at 40 °C on a Merck Purospher STAR column (RP-18e, 30 × 4 mm) using a flow rate of 3 mL/min in a 2 min gradient elution with detection at 254 nm. The mobile phase was a mixture of methanol (solvent A) and water containing formic acid at 0.1% (solvent B). Gradient elution was as follows: 1:9 (A/B) to 9:1 (A/B) over 1.25 min, 9:1 (A/B) for 0.5 min, and then reversion back to 1:9 (A/B) over 0.15 min, and finally 1:9 (A/B) for 0.1 min. For method B, analytical separation was carried out at 30 °C on a Merck Chromolith SpeedROD column (RP-18e, 50 × 4.6 mm) using a flow rate of 2 mL/min in a 4 min gradient elution with detection at 254 nm. The mobile phase was a mixture of methanol (solvent A) and water containing formic acid at 0.1% (solvent B). Gradient elution was as follows: 1:9 (A/B) to 9:1 (A/B) over 2.5 min, 9:1 (A/B) for 1 min, and then reversion back to 1:9 (A/B) over 0.3 min, and finally 1:9 (A/B) for 0.2 min. For method C, analytical separation was carried out at 30 °C on a Merck Purospher STAR column (RP-18e, 30 × 4 mm) using a flow rate of 1.5 mL/min in a 4 min gradient elution with detection at 254 nm. The following reference masses were used for HRMS analysis: caffeine [M + H]+ 195.087652; hexakis(1H,1H,3H-tetrafluoropentoxy)phosphazene [M + H]+ 922.009798 and hexakis(2,2-difluoroethoxy)phosphazene [M + H]+ 622.02896 or reserpine [M + H]+ 609.280657. All compounds submitted for biological testing were determined to be >95% pure by method A, B, or C.
Preparation of Compounds in Table 1 Exemplified by Compound 9
1-(3,5-Dichloropyridin-4-yl)piperidine-4-carboxamide (9)41
To a stirred suspension of 3,4,5-trichloropyridine (10) (500 mg, 2.74 mmol) and piperidine-4-carboxamide (350 mg, 2.73 mmol) in NMP (15 mL) at ambient temperature was added triethylamine (770 μL, 5.48 mmol) in one portion. The reaction was heated to 220 °C for 60 min under microwave irradiation. Once cooled, the reaction mixture was partitioned between EtOAc and sat. NaHCO3. The separated aqueous layer was extracted with EtOAc (2 × 100 mL), and the combined organic extracts were washed with water (50 mL) and brine (50 mL), dried over magnesium sulfate, filtered, and concentrated in vacuo to give a crude oily solid. Purification was accomplished by flash chromatography on silica gel eluting with CH2Cl2/1 M methanolic ammonia (95:5 to 92:8) to yield impure title compound (963 mg) as a white solid. The solid was triturated in hot Et2O (25 mL) to give the title compound 9 as a white solid (519 mg, 69% yield): 1H NMR (500 MHz, CDCl3) δ 1.89–1.97 (m, 4H), 2.33–2.40 (m, 1H), 3.27–3.33 (m, 2H), 3.38–3.44 (m, 2H), 5.62 (br s, 1H), 5.79 (br s, 1H), 8.32 (s, 2H); 13C NMR (126 MHz, CDCl3) δ 29.4, 42.4, 50.1, 128.6, 149.1, 151.7, 176.9; LC–MS (method A, ESI, m/z) tR = 1.16 min, 274/276/278 (M + H)+; ESI-HRMS calcd for C11H1435Cl2N3O (M + H)+ 274.0508, found 274.0509.
Preparation of Compounds in Table 3 Exemplified by Compounds 26 and 38
1-(3-Chloro-5-phenylpyridin-4-yl)piperidine-4-carboxamide (26)41
To a stirred suspension of 1-(3,5-dichloropyridin-4-yl)piperidine-4-carboxamide (9) (100 mg, 0.365 mmol), phenylboronic acid (49 mg, 0.401 mmol), and tetrakis(triphenylphosphine)palladium(0) (21 mg, 0.018 mmol) in MeCN (4 mL) was added 0.5 M aqueous sodium carbonate solution (1.0 mL, 0.50 mmol). The reaction was heated to 150 °C under microwave irradiation for 30 min. Once cooled, the reaction was concentrated in vacuo and azeotroped with toluene (2 × 50 mL) to give a gray, oily solid. Purification was accomplished by flash chromatography on silica gel eluting with CH2Cl2/MeOH (97/3) and by semipreparitive HPLC (gemini column 250 × 10, 15 min gradient, MeCN/H2O 10:90 to 90:10) to give the title compound 26 as a white solid (41 mg, 36% yield): 1H NMR (500 MHz, CDCl3) δ 1.73–1.80 (m, 4H), 2.12–2.20 (m, 1H), 2.62 (dt, J = 13.4, 7.1 Hz, 2H), 3.15–3.21 (m, 2H), 5.40 (s, 2H), 7.24–7.30 (m, 2H), 7.38–7.51 (m, 3H), 8.21 (s, 1H), 8.44 (s, 1H); 13C NMR (126 MHz, CDCl3) δ 29.1, 42.4, 50.9, 127.8, 128.3, 128.6, 129.3, 134.1, 137.6, 149.6, 150.7, 152.8, 176.8; LC–MS (method A, ESI, m/z) tR = 0.99 min, 316/318 (M + H)+; ESI-HRMS calcd for C17H1935ClN3O (M + H)+ 316.1211, found 316.1206.
1-(3-Chloro-5-(4-morpholin-4-ylphenyl)pyridin-4-yl)piperidine-4-carboxamide (38)
To a stirred suspension of 1-(3,5-dichloropyridin-4-yl)piperidine-4-carboxamide (9) (70 mg, 0.26 mmol), 4-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)morpholine (92 mg, 0.32 mmol), and tetrakis(triphenylphosphine)palladium(0) (15 mg, 5 mol %) in MeCN (3 mL), at ambient temperature, was added 0.5 M aqueous sodium carbonate solution (720 μL, 0.360 mmol). The reaction was heated to 150 °C for 45 min under microwave irradiation. Once cooled, the reaction was concentrated in vacuo and then dry-loaded onto silica. Purification was accomplished by flash chromatography on silica gel (CH2Cl2/EtOH 98:2 to 93:7) to yield the title compound 38 (53 mg, 52% yield) as a pale orange/pink solid: 1H NMR (500 MHz, DMSO-d6) δ 1.53–1.60 (m, 4H), 2.04–2.11 (m, 1H), 2.49–2.57 (m, 2H), 3.00–3.06 (m, 2H), 3.15–3.20 (m, 4H), 3.73–3.78 (m, 4H), 6.73 (br s, 1H), 7.03 (d, J = 8.7 Hz, 2H), 7.18 (d, J = 8.7 Hz, 3H), 8.13 (br s, 1H), 8.40 (br s, 1H); 13C NMR (126 MHz, CDCl3) δ 29.2, 42.4, 48.9, 51.0, 67.0, 115.4, 128.3 130.1, 134.0, 147.9, 149.7, 150.9, 153.6, 176.9, (1 Cq not observed); LC–MS (method B, ESI, m/z) tR = 1.96 min, 401/403 (M + H)+; ESI-HRMS calcd for C21H2635ClN4O2 (M + H)+ 401.1739, found 401.1746.
Preparation of Compounds in Table 4 Exemplified by Compounds 44 and 54
8-(3,5-Dichloropyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (43)
To a stirred suspension of 3,4,5-trichloropyridine (10) (509 mg, 2.79 mmol) and 2,8-diazaspiro[4,5]decan-1-one hydrochloride (410 mg, 2.66 mmol) in NMP (14.6 mL), at ambient temperature, was added triethylamine (1.10 mL, 7.89 mmol). The reaction mixture was heated to 220 °C under microwave irradiation for 60 min. Once cooled, the reaction mixture was partitioned between EtOAc and sat. NaHCO3, the separated aqueous layer was extracted with EtOAc (2 × 100 mL), and the combined organic extracts were washed with brine (50 mL), dried over magnesium sulfate, filtered, and concentrated in vacuo to give a crude pale yellow oil. Purification was accomplished by flash chromatography on silica gel eluting with CH2Cl2/EtOH (97.5:2.5 to 97:3) to give 8-(3,5-dichloropyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (43) (422 mg, 53% yield) as a white solid: 1H NMR (500 MHz, CDCl3) δ 1.53 (br d, J = 13.1 Hz, 2H), 2.07–2.18 (m, 4H), 3.33 (dt, J = 11.9, 2.6 Hz, 2H), 3.37–3.45 (m, 4H), 6.66 (br s, 1H), 8.33 (br s, 2H); LC–MS (method B, ESI, m/z) tR = 2.66 min, 300/302/304 (M + H)+.
8-(3-Chloro-5-(4-morpholin-4-ylphenyl)pyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (44)
To a stirred suspension of 8-(3,5-dichloropyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (43) (70 mg, 0.23 mmol), 4-morpholinophenylboronic acid (60 mg, 0.29 mmol), and tetrakis(triphenylphosphine)palladium(0) (14 mg, 0.012 mmol) in MeCN (3 mL), at ambient temperature, was added 0.5 M aqueous sodium carbonate solution (650 μL, 0.326 mmol). The reaction mixture was heated to 150 °C under argon in the microwave for 60 min. Once cooled, the reaction mixture was concentrated in vacuo and then dry-loaded onto silica. Purification was accomplished by flash chromatography on silica gel eluting with CH2Cl2/MeOH (98:2 to 96:5) to give the title compound 44 (19 mg, 19% yield) as a white solid: 1H NMR (500 MHz, CDCl3) δ 1.34–1.39 (m, 2H), 1.92–2.01 (m, 4H), 2.74–2.81 (m, 2H), 3.17 (dt, J = 13.0, 4.0 Hz, 2H), 3.21–3.25 (m, 4H), 3.29 (t, J = 6.9 Hz, 2H), 3.87–3.90 (m, 4H), 5.93 (br s, 1H), 6.98 (d, J = 8.7 Hz, 2H), 7.20 (d, J = 8.7 Hz 2H), 8.17 (br s, 1H), 8.39 (br s, 1H); 13C NMR (126 MHz, CDCl3) δ 31.8, 32.5, 38.7, 41.8, 47.7, 49.1, 67.0, 115.4, 128.3, 128.9, 130.2, 134.0, 149.2, 150.8, 151.0, 152.9, 181.7; LC–MS (method B, ESI, m/z) tR = 2.20 min, 427/429 (M + H)+; ESI-HRMS calcd for C23H2835ClN4O2 (M + H)+ 427.1895, found 427.1903.
8-(3,5-Dichloropyridin-4-yl)-2,8-diazaspiro[4.5]decane-1,3-dione (53)
To a stirred suspension of 3,4,5-trichloropyridine (10) (215 mg, 1.18 mmol) and 2,8-diazaspiro[4,5]decan-1,3-dione (180 mg, 1.07 mmol) in NMP (4.5 mL), at ambient temperature, was added triethylamine (450 μL, 3.20 mmol). The reaction mixture was heated to 220 °C under microwave irradiation for 60 min. Once cooled, the mixture was diluted with MeOH (20 mL) and placed on an SCX column (10 g). Elution was carried out with CH2Cl2/MeOH followed by 1 M methanolic ammonia. Concentration of the ammonia extract in vacuo gave a light orange solid. Recrystallization from EtOAc gave 8-(3,5-dichloropyridin-4-yl)-2,8-diazaspiro[4.5]decane-1,3-dione (53) (206 mg, 62% yield) as a cream solid: 1H NMR (500 MHz, CDCl3) δ 1.69 (d, J = 13.4 Hz, 2H), 2.28 (ddd, J = 13.4, 10.7, 4.1 Hz, 2H), 2.72 (s, 2H), 3.32 (ddd, J = 13.2, 10.7, 2.6 Hz, 2H), 3.48 (dt, J = 13.2, 4.1 Hz, 2H), 8.26 (br s, 1H), 8.37 (br s, 2H); LC–MS (method B, ESI, m/z) tR = 2.43 min, 314/316/318 (M + H)+.
8-(3-Chloro-5-(4-morpholin-4-ylphenyl)pyridin-4-yl)-2,8-diazaspiro[4.5]decan-1,3-dione (54)
Ten microwave vials were prepared as follows: To a stirred suspension of 8-(3,5-dichloropyridin-4-yl)-2,8-diazaspiro[4.5]decane-1,3-dione (53) (300 mg, 0.764 mmol), 4-morpholinophenylboronic acid (222 mg, 0.963 mmol), and tetrakis(triphenylphosphine)palladium (0) (100 mg, 0.0850 mmol) in MeCN (12 mL) at ambient temperature was added 0.5 M aqueous sodium carbonate solution (3 mL). The reaction was heated to 150 °C by microwave for 60 min. Once cooled, the reaction mixtures from the ten vials were combined, concentrated in vacuo, and dry-loaded onto silica. Purification was accomplished by flash chromatography (hexane/EtOAc 30:70) to obtain the title compound 54 (950 mg, 25% yield) as a white solid: 1H NMR (500 MHz, CDCl3) δ 1.48–1.54 (m, 2H), 2.07–2.14 (m, 2H), 2.55 (s, 2H), 2.75 (t, J = 11.7 Hz, 2H), 3.18–3.23 (m, 2H), 3.23–3.27 (m, 4H), 3.88–3.92 (m, 4H), 6.97–7.05 (m, 2H), 7.17–7.24 (m, 2H), 8.10 (s, 1H), 8.25 (s, 1H), 8.47 (s, 1H); 13C NMR (126 MHz, CDCl3) δ 33.3, 40.6, 44.2, 47.2, 48.7, 66.8, 115.2, 127.8, 130.0, 148.1, 150.0, 150.9, 153.0, 174.9, 181.4, (2 Cq not observed); LC–MS (method B, ESI, m/z) tR = 2.20 min, 441/443 (M + H)+; ESI-HRMS calcd for C23H2635ClN4O3 (M + H)+ 441.1688, found 441.1693.
Preparation of Compounds in Table 5 Exemplified by Compounds 56, 59, and 60
8-(3-Bromo-5-methylpyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (55)
A solution of 3-bromo-4-chloro-5-methylpyridine (300 mg, 1.45 mmol), triethylamine (610 μL, 4.36 mmol), and 2,8-diazaspiro[4.5]decan-1-one hydrochloride (291 mg, 1.53 mmol) in NMP (4.4 mL) was heated to 220 °C under microwave irradiation for 5 h at 220 °C. Once cooled, the reaction was partitioned between EtOAc and sat. NaHCO3 (100 mL each), the separated organic layer was extracted with EtOAc (2 × 75 mL), and the combined organic extracts were washed with brine (75 mL), dried over MgSO4, filtered, and concentrated in vacuo to give a crude yellow solid. The solid was recrystallized in hot EtOAc, filtered, washed with ether, and dried in vacuo to yield 8-(3-bromo-5-methylpyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (55) (44 mg, 9% yield) as an off white solid: 1H NMR (500 MHz, CDCl3) δ 1.51–1.58 (m, 2H), 2.06–2.15 (m, 2H), 2.16 (t, J = 6.8 Hz, 2H), 2.31 (s, 3H), 3.18 (br s, 2H), 3.33 (br s, 2H), 3.39 (t, J = 6.8 Hz, 2H), 6.18 (br s, 1H), 8.21 (br s, 1H), 8.45 (br s, 1H); LC–MS (method C, ESI, m/z) tR = 1.42 min, 324/326 (M + H)+.
8-(3-Methyl-5-(4-morpholin-4-ylphenyl)pyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (56)
To a stirred solution of 8-(3-bromo-5-methylpyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (55) (36 mg, 0.11 mmol), Pd(dppf)Cl2·CH2Cl2 (4.5 mg, 5.6 μmol), and 4-morpholinophenylboronic acid (25 mg, 0.12 mmol) in MeCN (830 μL), at ambient temperature, was added 0.5 M aqueous sodium carbonate solution (310 μL, 0.150 mmol). The reaction mixture was sealed in a microwave vial under argon and heated to 120 °C for 60 min under microwave irradiation. The reaction was concentrated in vacuo and then diluted with MeOH/DCM and dry-loaded onto silica. Purification was accomplished by flash chromatography on silica gel eluting with DCM/EtOH (99:1 to 90:10) to yield the title compound 56 (20 mg, 44% yield) as a clear colorless oil, which solidified on standing: 1H NMR (500 MHz, CDCl3) δ 1.28–1.34 (m, 2H), 1.88–1.96 (m, 4H), 2.31 (s, 3H), 2.63 (br t, J = 12.3 Hz, 2H), 2.91–2.97 (m, 2H), 3.20–3.24 (m, 4H), 3.28 (t, J = 6.9 Hz, 2H), 3.87–3.91 (m, 4H), 6.25 (br s, 1H), 6.97 (d, J = 8.7 Hz, 2H), 7.18 (d, J = 8.7 Hz, 2H), 8.17 (br s, 1H), 8.27 (br s, 1H); 13C NMR (126 MHz, CDCl3) δ 16.4, 31.0, 32.5, 38.7, 42.1, 47.6, 49.1, 66.9, 115.1, 130.3, 130.4, 150.4, 150.6, 150.7, 155.3, 181.8; LC–MS (method C, ESI, m/z) tR = 1.70 min, 407 (M + H)+; ESI-HRMS calcd for C24H31N4O2 (M + H)+ 407.2442, found 407.2447.
3-Bromo-4,5-dichloropyridine (11)42
n-Butyllithium (1.6 M in hexane, 7.15 mL, 11.4 mmol) was added to a solution of diisopropylamine (1.70 mL, 11.9 mmol) in THF (30 mL) at −78 °C. The reaction was stirred for 30 min and then 3-bromo-5-chloropyridine (2.00 g, 10.4 mmol) in THF (10 mL) was added dropwise over 7 min. The reaction was stirred for 45 min, resulting in a yellow/brown suspension. Hexachloroethane (4.92 g, 20.8 mmol) in THF (7 mL) was added at −78 °C and the dark brown reaction mixture was stirred at −78 °C for 75 min. The cooling bath was removed and the brown suspension was allowed to warm to rt (about 30 min). The clear brown solution was quenched with sat. aq NH4Cl (100 mL) and the aqueous layer was extracted with ether (3 × 70 mL). The combined organic layers were washed with water (2 × 100 mL) and brine (70 mL), dried over MgSO4, and filtered, and the solvent was evaporated in vacuo. The crude product was purified by flash chromatography (CH2Cl2/cyclohexane, 1:4 to 1:3) to give the 3-bromo-4,5-dichloropyridine (11) (2.05 g, 87% yield) as a white solid: 1H NMR (500 MHz, CDCl3) δ 8.54 (s, 1H), 8.64 (s, 1H); LC–MS (method B, ESI, m/z) tR = 3.01 min, 225/227/229 (M + H)+.
8-(3-Bromo-5-chloropyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (57)
3-Bromo-4,5-dichloropyridine (11) (1.00 g, 4.41 mmol) and boc-2,8-diazaspiro[4.5]decan-1-one (1.35 g, 5.29 mmol) were introduced into a microwave vial with 1-methoxy-2-propanol (11 mL) and triethylamine (1.80 mL, 13.2 mmol). The reaction mixture was heated under microwave irradiation for 2.5 h at 220 °C. The solvent was evaporated and the crude material was purified via Biotage column chromatography (CH2Cl2/EtOH, 99.8:0.2 to 97:3) to give 8-(3-bromo-5-chloropyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (57) (1.14 g, 75% yield) as a white solid: 1H NMR (500 MHz, CDCl3) δ 1.52–1.58 (m, 2H), 2.11–2.20 (m, 4H), 3.32–3.41 (m, 6H), 6.00 (br s, 1H), 8.35 (s, 1H), 8.49 (s, 1H); LC–MS (method B, ESI, m/z) tR = 2.68 min, 344/346/348 (M + H)+.
8-(3-Chloro-5-(prop-1-en-2-yl)pyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (58)
8-(3-Bromo-5-chloropyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (57) (200 mg, 0.580 mmol), isopropenylboronic acid (120 μL, 0.638 mmol), and Pd(dppf)Cl2·CH2Cl2 (24 mg, 0.029 mmol) were loaded in a microwave vial under argon, and then acetonitrile (10.4 mL) and 0.5 M aqueous sodium carbonate solution (1.60 mL, 0.812 mmol) were added. The reaction mixture was heated at 120 °C for 60 min under microwave irradiation, concentrated, and purified via Biotage column chromatography (DCM/EtOH 99:1 to 95:5) to give 8-(3-chloro-5-(prop-1-en-2-yl)pyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (58) as a colorless oil (122 mg, 69% yield): 1H NMR (500 MHz, CDCl3) δ 1.50–1.56 (m, 2H), 2.07–2.14 (m, 5H), 2.16 (t, J = 6.9 Hz, 2H), 3.15–3.20 (m, 2H), 3.36–3.42 (m, 4H), 4.97–4.98 (m, 1H), 5.26–5.28 (m, 1H), 5.81 (s, 1H), 8.15 (s, 1H), 8.39 (s, 1H); LC–MS (method C, ESI, m/z) tR = 1.78 min, 306/308 (M + H)+.
8-(3-(4-Morpholin-4-ylphenyl)-5-(prop-1-en-2-yl)pyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (59)
8-(3-Chloro-5-(prop-1-en-2-yl)pyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (58) (40 mg, 0.13 mmol), 4-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)morpholine (57 mg, 0.20 mmol), sodium carbonate (35 mg, 0.33 mmol), and dichlorobis(tricyclohexylphosphine)palladium(II) (9.7 mg, 0.013 mmol) were loaded in a microwave vial under argon, and then acetonitrile (1.0 mL) and water (260 μL) were added. The reaction mixture was heated at 150 °C for 35 min under microwave irradiation. The solvent was evaporated and the crude material was purified via Biotage column chromatography (DCM/EtOH 98:2 to 90:10) to give the title compound 59 as a colorless oil (24.5 mg, 44% yield): 1H NMR (500 MHz, CDCl3) δ 1.21–1.25 (m, 2H), 1.80–1.88 (m, 2H), 1.96 (t, J = 6.9 Hz, 2H), 2.13 (s, 3H), 2.72–2.79 (m, 2H), 3.11–3.16 (m, 2H), 3.21–3.24 (m, 4H), 3.27 (t, J = 6.9 Hz, 2H), 3.87–3.91 (m, 4H), 5.00–5.02 (m, 1H), 5.24–5.26 (m, 1H), 5.52 (s, 1H), 7.00 (d, J = 8.8 Hz, 2H), 7.30 (d, J = 8.8 Hz, 2H), 8.17 (s, 1H), 8.18 (s, 1H); LC–MS (method C, ESI, m/z) tR = 1.87 min, 433 (M + H)+; ESI-HRMS calcd for C26H33N4O2 (M + H)+ 433.2598, found 433.2595.
8-(3-Isopropyl-5-(4-morpholin-4-ylphenyl)pyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (60)
8-(3-(4-Morpholinophenyl)-5-(prop-1-en-2-yl)pyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (59) (5 mg, 0.01 mmol) was solubilized in ethanol (0.5 mL) and a few drops of DCM. Then 1 M HCl (35 μL, 0.035 mmol) and palladium 10% on carbon (3 mg, 0.01 mmol) were added. The reaction was hydrogenated under a balloon of hydrogen at rt for 2 days and filtered over Celite. The crude material was filtered on a flash NH2 column and concentrated to give the title compound 60 as a colorless oil (4 mg, 80% yield): 1H NMR (500 MHz, CDCl3) δ 1.28–1.33 (m, 8H), 1.88–2.00 (m, 4H), 2.58–2.68 (m, 2H), 2.85–2.92 (m, 2H), 3.21–3.25 (m, 4H), 3.25–3.29 (m, 2H), 3.37–3.45 (m, 1H), 3.88–3.92 (m, 4H), 5.64 (s, 1H), 6.96–6.99 (m, 2H), 7.20 (d, J = 8.3 Hz, 2H), 8.15 (s, 1H), 8.42 (s, 1H); 13C NMR (126 MHz, CDCl3) δ 24.0, 26.2, 30.5, 32.4, 38.6, 42.0, 48.5, 49.2, 66.9, 115.1, 130.4, 130.6, 134.3, 140.9, 147.9, 150.3, 150.5, 154.5, 181.6; LC–MS (method C, ESI, m/z) tR = 1.85 min, 435 (M + H)+; ESI-HRMS calcd for C26H35N4O2 (M + H)+ 435.2755, found 435.2747.
