

Abstract
The use of Xenopus embryonic skin as a model system for the development of ciliated epithelia is well established. This tissue is comprised of numerous cell types, most notably the multiciliated cells (MCCs) that each contain approximately 150 motile cilia. At the base of each cilium lies the centriole-based structure called the basal body. Centriole biogenesis is typically restricted to two new centrioles per cell cycle, each templating from an existing “mother” centriole. In contrast, MCCs are post-mitotic cells in which the majority of centrioles arise “de novo” without templating from a mother centriole, instead, these centrioles nucleate from an electron-dense structure termed the deuterostome. How centriole number is regulated in these cells and the mechanism by which the deuterosome templates nascent centrioles is still poorly understood. Here, we describe methods for regulating MCC cell fate as well as for visualizing and manipulating centriole biogenesis.
INTRODUCTION
The ability of cells to duplicate their centrioles each cell cycle is a critical step in proper chromosome segregation and cell division. During interphase, centrioles act as important microtubule organizing centers (MTOCs) creating inward-outward directionality to microtubule-based transports. Moreover, in many differentiated cells the older of the two centrioles, termed the “mother” centriole, acts as the basal body nucleating a primary cilium. When this process goes awry, supernumerary centrioles can form, which has been observed in numerous types of cancer and is considered to correlate with cancer progression (Vitre & Cleveland, 2012). Additionally, genetic disruption of numerous centriolar proteins results in a wide range of phenotypes, most notably microcephaly and dwarfism (Nigg, Cajanek, & Arquint, 2014). For the reasons above, the study of centriole biogenesis has been the focus of many labs and there are a number of elegant systems with which to study centriole formation. The focus of this chapter will be to highlight a variation on the main theme of centriole biogenesis. Specifically, in post-mitotic MCCs there is a massive centriole amplification process that results in the production of more than 100 centrioles that nucleate the numerous motile cilia that project from the apical surface of these cells (Figure 1(A) and (B)).
Figure 1.
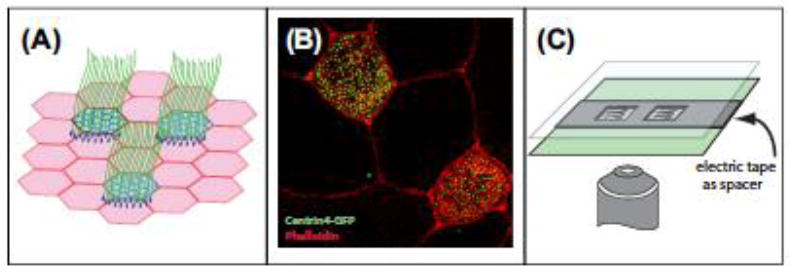
Xenopus MCCs.
(A) Illustration of the ciliated epithelia that covers the skin of Xenopus embryos. (B) Confocal image of multiciliated cells with centrin4-GFP marking centrioles/basal bodies (green) and rhodamine-phalloidin marking actin in the cell periphery (red). (C) Illustration of the mounting of Xenopus embryos using electrical tape as a spacer.
During cell cycle-regulated centriole duplication, the two centrioles that comprise the centrosome separate, and a new daughter centriole nucleates orthogonally from the side of each mother centriole (Figure 2(A)). In cells in which key regulators of centriole duplication (e.g., Plk4 or Cep152) are disregulated, it has been observed that numerous centrioles can simultaneously nucleate off of a single mother centriole (Figure 2(B)) (Brownlee & Rogers, 2013). Interestingly, in MCCs it has been observed that numerous centrioles simultaneously nucleate from the mother centriole, suggesting that the critical regulators might be differentially regulated. However, it has been suggested from the Electron-Microscopy literature that this form of centriole biogenesis accounts for only a small portion of the centrioles that are generated. In contrast, the vast majority of centrioles in MCCs arise “de novo” without a mother centriole to template from. Instead, these centrioles nucleate from a nondescript electron dense structure termed the deuterosome (Figure 2(C)) (Kalnins & Porter, 1969; Sorokin, 1968; Steinman, 1968). Recent papers have described several molecular components of the deuterosome; however, the functional regulation of this structure is still poorly understood (Klos Dehring et al., 2013; Zhao et al., 2013). The ciliated epithelium that lines the skin of Xenopus embryos represents an ideal system for characterizing this structure due to the facile nature of molecular manipulations and the ease of microscopic analysis (Figure 1(A) and (B)). Here, we present a detailed summary of the tools and techniques available to study centriole biogenesis in the MCCs of the Xenopus skin.
