

Abstract
Aim
We aimed to assess the analgesic efficacy, pharmacokinetics, tolerability and safety of a single dose of Δ9‐THC in patients with chronic abdominal pain resulting from chronic pancreatitis (CP).
Methods
This was a randomized, single dose, double‐blinded, placebo‐controlled, two way crossover study in patients suffering from abdominal pain as result of CP (n = 24), post hoc subdivided into opioid and non‐opioid users. Δ9‐THC (8 mg) or active placebo (5 mg/10 mg diazepam) was administered orally in a double dummy design.
Results
No treatment effect was shown for delta VAS pain scores after Δ9‐THC compared with diazepam. Δ9‐THC was well absorbed with a mean t max of 123 min. No significant differences were found between Δ9‐THC vs. diazepam for alertness, mood, calmness or balance. Feeling anxious and heart rate were significantly increased after Δ9‐THC compared with diazepam. The most frequently reported adverse events (AEs) after Δ9‐THC administration were somnolence, dry mouth, dizziness and euphoric mood.
Conclusions
A single dose of Δ9‐THC was not efficacious in reducing chronic pain resulting from CP, but was well tolerated with only mild or moderate AEs. The PK results in CP patients showed delayed absorption and an increased variability compared with healthy volunteers.
Keywords: chronic pain, chronic pancreatitis, delta‐9‐tetrahydrocannabinol, dronabinol, THC
What is Already Known About This Subject
Delta‐9‐tetrahydrocannabinol (Δ9‐THC) is the psychoactive substance of the Cannabis sativa plant and has been shown to be a promising analgesic in previous studies.
Namisol® is an oral tablet containing purified Δ9‐THC in a novel formulation.
Bioavailability and clinical effects of oral Δ9‐THC need to be studied in a chronic pain population.
What this Study Adds
Orally administered Δ9‐THC results in a reliable pharmacokinetic profile in patients with chronic pancreatitis.
A single 8 mg dose of Δ9‐THC is not efficacious in achieving pain relief compared with diazepam in patients with chronic pancreatic pain, but is generally well tolerated with mostly mild AEs.
Introduction
Chronic pancreatitis (CP) is a disease characterized by inflammation and progressive destruction of the pancreatic gland, which results in irreversible morphologic changes that typically cause endocrine and/or exocrine dysfunction 1. The most important symptom of CP is abdominal pain, present in 80–90% of patients during the disease course 2. Pancreatic pain is described by most patients as severe abdominal pain, frequently radiating to the back. The pain is typically recurrent, intense and long‐lasting, and is extremely difficult to treat 3. Initial treatment of CP consists of low fat diet and non‐narcotic analgesics, which can be supplemented by oral pancreatic enzymes and proton pump inhibitors. If an acceptable level of pain relief is not obtained with these drugs, opioids are the next stage in the management of pain. Opioids have a number of well‐known adverse effects including elevation of smooth muscle tone (affecting gastrointestinal motility), toxicity in the central nervous system, opioid‐induced hyperalgesia and tolerance, and risk of addiction 4, 5. Alternatives to medicinal treatment exist in the form of nerve blockade, lithotripsy and surgical treatment. However, results from studies of non‐medicinal treatment modalities are equivocal and these treatments are only applicable in a minority of patients. Therefore, medicinal analgesic therapy must still be considered as the first choice in the management of painful CP 6.
Underlying pain mechanisms of CP are poorly understood and multifactorial and, therefore, treatment is often empirical and insufficient. Several intra‐ and extrapancreatic causes of pain have been suggested. However, most research is focused alterations in pain processing, with peripheral causes including an increase in nerve fibres and neurogenic inflammation 7, and central causes including central sensitization and somatotopic reorganization 8, 9. Furthermore, Olesen et al. demonstrated activation of descending inhibition in early CP patients, and loss of diffuse noxious inhibitory control (DNIC) in more advanced CP patients 10. It should be noted in this context that when opioid treatment becomes less effective the more central sensitization an individual has 11. Thus there is a clear need for alternatives (or adjuvants) to opioid treatment in CP patients with pain, targeting changes in (central) pain processing.
Delta‐9‐tetrahydrocannabinol (Δ9‐THC) is the most potent psychoactive cannabinoid from the plant Cannabis sativa, and has been used to treat pain for many centuries. However, the therapeutic potential of cannabinoids in current pain management remains unclear. To date, a wide range of products containing Δ9‐THC are available for medicinal purposes, including (i) crude medicinal cannabis containing several active compounds, (ii) pharmaceutical products with standardized natural or synthetic Δ9‐THC content containing whole cannabis plant extract, a defined combination of Δ9‐THC and cannabidiol (CBD) or pure Δ9‐THC and (iii) synthetic analogues interacting with cannabinoid receptors 12. The pharmacokinetics (PK) of the different administration routes of herbal cannabis and cannabis‐based medicines are variable and dosing is difficult to regulate. The development of pharmaceutical products for oral administration with pure and defined Δ9‐THC content may offer a favourable alternative. Namisol® (Echo Pharmaceuticals, The Netherlands) is a novel formulation for oral administration, containing purified Δ9‐THC isolated from the Cannabis sativa plant, with a reliable PK profile as demonstrated in a phase I healthy volunteer study 13.
Δ9‐THC induces pharmacological effects by binding non‐selectively to cannabinoid receptors. Two cannabinoid receptors have been identified, the CB1 and CB2 receptor 14, 15, 16. CB1 receptors are most densely present in the brain, particularly in the hippocampus, cerebellum and striatum, and occur in several areas providing targets through which cannabinoids could modulate pain. These areas include the periaqueductal grey (PAG), the rostral ventrolateral medulla, the superficial layers of the spinal dorsal horn and the dorsal root ganglion from which they are transported to both central and peripheral terminals of primary afferent neurons 17, 18, 19. CB2 receptors are expressed in high quantities in human immune tissues and cells, e.g. in the spleen, tonsils and leucocytes.
Apart from potential direct analgesic effects, it is suggested that cannabis might further be useful to treat pain through possible synergistic interactions with opioid analgesics or by improving the efficacy of pain treatment in patients with a tolerance to opioids 20.
In this phase 2 study, we aimed to study the analgesic efficacy, PK, pharmacodynamics (PD) and safety of a single oral dose of Δ9‐THC in patients with chronic abdominal pain resulting from CP, subdivided into opioid and non‐opioid users.
Methods
This was an equally randomized (1 : 1 ratio), single dose, double‐blind, placebo‐controlled, crossover study to evaluate the analgesic efficacy, PK, PD, pharmacogenetics and safety of a single dose of Δ9‐THC. The study population consisted of 24 subjects with CP, subdivided into daily opioid (n = 12) and non‐opioid users (n = 12). The Medical Ethical Committee and Competent Authority approved the study (2011/114). The study was conducted according to the principles of the Declaration of Helsinki, and in accordance with the International Conference on Harmonization guidelines of Good Clinical Practice. All subjects provided oral and written consent before conduct of any protocol‐related procedures. The Clinicaltrials.gov identification number was NCT01318369.
Study population
Eligible patients were adults (age > 18 years) diagnosed with CP according to the Marseille and Cambridge Classification System. All patients had chronic abdominal pain, persistent or intermittent on a daily basis during the past 3 months, and considered their pain as severe enough for medical treatment (numeric rating scale (NRS) ≥ 3). Patients in the opioid subgroup took stable doses of prescribed opioids, whereas patients in the non‐opioid subgroup had not taken opioids or only occasionally for pain flares in the past 2 months. The study took place at the Radboud University Medical Centre, The Netherlands, from October 2011 to May 2013. Patients were recruited by their physician or by advertisement.
Key exclusion criteria were cannabis use in previous year, history of hypersensitivity to THC, BMI <18.0 or >31.2 kg m−2, serious painful conditions other than CP, significant medical disorder or concomitant medication that may interfere with the study or may pose a risk for the patient, major psychiatric illness in history, epileptic seizure in history, diabetic neuropathy, significant exacerbation in illness within 2 weeks, more than 1 daily defined dose (DDD) benzodiazepines 6 h prior to or following intake of study medication in the opioid subgroup or more than 1 DDD benzodiazepines according to prescription in the non‐opioid subgroup (1 DDD was defined as 20 mg oxazepam), positive urine drug screen or alcohol test at screening or on study days, clinically relevant abnormalities in ECG or laboratory results, pregnant or breastfeeding females, intending to conceive a child or participation in another investigational drug study within 90 days before study entry.
Randomization
Eligible patients were stratified into opioid and non‐opioid users, then randomly assigned to one of two treatment sequences in a 1 : 1 ratio using a computer‐generated list of random numbers. Patients, staff and investigators were all blinded by a double dummy design. Each study day, patients were given either a single dose of Δ9‐THC (Namisol® 8 mg simultaneously with placebo diazepam) or a single dose diazepam (placebo Namisol® simultaneously with diazepam (5 mg non‐opioid group/10 mg opioid group)). Each patient subsequently received the alternative after at least a 14 day washout period. Namisol® or matching placebos were taken in three tablets (1 × 5mg + 2 × 1.5 mg). The previous phase I study demonstrated that the maximal tolerable dosage with acceptable adverse events was 8 mg Namisol®. With respect to the expected THC‐mediated sedative effects of cannabis, as demonstrated by frequently reported AEs such as somnolence and fatigue 21, low dose diazepam was used as ‘active placebo’ to prevent unblinding of patient and investigator. A study in healthy male subjects found no central effects after a single oral dose of 2 mg diazepam, but intermediate effects after 5 mg and highly significant effects after 10 mg diazepam 22. Opioid users are generally more used to sedative (side) effects due to their regular medication use. Therefore, a dosage of 10 mg diazepam was chosen in order to induce similar sedative effects in this subgroup. Diazepam was packaged in capsules, which were identically prepared for the placebo diazepam. Oral administration was performed using 200 ml of water.
Study procedures
The study consisted of 1 screening and 2 treatment days, with a telephone follow‐up after each study day. Screening included demographics, medical history, NRS pain score, physical examination, 12‐lead electrocardiogram (ECG), standard laboratory tests and urine drug screening in order to assess the overall eligibility of the patient. Screening was carried out a maximum of 40 days before the first day of drug administration. All patients received a pain diary to fill in 5 days in a row, starting on the first day after screening in order to obtain a more convenient description of the pain status of the study population.
Use of illicit drugs and use of opioids were both tested using urine drug screening tests prior to drug administration. In addition, patients were not allowed to consume alcohol within 24 h or caffeine within 6 h prior drug administration. Urine pregnancy tests and saliva alcohol tests were performed at the beginning of both study days.
Study days were carried out at the research department of the hospital, where each patient stayed in a separate quiet room. Patients consumed as much as they preferred from a standardized menu on the first study day, but had to consume exactly the same on the second study day. The same applied to co‐medication. Patients used their regular medication, including painkillers, according to prescription on both study days. Every food and medication intake was recorded.
Analgesic efficacy
A visual analogue scale (VAS) was used to quantify pain intensity. VAS scores at rest and on movement after five sit‐ups were marked on a 10 cm line. The boundaries of these lines were ‘no pain’ on the most left hand side and ‘unbearable pain’ on the most right hand side. The VAS was measured pre‐dose and post‐dose at 35 min, 1 h 5 min, 1 h 40 min, 2 h 5 min, 3 h 5 min, 4 h 10 min and 5 h after administration of study medication.
Pharmacokinetics
Plasma concentrations of THC and its active metabolite 11‐OH‐THC were determined in serial venous blood samples, which were collected in 4 ml EDTA tubes pre‐dose at −15 min and 10 min, 30 min, 45 min, 1 h, 1.5 h, 2 h, 3 h, 4 h and 6 h post‐dose. Immediately after collection, samples were wrapped in aluminum foil and kept on ice. Samples were centrifuged within 30 min at 2000 g for 10 min at 4°C. The handling of THC samples was done avoiding direct light. The separated plasma was divided into primary and backup samples, and stored at −80°C until bioanalysis. Bioanalysis (Analytisch Biochemisch Laboratorium b.v., Assen, The Netherlands) was performed using a validated liquid chromatography/mass spectrometry/mass spectrometry (LC/MS/MS) assay method according to good laboratory practice procedures. The lower limit of quantification for THC and 11‐OH‐THC was 0.100 ng ml−1.
Non‐compartmental analysis to determine plasma PK parameters of the active compounds, THC and 11‐OH‐THC, was performed using the WinNonlin modeling and analysis software (version 2.1 a; Pharsight Inc., Apex, NC, USA). The maximum plasma concentration (C max), the time to reach C max (t max), and the AUC from 0 up to the last measurement (AUC(0,6 h), using the linear log trapezoidal rule) were calculated from the individual plasma concentration vs. time profiles. The terminal half‐life (t 1/2) was calculated only if there were two or more points (excluding C max) in the elimination phase of the plasma concentration–time curve with r 2 > 0.80. For that reason, five patients were excluded from this part of the analysis for both THC and 11‐OH‐THC. Subsequently, the areas under the plasma concentration curves extrapolated to infinity (AUC(0,∞)) were calculated using the linear log trapezoidal rule and extrapolation to zero.
Pharmacogenetics
Genotyping of cytochrome P450 enzymes CYP2C9 and CYP2C19 was performed in order to investigate the effect of genetic polymorphisms on the pharmacokinetics of Δ9‐THC 23, 24. Two variants in genetic CYP2C9 polymorphisms (CYP2C9*2 and CYP2C9*3) and three variants in genetic CYP2C19 polymorphisms (CYP2C19*2, CYP2C19*3 and CYP2C19*17) were genotyped. To this end, saliva from 21 participating subjects was collected from which DNA extraction and genotyping was done.
Pharmacodynamics
Pre‐dose and post‐dose at 1 h 5 min, 2 h 5 min, 3 h 5 min, and 5 h, drug effects on mood and behavior were explored with a set of 16 individual Bond & Lader visual analogue scales. Three main factors were calculated as described by Bond and Lader: alertness (from nine scores), mood (from five scores) and calmness (from two scores) 25. Potential subjective psychotomimetic (psychedelic) effects were evaluated using the Bowdle questionnaire. The Bowdle questionnaire consists of 13 visual analogue lines ranging from ‘not at all’ to ‘extremely’ quantifying psychedelic effects 26. Subjects were asked to fill in both questionnaires pre‐dose and post‐dose at 1 h 5 min, 2 h 5 min, 3 h 5 min, and 5 h after drug administration.
Left‐right (roll) and anterior‐posterior (pitch) postural oscillations were measured using a gyroscope‐based measurement system (SwayStar™, Balance International Innovations GmbH, Switzerland), which was attached to the waist of the patient. Patients stood, without shoes, as still as possible in a standardized base of support with their arms hanging at both sides of their body. Body sway was measured pre‐dose and at 1 h 25 min, 2 h 25 min, 3 h 25 min and 5.5 h post‐dose for 1 min with eyes open and 1 min with eyes closed. During the task with eyes open patients were asked to fixate at one point. The computerized measures used for analysis reflect the 90% range roll and pitch excursion in degrees from the centre of gravity.
Safety and tolerability
Safety and tolerability were evaluated using spontaneously reported adverse events (AEs) recorded at study days until follow‐up, measurements of vital functions, ECG and laboratory tests. Blood pressure and heart rate were measured at screening and on both treatment days (pre‐dose and repeatedly post‐dose). ECG was recorded at screening, pre‐dose and at the end of each treatment day. Haematology, blood chemistry and urinalysis were performed at screening and at the end of the study.
Statistical methods
This was an exploratory study for which no sample size calculation was performed. Patients withdrawn prior to the first study day were replaced in order to have a total number of 24 evaluable patients for the analysis. The placebo treatment was considered as equal between opioid and non‐opioid users, despite the distinction in dose treatment across both groups. For statistical analysis SPSS software for Windows v.20 was used. All statistical tests were performed two‐tailed, and the limit for statistical significance was set at P < 0.05.
Differences between Δ9‐THC vs. diazepam in VAS scores at rest at time point 2 h 5 min were the primary outcome of this study. This was based on the assumption that C max is reached within 2 h after medication intake. Differences between both treatments were statistically analyzed using a linear mixed model analysis with two fixed factors (period and treatment) and a random subject effect (random intercept). A period × treatment interaction was absent. The effect of treatment (Δ9‐THC vs. placebo) was exploratory post hoc evaluated for both subgroups (opioid vs. non‐opioid).
Statistics of repeated measures data were analyzed using the area under the curve (AUC) of difference with baseline as summary measure. The AUC was computed using the trapezoid rule, ΔX × (Y1 + Y2)/2, repeatedly for each adjacent pair of points defining the curve from zero until the last measurement. Differences between Δ9‐THC versus diazepam were statistically analyzed using a linear mixed model analysis. Opioid users and non‐opioid users were compared in a subgroup analysis. The pharmacokinetics of patients with genetic polymorphisms were compared observationally.
Results
Twenty‐five patients were enrolled according to the flowchart in Figure 1. One patient was not treated because of a positive drug screening on the first study day and was replaced. Two patients in the opioid subgroup were lost to crossover after the first study day, one female patient due to mild AEs and one male patient after withdrawal of consent. Consequently, 24 patients received a single dose of Δ9‐THC and 22 patients received a single dose of diazepam.
Figure 1.
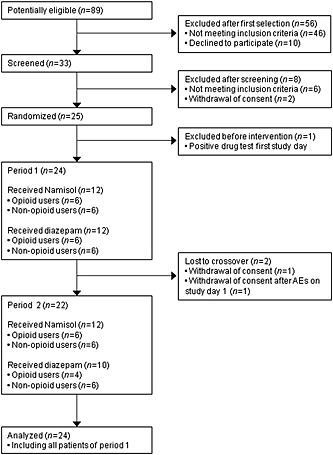
Participant flowchart
Patient demographics and baseline characteristics are described in Table 1. The mean age at screening was 52 years, mean BMI was 23.0 kg m−2 and nine of 24 patients were female. Patients reported a mean NRS at screening of 6.0, whereas the mean VAS reported in the pain diary was 3.9. The average abdominal pain duration was 8.3 years at screening.
Table 1.
Baseline demographics and disease characteristics
| Gender (M/F) | Age (years) | BMI (kg m −2 ) | Aetiology CP | Pain screen (NRS) | Pain diary (VAS) | Pain duration (years) | Concomitant medication | |
|---|---|---|---|---|---|---|---|---|
| Opioid subgroup | ||||||||
| 1 | M | 54 | 25.7 | Post‐ERCP | 6 | 4.2 | 5 | SOPI, PCM, AC |
| 2 | M | 48 | 26.9 | Idiopathic | 4 | 4.5 | 2 | SOPI, PCM, AC |
| 3 | M | 46 | 26 | Idiopathic | 7 | 2.2 | 21 | SOPI, AC, PE |
| 4 | F | 61 | 26.6 | Idiopathic | 5 | 5.2 | 15 | SOPI, PE |
| 5 | M | 44 | 18.8 | Neoplasm | 3 | 4.5 | 0 | SOPI, PE |
| 6 | M | 42 | 22.5 | Alcohol | 6 | 5.1 | 14 | WOPI, PCM, PE |
| 7 | M | 45 | 22 | Idiopathic | 6 | 7.2 | 4 | SOPI, WOPI, PCM |
| 8 | F | 42 | 21.5 | Hereditary | 6 | 4.9 | 13 | SOPI, PCM |
| 9 | M | 52 | 22.2 | Alcohol | 5 | 4.4 | 1 | SOPI, NSAID, PCM |
| 10 | M | 50 | 26.2 | Idiopathic | 8 | 2.5 | 2 | SOPI, PE |
| 11 | F | 34 | 19.5 | Idiopathic | 4 | 4.0 | 11 | SOPI, PCM |
| 12 | F | 52 | 19.2 | Idiopathic | 8 | 4.6 | 8 | SOPI, AC |
| mean (SD) | 8/4 | 47.5 (7.0) | 23.1 (3.1) | 5.7 (1.6) | 4.4 (1.3) | 8.0 (6.7) | ||
| Non‐opioid subgroup | ||||||||
| 13 | F | 52 | 26.2 | Idiopathic | 8 | 6.9 | 11 | PCM, AC |
| 14 | M | 69 | 26.2 | Hereditary | 6 | 5.1 | 4 | – |
| 15 | M | 56 | 20.6 | Neoplasm | 8 | 4.0 | 8 | AC, PE |
| 16 | M | 71 | 23.6 | Idiopathic | 5 | 2.1 | 6 | PCM, PE |
| 17 | M | 51 | 26.3 | Idiopathic | 7 | 4.7 | 3 | NSAID, PCM, PE |
| 18 | M | 53 | 24.2 | Idiopathic | 3 | 2.5 | 9 | PE |
| 19 | M | 39 | 18.4 | Idiopathic | 7 | 2.5 | 6 | NSAID, PCM, PE |
| 20 | F | 54 | 18.1 | Idiopathic | 6 | 3.0 | 22 | PCM, PE |
| 21 | F | 57 | 23.8 | Idiopathic | 6 | 1.0 | 6 | PE |
| 22 | M | 44 | 18.5 | Alcohol | 9 | 2.1 | 6 | PCM, PE |
| 23 | F | 62 | 23.3 | Alcohol | 5 | 3.2 | 15 | PE |
| 24 | F | 65 | 26.3 | Idiopathic | 5 | 2.7 | 7 | PCM |
| mean (SD) | 7/5 | 56.1 (9.5) | 23.0 (3.2) | 6.3 (1.7) | 3.3 (1.6) | 8.6 (5.3) | ||
| Total mean (SD) | 15/9 | 51.8 (9.3) | 23.0 (3.1) | 6.0 (1.6) | 3.9 (1.5) | 8.3 (5.9) | ||
SOPI, strong opioids including pethidine; WOPI, weak opioids including tramadol and codeine; NSAID, non‐steroidal anti‐inflammatory drugs including diclofenac and ibuprofen; PCM, paracetamol; AC, anticonvulsants including pregabalin and gabapentin; AD, antidepressants; PA, pancreatic enzymes
Analgesic efficacy
Primary linear mixed model analysis at time point 2 h 5 min showed no treatment effect of Δ9‐THC compared with diazepam on delta VAS pain at rest (mean difference Δ9‐THC ‐ diazepam −.17, 95% CI of the difference −0.95, 0.61, P = 0.65). Figure 2 shows the VAS pain at rest and on movement compared with baseline from 35 min until 5 h after administration of Δ9‐THC as well as diazepam. The AUC VAS pain at rest (mean difference 18.37, 95% CI of the difference −60.49, 97.23, P = 0.63) and AUC VAS pain on movement (mean difference −18.14, 95% CI of the difference −168.31, 132.03, P = 0.80) after Δ9‐THC were both not significantly decreased compared with diazepam. These parameters were similar for opioid vs. non‐opioid users.
Figure 2.
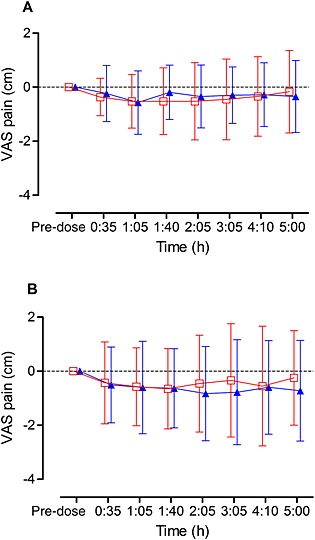
VAS pain. Differences (mean and SD) in VAS pain compared with baseline are shown for Δ9‐THC and diazepam
and diazepam  measured at rest (A) and on movement (B) in patients with pancreatic pain (n = 24). Pre‐dose, maximal 1 h prior drug administration
measured at rest (A) and on movement (B) in patients with pancreatic pain (n = 24). Pre‐dose, maximal 1 h prior drug administration
Pharmacokinetics
Mean plasma concentration vs. time curves of THC and 11‐OH‐THC are shown in Figure 3 and Table 2 summarizes the PK of THC and its active metabolite 11‐OH‐THC. The PK parameters were similar between opioid and non‐opioid users. One patient demonstrated a clearly enhanced C max compared with the rest of the population, which could not be explained by genetic polymorphism.
Figure 3.
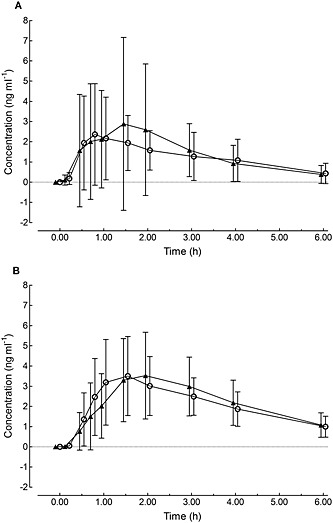
Mean plasma concentration−time curves of THC (A) and 11‐OH‐THC (B) after a single dose of Δ9‐THC in CP patients subdivided into opioid  (n = 12) and non‐opioid
(n = 12) and non‐opioid (n = 12) users. Error bars represent standard deviation (SD)
(n = 12) users. Error bars represent standard deviation (SD)
Table 2.
Pharmacokinetic parameters of THC and 11‐OH‐THC
| THC | 11‐OH‐THC | ||||
|---|---|---|---|---|---|
| Mean | SD | Mean | SD | ||
| C max | Group (n = 24) | 4,01 | 3,39 | 4,38 | 1,50 |
| (ng ml ‐1 ) | Opioid (n = 12) | 4,44 | 4,40 | 4,51 | 1,62 |
| Non‐opioid (n = 12) | 3,58 | 2,08 | 4,25 | 1,44 | |
| t max | Group (n = 24) | 122,80 | 87,99 | 135,70 | 77,50 |
| (min) | Opioid (n = 12) | 126,60 | 90,49 | 142,10 | 86,66 |
| Non‐opioid (n = 12) | 119,10 | 89,26 | 129,30 | 70,44 | |
| AUC(0,t last ) | Group (n = 24) | 477,50 | 381,80 | 764,90 | 241,30 |
| (ng ml ‐1 min) | Opioid (n = 12) | 507,90 | 506,70 | 777,70 | 298,10 |
| Non‐opioid (n = 12) | 447,20 | 214,70 | 752,20 | 180,50 | |
| AUC(0,∞) | Group (n = 24) | 532,20 | 442,50 | 920,70 | 316,40 |
| (observed) | Opioid (n = 11) | 577,70 | 571,10 | 954,00 | 400,00 |
| (ng ml ‐1 min) | Non‐opioid (n = 8) | 469,70 | 173,20 | 883,70 | 205,90 |
| t 1/2, term | Group (n = 24) | 67,12 | 20,37 | 110,10 | 26,57 |
| (min) | Opioid (n = 11) | 67,89 | 19,71 | 111,70 | 29,51 |
| Non‐opioid (n = 8) | 66,05 | 22,57 | 108,40 | 24,55 | |
Pharmacogenetics
Several genetic polymorphisms were observed. Two patients were heterozygote carriers of CYP2C9*2 (C > T) and four patients were heterozygote carriers of CYP2C9*3 (A > C). One patient was found to be AA homozygote and four patients GA heterozygote for CYP2C19*2 (G > A). No CYP2C19*3 (G > A) polymorphisms were observed. Genetic polymorphisms in CYP2C19*17 (C > T) were found for five subjects who were heterozygote CT carriers. Genetic CYP2C9 and CYP2C19 polymorphisms did not evidently affect the pharmacokinetics of Δ9‐THC.
Pharmacodynamics
Figure 4 shows the effects of Δ9‐THC and diazepam for alertness, mood and calmness obtained by the VAS Bond & Lader questionnaire. No significant differences were found between Δ9‐THC vs. diazepam. Feeling anxious obtained by the VAS Bowdle questionnaire was significantly increased after Δ9‐THC compared with diazepam (mean difference 166,92, 95% CI of the difference 10,86, 322,97, P = 0.037).
Figure 4.
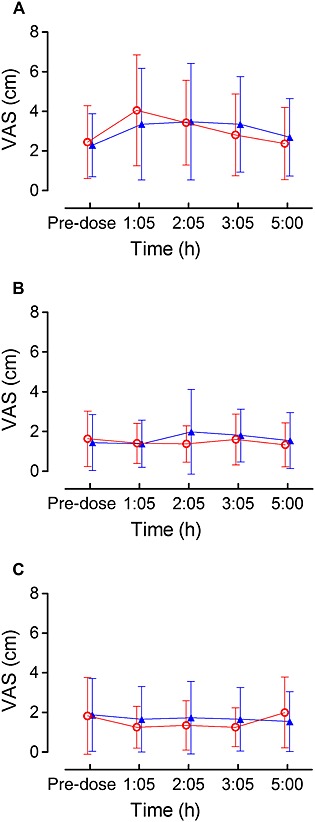
VAS Bond & Lader questionnaire. Mean scores for alertness (A), mood (B), and calmness (C) were shown for Δ9‐THC and diazepam
and diazepam  in CP patients (n = 24). Error bars represent standard deviation (SD)
in CP patients (n = 24). Error bars represent standard deviation (SD)
Overall 10 body sway measurements (4% of all measurements), from which six in the eyes closed condition and eight after Δ9‐THC administration, could not be conducted due to adverse events at that particular moment. There were no group differences in balance outcomes in both the eyes open and eyes closed condition between Δ9‐THC and diazepam. However, balance performance was considerably disturbed in certain individuals after both Δ9‐THC and diazepam. These individuals were found in both subgroups. Heart rate was significantly enhanced after Δ9‐THC compared with diazepam (at time point 1 h 40 min mean difference −5.5 beats min−1, 95% CI of the difference −9.0, −1.9, P = .004). In one patient, heart rate at rest was measured above 100 beats min−1 after Δ9‐THC intake. Δ9‐THC and diazepam did not affect diastolic or systolic blood pressure. Alterations in heart rate were not associated with PK parameters such as C max and AUC(0,∞). All pharmacodynamic parameters were similar for opioid vs. non‐opioid users and did not affect the treatment effect.
Safety and tolerability
All related, probably related and possibly related AEs are presented in Table 3. Overall, there was a higher frequency of AEs following Δ9‐THC administration compared with diazepam (54 AEs in 24 patients vs. 36 AEs in 22 patients, respectively), although fewer patients reported at least one AE after Δ9‐THC administration compared with diazepam (71% vs. 91% respectively). The most frequently reported AEs after Δ9‐THC administration were somnolence, dry mouth, dizziness and euphoric mood. Somnolence, dizziness and fatigue were most commonly related or possibly related to diazepam administration. All AEs were mild or moderate, and equally divided between opioid and non‐opioid users. The number of AEs was not associated with PK parameters such as C max and AUC(0,∞). However, the subject showing the highest C max also had the greatest number of AEs. There were no serious AEs during the study. One patient was withdrawn after the administration of Δ9‐THC on the first study day due to somnolence, dizziness, increased heart rate, nausea, paraesthesia and feelings of tension. There were no clinically relevant changes in vital signs, ECG parameters or safety laboratory parameters (haematology, biochemistry and urinalysis).
Table 3.
Summary of adverse events
| Adverse event | Diazepam (n = 22) | Δ9‐THC (n = 24) | ||
|---|---|---|---|---|
| n | % | n | % | |
| General | ||||
| Fatigue | 8 | 36% | 7 | 29% |
| Nervous system symptoms | ||||
| Somnolence | 11 | 50% | 8 | 33% |
| Dizziness | 6 | 27% | 4 | 17% |
| Headache | 3 | 14% | 2 | 8% |
| Balance disorder | 0 | 0% | 2 | 8% |
| Amnesia | 0 | 0% | 1 | 4% |
| Paraesthesia | 1 | 5% | 2 | 8% |
| Depressed level of conciousness | 1 | 5% | 0 | 0% |
| Psychiatric symptoms | ||||
| Confusional state | 0 | 0% | 2 | 8% |
| Indifference | 0 | 0% | 1 | 4% |
| Euphoric mood | 2 | 9% | 4 | 17% |
| Derealization | 0 | 0% | 1 | 4% |
| Disorientation | 0 | 0% | 1 | 4% |
| Tension | 0 | 0% | 1 | 4% |
| Gastro‐intestinal system symptoms | ||||
| Nausea | 1 | 5% | 3 | 13% |
| Vomiting | 0 | 0% | 1 | 4% |
| Steatorrhoea | 0 | 0% | 1 | 4% |
| Constipation | 1 | 5% | 0 | 0% |
| Abdominal discomfort | 0 | 0% | 1 | 4% |
| Dry mouth | 0 | 0% | 5 | 21% |
| Throat irritation | 0 | 0% | 1 | 4% |
| Vision symptoms | ||||
| Visual impairment | 1 | 5% | 3 | 13% |
| Cardiac symptoms | ||||
| Heart rate increased | 1 | 5% | 1 | 4% |
| Eye symptoms | ||||
| Dry eye | 0 | 0% | 1 | 4% |
| Photophobia | 0 | 0% | 1 | 4% |
| Total | 36 | 54 | ||
Discussion
Our study investigated the analgesic efficacy, pharmacokinetics, pharmacodynamics and safety of a single dose of Δ9‐THC in patients with chronic abdominal pain related to CP. We demonstrated in an exploratory study, that a single dose of 8 mg Δ9‐THC was not efficacious in reducing chronic pancreatic pain compared with the active placebo diazepam. Δ9‐THC was absorbed with an average t max of 123 min, which was similar for opioid and non‐opioid users, but slower than observed in a previous study in healthy subjects 13. We observed a small, but significant, increase in feeling anxious after Δ9‐THC compared with diazepam. Other pharmacodynamic outcomes did not differ between Δ9‐THC and diazepam. A single dose of Δ9‐THC was well tolerated resulting in mild to moderate AEs.
Analgesic efficacy
Several randomized controlled trials investigated the analgesic efficacy of different products containing THC in various pain states 12, 27, 28, 29, 30. In a majority of these studies, THC treatment resulted in pain reduction in chronic pain, whereas the data for acute pain were less conclusive. Most studies in chronic non‐malignant pain conditions demonstrated analgesic efficacy in chronic non‐malignant pain using a single dose or treatment periods of 2 to 15 weeks 20, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42. The majority of studies with cannabis‐based medicines were conducted in patients suffering from central neuropathic pain in multiple sclerosis. Ours is the first study in patients with chronic abdominal pain resulting from CP, which is generally recognized as difficult to treat and associated with high opioid use.
Narang et al. demonstrated that patients who received a single dose of THC experienced decreased pain intensity compared with placebo in patients taking opioids for chronic non‐malignant pain of various origins (e.g. low back, lower extremity, cervical and abdominal/pelvic pain), suggesting that THC may have an additive effect on pain relief 20. Preclinical evidence also suggests that THC may act synergistically with opioids 43, 44. However, in the present study we did not observe any analgesic effect of Δ9‐THC compared with diazepam nor a difference between opioid users and non‐opioid users. Although pain was decreased after Δ9‐THC administration, the same effect was observed after diazepam administration. As for diazepam no analgesic efficacy is described and it is used in other pain studies as an active placebo 45 It is assumed that the pain relief after diazepam is a placebo effect. It is well known that placebo and nocebo effects are present in chronic pain populations 46.
Several explanations for the lack of analgesic effect in our study can be proposed:
a single dose of Δ9‐THC is insufficient to achieve adequate exposure duration. THC is lipophilic and will diffuse to the fatty tissues immediately. The question is whether the THC concentration at target site is sufficient to modulate pain. Therefore, long term treatment studies are necessary to achieve sufficient exposure duration and evaluate the efficacy of Δ9‐THC.
the dosage of 8 mg Δ9‐THC is inadequate for each individual patient. The dosage should be adjusted for individual patients according to genetic, mechanistic and other patient‐related factors that potentially influence the PK and clinical effects 47, 48.
Δ9‐THC is effective only in certain types of pain, e.g. chronic vs. acute or visceral vs. neuropathic. It is difficult to specify responders, because the working mechanism of how THC potentially modulates pain is unclear. It should be noted that several previous clinical trials demonstrated analgesic efficacy in chronic pain, particularly in multiple sclerosis, whereas the data in acute pain were less conclusive 12.
sensitization of nociceptive pathways (e.g. central sensitization) and alterations in central cognitive and autonomic processing, which are all associated with chronic pancreatic pain, 49, 50, 51 impedes analgesic efficacy in this particular research population.
THC in general is ineffective for pain relief. However, the absence of significant pain relief in current study, after only one single dose, does not give evidence that supports this suggestion.
Pharmacokinetics
The mean plasma concentration curves demonstrated that THC was generally well absorbed and further metabolized to 11‐OH‐THC in this group of CP patients. However, it should be noted that, according to the mean plasma concentration curve of THC, the time to reach maximal THC concentration was 45–90 min, whereas the computed mean t max of THC was 119–127 min. This phenomenon can be explained by the observation that subjects with an early t max have a much higher C max compared with those subjects with a late t max which show a relatively low C max. The previously mentioned phase I study reported a time to reach maximal THC concentration of 39–56 min, but these subjects were young, healthy and fasted before Δ9‐THC administration 13. Thus, the absorption of Δ9‐THC was delayed in a subgroup of CP patients, resulting in an increased variability.
CP is associated with malabsorption 52, 53, which potentially affects drug absorption and could explain the inter‐individual PK variation in patients with CP 54. Drug absorption in CP patients might further be affected by alterations in gastrointestinal intraluminal pH, gastrointestinal motility, bacterial overgrowth and changed pancreatic gland secretion 54. In addition, bowel dysfunction is a common adverse effect of prolonged opioid use 55, which may affect the absorption of drugs as well. Therefore, the role of these factors in modulating the pharmacokinetic profile of THC should be further studied.
Pharmacogenetics
We aimed to evaluate the effects of CYP2C9 and CYP2C19 polymorphism on the pharmacokinetics of Δ9‐THC, which is subsequently relevant for its efficacy and adverse effects. Sachse‐Seeboth et al. found that the homozygous CYP2C9*3 variant affected the pharmacokinetics of THC, resulting in a three fold area under the plasma concentration curve of THC, as well as a trend towards increased sedation after oral administration of THC 23. In the current study, we did not observe significant differences between wild‐type subjects and subjects with homozygous or heterozygous CYP polymorphisms. This can be explained by the small number of subjects with a genetic variant. However, it cannot be precluded that genetic polymorphisms may have contributed to the inter‐individual variation in the pharmacokinetics of Δ9‐THC.
Pharmacodynamics
Several psychological outcomes such as alertness, feelings of unreality, control of thoughts, feeling high and feeling drowsy seem to be affected after administration of both Δ9‐THC and diazepam. Feeling anxious was the only outcome with a significant difference between Δ9‐THC and diazepam, which is not surprising considering the anxiolytic properties of diazepam. Similar results were observed for the body sway measurements. Balance disturbances were found in several individuals after both Δ9‐THC as well as diazepam. After 1 h 40 min post‐dose, heart rate was significantly enhanced by 5.5 beats min−1 after Δ9‐THC compared with diazepam. This is in line with previous studies and for most patients not clinically relevant 13.
Adverse effects
Δ9‐THC was generally well tolerated resulting in only mild to moderate adverse events, which were very similar compared with those observed in healthy volunteers 13. However, we observed an inter‐individual variation with certain subjects experiencing no single side effect while others experienced several side effects at the same time. This could not be explained by subgroups of (non)opioid users or pharmacogenetic polymorphisms, and could not be associated with pharmacokinetic parameters such as C max or AUC(0,t last) However, side effects of THC are considered to be dose‐related, 13 and therefore, adverse events should be avoidable by adjusting the dosage or by adequate dosage titration.
Methodological considerations
The similarities in the pharmacodynamics of Δ9‐THC compared with diazepam clearly demonstrate that we succeeded in adequate blinding of subjects by giving the impression of an active psychotropic drug in both periods. Additionally, with respect to the sedative effects of THC, diazepam was used to control for indirect pain relief through the sedative effects on the experienced pain. Diazepam is more often chosen as active placebo for THC and other central working analgesics 45, 56. However, it should be mentioned that the role of GABA in mediating the transmission and perception of pain is not evidently clear. GABAergic neurons are widely distributed throughout the central nervous system, including regions of the spinal cord dorsal horn known to be important for transmitting pain impulses to the brain 57. GABA receptor agonists demonstrated antinociceptive properties in a variety of pain models in animal studies 58, and showed possible anti‐hyperalgesic effects in experimental human pain models 59. However, benzodiazepines largely lack clear analgesic efficacy in humans 57, 60, and diazepam is thus unlikely to affect the primary outcome. The comparison with diazepam, however, may have complicated the evaluation of the PD effects of Δ9‐THC. Several psychedelic outcomes such as alertness, feelings of unreality, control of thoughts, feeling high, feeling drowsy and feeling anxious were affected after administration of both drugs.
In conclusion, this study demonstrates that a single dose of 8 mg Δ9‐THC was not efficacious in achieving pain relief. At this dose, Δ9‐THC was generally well tolerated with mostly mild AEs. The PK results in CP patients showed delayed absorption and an increased variability compared with healthy volunteers, most probably due to underlying pathology and concomitant medication use. Further long term treatment studies are necessary to evaluate the efficacy and tolerability of Δ9‐THC in CP and other chronic visceral pain conditions.
Competing Interests
All authors have completed the Unified Competing Interest form at www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare HvG received a grant from the European Union, the European Fund for Regional Development (EFRO, ‘Here is an investment in your future’) for the submitted work, no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years and no other relationships or activities that could appear to have influenced the submitted work.
The authors thank all participating patients and Simone Hins‐deBree (research nurse) for all kinds of study‐related work.
de Vries, M. , Van Rijckevorsel, D. C. M. , Vissers, K. C. P. , Wilder‐Smith, O. H. G. , and Van Goor, H. (2016) Single dose delta‐9‐tetrahydrocannabinol in chronic pancreatitis patients: analgesic efficacy, pharmacokinetics and tolerability. Br J Clin Pharmacol, 81: 525–537. doi: 10.1111/bcp.12811.
References
- 1. Sarles H. Definitions and classifications of pancreatitis. Pancreas 1991; 6: 470–4. [DOI] [PubMed] [Google Scholar]
- 2. Andren‐Sandberg A, Hoem D, Gislason H. Pain management in chronic pancreatitis. Eur J Gastroenterol Hepatol 2002; 14: 957–70. [DOI] [PubMed] [Google Scholar]
- 3. Warshaw ALBP, Fernandez‐Del Castillo C. American gastroenterological association medical position statement:treatment of pain in chronic pancreatitis. Gastroenterology 1998; 115: 763–4. [DOI] [PubMed] [Google Scholar]
- 4. Fishbain DA, Cole B, Lewis JE, Gao J, Rosomoff RS. Do opioids induce hyperalgesia in humans? An evidence‐based structured review. Pain Med 2009; 10: 829–39. [DOI] [PubMed] [Google Scholar]
- 5. Tompkins DA, Campbell CM. Opioid‐Induced Hyperalgesia: Clinically Relevant or Extraneous Research Phenomenon? Curr Pain Headache Rep 2011; 15: 129–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chauhan S, Forsmark CE. Pain management in chronic pancreatitis: a treatment algorithm. Best Pract Res Clin Gastroenterol 2010; 24: 323–35. [DOI] [PubMed] [Google Scholar]
- 7. Di Sebastiano P, di Mola FF, Bockman DE, Friess H, Buchler MW. Chronic pancreatitis: the perspective of pain generation by neuroimmune interaction. Gut 2003; 52: 907–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Buscher HC, Wilder‐Smith OH, van Goor H. Chronic pancreatitis patients show hyperalgesia of central origin: a pilot study. Eur J Pain 2006; 10: 363–70. [DOI] [PubMed] [Google Scholar]
- 9. Drewes AM, Gratkowski M, Sami SA, Dimcevski G, Funch‐Jensen P, Arendt‐Nielsen L. Is the pain in chronic pancreatitis of neuropathic origin? Support from EEG studies during experimental pain. World J Gastroenterol 2008; 14: 4020–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Olesen SS, Brock C, Krarup AL, Funch‐Jensen P, Arendt‐Nielsen L, Wilder‐Smith OH, Drewes AM. Descending inhibitory pain modulation is impaired in patients with chronic pancreatitis. Clin Gastroenterol Hepatol 2010; 8: 724–30. [DOI] [PubMed] [Google Scholar]
- 11. Edwards RR, Ness TJ, Weigent DA, Fillingim RB. Individual differences in diffuse noxious inhibitory controls (DNIC): association with clinical variables. Pain 2003; 106: 427–37. [DOI] [PubMed] [Google Scholar]
- 12. de Vries M, van Rijckevorsel DC, Wilder‐Smith OH, van Goor H. Dronabinol and chronic pain: importance of mechanistic considerations. Expert Opin Pharmacother 2014; 15: 1–10. [DOI] [PubMed] [Google Scholar]
- 13. Klumpers LE, Beumer TL, van Hasselt JG, Lipplaa A, Karger LB, Kleinloog HD, Freijer JI, de Kam ML, van Gerven JM. Novel delta(9) ‐tetrahydrocannabinol formulation Namisol(R) has beneficial pharmacokinetics and promising pharmacodynamic effects. Br J Clin Pharmacol 2012; 74: 42–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Munro S, Thomas KL, Abu‐Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993; 365: 61–5. [DOI] [PubMed] [Google Scholar]
- 15. Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990; 346: 561–4. [DOI] [PubMed] [Google Scholar]
- 16. Alexander SP, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 5th edition. Br J Pharmacol 2011; 164 (Suppl 1): S1–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hohmann AG, Herkenham M. Regulation of cannabinoid and mu opioid receptors in rat lumbar spinal cord following neonatal capsaicin treatment. Neurosci Lett 1998; 252: 13–6. [DOI] [PubMed] [Google Scholar]
- 18. Hohmann AG, Herkenham M. Localization of central cannabinoid CB1 receptor messenger RNA in neuronal subpopulations of rat dorsal root ganglia: a double‐label in situ hybridization study. Neuroscience 1999; 90: 923–31. [DOI] [PubMed] [Google Scholar]
- 19. Hohmann AG, Briley EM, Herkenham M. Pre‐ and postsynaptic distribution of cannabinoid and mu opioid receptors in rat spinal cord. Brain Res 1999; 822: 17–25. [DOI] [PubMed] [Google Scholar]
- 20. Narang S, Gibson D, Wasan AD, Ross EL, Michna E, Nedeljkovic SS, Jamison RN. Efficacy of dronabinol as an adjuvant treatment for chronic pain patients on opioid therapy. J Pain 2008; 9: 254–64. [DOI] [PubMed] [Google Scholar]
- 21. Whiting PF, Wolff RF, Deshpande S, Di Nisio M, Duffy S, Hernandez AV, Keurentjes JC, Lang S, Misso K, Ryder S, Schmidlkofer S, Westwood M, Kleijnen J. Cannabinoids for medical use: a systematic review and meta‐analysis. JAMA 2015; 313: 2456–73. [DOI] [PubMed] [Google Scholar]
- 22. Friedman H, Greenblatt DJ, Peters GR, Metzler CM, Charlton MD, Harmatz JS, Antal EJ, Sanborn EC, Francom SF. Pharmacokinetics and pharmacodynamics of oral diazepam: effect of dose, plasma concentration, and time. Clin Pharmacol Ther 1992; 52: 139–50. [DOI] [PubMed] [Google Scholar]
- 23. Sachse‐Seeboth C, Pfeil J, Sehrt D, Meineke I, Tzvetkov M, Bruns E, Poser W, Vormfelde SV, Brockmoller J. Interindividual variation in the pharmacokinetics of Delta9‐tetrahydrocannabinol as related to genetic polymorphisms in CYP2C9. Clin Pharmacol Ther 2009; 85: 273–6. [DOI] [PubMed] [Google Scholar]
- 24. Kirchheiner J, Brockmoller J. Clinical consequences of cytochrome P450 2C9 polymorphisms. Clin Pharmacol Ther 2005; 77: 1–16. [DOI] [PubMed] [Google Scholar]
- 25. Bond A, Lader M. The use of analogue scales in rating subjective feelings. Br J Med Psychol 1974; 47: 211–8. [Google Scholar]
- 26. Bowdle TA, Radant AD, Cowley DS, Kharasch ED, Strassman RJ, Roy‐Byrne PP. Psychedelic effects of ketamine in healthy volunteers: relationship to steady‐state plasma concentrations. Anesthesiology 1998; 88: 82–8. [DOI] [PubMed] [Google Scholar]
- 27. Lynch ME, Campbell F. Cannabinoids for treatment of chronic non‐cancer pain: a systematic review of randomized trials. Br J Clin Pharmacol 2011; 72: 735–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Russo EB. Cannabinoids in the management of difficult to treat pain. Ther Clin Risk Manag 2008; 4: 245–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Beaulieu P, Ware M. Reassessment of the role of cannabinoids in the management of pain. Curr Opin Anaesthesiol 2007; 20: 473–7. [DOI] [PubMed] [Google Scholar]
- 30. Ben AM. Cannabinoids in medicine: a review of their therapeutic potential. J Ethnopharmacol 2006; 105: 1–25. [DOI] [PubMed] [Google Scholar]
- 31. Notcutt W, Price M, Miller R, Newport S, Phillips C, Simmons S, Sansom C. Initial experiences with medicinal extracts of cannabis for chronic pain: results from 34 'N of 1' studies. Anaesthesia 2004; 59: 440–52. [DOI] [PubMed] [Google Scholar]
- 32. Langford RM, Mares J, Novotna A, Vachova M, Novakova I, Notcutt W, Ratcliffe S. A double‐blind, randomized, placebo‐controlled, parallel‐group study of THC/CBD oromucosal spray in combination with the existing treatment regimen, in the relief of central neuropathic pain in patients with multiple sclerosis. J Neurol 2013; 260: 984–97. [DOI] [PubMed] [Google Scholar]
- 33. Nurmikko TJ, Serpell MG, Hoggart B, Toomey PJ, Morlion BJ, Haines D. Sativex successfully treats neuropathic pain characterised by allodynia: a randomised, double‐blind, placebo‐controlled clinical trial. Pain 2007; 133: 210–20. [DOI] [PubMed] [Google Scholar]
- 34. Rintala DH, Fiess RN, Tan G, Holmes SA, Bruel BM. Effect of dronabinol on central neuropathic pain after spinal cord injury: a pilot study. Am J Phys Med Rehabil 2010; 89: 840–8. [DOI] [PubMed] [Google Scholar]
- 35. Selvarajah D, Gandhi R, Emery CJ, Tesfaye S. Randomized placebo‐controlled double‐blind clinical trial of cannabis‐based medicinal product (sativex) in painful diabetic neuropathy: depression is a major confounding factor. Diabetes Care 2010; 33: 128–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Svendsen KB, Jensen TS, Bach FW. Does the cannabinoid dronabinol reduce central pain in multiple sclerosis? Randomised double blind placebo controlled crossover trial. BMJ 2004; 329: 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Berman JS, Symonds C, Birch R. Efficacy of two cannabis based medicinal extracts for relief of central neuropathic pain from brachial plexus avulsion: results of a randomised controlled trial. Pain 2004; 112: 299–306. [DOI] [PubMed] [Google Scholar]
- 38. Blake DR, Robson P, Ho M, Jubb RW, McCabe CS. Preliminary assessment of the efficacy, tolerability and safety of a cannabis‐based medicine (sativex) in the treatment of pain caused by rheumatoid arthritis. Rheumatology 2006; 45: 50–2. [DOI] [PubMed] [Google Scholar]
- 39. Rog DJ, Nurmikko TJ, Young CA. Oromucosal delta9‐tetrahydrocannabinol/cannabidiol for neuropathic pain associated with multiple sclerosis: an uncontrolled, open‐label, 2‐year extension trial. Clin Ther 2007; 29: 2068–79. [DOI] [PubMed] [Google Scholar]
- 40. Wade DT, Makela P, Robson P, House H, Bateman C. Do cannabis‐based medicinal extracts have general or specific effects on symptoms in multiple sclerosis? A double‐blind, randomized, placebo‐controlled study on 160 patients. Mult Scler 2004; 10: 434–41. [DOI] [PubMed] [Google Scholar]
- 41. Wade DT, Robson P, House H, Makela P, Aram J. A preliminary controlled study to determine whether whole‐plant cannabis extracts can improve intractable neurogenic symptoms. Clin Rehabil 2003; 17: 21–9. [DOI] [PubMed] [Google Scholar]
- 42. Zajicek J, Fox P, Sanders H, Wright D, Vickery J, Nunn A, Thompson A, Group UMR. Cannabinoids for treatment of spasticity and other symptoms related to multiple sclerosis (CAMS study): multicentre randomised placebo‐controlled trial. Lancet 2003; 362: 1517–26. [DOI] [PubMed] [Google Scholar]
- 43. Cichewicz DL. Synergistic interactions between cannabinoid and opioid analgesics. Life Sci 2004; 74: 1317–24. [DOI] [PubMed] [Google Scholar]
- 44. Roberts JD, Gennings C, Shih M. Synergistic affective analgesic interaction between delta‐9‐tetrahydrocannabinol and morphine. Eur J Pharmacol 2006; 530: 54–8. [DOI] [PubMed] [Google Scholar]
- 45. Kraft B, Frickey NA, Kaufmann RM, Reif M, Frey R, Gustorff B, Kress HG. Lack of analgesia by oral standardized cannabis extract on acute inflammatory pain and hyperalgesia in volunteers. Anesthesiology 2008; 109: 101–10. [DOI] [PubMed] [Google Scholar]
- 46. Capurso G, Cocomello L, Benedetto U, Camma C, Delle FG. Meta‐analysis: the placebo rate of abdominal pain remission in clinical trials of chronic pancreatitis. Pancreas 2012; 41: 1125–31. [DOI] [PubMed] [Google Scholar]
- 47. Lesko LJ. Personalized medicine: elusive dream or imminent Reality? Clin Pharmacol Ther 2007; 81: 807–16. [DOI] [PubMed] [Google Scholar]
- 48. Bruehl S, Apkarian AV, Ballantyne JC, Berger A, Borsook D, Chen WG, Farrar JT, Haythornthwaite JA, Horn SD, Iadarola MJ, Inturrisi CE, Lao L, Mackey S, Mao J, Sawczuk A, Uhl GR, Witter J, Woolf CJ, Zubieta JK, Lin Y. Personalized medicine and opioid analgesic prescribing for chronic pain: opportunities and challenges. J Pain 2013; 14: 103–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Apkarian AV, Hashmi JA, Baliki MN. Pain and the brain: specificity and plasticity of the brain in clinical chronic pain. Pain 2011; 152: S49–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Baliki MN, Petre B, Torbey S, Herrmann KM, Huang L, Schnitzer TJ, Fields HL, Apkarian AV. Corticostriatal functional connectivity predicts transition to chronic back pain. Nat Neurosci 2012; 15: 1117–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Woolf CJ. Central sensitization: implications for the diagnosis and treatment of pain. Pain 2011; 152: S2–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Forsmark CE. Chronic pancreatitis and malabsorption. Am J Gastroenterol 2004; 99: 1355–7. [DOI] [PubMed] [Google Scholar]
- 53. Pezzilli R. Chronic pancreatitis: maldigestion, intestinal ecology and intestinal inflammation. World J Gastroenterol 2009; 15: 1673–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Olesen AE, Brokjaer A, Fisher IW, Larsen IM. Pharmacological challenges in chronic pancreatitis. World J Gastroenterol 2013; 19: 7302–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pappagallo M. Incidence, prevalence, and management of opioid bowel dysfunction. Am J Surg 2001; 182: 11S–8S. [DOI] [PubMed] [Google Scholar]
- 56. Kaufmann RM, Kraft B, Frey R, Winkler D, Weiszenbichler S, Backer C, Kasper S, Kress HG. Acute psychotropic effects of oral cannabis extract with a defined content of delta9‐tetrahydrocannabinol (THC) in healthy volunteers. Pharmacopsychiatry 2009; 43: 24–32. [DOI] [PubMed] [Google Scholar]
- 57. Enna SJ, McCarson KE. The role of GABA in the mediation and perception of pain. Adv Pharmacol 2006; 54: 1–27. [DOI] [PubMed] [Google Scholar]
- 58. Knabl J, Zeilhofer UB, Crestani F, Rudolph U, Zeilhofer HU. Genuine antihyperalgesia by systemic diazepam revealed by experiments in GABAA receptor point‐mutated mice. Pain 2009; 141: 233–8. [DOI] [PubMed] [Google Scholar]
- 59. Vuilleumier PH, Besson M, Desmeules J, Arendt‐Nielsen L, Curatolo M. Evaluation of anti‐hyperalgesic and analgesic effects of two benzodiazepines in human experimental pain: a randomized placebo‐controlled study. PLoS One 2013; 8: e43896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chapman CR, Feather BW. Effects of diazepam on human pain tolerance and pain sensitivity. Psychosom Med 1973; 35: 330–40. [DOI] [PubMed] [Google Scholar]