Preparation of Compounds in Table 8 Exemplified by Compounds 68 and 74
8-(3-Chloro-5-(4-(4-methylpiperazin-1-yl)phenyl)pyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (68)
8-(3-Bromo-5-chloropyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (57) (35 mg, 0.10 mmol), 1-methyl-4-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)piperazine (34 mg, 0.11 mmol), and Pd(dppf)Cl2·CH2Cl2 (3.7 mg, 5.1 μmol) were loaded in a microwave vial under argon, and then degassed MeCN (1.8 mL) and 0.5 M aqueous sodium carbonate solution (284 μL, 0.142 mmol) were added. The reaction mixture was heated under microwave irradiation at 120 °C for 60 min. The solvent was evaporated and the crude material was dry-loaded and purified via Biotage column chromatography (CH2Cl2/MeOH 97/3 to 85/15) to give the title compound 68 (38 mg, 85% yield) as a cream solid: 1H NMR (500 MHz, CDCl3) δ 1.32–1.38 (m, 2H), 1.94–2.02 (m, 4H), 2.39 (s, 3H), 2.60–2.65 (m, 4H), 2.70–2.78 (m, 2H), 3.11–3.17 (m, 2H), 3.27–3.31 (m, 6H), 5.54 (br s, 1H), 7.00 (d, J = 8.7 Hz, 2H), 7.19 (d, J = 8.7 Hz, 2H), 8.18 (s, 1H), 8.39 (s, 1H); 13C NMR (126 MHz, CDCl3) δ 31.4, 32.4, 38.8, 42.0, 46.1, 47.7, 48.6, 55.0, 115.7, 128.3, 128.7, 130.1, 133.9, 149.1, 150.6, 151.0, 152.9, 181.8; LC–MS (method C, ESI, m/z) tR = 1.43 min, 440/442 (M + H)+; ESI-HRMS calcd for C24H3135ClN5O (M + H)+ 440.2212, found 440.2205.
8-(3-Chloro-5-(4-(1-methyl-1H-pyrazol-4-yl)phenyl)pyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (74)
8-(3-Bromo-5-chloro-pyridin-4-yl)-2,8-diaza-spiro[4.5]decan-1-one (57) (250 mg, 0.725 mmol), 1-methyl-4-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)-1H-pyrazole (82) (268 mg, 0.943 mmol), and Pd(dppf)Cl2·CH2Cl2 (30 mg, 0.036 mmol) were introduced into a microwave vial, and then MeCN (13 mL) and aqueous sodium carbonate solution (0.5 M, 2.00 mL, 1.02 mmol) were added. The reaction mixture was heated under microwave irradiation at 120 °C for 1 h and concentrated. The crude material was purified via Biotage column chromatography (CH2Cl2/EtOH, 96:4 to 91:9), and trituration with hot EtOAc gave the title compound 74 (700 mg, 76% yield) as a white solid: 1H NMR (500 MHz, CDCl3) δ 1.33–1.39 (m, 2H), 1.94–2.01 (m, 4H), 2.71–2.79 (m, 2H), 3.14–3.19 (m, 2H), 3.28 (t, J = 6.8 Hz, 2H), 3.97 (s, 3H), 5.58 (br s, 1H), 7.29 (d, J = 8.3 Hz, 2H), 7.56 (d, J = 8.3 Hz, 2H), 7.68 (s, 1H), 7.82 (s, 1H), 8.22 (s, 1H), 8.44 (s, 1H); 13C NMR (126 MHz, CDCl3) δ 31.6, 32.4, 38.7, 39.3, 41.8, 47.8, 122.7, 125.6, 127.2, 128.3, 129.8, 132.3, 133.7, 135.7, 136.9, 149.6, 150.9, 152.9, 181.6 ; LC–MS (method C, ESI, m/z) tR = 2.37 min, 422/424 (M + H)+; ESI-HRMS calcd for C23H2535ClN5O (M + H)+ 422.1742, found 422.1730.
Preparation of Compounds in Table 9 Exemplified by Compound 81
8-(3-Chloro-5-(4-(1-methyl-1H-pyrazol-4-yl)phenyl)pyridin-4-yl)-2,8-diazaspiro[4.5]decane-1,3-dione (81)
Two microwave vials were prepared as follows: 1-methyl-4-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)-1H-pyrazole (82) (52 mg, 0.18 mmol), 8-(3-bromo-5-chloropyridin-4-yl)-2,8-diazaspiro[4.5]decane-1,3-dione (84) (52 mg, 0.18 mmol), and Pd(dppf)Cl2·CH2Cl2 (5.7 mg, 7.0 μmol) were loaded into a microwave vial, and then 0.5 M aqueous sodium carbonate solution (390 μL, 0.195 mmol) and MeCN (2.5 mL) were added. The reaction mixture was heated at 120 °C for 60 min under microwave irradiation. The reaction mixtures from the two vials were combined and the volatiles were evaporated. The crude material was purified via Biotage column chromatography (CH2Cl2/EtOH, 97:3 to 92:8) to give the title compound 81 (70 mg, 58% yield) as a white solid: 1H NMR (500 MHz, DMSO-d6) δ 1.46–1.52 (m, 2H), 1.77–1.84 (m, 2H), 2.47 (s, 2H), 2.62–2.69 (m, 2H), 3.02–3.08 (m, 2H), 3.89 (s, 3H), 7.34 (d, J = 8.2 Hz, 2H), 7.69 (d, J = 8.2 Hz, 2H), 7.95 (d, J = 0.8 Hz, 1H), 8.22 (s, 1H), 8.23 (s, 1H), 8.48 (s, 1H), 11.12 (br s, 1H); 13C NMR (126 MHz, DMSO-d6) δ 32.9, 39.2, 39.7, 44.0, 47.7, 121.8, 125.4, 127.9, 128.5, 130.1, 132.6, 134.1, 134.9, 136.6, 149.2, 151.1, 152.6, 177.4, 183.7; LC–MS (method C, ESI, m/z) tR = 2.30 min, 436/438 (M + H)+; ESI-HRMS calcd for C23H2335ClN5O2 (M + H)+ 436.1535, found 436.1524.
1-Methyl-4-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)-1H-pyrazole (82)
1-Chloro-4-iodobenzene (6.39 g, 26.8 mmol), 1-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (5.58 g, 26.8 mmol), sodium carbonate (6.25 g, 59.0 mmol), and Pd(dppf)Cl2·CH2Cl2 (2.20 g, 2.68 mmol) were loaded into a flask, and then a mixture of THF/H2O 3/1 (117 mL) was added. The reaction mixture was heated in an oil bath at 80 °C overnight. The solvents were evaporated, and the residue was purified by column chromatography (cyclohexane/EtOAc 99:1 to 70:30) to afford 4-(4-chlorophenyl)-1-methyl-1H-pyrazole as a white solid (3.80 g, 74% yield): 1H NMR (500 MHz, CDCl3) δ 3.93 (s, 3H), 7.31 (d, J = 8.7 Hz, 2H), 7.38 (d, J = 8.7 Hz, 2H), 7.57 (s, 1H), 7.72 (s, 1H); LC–MS (method B, ESI, m/z) tR = 2.88 min, 193/195 (M + H)+.
4-(4-Chlorophenyl)-1-methyl-1H-pyrazole (3.30 g, 17.1 mmol), bis(pinacolato)diboron (5.20 g, 20.6 mmol), potassium acetate (5.00 g, 51.4 mmol), Xphos (650 mg, 1.37 mmol), and Pd2dba3 (310 mg, 0.343 mmol) were loaded in a flask, and dioxane (34 mL) was added. The reaction mixture was stirred in an oil bath at 85 °C overnight. The solvent was evaporated and the crude product purified by column chromatography (cyclohexane/EtOAc, 90:10 to 70:30) to afford 1-methyl-4-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)-1H-pyrazole (82) as a white solid (3.90 g contaminated by 10% of 1-methyl-4-phenyl-1H-pyrazole, corrected yield 75%): 1H NMR (500 MHz, CDCl3) δ 1.35 (s, 12H), 3.93 (s, 3H), 7.47 (d, J = 8.3 Hz, 2H), 7.64 (s, 1H), 7.79 (d, J = 8.3 Hz, 2H), 7.79 (s, 1H); LC–MS (method B, ESI, m/z) tR = 3.06 min, 284/285 (M + H)+.
2,8-Diazaspiro[4.5]decane-1,3-dione (83):43
To a suspension of 8-benzyl-2,8-diazaspiro[4.5]decane-1,3-dione (1.20 g, 4.65 mmol) and concentrated acetic acid (270 μL, 4.65 mmol) in ethanol (20 mL) was added palladium hydroxide (20 wt % on carbon, wet, 360 mg, 4.65 mmol) and the mixture stirred under a H2 atmosphere for 24 h at rt. The mixture was filtered over Celite and washed with EtOH and 1 M NH3 in MeOH. The filtrate was concentrated and purified on an SCX2 cartridge (loading with DCM, byproduct elution with DCM/MeOH 9/1, elution of product with DCM/MeOH/NH3 9/1/0.01) to give 2,8-diazaspiro[4.5]decane-1,3-dione (83) (754 mg, 97%) as a white solid: 1H NMR (500 MHz, DMSO-d6) δ 1.41 (dd, J = 12.5, 1.4 Hz, 2H), 1.65 (td, J = 12.5, 3.9 Hz, 2H), 2.44–2.53 (m, 2H), 2.56 (s, 2H), 2.86 (dt, J = 12.5 Hz, 3.9, 2H); LC–MS (method B, ESI, m/z) tR = 0.17 min, 169 (M + H)+.
8-(3-Bromo-5-chloropyridin-4-yl)-2,8-diazaspiro[4.5]decane-1,3-dione (84)
3-Bromo-4,5-dichloropyridine (11) (1.00 g, 4.41 mmol) and 2,8-diazaspiro[4.5]decane-1,3-dione (83) (1.10 g, 6.61 mmol) were introduced in a microwave vial, and then NMP (11 mL) and triethylamine (1.90 mL, 13.2 mmol) were added. The reaction mixture was heated under microwave irradiation for 1 h at 220 °C. The reaction mixture was poured into water (110 mL) and the precipitate was filtered to give 8-(3-bromo-5-chloropyridin-4-yl)-2,8-diazaspiro[4.5]decane-1,3-dione (84) (1.08 g, 68% yield) as a white solid: 1H NMR (500 MHz, DMSO-d6) δ 1.64–1.70 (m, 2H), 1.94–2.01 (m, 2H), 2.69 (s, 2H), 3.24–3.30 (m, 4H), 8.47 (s, 1H), 8.57 (s, 1H), 11.18 (br s, 1H); LC–MS (method B, ESI, m/z) tR = 2.50 min, 358/360/362 (M + H)+.
Mouse Liver Microsomal Stability
Compounds (10 μM) were incubated with male CD1 mouse liver microsomes (1 mg/mL) protein in the presence of 1 mM NADPH, 2.5 mM UDP-glucuronic acid (UDPGA), and 3 mM MgCl2 in 10 mM PBS at 37 °C. Incubations were conducted for 0 and 30 min. Control incubations were generated by the omission of NADPH and UDPGA from the incubation reaction. The percentage compound turned over was determined after analysis by LC–MS.
Human Liver Microsomal Stability
Compounds (10 μM) were incubated with mixed-gender, pooled human liver microsomes (1 mg/mL) protein in the presence of 1 mM NADPH, 2.5 mM UDPGA, and 3 mM MgCl2 in 10 mM PBS at 37 °C. Incubations were conducted for 0 and 30 min. Control incubations were generated by the omission of NADPH and UDPGA from the incubation reaction. The percentage compound turned over was determined after analysis by LC–MS.
Mouse, Rat, and Human Clint Determination
Microsomes (final concentration 0.5 mg/mL), 50 mM phosphate buffer pH 7.4, and compound (final concentration 1 μM) were added to the assay plate and allowed to preincubate for 5 min at 37 °C. The reaction was initiated by the addition of NADPH (final concentration 1.5 mM) and the plate was shaken at 800 rpm at 37 °C. After 0, 5, 10, 20, and 30 min, aliquots were taken, and the reaction was stopped using cold acetonitrile. The samples were centrifuged at 4000 rpm for 30 min at 4 °C and analyzed by LC–MS/MS. Four test compounds were pooled for analysis. The in vitro intrinsic clearance was calculated from the rate of compound disappearance.
Caco-2 Papp Determination
Caco-2 cells (TC7 clone) were maintained in DMEM in an atmosphere of 8.5% CO2. For transport experiments, 0.125 × 106 cells/well were seeded on polycarbonate filter inserts and allowed to grow and differentiate for 10–14 days before the cell monolayers were used for experiments. Drug transport experiments were carried out using a cocktail approach in a 4-dimensional setting. Apparent permeability coefficients were determined for A > B and B > A directions with and without the presence of cyclosporine A as a transporter inhibitor. Up to five test items and reference compounds were dissolved in Hank’s balanced salt solution at pH 7.4 to yield a final concentration of 1 μM. The assays were performed in HBSS containing 25 mM HEPES (pH 7.4) in an atmosphere of 5% CO2 at 37 °C. Prior to the study, the monolayers were washed in prewarmed HBSS. At the start of the experiments, prewarmed HBSS containing the test items was added to the donor side of the monolayer and HBSS without test items was added to the receiver side. The plates were shaken at 150 rpm at 37 °C during the experiment. After 2 h, the transwell insert containing the monolayer was carefully removed and placed in a new plate, and aliquots of both the receiver and donor sides were taken and diluted with an equal volume of ACN containing the internal standard. The mixture was centrifuged and the supernatant analyzed by LC–MS/MS. The apparent permeability coefficients (Papp) were calculated using the formula Papp = (Vrec/A × C0donor) × dCrec/dt × 106 with dCrev/dt being the change in concentration in the receiver compartment with time, Vrec the volume of the sample in the receiver compartment, C0donor the concentration in the donor compartment at time 0, and A the area of the compartment with the cells.
Protein Binding Assays
Protein binding of test compounds was determined by ultrafiltration using serum from mouse, rat, and human. The test items (final concentration 5 μM) were incubated in triplicate with three different serum dilutions (1:2, 1:5, and 1:10) for 30 min at 37 °C using slight agitation. After the incubation, the 96-well filter plates were centrifuged for 45 min at 3500 rpm and 37 °C; 25 μL portions of filtrate samples were treated with 50 μL of ethanol and 50 μL of internal standard solution and analyzed by LC–MS/MS. The fraction unbound was calculated from the drug concentrations in the filtrate samples.
In Vivo Mouse PK in-Life Phase
Female NMRI mice (n = 5) received either a single intravenous (bolus) injection or a single oral administration (by gavage) of the compound in a cocktail preparation. Doses of 0.2 mg/kg (per compound, intravenous) and 0.5 mg/kg (per compound, orally) were given as solutions in DMSO/PEG 200/water. Consecutive blood samples were taken retroorbitally under isofluorane inhalation from n = 3 animals per route of administration after 0.1 (iv only), 0.25 (po only), 0.5, 1, 2, 4, and 6 h and were further processed to obtain plasma. In addition, feces of two mice per route of administration were collected over the time interval from 0 to 24 (pooled for analysis, no blood sampling).
In Vivo Rat PK in-Life Phase
Male Wistar rats (n = 6) received either a single intravenous (bolus) injection or a single oral administration (by gavage) of the compound in a cocktail preparation. Doses of 0.2 mg/kg (per compound, intravenous) and 0.5 mg/kg (per compound, orally) were given as solutions in DMSO/PEG 200/water. Consecutive blood samples were taken sublingually under isofluorane inhalation from n = 3 animals per route of administration after 0.1 (iv only), 0.25 (po only), 0.5, 1, 2, 4, and 6 h and were further processed to obtain plasma. In addition, feces and urine of three rats per route of administration were collected over the time interval from 0 to 24 (pooled for analysis, no blood sampling).
Bioanalytics
The concentrations of compound in plasma and feces were quantified using an UPLC method with tandem mass spectrometric detection (LC–MS/MS). The LC–MS system consisted of a Waters Acquity UPLC coupled to an AB Sciex mass spectrometer API 5500 Q-trap. The UPLC separation was carried out on a reversed-phase column (Acquity UPLC BEC C8, 1.7 μM, 2.1 × 50 mm) using a mobile phase gradient with 0.1% formic acid and acetonitrile as eluents. The detection of the drug was performed using multiple reaction monitoring in the positive ionization mode. Plasma samples were spiked with internal standard, and the analyte was extracted from the matrix using tert-butyl methyl ether (tBME). The organic phase was evaporated to dryness under a stream of nitrogen. The residue was dissolved in acetonitrile/0.1% formic acid for LC–MS/MS analysis. Feces samples were homogenized with 4 times their volume of ethanol/water (4:1). Aliquots of the aqueous ethanolic extracts were further diluted with acetonitrile/0.1% formic acid, spiked with internal standard, and directly injected into the system.
Biochemical Activity Testing of TNKS1 and -2 (Autoparsylation Assay)
The autoparsylation assay was run in two steps: first, the enzymatic reaction in which GST-tagged TNKS1 or TNKS2 transferred biotinylated ADP-ribose to itself from biotinylated NAD as the cosubstrate and, second, the detection reaction where a time-resolved FRET between cryptate-labeled anti-GST bound to the GST tag of the enzyme and Xlent-labeled streptavidin bound the biotin-parsylation residue was analyzed. The autoparsylation activity was detectable directly via the increase in HTRF signal.
The autoparsylation assay was performed in 384-well HTRF (Cisbio, Codolet, France) assay format in Greiner low-volume 384-well microtiter plates; 250 nM GST-tagged TNKS1 (1023–1327 aa) or 250 nM GST-tagged TNKS2 (873–1166 aa) and 5 μM bio-NAD (Biolog, Life science Inst., Bremen, Germany) as cosubstrate were incubated in a total volume of 5 μL (50 mM HEPES, 4 mM MgCl2, 0.05% Pluronic F-68, 1.4 mM DTT, 0.5% DMSO, pH 7.7) in the absence or presence of the test compound (10 dilution concentrations) for 90 min at 30 °C. The reaction was stopped by the addition of 1 μL of 50 mM EDTA solution, and 2 μL of the detection solution [1.6 μM SA-Xlent (Cisbio, Codolet, France), 7.4 nM Anti-GST-K (Eu-labeled anti-GST, Cisbio, Codolet, France) in 50 mM HEPES, 800 mM KF, 0.1% BSA, 20 mM EDTA, 0.1% CHAPS, pH 7.0)] was added. After 1 h of incubation at rt the HTRF was measured with an Envision multimode reader (PerkinElmer LAS Germany GmbH) at an excitation wavelength of 340 nm (laser mode) and emission wavelengths of 615 and 665 nm. The ratio of the emission signals was determined. The full value used was the inhibitor-free reaction. The pharmacological zero value used was XAV-939 (Tocris) in a final concentration of 5 μM. The inhibitory values (IC50) were determined using either the program Symyx Assay Explorer or Condosseo from GeneData.
Taqman Assays
7dF3 cells were seeded at 20 000 cells/well into 96-well cell culture plates. After overnight incubation the cells were treated with 74 at concentrations ranging from 9.1 μM to 0.068 nM. After 2 or 6 h of incubation, cells were washed with PBS and stored at −80 °C. Cells were lysed in Cells-to-cDNA II Cell lysis buffer (Life Technologies Ltd., Paisley, UK) and transferred to PCR plates. Lysis was completed by heating at 75 °C for 15 min. Lysates were DNase treated (1 μL/well Life Technologies) at 37 °C for 15 min and heat inactivated at 75 °C for 5 min. cDNA synthesis was performed using the High-Capacity cDNA Reverse Transcription Kit (Life Technologies). Taqman reactions were performed in 384-well plates using 1.5 μL cDNA and 1× FAM-labeled primer probe for the measurement of target gene and 1× VIC-labeled primer probe for LRP0 and were multiplexed in each well as the internal reference control with TaqMan Universal Master Mix (Life Technologies). Plates were run for 40 cycles using an ABI Via7 Sequence Detection System and software. Ct values for target genes were normalized relative to the LRP0 control before being expressed relative to the vehicle treated control sample.
In Vitro Cell-Based Reporter Assays
7Df3 Luciferase Reporter Assay
7dF3 cells were seeded at 5000 per well in 50 μL of Dulbecco’s modified Eagle’s medium (DMEM) with 4 mM supplemental l-glutamine in 384-well white tissue culture plates (Corning 3570). After incubating overnight at 37 °C/5% CO2, compounds ranging in final concentration from 90 μM to 0.3 nM were added in duplicate to the wells. After 2 h of further incubation as above, β-oestradiol was added to a final concentration of 10 μM. The cells were incubated for 24 h at 37 °C/5% CO2 and then 25 μL of luciferase reagent (SteadyGlo, Promega) was added, and the cells were mixed. After leaving the plate for 60 min at room temperature, luminescence was read on a plate luminescence reader (TopCount, PerkinElmer). The percentage inhibition for each compound concentration was calculated using total counts (no compound) and low counts (no β-oestradiol) as highs and lows, respectively. IC50 was subsequently determined using a curve-fitting software package in Excel (Excelfit, IDBS).
Tet-O-WNT Reporter Lines
T-Rex HEK293 Tet-O cells (Life Technologies) were transfected with the TCF-firefly luciferase-IRES-GFP (TLIG) reporter, and a highly responsive reporter clone was derived using an analogous process to that previously used to derive the HEK293-based 7df3 cell line.30 Expression constructs coding for WNT inducers that act at different levels of the pathway30 were introduced downstream of the Tet-O promoter by FLP-FRT-site-directed recombination. This generated a set of cell lines that contain an identical TCF-luciferase reporter in combination with Tet-inducible cDNAs encoding ΔN-LRP6, Dvl2, Axin-GID, ΔN-β-catenin, and VP16-TCF4.
Assay Protocol for Tet-O-WNT Reporter Cell Lines
Cells were seeded from frozen at 40 000/well into 96-well plates in 80 μL of medium. On day 3, eight compound concentrations ranging from 30 μM to 14 nM were prepared at 333× final concentration in DMSO and then diluted to 5× final concentration in OptiMEM (Life Technologies) supplemented with 0.05 μg/mL doxycycline hyclate (Sigma-Aldrich). These solutions were then pipetted at 20 μL/well into each well of the plate. On day 4, medium was removed, and cells were lysed in 30 μL/well GLOlysis buffer (Promega) for 30 min. BrightGLO firefly luciferase substrate (Promega) was pipetted at 30 μL/well. Firefly luciferase activity was measured on a Fluostar plate reader (BMG) in luminescence mode.
Generation of Other Cell-Based Reporter Cell Lines
SW480 human colorectal cancer cells were cotransfected with Topflash and CMV-lacZ reporter plasmids as described previously.30 RKO (ATCC CRL-2577) cells were transfected with plasmid F1522 encoding PEST-destabilized firefly luciferase (luc2P, designated Fluc2P; Promega) and IRES-driven PEST-destabilized renilla (hRlucP; Promega) downstream of the EF-1-α promoter.44 LS174T (ATCC CL-188) and COLO205 (ATCC, #CCL-222) human colorectal cancer cells were transduced with a TCF/LEF lentiviral firefly luciferase reporter construct. This construct was generated by synthesizing and cloning 16 repeats of the TCF/LEF responsive element (5′-AGATCAAAGG-3′) upstream of the minimal pTA promoter driving PEST-destabilized firefly luciferase Fluc2P (t1/2 < 1 h) from plasmid pGL4.24 (Promega). The TCF/LEF-pTA-Fluc2P cassette was subcloned into lentiviral vector pCDF1-MCS2-EF1-Puro (Systems Biosciences) to generate the final reporter construct F1756, which contained a puromycin resistance gene for selection purposes. F1756 was cotransfected with pPackF1 packaging mix (Systems Biosciences) into the HEK293 cell line. F1756 reporter lentivirus was recovered in cell media after 48 h, purified, and used to transduce cell lines. After 72 h, successfully transduced cells were selected using 5 μg/mL puromycin and stable colonies left to grow for 2 weeks. Colonies showing high-level basal luciferase expression on addition of luciferin to the flask and imaging on an IVIS 200 (PerkinElmer Inc.) were picked and expanded. Subsequent studies identified clone 4 as a suitable candidate for further studies. Reporter activity (and its response to inhibition by compounds) was measured in vitro in 384-well plates using Steady-Glo luciferase reagent (Promega) for firefly luciferase. Cells were exposed to compound for 24 h before reporter assays were conducted. Cell viability was assayed in parallel using Alamar blue (Promega). PA-1 human teratocarcinoma cells were stably transfected with a TOPflash TCF reporter plasmid (Merck Millipore) that was modified by introduction of a neomycin resistance marker. Single clones were selected and tested for WNT-driven reporter pathway activation by addition of Wnt-3A supernatant produced by L Wnt-3A cells (ATCC, CRL-2647). Reporter activity was measured in 96-well plates using Steady-Glo luciferase reagent (Promega). Cells were treated with compound for 6 h and subsequently stimulated with Wnt-3A supernatant for 24 h before reporter assays were conducted.
COLO205-F1756 Clone 4 Human Tumor Xenograft Model Expressing a Modified Luciferase-Based Wnt Reporter Construct
Female athymic CrTac:NCr-Foxn1nu mice from Taconic (6–8 weeks of age) were injected subcutaneously in the flank with 3 million COLO205-F1756 clone 4 cells, and when tumor xenografts reached a mean diameter of 5–6 mm (equating to a mean volume of about 100 mm3), mice (n = 8 per group) were dosed orally by gavage with compound 74 (70 mg/kg bid for 8 days) or vehicle (10% dimethyl sulfoxide; 5% Tween 20 in sterile saline). Luminescent flux as a measure of WNT pathway activity was quantified by whole body imaging in cohorts of four animals at 2 and 6 h after the final dose. This was achieved by sc injection of 100 μL of 30 mg/mL D-Luciferin firefly substrate (XR-1001, PerkinElmer) into anaesthetized mice and imaging with a Xenogen IVIS 200 (XR-1001, PerkinElmer), utilizing the software Living Image (version 4). At these time points, heparinized blood samples were collected by cardiac puncture under terminal anesthesia, and plasma was separated by centrifugation. Tumors were excised and weighed, and both plasma and tumor samples were snap frozen for PK analyses.
Bioanalytics
The concentrations of compound in plasma and tumor were quantified using an UPLC method with tandem mass spectrometric detection (LC–MS/MS). The LC–MS system consisted of a 1290 binary pump from Agilent coupled to an Agilent 6420 triple quadrupole mass spectrometer. The UPLC separation was carried out on a reversed-phase column (Kinetic column C18, 2.6 μM, 2.1 × 50 mm purchased from Phenomenex) using a mobile phase gradient with 0.1% formic acid and methanol as eluent. Detection of the drug was performed using multiple reaction monitoring in the positive ionization mode. Plasma samples were spiked with internal standard, and the analyte was extracted from the matrix (plasma and tumor homogenized with 3 v/w PBS) with 3–4 volumes of methanol and directly injected into the system.
Acknowledgments
This work was supported by Cancer Research UK (grant number C309/A11566). We acknowledge NHS funding to the NIHR Biomedical Research Centre at The Institute of Cancer Research and The Royal Marsden. We thank Dr. Amin Mirza, Mr. Meirion Richards, and Dr Maggie Liu for their assistance with NMR, mass spectrometry, and HPLC. We thank Prof. Dr. H. Fueß of the TU Darmstadt for the X-ray crystal structure of compound 9 and Dr. Mark Honey for resynthesis of selected compounds.
Glossary
Abbreviations Used
- Cl
clearance
- Clint
intrinsic clearance
- DSH
Dishevelled
- ER
efflux ratio
- GSK3β
glycogen synthase kinase 3β
- GFP
green fluorescent protein
- GID
GSK3β-interaction domain
- HLM
human liver microsomes
- IRES
internal ribosome entry site
- MLM
mouse liver microsomes
- Papp
apparent permeability coefficient
- Vss
apparent volume of distribution at steady state
Supporting Information Available
1H NMR spectra for all final compounds; preparation of compounds 15–25, 27–37, 39–42, 45–52, 61–67, 69–73, and 75–80; CYP450 inhibition for compounds 9 and 74 (Supplementary Table S1), summaries of CEREP and kinase profiles (Supplementary Tables S2−S5); Supplementary Figures S1 and S2; and small-molecule X-ray crystallographic data analysis for compound 9. This material is available free of charge via the Internet at http://pubs.acs.org
The authors declare the following competing financial interest(s): A.M., S.C., M.P., D.W., M.S., O.A-P., E.S., C.T., A.T.H., W.C., S.H., G.B., M.-J.O.R., M.V., A.D.H.B., R.T., P.W., W.A., F.R., P.A.C., S.A.E., and J.B. are current or former employees of The Institute of Cancer Research, which has a commercial interest in the development of WNT pathway inhibitors. K.G., O.P., D.W., C.E., F.R., F.S., and K.S. are current employees of Merck Serono.
Supplementary Material
References
- Angers A.; Moon R. T. Proximal events in Wnt signal transduction. Nat. Rev. Mol. Cell. Biol. 2009, 10, 468–477. [DOI] [PubMed] [Google Scholar]
- Ewan K. B.; Dale T. C. The potential for targeting oncogenic Wnt/β-catenin signalling in therapy. Curr. Drug Targets 2008, 9, 532–547. [DOI] [PubMed] [Google Scholar]
- Bienz M.; Clevers H. Linking colorectal cancer to Wnt signalling. Cell 2000, 103, 311–320. [DOI] [PubMed] [Google Scholar]
- Vlad A.; Rohrs S.; Klein-Hitpass L.; Muller O. The first five years of the Wnt targetome. Cell Signalling 2008, 20, 795–802. [DOI] [PubMed] [Google Scholar]
- MacDonald B. T.; Tamai K.; He X. Wnt/β-catenin signalling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker N.; Clevers H. Mining the Wnt pathway for cancer therapeutics. Nat. Rev. Drug Discovery 2007, 5, 997–1014. [DOI] [PubMed] [Google Scholar]
- Levy D. B.; Smith K. J.; Beazer-Barclay Y.; Hamilton S. R.; Vogelstein B.; Kinzler K. W. Inactivation of both APC alleles in human and mouse tumours. Cancer Res. 1994, 54, 5953–5958. [PubMed] [Google Scholar]
- Smith K. J.; Johnson K. A.; Bryan T. M.; Hill D. E.; Markowitz S.; Willson J. K.; Paraskeva C.; Petersen G. M.; Hamilton S. R.; Vogelstein B. The APC gene product in normal and tumour cells. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 2846–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks A. B.; Morin P. J.; Vogelstein B.; Kinzler K. W. Mutational analysis of the APC/catenin/Tcf pathway in colorectal cancer. Cancer Res. 1998, 58, 1130–1134. [PubMed] [Google Scholar]
- Cowin P.; Rowlands T. M.; Hatsell S. J. Cadherins and catenins in breast cancer. Curr. Opin. Cell. Biol. 2005, 17, 499–508. [DOI] [PubMed] [Google Scholar]
- Kim S.-J.; Crooks H.; Foxworth A.; Waldman T. Proof-of-principle: Oncogenic β-catenin is a valid molecular target for the development of pharmacological inhibitors. Mol. Cancer Ther. 2002, 1, 1355–1359. [PubMed] [Google Scholar]
- Akiri G.; Cherian M. M.; Vijayakumar S.; Liu G.; Bafico A.; Aaronson S. A. Wnt pathway abberations including autocrine Wnt activation occur at high frequency in human non-small-cell lung carcinoma. Oncogene 2009, 28, 2163–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto M.; Mitra R. S.; Liu M.; Lee J.; Henson B. S.; Carey T.; Bradford C.; Prince M.; Wang C.-Y.; Fearon E. R.; D’Silva N. J. Rap1 stabilises β-catenin and enhances β-catenin dependent transcription and invasion in squamous cell carcinoma of the head and neck. Clin. Cancer Res. 2010, 16, 65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi-Yanaga F.; Kahn M. Targeting Wnt signalling: Can we safely eradicate cancer stem cells. Clin. Cancer Res. 2010, 16, 3153–3162. [DOI] [PubMed] [Google Scholar]
- Luu H. H.; Zhang R.; Haydon R. C.; Rayburn E.; Kang Q.; Si W.; Park J. K.; Wang H.; Peng Y.; Jiang W.; He T.-C. Wnt/β-catenin signalling pathway as novel cancer drug targets. Curr. Cancer Drug Targets 2004, 4, 653–671. [DOI] [PubMed] [Google Scholar]
- Kahn M. Can we safely target the WNT pathway?. Nat. Rev. Drug Discovery 2014, 13, 513–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney A.; Axelrod F.; Bond C. J.; Cain J.; Chartier C.; Donigan L.; Fischer M.; Chaudhari A.; Ji M.; Kapoun A. M.; Lam A.; Lazetic S.; Ma S.; Mitra S.; Park I. K.; Pickell K.; Sato A.; Satyal S.; Stroud M.; Tran H.; Yen W. C.; Lewicki J.; Hoey T. Wnt pathway inhibition via the targeting of frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 11717–11722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D. C.; Rosen L. S.; Chugh R.; Goldman J. W.; Xu L.; Kapoun A.; Brachmann R. K.; Dupont J.; Stagg R. J.; Tolcher A. W.; Papadopoulos K. P. First-in-human evaluation of the human monoclonal antibody vantictumab (OMP-18R5; anti-Frizzled) targeting the WNT pathway in a phase I study for patients with advanced solid tumors. J. Clin. Oncol. 2013, 31 (Suppl. Abstr.), 2540.23733781 [Google Scholar]
- Emami K. H.; Nguyen C.; Ma H.; Kim D. H.; Jeong K. W.; Eguchi M.; Moon R. T.; Teo J. L.; Kim H. Y.; Moon S. H.; Ha J. R.; Kahn M. A small molecule inhibitor of beta-catenin/CREB-binding protein transcription. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 12682–12687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyabayashi T.; Teo J. L.; Yamamoto M.; McMillan M.; Nguyen C.; Kahn M. Wnt/beta-catenin/CBP signalling maintains long-term murine embryonic stem cell pluripotency. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 5668–5673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El–Khoueiry A. B. A phase I first in human study of PRI-724 in patients with advanced solid tumors. J. Clin. Oncol. 2013, 31 (Suppl. Abstr.), 2501. [Google Scholar]
- Huang S. M.; Mishina Y. M.; Liu S.; Cheung A.; Stegmeier F.; Michaud G. A.; Charlat O.; Wiellette E.; Zhang Y.; Wiessner S.; Hild M.; Shi X.; Wilson C. J.; Mickanin C.; Myer V.; Fazal A.; Tomlinson R.; Serluca F.; Shao W.; Cheng H.; Shultz M.; Rau C.; Schirle M.; Schlegl J.; Ghidelli S.; Fawell S.; Lu C.; Curtis D.; Kirschner M. W.; Lengauer C.; Finan P. M.; Tallarico J. A.; Bouwmeester T.; Porter J. A.; Bauer A.; Cong F. Tankyrase inhibition stabilises axin and antagonises Wnt signalling. Nature 2009, 461, 614–620. [DOI] [PubMed] [Google Scholar]
- Chen B.; Dodge M. E.; Tang W.; Lu J.; Ma Z.; Fan C. W.; Wei S.; Hao W.; Kilgore J.; Williams N. S.; Roth M. G.; Amatruda J. F.; Chen C.; Lum L. Small molecule-mediated disruption of Wnt-dependent signalling in tissue regeneration and cancer. Nat. Chem. Biol. 2009, 5, 100–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtiö L.; Chi N. W.; Krauss S. Tankyrases as drug targets. FEBS J. 2013, 280, 3576–3593. [DOI] [PubMed] [Google Scholar]
- Shultz M. D.; Cheung A. K.; Kirby C. A.; Firestone B.; Fan J.; Chen C. H.; Chen Z.; Chin D. N.; Dipietro L.; Fazal A.; Feng Y.; Fortin P. D.; Gould T.; Lagu B.; Lei H.; Lenoir F.; Majumdar D.; Ochala E.; Palermo M. G.; Pham L.; Pu M.; Smith T.; Stams T.; Tomlinson R. C.; Touré B. B.; Visser M.; Wang R. M.; Waters N. J.; Shao W. Identification of NVP-TNKS656: The use of structure–efficiency relationships to generate a highly potent, selective, and orally active tankyrase inhibitor. J. Med. Chem. 2013, 56, 6495–6511. [DOI] [PubMed] [Google Scholar]
- Lau T.; Chan E.; Callow M.; Waaler J.; Boggs J.; Blake R. A.; Magnuson S.; Sambrone A.; Schutten M.; Firestein R.; Machon O.; Korinek V.; Choo E.; Diaz D.; Merchant M.; Polakis P.; Holsworth D. D.; Krauss S.; Costa M. A novel tankyrase small-molecule inhibitor suppresses APC mutation-driven colorectal tumor growth. Cancer Res. 2013, 73, 3132–3144. [DOI] [PubMed] [Google Scholar]
- Hua Z.; Bregman H.; Buchanan J. L.; Chakka N.; Guzman-Perez A.; Gunaydin H.; Huang X.; Gu Y.; Berry V.; Liu J.; Teffera Y.; Huang L.; Egge B.; Emkey R.; Mullady E. L.; Schneider S.; Andrews P. S.; Acquaviva L.; Dovey J.; Mishra A.; Newcomb J.; Saffran D.; Serafino R.; Strathdee C. A.; Turci S. M.; Stanton M.; Wilson C.; DiMauro E. F. Development of novel dual binders as potent, selective, and orally bioavailable tankyrase inhibitors. J. Med. Chem. 2013, 56, 10003–10015. [DOI] [PubMed] [Google Scholar]
- Liu J.; Pan S.; Hsieh M. H.; Ng N.; Sun F.; Wang T.; Kasibhatla S.; Schuller A. G.; Li A. G.; Cheng D.; Li J.; Tompkins C.; Pferdekamper A.; Steffy A.; Cheng J.; Kowal C.; Phung V.; Guo G.; Wang Y.; Graham M. P.; Flynn S.; Brenner J. C.; Li C.; Villarroel M. C.; Schultz P. G.; Wu X.; McNamara P.; Sellers W. R.; Petruzzelli L.; Boral A. L.; Seidel H. M.; McLaughlin M. E.; Che J.; Carey T. E.; Vanasse G.; Harris J. L. Targeting Wnt-driven cancer through the inhibition of porcupine by LGK974. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 20224–20229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu P. B.; Hong C. C.; Sachidanandan C.; Babitt J. L.; Deng D. Y.; Hoyng S. A.; Lin H. Y.; Bloch K. D.; Peterson R. T. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat. Chem. Biol. 2008, 4, 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewan K.; Stubbs M.; Wildish H.; Pajak B.; Barbeau O.; Quevedo C.; Botfield H.; Young R.; Ruddle R.; Samuel L.; Battersby A.; Raynaud F.; Allen N.; Wilson S. W.; Latinkic B.; Workman P.; McDonald E.; Blagg J.; Aherne W.; Dale T. A useful approach to identify novel small-molecule inhibitors of Wnt-dependent transcription. Cancer Res. 2010, 70, 5963–5973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichowicz M.; Crumpler S. R.; McDonald E.; Blagg J. Microwave assisted synthesis of 4-amino-3,5-dihalopyridines. Tetrahedron 2010, 66, 2398–2403. [Google Scholar]
- pKa measurement was conducted at Cyprotex according to their published protocol: http://www.cyprotex.com.
- Angyal S. J.; Angyal C. L. The tautomerism of N-hetero-aromatic amines. Part I. J. Chem. Soc. 1952, 1461–1466. [Google Scholar]
- Ife R. J.; Dyke C. A.; Keeling D. J.; Meenan E.; Meeson M. L.; Parsons M. E.; Price C. A.; Theobald C. J.; Underwood A. H. 2-[[(4-Amino-2-pyridyl)methyl]sulfinyl]benzimidazole H+/K+-ATPase inhibitors. The relationship between pyridine basicity, stability and activity. J. Med. Chem. 1989, 32, 1970–1977. [DOI] [PubMed] [Google Scholar]
- Caballero N. A.; Melendez F. J.; Muñoz-Caro C.; Niño A. Theoretical prediction of relative and absolute pKa values of aminopyridines. Biophys. Chem. 2006, 124, 155–160. [DOI] [PubMed] [Google Scholar]
- Cambridge Small Molecule Crystallographic Database: http://webcsd.ccdc.cam.ac.uk/ (reference codes BEBKID, QETPIO).
- Bouchard R.; Fedida D. Closed and open state binding of 4-aminopyridine to the cloned human potassium channel Kv1.5. J. Pharmacol. Exp. Ther. 1995, 275, 864–876. [PubMed] [Google Scholar]
- Gramec D.; Mašič L. P.; Dolenc S. P. Bioactivation potential of thiophene-containing drugs. Chem. Res. Toxicol. 2014, 27, 1344–1358. [DOI] [PubMed] [Google Scholar]
- Bolleddula J.; DeMent K.; Driscoll J. P.; Worboys P.; Brassil P. J.; Bourdet D. L. Biotransformation and bioactivation reactions of alicyclic amines in drug molecules. Drug Met. Rev. 2014, 46, 379–419. [DOI] [PubMed] [Google Scholar]
- Van de Wetering M.; Sancho E.; Verweij C.; de Lau W.; Oving I.; Hurlstone A.; van der Horn K.; Batlle E.; Coudreuse D.; Haramis A. P.; Tjon-Pon-Fong M.; Moerer P.; van den Born M.; Soete G.; Pals S.; Eilers M.; Medema R.; Clevers H. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 2002, 111, 241–250. [DOI] [PubMed] [Google Scholar]
- McDonald E.; Blagg J.; Pichowicz M.; Crumpler S. R.. Pyridine and pyrimidine based compounds as WNT signalling pathway inhibitors for the treatment of cancer. WO2010041054, 2010.
- Govek S. P.; Shiau A. K.; Noble S. A.; Thomas D. J.. Preparation of bicyclic heteroaryl inhibitors of PDE4. WO2008006051, 2008.
- Teno N.; Masuya K.; Ehara T.; Kosaka T.; Miyake T.; Irie T.; Hitomi Y.; Matsuura N.; Umemura I.; Iwasaki G.; Fukaya H.; Toriyama K.; Uchiyama N.; Nonomura K.; Sugiyama I.; Kometani M. Effect of cathepsin K inhibitors on bone resorption. J. Med. Chem. 2008, 51, 5459–5462. [DOI] [PubMed] [Google Scholar]
- Hobbs S.; Jitrapakdee S.; Wallace J. C. Development of a bicistronic vector driven by the human polypeptide chain elongation factor 1alpha promoter for creation of stable mammalian cell lines that express very high levels of recombinant proteins. Biochem. Biophys. Res. Commun. 1998, 252, 368–372. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.