Figure 2.
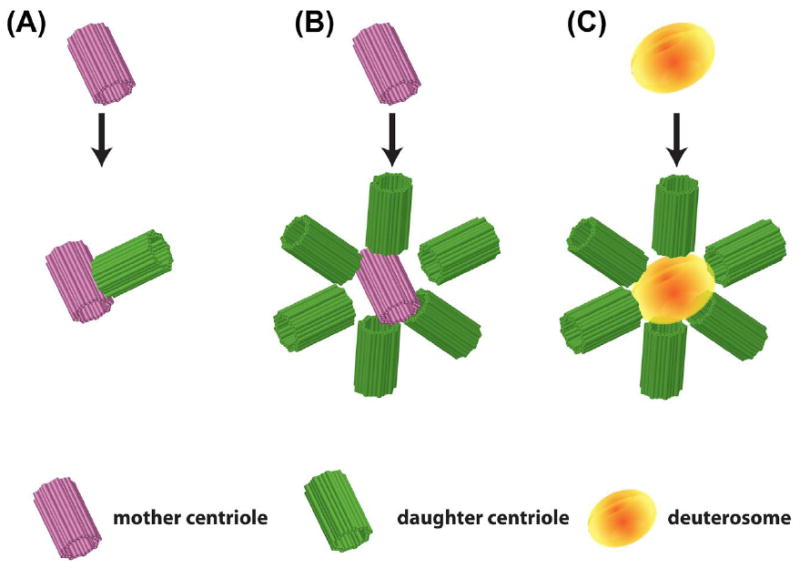
Centriole biogenesis.
(A) Standard centriole duplication with a single daughter (green (light gray in print versions)) nucleating from a mother centriole (pink (dark gray in print versions)). (B) Mother centriole-derived centriole amplification with numerous daughters (green (light gray in print versions)) simultaneously nucleating from a mother centriole (magenta (dark gray in print versions)). (C) Deuterosome-mediated centriole amplification with numerous daughters (green (light gray in print versions)) simultaneously nucleating from the deuterosome (orange (gray in print versions)).
The well-characterized centriole duplication regulatory proteins Plk4 and Cep152, as well as the core structural protein Sas6, have all been observed at the deuterosome suggesting that key elements between centriole-centriole and deuterosome-centriole biogenesis are maintained (Klos Dehring et al., 2013). However, there are obvious fundamental differences that account for the abilities to (1) generate centrioles in the G0 phase of the cell cycle and (2) generate greater than a hundred centrioles. A significant milestone in the study of deuterosomes in Xenopus was the observation that a green fluorescent protein (GFP)-tagged version of the protein CCDC78 localizes specifically to the deuterosome (Klos Dehring et al., 2013). This allowed the first detailed characterization of the structure. Importantly, the depletion of CCDC78 leads to a loss of deuterosome localization of Cep152 and subsequently a severe reduction in centriole amplification. Another deuterosome-specific protein CCDC67, which has been named Deup1, also localizes to the deuterosome in mouse (Zhao et al., 2013). Deup1 is thought to provide a counterbalance to the mother centriole-driven centriole biogenesis from its paralogue Cep63 (Zhao et al., 2013). This again suggests that there are both similarities and unique features that distinguish centriole-centriole and deuterosome-centriole biogenesis. Finally, it has been recently proposed from work in the MCCs of mouse ependymal cells that deuterosomes nucleate exclusively from the daughter centrioles (Al Jord et al.,2014). This suggests that centriole biogenesis is not entirely “de novo” but, instead, deuterosomes act as amplifiers that acquire some signal from existing centrioles to increase the centriole nucleating capacity. While these recent studies have begun to provide a better picture on this structure, clearly there is still much to be learned. Many of the techniques used to study centriole biology in Xenopus rely on standard approaches that have been described in detail elsewhere (Mimoto & Christian, 2011;Sive, Grainger, & Harland, 1998, 2007c, 2007e, 2010a; Werner & Mitchell, 2013). Here, we aim to highlight specific tools, reagents, and experimental conditions that will facilitate the study of centriole amplification in MCCs.
1. VISUALIZATION OF CENTRIOLES AND RELATED STRUCTURES IN XENOPUS MCCs
1.1 VISUALIZATION OF CENTRIOLES VIA EXPRESSION OF FLUORESCENTLY LABELED CENTRIOLE-RELATED PROTEIN FROM EXOGENOUS MESSENGER RNA
One significant advantage of using Xenopus as a model organism is the ease of inducing localized protein expression via direct injection of in vitro synthesized messenger RNA (mRNA) without the requirement of genome-level manipulations (Mimoto & Christian, 2011; Sive et al., 2007e, 2010a; Sive, Grainger, & Harland, 2010b). The detailed protocols for manipulating early Xenopus embryos using mRNA injection method has been extensively described and profiled. Here, we will briefly describe these methods and focus on the slight modifications that we have found useful. These methods can be used to introduce proteins that will modulate developmental processes (e.g., transcription factors or signaling molecules) or simply to introduce fluorescently tagged proteins for visualization. In MCCs, the ability to fluorescently tag centrioles and deuterosomes allows the initiation of analysis of the process of centriole amplification.
Prior to mRNA synthesis, it is recommended to use column-based DNA purification of linearized plasmid DNA to prevent carry-over contamination of residual phenol-chloroform that may severely inhibit mRNA synthesis (e.g., Qiagen PCR Purification kit; Cat. No. 28104). The quality of mRNA for injections is critical for reproducibility. Techniques for generating mRNA are well established and there are numerous commercial kits available to facilitate optimal production (e.g., Life Technologies mMessage mMachine kits Cat. No. #1340/1341/1342). Additionally, column-based mRNA clean-up kits such as Life Technologies MegaClear kit (Cat. No. #AM1908) or Qiagen minElute clean-up kit (Cat. No. 74204) are preferable over the traditional phenol-chloroform method to minimize loss and prevent carry-over contamination. Column-based purification is essential if mRNA is to be combined with morpholino oligos (MOs) for injection. While these purification methods are not always essential and they do slightly increase the cost of mRNA synthesis, it is our experience that quality, yield, and reproducibility are all significantly enhanced with these techniques.
Eggs collected from mature adult females are in vitro fertilized with isolated testes (Sive et al., 1998, 2007e; Sive, Grainger, & Harland, 2007b, 2007d). Typically, injections are performed at the two or four cell stage. This is done so that the quality of the eggs can be evaluated by the quality of the first and second cell divisions. It is not recommended to inject embryos with poor cleavage planes as these rarely develop properly. Since the ectoderm gives rise to the ciliated epithelium, injection sites should be located on the animal hemisphere of the embryo, away from the midline and towards the center of the embryo within each blastomere. While the proper amount of mRNA to inject is empirical and requires titration, a reasonable starting point is a 10e20 pL injection of 50e200 pg mRNA. (Helpful hint: if using fluorescent proteins within the blue spectrum [BFP/CFP] it is preferable to use albino frogs since pigment granules exhibit strong autofluorescence in that range.)
Injected embryos are allowed to develop until the desired stage. The developmental rate of embryos can be artificially controlled by incubating embryos under different temperatures (Khokha et al., 2002). Generally, the earliest expression of MCC-specific genes initiates at stage 11.5, with high levels of expression observed at stage 14. Radial intercalation, the process by which MCCs interdigitate from an inner layer to the superficial layer, starts at stage 14 and can be optimally visualized at stage 17e18 (Stubbs, Davidson, Keller, & Kintner, 2006; Werner et al., 2014). At this stage, the formation and dynamics of centrioles, as well as the acentriolar structure, the deuterosome, can be observed. Centriole amplification typically occurs between stage 15 and 18 and ciliogenesis occurs after cells have completed radial intercalation (stage 19e20). While cilia are formed around stage 20, the process of properly polarizing these cilia occurs over time based on a combination of planar polarity signaling and a flow-mediated positive feedback loop, both of which impinge on the regulation of cytoskeletal dynamics (Mitchell, Jacobs, Li, Chien, & Kintner, 2007; Mitchell et al., 2009; Park, Mitchell, Abitua, Kintner, & Wallingford, 2008; Werner et al., 2011). Generally, the structure and formation of centrioles in Xenopus MCCs can be conveniently observed using fluorescently tagged centriolar proteins.
A list of molecular markers for centrioles and related structures as well as fixation conditions and optimal concentrations has been provided in Table 1.
Table 1.
Useful Molecular Markers for the Study of Multiciliated Cells
| Marker Name | Description | Labeled Structure | Fixatives | Suggested Concentration |
|---|---|---|---|---|
|
| ||||
| Centrioles and basal bodies | ||||
|
| ||||
| g-tubulin | Antibody sigma #T-6557 | Label basal bodies in fixed tissue (Park et al., 2008) | 100% MeOH; Dent’s | 1:300e1:500 |
| Variable results: 3% PFA/EtOH | ||||
| Centrin-RFP | Tagged centrin4 (Xl. 50473) | Strongly labels basal bodies (Mitchell et al., 2009; Park et al., 2008) | 3% PFA or 3% PFA/EtOH | 40 pg mRNA |
| Variable results: 100% MeOH; Dent’s | ||||
| GFP-HYLS | Tagged HYLS (Xl. 13199) | Strongly labels basal bodies (Dammermann et al., 2009) | Dent’s | 250 pg mRNA; 20 pg DNA |
| 3% PFA/EtOH | ||||
| 3% PFA | ||||
| Sas6-RFP | Tagged SAS6 (Xl. 33005) | Labels active deuterosomes and centrioles (Klos Dehring et al., 2013) | Dent’s | 250 pg mRNA |
| 3% PFA/EtOH | ||||
| 3% PFA | ||||
| RFP-Cep152 | Tagged Cep152 (Xl. 13956) | Labels deuterosomes and nascent centrioles (Klos Dehring et al., 2013) | Dent’s | 250 pg mRNA |
| 3% PFA/EtOH | ||||
| 3% PFA | ||||
| GFP-Plk4 | Tagged Plk4 (Xl. 5660) | Labels deuterosomes and centrioles (Klos Dehring et al., 2013) | Dent’s | 250 pg mRNA |
| 3% PFA/EtOH | ||||
| 3% PFA | ||||
| GFP-Ccdc78 | N-terminal GFP tagged ccdc78 (Xl. 4890) | Specifically labels deuterosomes (Klos Dehring et al., 2013) | Dent’s | 200 pg mRNA |
| 3% PFA/EtOH | ||||
| 3% PFA | ||||
| Ccdc78-GFP | C-terminal GFP tagged ccdc78 (Xl. 4890) | Labels both deuterosomes and centrioles (Klos Dehring et al., 2013) | Dent’s | 200 pg mRNA |
| 3% PFA/EtOH | ||||
| 3% PFA | ||||
| GFP-Deup1 | N-terminal GFP tagged Deup1 (CCDC67 cloned from Xenopus laevis) | Strongly labels deuterosomes and weakly labels centrioles (unpublished observation) | 3% PFA | 200pg mRNA |
| GFP/RFP CLAMP (Spef1) | Tagged CLAMP (Xl. 26316) | Labels striated rootlets. Can be used to score cilia orientation (Mitchell et al., 2009; Park et al., 2008) | 3% PFA or 3% PFA/EtOH | 250 pm mRNA; 20 pg DNA |
| Variable results: 100% MeOH; Dent’s | ||||
| GFP-mid1ip1 (Mig12) | Tagged mig12 (Xl. 47645) | Labels rootlet, striated. Can be used to score cilia orientation (Park et al., 2008) | 3% PFA or 3% PFA/EtOH | 250 pm mRNA; 30 pg DNA |
| Variable results: 100% MeOH; Dent’s | ||||
| Vinculin | GFP-tagged vinculin (Xl. 21637) | Labels rootlets in ciliated cells (Antoniades, Stylianou, & Skourides, 2014) | Live imaging is recommended | 250 pg mRNA |
| 3% PFA if fixation is required | ||||
| FAK | GFP-tagged FAK (Xl. 6819) | Labels rootlets in ciliated cells (Park, Shen, Chien, & Guan, 2009) | Live imaging is recommended | 250 pg mRNA |
| 3% PFA if fixation is required | ||||
| Paxilin | GFP-tagged Paxilin (Xl. 724) | Labels rootlets in ciliated cells (Antoniades et al., 2014) | Live imaging is recommended | 250 pg mRNA |
| 3% PFA if fixation is required | ||||
| Dvl2 | GFP-tagged Dvl2, (Xl. 7670) | Labels adjacent to the basal body (Park et al., 2008) | 3% PFA | 250 pg mRNA |
| Bbof1 | GFP-tagged Bbof1 (Xl. 66678) | Transiently associated with basal bodies (Chien et al., 2013) | 3.7% Formaldehyde þ 0.25% glutaraldehyde | 250 pg mRNA |
1.2 VISUALIZATION OF CENTRIOLES IN FIXED EMBRYOS
Several techniques have been described for fluorescent imaging in whole Xenopus embryos (Kieserman, Lee, Gray, Park, & Wallingford, 2010; Lee, Kieserman, Gray, Park, & Wallingford, 2008). A simple method of mounting the relative-flat embryos for imaging can be achieved by sandwiching the fixed embryos between two pieces of coverglass, while using several layers of electrical tape as a spacer (Figure 1(C)) (Werner & Mitchell, 2013). It should be noted that for optimal imaging of samples fixed in MeOH or Dent’s, sufficient rehydration time (approximately 4 h) should be allowed for fluorescent proteins to return to their native conformations.
For successful observation of centrioles in mounted embryos, appropriate microscope configuration for confocal microscopy is highly recommended. Traditional epifluorescent microscopy may be challenging due to excessive fluorescence emitted from the underlying tissue. As a general guideline, a 60e100x oil objective (1.4 n. a.) with sufficient working distance is recommended for taking images on centrioles and subcortical structures. In order to obtain clear images on centrioles without interference from background and cytosolic particles, it is recommended to use Z-stacks (10e15 images) with a step size of 0.2e0.5 mm. Collecting too few images may result in decreased Z-axis resolution on centrioles and associated subcortical structures, while collecting too many stacks may risk excessive photobleaching to fluorescent proteins, especially red fluorescent protein (RFP). This balance is particularly challenging while visualizing MCCs amplifying their centrioles as this occurs below the surface of the embryo (see below).
1.3 VISUALIZATION OF CENTRIOLES IN LIVE EMBRYOS
Batches of injected embryos should be prescreened under a fluorescent dissecting microscope to identify embryos with sufficient fluorescence intensities. After which,embryos can be mounted for live imaging. Mounting techniques and methods in Xenopus embryos vary between stages of the embryos, as well as the desired timespan in which the embryos are to be imaged. Generally, a detailed and comprehensive review of over mounting Xenopus embryos has been published (Kieserman et al., 2010). Essentially, mounted embryos are pressed against a flat coverglass to maintain a flat field of view for live imaging, while maintaining sufficient level of access to the culture media to ensure embryo survival and growth, especially for extended imaging during time course analysis. Alternatively, instead of whole embryos, explanted tissue such as animal cap explants can also be used for imaging, especially if the observed tissue is expected to undergo growth, extension, or movement within one or more dimensions (Figure 3(A)). For a detailed protocol on customized chamber making and embryo mounting, please refer to Ref. