

Abstract
BACKGROUND
It is unclear whether therapy for human immunodeficiency virus type 1 (HIV-1) should be initiated with a four-drug or two sequential three-drug regimens.
METHODS
In this multicenter trial we compared initial therapy involving four-drug regimens containing efavirenz and nelfinavir in combination with either didanosine and stavudine or zidovudine and lamivudine with therapy involving two consecutive three-drug regimens the first of which contained either efavirenz or nelfinavir.
RESULTS
A total of 980 subjects were followed for a median of 2.3 years. There was no significant difference in the occurrence of regimen failures between the group that received the four-drug regimen containing didanosine, stavudine, nelfinavir, and efavirenz and the groups that received the three-drug regimens beginning with didanosine, stavudine, and nelfinavir (hazard ratio for regimen failure, 1.24) or didanosine, stavudine, and efavirenz (hazard ratio, 1.01). There was no significant difference between the group that received the four-drug regimen containing zidovudine, lamivudine, nelfinavir, and efavirenz and the groups that received the three-drug regimens beginning with zidovudine, lamivudine, and nelfinavir (hazard ratio, 1.06) or zidovudine, lamivudine, and efavirenz (hazard ratio, 1.45). A four-drug regimen was associated with a longer time to the first regimen failure than the three-drug regimens containing didanosine, stavudine, and nelfinavir (hazard ratio for a first regimen failure, 0.55); didanosine, stavudine, and efavirenz (hazard ratio, 0.63); or zidovudine, lamivudine, and nelfinavir (hazard ratio, 0.49), but not the three-drug regimen containing zidovudine, lamivudine, and efavirenz (hazard ratio, 1.21).
CONCLUSIONS
There was no significant difference in the duration of successful HIV-1 treatment between a single four-drug regimen and two consecutive three-drug regimens. Among these treatment strategies, initiating therapy with the three-drug regimen of zidovudine, lamivudine, and efavirenz is the optimal choice.
Although many drugs are approved for the treatment of human immunodeficiency virus type 1 (HIV-1) infection, the cause of the acquired immunodeficiency syndrome (AIDS), they belong to just four different classes: nucleoside or nucleotide reverse-transcriptase inhibitors (nucleoside analogues), nonnucleoside reverse-transcriptase inhibitors, protease inhibitors, and fusion inhibitors. Because cross-resistance within a class is common, the failure of the initial regimen used may compromise the success of future regimens. Indeed, the results of cohort studies suggest that a patient’s first treatment regimen has the greatest chance of success.1–3
Four-drug antiretroviral regimens containing drugs from three drug classes have the potential for increased potency but may also be associated with increased toxicity, decreased adherence, and the limitation of subsequent treatment options.4,5 Three-drug regimens containing drugs from only two classes may be less potent but more tolerable, and patients treated with regimens that exclude some classes of drugs may have a better response to subsequent treatments. We compared the use of four-drug regimens with the use of sequential pairs of three-drug regimens. Comparisons among the pairs of three-drug regimens are presented by Robbins et al. elsewhere in this issue of the Journal.6
METHODS
STUDY DESIGN
AIDS Clinical Trials Group (ACTG) study 384 was a multicenter, randomized, partially double-blind, controlled trial in which subjects were randomly assigned to six treatment groups. Groups 1, 2, 3, and 4 received two consecutive three-drug regimens. Groups 5 and 6 each received a four-drug regimen (Fig. 1). The criteria for entry, study treatments, and procedures for informed consent, enrollment, and monitoring are described by Robbins et al.6
Figure 1. Study Design and Disposition of Subjects.

Panel A shows the factorial design of the trial, with the treatment factors represented in terms of the initial treatment regimens. Panel B shows the numbers of subjects who were randomly assigned to each group, the numbers of virologic and toxicity-related regimen failures, and the numbers of premature discontinuations of study treatment. The primary study end point was defined as the failure of two consecutive regimens for subjects in groups 1, 2, 3, and 4 and the failure of a single regimen for subjects in groups 5 and 6. The premature discontinuation of all study treatment for any reason contributed to the primary study end point but not to the secondary end point of the first regimen failure. Panel C lists the criteria for regimen failure.
The primary objective of this portion of the study was the comparison of the use of four-drug regimens with the use of two sequential three-drug regimens; comparisons were performed separately for the three-drug strategies starting with a regimen containing nelfinavir and those starting with a regimen containing efavirenz. The primary study end point was the failure of two consecutive three-drug regimens (groups 1, 2, 3, and 4), the failure of a single four-drug regimen (groups 5 and 6), or the premature discontinuation of the study treatment for any reason (Fig. 1). The secondary end points reported here are the length of time to the failure of the initial regimen, the time to virologic failure, the time to viral suppression, the time to the first severe toxic effect or toxic effect resulting in dose modification, the change in the CD4 cell count at weeks 48, 96, and 144, the self-reported level of adherence during the four days before selected clinic visits,7 and the occurrence of virologic failure accompanied by genotypic drug resistance.
The criterion for regimen failure due to virologic failure was a lack of adequate viral suppression or virologic rebound. The criterion for regimen failure due to toxicity was the occurrence of a toxic effect related to the study medications that could not be managed with the use of the permitted drug substitutions (stavudine for zidovudine or lamivudine for didanosine), reductions in the doses, or the temporary discontinuation of the use of a drug.6,8 Peripheral neuropathy was assessed on the basis of a constellation of moderately severe symptoms (of grade 2 or higher) typically beginning in the distal portion of the legs, including pain, burning sensations, numbness, and paresthesias.
Testing for genotypic resistance was performed at the time of regimen failure, provided that the plasma HIV-1 RNA level was 1000 copies per milliliter or higher, with the use of the Trugene HIV-1 genotyping test (Visible Genetics). Genotypic resistance was defined by the acquisition of mutations that conferred definite resistance to a drug according to the manufacturer’s guidelines (version 4.0) at the time of the failure of the first or second regimen in groups 1, 2, 3, and 4 or the first regimen in groups 5 and 6.
STATISTICAL ANALYSIS
The general methods for the comparisons with respect to the primary end point and related time-to-event secondary end points are described by Robbins et al.6 In the comparison of the three-drug strategies, there was a significant interaction between the combination of nucleoside analogues used in the initial regimen and the use of nelfinavir or efavirenz in the initial regimen. For this reason, we report separate comparisons between the four-drug regimens and the three-drug strategies starting with regimens containing either efavirenz or nelfinavir according to the combination of nucleoside analogues used. We noted significant interactions. Therefore, the significance level for the testing of hypotheses and the estimation of confidence intervals was adjusted to 0.0125, reflecting the performance of four different comparisons addressing the same primary objective.9 Individual confidence intervals were calculated at 98.75 percent and are reported as adjusted 95 percent confidence intervals.
Post hoc comparisons were made between the groups receiving four drugs and those receiving two three-drug regimens with respect to the time to viral suppression and the occurrence of regimen failure accompanied by genotypic drug resistance. Additional post hoc analyses considered toxic effects during treatment with the first two study regimens in groups 1, 2, 3, and 4 and during treatment with the single study regimen in groups 5 and 6. Post hoc assessments of whether the hazard of a first toxic effect changed over time were performed with the use of likelihood-ratio tests comparing Weibull models with exponential models of survival. Exploratory subgroup analyses of the rates of the primary and related secondary end points according to the base-line CD4 cell count were performed in the following previously described subgroups: subjects with fewer than 200 CD4 cells per cubic millimeter, those with 200 to 350 cells per cubic millimeter, and those with more than 350 cells per cubic millimeter.4,5 Self-reported adherence was summarized as the percentage of the prescribed regimen taken during the scheduled sampling periods within the first 32 weeks of study treatment.
RESULTS
BASE-LINE CHARACTERISTICS
There were 980 subjects included in the analysis: 155 each in groups 1, 2, 3, and 4, 178 in group 5, and 182 in group 6. A total of 898 subjects were enrolled in the United States, and 82 in Italy. The median age was 36 years, and 82 percent of the subjects were male. The median base-line plasma HIV-1 RNA level was 4.9 log10 copies per milliliter, and the median CD4 cell count was 278 per cubic millimeter. The treatment groups were well balanced in terms of base-line characteristics (see Supplementary Appendix 1, available with the full text of this article at www.nejm.org).
STUDY FOLLOW-UP AND ADHERENCE
The median duration of follow-up was 28 months, and the median duration of treatment with study medication was 27 months. The mean self-reported level of adherence ranged from 97.5 to 98.8 percent, with no significant differences among the six treatment groups. Stavudine was substituted for zidovudine in 25 subjects, and lamivudine was substituted for didanosine in 78 subjects.
PRIMARY END POINT
The primary end point was reached in 441 subjects (45.0 percent) including 272 in groups 1, 2, 3, and 4 (43.9 percent) and 169 in groups 5 and 6 (46.9 percent) (Fig. 1). Among the 620 subjects in the three-drug groups, the primary study end point was reached in 80 because of virologic or toxicity-related failure of the second three-drug regimen, and 192 subjects discontinued treatment prematurely for reasons other than protocol-defined virologic or toxicity-related failure.6 These 192 subjects included 82 who had virologic or toxicity-related failure of their first regimen. Among the 360 subjects in the four-drug groups, the primary study end point was reached in 98 because of virologic failure (in 63 subjects) or toxicity-related failure (in 35 subjects), and 71 discontinued treatment prematurely for reasons other than protocol-defined virologic or toxicity-related failure.
There was no significant difference in the time to the primary end point between the group that received the four-drug regimen containing didanosine and stavudine and the group that received sequential three-drug regimens beginning with nelfinavir, didanosine, and stavudine (hazard ratio for the primary end point in the four-drug group as compared with the three-drug group, 1.24; adjusted 95 percent confidence interval, 0.82 to 1.87) or between the four-drug group that received zidovudine and lamivudine and the group that received sequential three-drug regimens beginning with nelfinavir, zidovudine, and lamivudine (hazard ratio, 1.06; adjusted 95 percent confidence interval, 0.71 to 1.58) (Fig. 2A, 2B, and 2C). Similarly, there was no significant difference in the time to the primary end point between the group that received the four-drug regimen containing didanosine and stavudine and the group that received sequential three-drug regimens beginning with efavirenz, didanosine, and stavudine (hazard ratio, 1.01; adjusted 95 percent confidence interval, 0.68 to 1.50) or between the four-drug group that received zidovudine and lamivudine and the group that received sequential three-drug regimens beginning with efavirenz, zidovudine, and lamivudine (hazard ratio, 1.45; adjusted 95 percent confidence interval, 0.94 to 2.23) (Fig. 2A, 2D, and 2E).
Figure 2. Time to the Primary End Point.
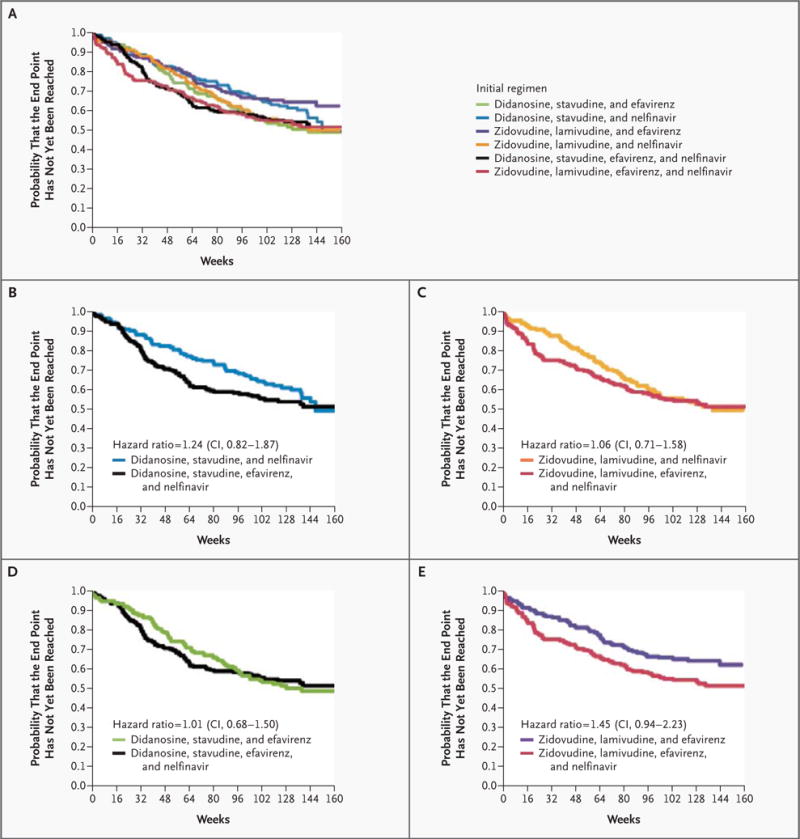
The Kaplan–Meier curves show the time to the primary study end point — failure of two consecutive three-drug regimens or a single four-drug regimen — in all treatment groups (Panel A) and in individual four-drug groups as compared with individual three-drug groups according to the drugs included in the initial regimen (Panels B, C, D, and E). The hazard ratios given are for the primary end point in the four-drug group as compared with the three-drug group; adjusted 95 percent confidence intervals (CIs) were calculated in order to adjust the significance level to 0.0125 to account for the performance of four comparisons.
FIRST REGIMEN FAILURE
The use of a four-drug regimen was associated with a longer time to the first regimen failure than the three-drug regimens containing nelfinavir, whether the subjects were assigned to start treatment with didanosine and stavudine (hazard ratio for first regimen failure in the four-drug group as compared with the three-drug group, 0.55; adjusted 95 percent confidence interval, 0.36 to 0.86) or with zidovudine and lamivudine (hazard ratio, 0.49; adjusted 95 percent confidence interval, 0.30 to 0.81) (Fig. 3A, 3B, and 3C). However, the results of the comparisons between the four-drug regimens and the three-drug regimens containing efavirenz depended on which nucleoside analogues were used (P=0.02 for the interaction between the number of drugs and the nucleoside analogues received). The four-drug regimen was associated with a longer time to the first regimen failure than the three-drug regimen containing efavirenz among the subjects who were randomly assigned to didanosine and stavudine (hazard ratio, 0.63; adjusted 95 percent confidence interval, 0.40 to 0.98), but not among those who were assigned to zidovudine and lamivudine (hazard ratio, 1.21; adjusted 95 percent confidence interval, 0.67 to 2.20) (Fig. 3A, 3D, and 3E).
Figure 3. Time to the First Regimen Failure.

The Kaplan–Meier curves show the time to the first regimen failure in all treatment groups (Panel A) and in individual four-drug groups as compared with individual three-drug groups according to the drugs included in the initial regimen (Panels B, C, D, and E). Because most of the first regimen failures were due to virologic failure, the Kaplan–Meier curves for the time to the first virologic failure were similar to those shown here. The hazard ratios given are for the first regimen failure in the four-drug group as compared with the three-drug group; adjusted 95 percent confidence intervals (CIs) were calculated in order to adjust the significance level to 0.0125 to account for the performance of four comparisons.
FIRST VIROLOGIC FAILURE
The four-drug regimens were associated with a longer time to the first virologic failure than the three-drug strategies beginning with a regimen containing nelfinavir, both among subjects who were assigned to begin treatment with didanosine and stavudine (hazard ratio for first virologic failure in the four-drug group as compared with the three-drug group, 0.49; adjusted 95 percent confidence interval, 0.31 to 0.78) and among those assigned to begin treatment with zidovudine and lamivudine (hazard ratio, 0.41; adjusted 95 percent confidence interval, 0.25 to 0.68). However, the results of the comparisons between the four-drug regimens and the three-drug strategies beginning with regimens containing efavirenz depended on which nucleoside analogues were used (P=0.02 for the interaction). The four-drug regimen was associated with a longer time to the first virologic failure than the three-drug strategy beginning with a regimen containing efavirenz among subjects randomly assigned to receive didanosine and stavudine (hazard ratio, 0.57; adjusted 95 percent confidence interval, 0.35 to 0.93), but not among those assigned to receive zidovudine and lamivudine (hazard ratio, 1.16; adjusted 95 percent confidence interval, 0.63 to 2.14).
TIME TO VIRAL SUPPRESSION
Suppression of plasma HIV-1 RNA levels to a level below 50 copies per milliliter occurred faster in subjects randomly assigned to receive zidovudine, lamivudine, and efavirenz than among those assigned to the four-drug regimen containing zidovudine, lamivudine, efavirenz, and nelfinavir, but only among subjects in the highest stratum of HIV-1 RNA (≥5.0 log10 copies per milliliter) (P<0.001). This effect was not observed within other HIV-1 RNA strata or among the subjects randomly assigned to receive didanosine and stavudine; in fact, there was a significant interaction among the treatment factors (the use of four drugs or two three-drug regimens and the choice of nucleoside analogues) and the HIV-1 RNA strata (P=0.002). The cumulative proportions of subjects in whom viral suppression had been achieved by weeks 16 and 24 were 82 percent and 94 percent, respectively, among subjects randomly assigned to begin treatment with zidovudine, lamivudine, and efavirenz, as compared with 68 percent and 84 percent, respectively, among subjects randomly assigned to receive zidovudine, lamivudine, efavirenz, and nelfinavir (see Supplementary Appendix 2, available with the full text of this article at www.nejm.org). The higher rate of viral suppression among the subjects who received zidovudine, lamivudine, and efavirenz did not appear to result from differential dropout rates among the treatment groups (data not shown).
CD4 CELL COUNTS
The median increase from base line in the CD4 cell count was 168 cells per cubic millimeter at week 48 (among 795 subjects), 251 cells per cubic millimeter at week 96 (712 subjects), and 295 cells per cubic millimeter at week 144 (401 subjects). There were no significant differences in the changes in the CD4 cell count according to the number of drugs used or the combination of nucleoside analogues used (data not shown).
DEATHS, SERIOUS ADVERSE EVENTS, AND TOXIC EFFECTS OF DRUGS
There were six deaths in the three-drug groups and six deaths in the four-drug groups. Three subjects died from myocardial infarction, two from opportunistic infection, two from cancer, one from cirrhosis, and one from trauma; the cause of death was uncertain in three subjects. There were 44 reported AIDS-defining events in 35 subjects; the incidence of a new diagnosis of AIDS or death was 2.1 per 100 person-years of follow-up. A lower base-line CD4 cell count and older age (but not a higher HIV-1 RNA stratum at screening or any treatment-group assignment) were independently associated with an increased risk of development of AIDS or death (data not shown).
Between 65 and 104 subjects from each of the six groups (42 to 58 percent) had a serious toxic effect (a severe toxic effect or a toxic effect resulting in dose modification) during treatment with their first regimen, and there was a significant difference across the six groups (P<0.001 by the log-rank test). Each of the three groups that began therapy with didanosine and stavudine had a higher incidence of serious toxic effects than the groups that began therapy with zidovudine and lamivudine. This observation prompted a post hoc comparison of the combinations of nucleoside analogues, which showed that the time to a serious toxic effect was shorter among subjects who began therapy with didanosine and stavudine than among those who began therapy with zidovudine and lamivudine (P<0.001).
Table 1 summarizes the incidence of selected toxic effects occurring before the primary end point according to the assigned treatment group. Peripheral neuropathy developed in 154 subjects and was more common in the groups that began therapy with didanosine and stavudine than among those that began therapy with zidovudine and lamivudine (P<0.001). The risk of peripheral neuropathy was inversely correlated with the base-line CD4 cell count (hazard ratio for peripheral neuropathy, 0.86 for every increment of 100 cells per cubic millimeter in the CD4 cell count; 95 percent confidence interval, 0.79 to 0.95). Among the subjects who began therapy with didanosine and stavudine, the risk of a first serious toxic effect decreased over time (P=0.001).
Table 1.
Selected Toxic Effects According to Treatment Group.*
| Toxic Effects | All Groups (N=980) | Group 1 (N=155) | Group 2 (N=155) | Group 3 (N=155) | Group 4 (N=155) | Group 5 (N=178) | Group 6 (N=182) |
|---|---|---|---|---|---|---|---|
| number of subjects (percent) | |||||||
|
| |||||||
| Pancreatitis | 18 (1.8) | 7 (4.5) | 6 (3.9) | 1 (0.6) | 1 (0.6) | 2 (1.1) | 1 (0.5) |
|
| |||||||
| Elevated serum lipase level | 99 (10.1) | 23 (14.8) | 20 (12.9) | 13 (8.4) | 9 (5.8) | 20 (11.2) | 14 (7.7) |
|
| |||||||
| Peripheral neuropathy | 154 (15.7) | 41 (26.5) | 29 (18.7) | 10 (6.5) | 16 (10.3) | 42 (23.6) | 16 (8.8) |
|
| |||||||
| Rash | 113 (11.5) | 22 (14.2) | 17 (11.0) | 16 (10.3) | 16 (10.3) | 19 (10.7) | 23 (12.6) |
|
| |||||||
| Central nervous system effects | 122 (12.4) | 18 (11.6) | 22 (14.2) | 20 (12.9) | 21 (13.5) | 16 (9.0) | 25 (13.7) |
|
| |||||||
| Gastrointestinal effects | 68 (6.9) | 10 (6.5) | 20 (12.9) | 6 (3.9) | 11 (7.1) | 11 (6.2) | 10 (5.5) |
|
| |||||||
| Hepatic effects | 62 (6.3) | 12 (7.7) | 16 (10.3) | 4 (2.6) | 7 (4.5) | 14 (7.9) | 9 (4.9) |
|
| |||||||
| Hematologic effects | 34 (3.5) | 3 (1.9) | 7 (4.5) | 8 (5.2) | 6 (3.9) | 3 (1.7) | 7 (3.8) |
Group 1 initially received didanosine, stavudine, and efavirenz; group 2 initially received didanosine, stavudine, and nelfinavir; group 3 initially received zidovudine, lamivudine, and efavirenz; group 4 initially received zidovudine, lamivudine, and nelfinavir; group 5 received didanosine, stavudine, efavirenz, and nelfinavir; and group 6 received zidovudine, lamivudine, efavirenz, and nelfinavir. Pancreatitis was defined clinically. Data were collected for elevated serum lipase levels (>1.5 times the upper limit of normal), peripheral neuropathy, rash, and central nervous system toxic effects, and data on effects of grade 2 or higher are shown. Data on gastrointestinal toxic effects, hepatic toxic effects (elevated serum aspartate aminotransferase, serum alanine aminotransferase, or total bilirubin levels), and hematologic toxic effects of grade 3 or higher are shown. Central nervous system symptoms included agitation, confusion, depression, lightheadedness, abnormal level of consciousness, and sleeping abnormalities. Gastrointestinal symptoms included nausea, vomiting, and diarrhea. Peripheral neuropathy, pancreatitis, elevated lipase, and hepatic abnormalities were significantly more likely to occur in groups that began therapy with didanosine and stavudine than in groups that began therapy with zidovudine and lamivudine (P<0.001 for peripheral neuropathy, P=0.005 for pancreatitis, P=0.003 for elevated serum lipase levels, and P=0.004 for hepatic abnormalities).
Post hoc analyses showed a greater risk of pancreatitis (P=0.005), elevated serum lipase levels (P=0.003), and hepatic abnormalities (P=0.004) among subjects who were randomly assigned to begin therapy with didanosine and stavudine than among those assigned to begin therapy with zidovudine and lamivudine (Table 1). The occurrence of lactic acidosis during treatment with study medications was reported in nine subjects, all of whom were receiving didanosine and stavudine. There was no significant difference in the time to a toxic effect according to whether subjects began therapy with nelfinavir, efavirenz, or both.
GENOTYPIC DRUG RESISTANCE
Mutations conferring resistance to nucleoside analogues were detected at positions 184 (in 80 percent of cases), 74 (in 5 percent), 65 (in 3 percent), 215 (in 3 percent), 75 (in 1 percent), and 151 (in 1 percent) and at more than one of these positions in 5 percent of subjects with nucleoside-analogue resistance. Resistance to nucleoside analogues developed in significantly fewer subjects in the four-drug group than in the three-drug group that began therapy with nelfinavir among subjects who began therapy with didanosine and stavudine (0.6 percent vs. 7.1 percent, P=0.001) and among those who began therapy with zidovudine and lamivudine (3.8 percent vs. 22.6 percent, P<0.001) (Fig. 4A). The effect of starting treatment with a four-drug regimen or a three-drug regimen containing efavirenz depended on the combination of nucleoside analogues used (P=0.003 for the interaction): resistance to nucleoside analogues developed in significantly fewer subjects in the four-drug group than in the three-drug group that began therapy with efavirenz among subjects who began therapy with didanosine and stavudine (0.6 percent vs. 16.1 percent, P<0.001) but not among those who began therapy with zidovudine and lamivudine (3.8 percent vs. 7.7 percent, P=0.12) (Fig. 4A). Among subjects in three-drug groups, those who began therapy with zidovudine, lamivudine, and efavirenz had fewer treatment failures with resistance to nucleoside analogues (7.7 percent) than those who began therapy with zidovudine, lamivudine, and nelfinavir (22.6 percent, P<0.001) or those who began therapy with didanosine, stavudine, and efavirenz (16.1 percent, P=0.02) (Fig. 4A).
Figure 4. Proportions of Subjects Who Had Regimen Failure with Resistance to Nucleoside Analogues (Panel A), Nonnucleoside Reverse-Transcriptase Inhibitors (Panel B), and Protease Inhibitors (Panel C) According to Treatment Group.

For subjects in groups 1, 2, 3, and 4, the bars show the cumulative proportions of subjects with drug resistance at the time of the first or second regimen failure; for subjects in groups 5 and 6, the bars show the proportions of subjects with new drug-resistance mutations after the failure of their first regimen. Drug-resistance mutations were identified through the sequencing of the HIV-1 reverse transcriptase and protease of virus isolated from subjects with virologic failure or toxicity-related failure who had plasma HIV-1 RNA levels of at least 1000 copies per milliliter.
Mutations conferring resistance to nonnucleoside reverse-transcriptase inhibitors were detected at positions 103 (in 72 percent of cases), 190 (in 6 percent), and 188 (in 3 percent) and at more than one of these positions in 19 percent of subjects with resistance to nonnucleoside reverse-transcriptase inhibitors. Resistance to nonnucleoside reverse-transcriptase inhibitors developed in significantly fewer subjects in the four-drug group than in the three-drug group that began therapy with efavirenz among subjects who began therapy with didanosine and stavudine (10.7 percent vs. 23.9 percent, P=0.001) but not among those who began therapy with zidovudine and lamivudine (8.2 percent vs. 7.1 percent, P=0.69) (Fig. 4B). There was no significant difference in the rate of resistance to nonnucleoside reverse-transcriptase inhibitors between the four-drug groups and the three-drug groups that began therapy with nelfinavir (Fig. 4B). The results of comparisons among the three-drug groups depended on the combination of nucleoside analogues used (P=0.006 for the interaction). Among the subjects in three-drug groups, resistance to non-nucleoside reverse-transcriptase inhibitors developed in a higher proportion of those who began therapy with didanosine, stavudine, and efavirenz (23.9 percent) than of those who began therapy with didanosine, stavudine, and nelfinavir (5.2 percent, P<0.001) or those who began therapy with zidovudine, lamivudine, and efavirenz (7.1 percent, P< 0.001) (Fig. 4B).
Mutations conferring resistance to protease inhibitors were detected at positions 30 (in 61 percent of cases), 90 (in 17 percent), 46 (in 7 percent), and 84 (in 3 percent) and at more than one of these positions in 12 percent of subjects with resistance to protease inhibitors. Resistance to protease inhibitors developed in fewer subjects in the four-drug group than in the three-drug group that began therapy with nelfinavir among subjects who began therapy with didanosine and stavudine (2.2 percent vs. 11.0 percent, P=0.001) and among those who began therapy with zidovudine and lamivudine (0.5 percent vs. 7.7 percent, P<0.001) (Fig. 4C). There was no significant difference in the rate of resistance to protease inhibitors between the four-drug groups and the three-drug groups that began therapy with efavirenz (Fig. 4C).
DISCUSSION
This study compared different strategies for initiating antiretroviral therapy with four-drug and sequential three-drug regimens, with use of a composite primary end point of virologic rebound, toxic effects, or premature discontinuation of treatment, as well as seven secondary end points, including the change in the CD4 cell count, the first regimen failure (due to virologic rebound or toxic effects), the first virologic failure, the time to an undetectable HIV-1 RNA level, toxic effects, the level of adherence, and the development of drug resistance. Because no single surrogate end point has been validated as capturing the clinical benefit of prolonged treatment and because low rates of clinical events preclude the use of such end points for the investigation of first-line therapy, we looked for consistent results among the end points.
There was no significant difference in the duration of successful treatment, as measured in terms of the time to the primary end point, between the groups receiving a single four-drug regimen and those receiving two consecutive three-drug regimens, regardless of the combination of nucleoside analogues used in the initial regimen. There were also no significant differences between the four-drug groups and the three-drug groups in the changes in the CD4 cell count. The definition of the primary end point was designed to capture all possible reasons for the premature discontinuation of treatment in addition to virologic rebound and protocol-specified toxic effects. Because the primary end point was the time to the failure of drugs from all three classes, subjects receiving a four-drug regimen reached the primary end point when they had a single regimen failure (involving the use of only a single combination of nucleoside analogues).
In terms of the time to the secondary end points of the first regimen failure and the first virologic failure, the four-drug regimens were superior both to the three-drug strategies beginning with regimens containing nelfinavir and to therapy beginning with didanosine, stavudine, and efavirenz, but not to therapy beginning with zidovudine, lamivudine, and efavirenz. There were relatively few first regimen failures during the three-year study in the group that began therapy with zidovudine, lamivudine, and efavirenz. The potency of this regimen was further demonstrated by the finding that the use of this regimen resulted in a shorter time to viral suppression than either of the four-drug regimens among subjects with the highest levels of HIV-1 RNA. Although the addition of nelfinavir to these three drugs provided no further benefit with respect to any end point, we cannot rule out the possibility that a different protease inhibitor might have been more effective.
There was no significant difference in the risk of toxic effects between the four-drug groups and the three-drug groups. Only the combination of nucleoside analogues used in the initial regimen significantly influenced the risk of toxic effects, with a higher incidence of peripheral neuropathy, pancreatitis, and hepatic enzyme abnormalities among subjects who began therapy with didanosine and stavudine than among those who began therapy with zidovudine and lamivudine. Preliminary data from a subgroup of subjects have also shown a greater degree of fat loss in the limbs among subjects who began therapy with didanosine and stavudine.10 All these toxic effects may result from mitochondrial dysfunction induced by nucleoside analogues.11–13
Antiretroviral regimens containing drugs from more than two classes are not routinely recommended for patients beginning antiretroviral therapy because of concern that patients in whom a three-class regimen fails may be at higher risk for drug resistance should virologic failure occur, leaving them with fewer options for subsequent therapy. However, we found that the cumulative proportion of regimen failures with genotypic resistance was often lower, and never higher, in the four-drug groups than in the three-drug groups.
Reported adherence was high in each of the six groups, suggesting that the study’s findings result from the intrinsic activity of the treatment regimens rather than from the patterns of adherence. Our findings, however, may not be applicable to persons who do not maintain high levels of adherence.14 Treatment failure and resistance to efavirenz and lamivudine are usually caused by mutations of a single amino acid,15,16 whereas resistance to protease inhibitors frequently requires multiple amino acid substitutions.4,17–20 In this study, subjects in whom regimens containing efavirenz failed often had amino acid substitutions that would also confer resistance to other nonnucleoside reverse-transcriptase inhibitors. In contrast, resistance to protease inhibitors was often associated with a mutation at position 30 of the protease gene that confers resistance to nelfinavir alone.
Although four-drug regimens were effective and did not lead to an increased risk of toxic effects or drug resistance, the simpler three-drug regimen consisting of zidovudine, lamivudine, and efavirenz emerged as the optimal choice for the initiation of therapy. This three-drug regimen was effective in achieving and maintaining viral suppression throughout the study and allowed most subjects who began therapy with this regimen to reserve protease-inhibitor therapy for the future. The fact that the addition of nelfinavir to this three-drug regimen did not lead to more rapid viral suppression attests to the potency of this regimen; possible mechanisms underlying this potency are being explored in a pharmacologic substudy. Zidovudine, lamivudine, and efavirenz was the most effective of the initial three-drug regimens in terms of delaying the first regimen failure,6 and this was also the only three-drug regimen that was not significantly different from the four-drug regimens in terms of the lengths of time to the first regimen failure and the first virologic failure.
Supplementary Material
Acknowledgments
Supported in part by grants (AI38858 and AI38855) from the National Institute of Allergy and Infectious Diseases; by the General Clinical Research Center Units funded by grants (RR00070, RR00052, RR00044, and RR05096) from the National Center for Research Resources; by a grant (to the Istituto Superiore di Sanita) from the HIV Clinical Research Programme; and by virology laboratory contracts (201VC001 with Vanderbilt University, 200VC001 with the University of Alabama at Birmingham, and 200VC011 with Stanford University).
Dr. D’Aquila reports having served as a consultant for or having received honorariums from Agouron, Bristol-Myers Squibb, Gilead, GlaxoSmithKline, ViroLogic, and Visible Genetics; Dr. De Gruttola having served as a consultant for GlaxoSmithKline and Tibotec–Virco; Dr. Haubrich having served as a consultant for or having received honorariums from Agouron, Bristol-Myers Squibb, and Glaxo-SmithKline; Dr. Johnson having served as a consultant for or having received honorariums from Abbott, Bristol-Myers Squibb, Glaxo-SmithKline, and ViroLogic; Dr. Gripshover having served as a consultant for or having received honorariums from Abbott, Bristol-Myers Squibb, Gilead, and Roche and having received grant support from Boehringer–Ingleheim; Dr. Hirsch having served as a consultant for Bristol-Myers Squibb and GlaxoSmithKline; Dr. Merigan holding equity in Bristol-Myers Squibb and Pfizer and having received grant support from Hoffmann–LaRoche; Dr. Morse having served as a consultant for or having received honorariums or grant support from Agouron, Amgen, Bristol-Myers Squibb, GlaxoSmith-Kline, and Roche; Dr. Robbins having served as a consultant for or having received honorariums from Agouron, Bristol-Myers Squibb, and GlaxoSmithKline; and Dr. Shafer having served as a consultant for or having received honorariums from Bristol-Myers Squibb, and GlaxoSmithKline and having received grant support from Agouron, Bristol-Myers Squibb, and GlaxoSmithKline.
APPENDIX
Other members of the ACTG 384 team were M. Fischl (Miami University), R. Pollard (University of Texas, Galveston), M. Dubé (Indiana University), C. Pettinelli (National Institutes of Health), R. Delapenha (Howard University), B. Putnam (University of Colorado), M. Klebert (Washington University), M. Testa (Harvard School of Public Health), A. Chiesi, C. Tomino (Istituto Superiore de Sanita), S. Deeks (University of California, San Francisco), T. Nevin (Social & Scientific Systems), J. Levin, V. French, O. Fennell (Adult AIDS Clinical Trials Group Community Constituency Group), M. Stevens, R. Grosso, B. Dusak, S. Hodder (Bristol-Myers Squibb), J. Tolson, C. Brothers (GlaxoSmith-Kline), R. Leavitt (Merck), D. Manion, N. Ruiz, K. Morrissey (DuPont Pharmaceuticals), M. Becker, B. Quart (Agouron), C. Jennings (Northwestern University), L. Gedeon, S. Dascomb, M. Cooper, M. Murphy, K. Blakelock (Frontier Science and Technology Foundation), A. Doolan (Massachusetts General Hospital).
The following persons and institutions participated in the conduct of this trial: T. Powell, L. Simmermacher, J. Robinson (University of Cincinnati), L. Alexis, J. Brown, Y. Butler, C. St. Paul (Howard University), L. Meixner, T. Gilbert (University of California, San Diego), J. Kaufman, A. Chall, S. Pedersen, J. Horton (Carolinas Medical Center), T. Lane (Moses Cone Hospital), J. Castro, L. Thompson, L. Colon (University of Miami), M. Chance, J. Baum (Case Western Reserve University), D. Wininger, D. Gochnour (Ohio State University), C. Funk, D. Johnson (University of Southern California), I. Matozzo, I. Frank, K. Maffei, D. Kim (University of Pennsylvania), S. Marrero, O. Mendez, I. Torres, N. Rabell (University of Puerto Rico), G. Casey, C. Derkowski (University of Texas, Galveston), C. Fietzer, R. Nelson, K. Fox, R. Sanders (University of Minnesota), W. Wallace (University of Iowa), S. Swindells (University of Nebraska), H. Rominger, J. Richardson (Indiana University Hospital), P. Tebas, D. DeMarco, M. Royal (Washington University), B. Berzins (Northwestern University), H. Kessler (Rush–Presbyterian–St. Luke’s Medical Center), J. Forcht, D. Tolenaar, C. Talabucon, C. Gonzalez (New York University/Bellevue), S. Wright, J. Graham (University of Alabama, Birmingham), C. Basler, S. Canmann (University of Colorado Health Sciences Center), P. Cain, S. Stoudt, J. Norris, S. Valle (Stanford University), S. Tedder, C. Hamilton (Duke University Medical Center), T. Flynn, K. Hartman (Massachusetts General Hospital), H. Fitch (Beth Israel Deaconess Medical Center), T. Cooley (Boston Medical Center and Harvard University), J. Frederick, S. Souza (University of Hawaii), D. Baker, A. Weiss, B. Becker, I. Wiggins (Johns Hopkins University), R. Reichman, M. Shoemaker, R. Hewitt, T. O’Hara (University of Rochester Medical Center), J. Lertora, R. Clark, M. Beilke, J. Dumestre (Tulane University), A. Moe, M. Witt, M. Guerrero, T. Maldonado (University of California, Los Angeles), A. Collier, B. Royer, J. Stekler, J. Conley (University of Washington), M. Dolan, E. Chusid, H. Sacks, I. Lowy (Mt. Sinai Medical Center), M. Jacobson, J. Volinski (San Francisco General Hospital), T. Stroberg, R. Gulick, M. Glesby, V. Hughes (Cornell University), R. Arcieri, M. Pirillo, C. Galluzzo, E. Germinario, R. Amici, M. Marzi, A. Nobile, R. Di Nallo, C. Polizzi (Istituto Superiore di Sanita), O. Coronado, G. Fasulo (Ospedale Maggiore), G. Carosi, F. Castelli (Università degli Studi di Brescia Spedali Civili di Brescia), M. Di Pietro, F. Vichi (Ospedale Santa Maria Annunziata), G. Sterrantino, S. Ambu (Azienda Ospedaliera Careggi), M. Cargnel, P. Meraviglia, F. Niero, A. Capetti, A. D’Arminio Monforte, S. Sollima, C. Balotta (Ospedale Luigi Sacco), S. Delia, M. Ciardi, G. d’Ettore, G. Forcina (Università degli Studi di Roma La Sapienza Policlinico Umberto I), M. Soranzo, A. Macor (Ospedale Amadeo Di Savoia), D. Bassetti, A. Di Biagio (Università di Genova), L. Minoli, R. Maserati (Istituto di Recovero e Cura a Caratterre Scientifico Policlinico S. Matteo), F. Ghinelli, L. Sighinolfi (Azienda Arcispedale Sant’ Anna), A. Riva, G. Scalise (Università degli Studi di Ancona Ospedale Umberto I), D. Santoro, E. Rinaldi (Ospedale Sant’ Anna), F. Chiodo, M. Borderi (Policlinico Sant’ Orsola Malpighi), G. Guaraldi, R. Esposito (Università degli Studi di Modena), C. Ferrari, G. Pasetti (Azienda Ospedaliera di Parma), N. Abrescia, A. Busto, A. Chirianni, M. Gargiulo, C. Izzo, C. Sbreglia (Azienda Ospedaliera D. Cotugno), F. Alberici, D. Sacchini (Azienda Unitá Sanitaria Locale di Piacenza Ospedale Civile), G. Magnani, G. Zoboli (Arcispedale Santa Maria Nuova).
References
- 1.Paredes R, Mocroft A, Kirk O, et al. Predictors of virological success and ensuing failure in HIV-positive patients starting highly active antiretroviral therapy in Europe: results from the EuroSIDA study. Arch Intern Med. 2000;160:1123–32. doi: 10.1001/archinte.160.8.1123. [DOI] [PubMed] [Google Scholar]
- 2.Ledergerber B, Egger M, Opravil M, et al. Clinical progression and virological failure on highly active antiretroviral therapy in HIV-1 patients: a prospective cohort study. Lancet. 1999;353:863–8. doi: 10.1016/s0140-6736(99)01122-8. [DOI] [PubMed] [Google Scholar]
- 3.Grabar S, Pradier C, Le Corfec E, et al. Factors associated with clinical and virological failure in patients receiving a triple therapy including a protease inhibitor. AIDS. 2000;14:141–9. doi: 10.1097/00002030-200001280-00009. [DOI] [PubMed] [Google Scholar]
- 4.Department of Health and Human Services Panel on Clinical Practices for Treatment of HIV Infection. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. doi: 10.1310/hct.2000.1.1.008. (Accessed November 12, 2003, at http://www.aidsinfo.nih.gov/guidelines/adult/archive/AA_071403.html.) [DOI] [PubMed]
- 5.Yeni PG, Hammer SM, Carpenter CC, et al. Antiretroviral treatment for adult HIV infection in 2002: updated recommendations of the International AIDS Society–USA Panel. JAMA. 2002;288:222–35. doi: 10.1001/jama.288.2.222. [DOI] [PubMed] [Google Scholar]
- 6.Robbins GK, De Gruttola V, Shafer RW, et al. Comparison of sequential three-drug regimens as initial therapy for HIV-1 infection. N Engl J Med. 2003;349:2291–301. doi: 10.1056/NEJMoa030264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chesney MA, Ickovics JR, Chambers DB, et al. Self-reported adherence to antiretroviral medications among participants in HIV clinical trials: the AACTG adherence instruments. AIDS Care. 2000;12:255–66. doi: 10.1080/09540120050042891. [DOI] [PubMed] [Google Scholar]
- 8.Division of AIDS table for grading severity of adult adverse experiences. Rockville, Md: National Institute of Allergy and Infectious Diseases; Aug, 1992. [Google Scholar]
- 9.Cox DR, Oakes D, editors. Analysis of survival data. New York: Chapman and Hall; 1984. Proportional hazards model; pp. 97–110. [Google Scholar]
- 10.Dube MP, Zackin R, Tebas P, et al. Prospective study of regional body composition in antiretroviral-naive subjects randomized to receive zidovudine+lamivudine or didanosine+stavudine combined with nelfinavir, efavirenz, or both: A5005S, a sub-study of ACTG 384. Antiviral Ther. 2002;7:L18. abstract. [Google Scholar]
- 11.Brinkman K, Smeitink JA, Romijn JA, Reiss P. Mitochondrial toxicity induced by nucleoside-analogue reverse-transcriptase inhibitors is a key factor in the pathogenesis of antiretroviral-therapy-related lipodystrophy. Lancet. 1999;354:1112–5. doi: 10.1016/S0140-6736(99)06102-4. [DOI] [PubMed] [Google Scholar]
- 12.Squires KE. An introduction to nucleoside and nucleotide analogues. Antiviral Ther. 2001;6(Suppl 3):1–14. [PubMed] [Google Scholar]
- 13.Cote HC, Brumme ZL, Craib KJ, et al. Changes in mitochondrial DNA as a marker of nucleoside toxicity in HIV-infected patients. N Engl J Med. 2002;346:811–20. doi: 10.1056/NEJMoa012035. [DOI] [PubMed] [Google Scholar]
- 14.Ickovics JR, Cameron A, Zackin R, et al. Consequences and determinants of adherence to antiretroviral medication: results from Adult AIDS Clinical Trials Group protocol 370. Antiviral Ther. 2002;7:185–93. doi: 10.1177/135965350200700308. [DOI] [PubMed] [Google Scholar]
- 15.Boucher CA, Cammack N, Schipper P, et al. High-level resistance to (–) enantiomeric 2′-deoxy-3′-thiacytidine in vitro is due to one amino acid substitution in the catalytic site of human immunodeficiency virus type 1 reverse transcriptase. Antimicrob Agents Chemother. 1993;37:2231–4. doi: 10.1128/aac.37.10.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bacheler L, Jeffrey S, Hanna G, et al. Genotypic correlates of phenotypic resistance to efavirenz in virus isolates from patients failing nonnucleoside reverse transcriptase inhibitor therapy. J Virol. 2001;75:4999–5008. doi: 10.1128/JVI.75.11.4999-5008.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murphy RL, Gulick RM, DeGruttola V, et al. Treatment with amprenavir alone or amprenavir with zidovudine and lamivudine in adults with human immunodeficiency virus infection. J Infect Dis. 1999;179:808–16. doi: 10.1086/314668. [DOI] [PubMed] [Google Scholar]
- 18.Descamps D, Flandre P, Calvez V, et al. Mechanisms of virologic failure in previously untreated HIV-infected patients from a trial of induction-maintenance therapy. JAMA. 2000;283:205–11. doi: 10.1001/jama.283.2.205. [DOI] [PubMed] [Google Scholar]
- 19.Havlir DV, Hellmann NS, Petropoulos CJ, et al. Drug susceptibility in HIV infection after viral rebound in patients receiving indinavir-containing regimens. JAMA. 2000;283:229–34. doi: 10.1001/jama.283.2.229. [DOI] [PubMed] [Google Scholar]
- 20.Gallego O, de Mendoza C, Perez-Elias MJ, et al. Drug resistance in patients experiencing early virological failure under a triple combination including indinavir. AIDS. 2001;15:1701–6. doi: 10.1097/00002030-200109070-00014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.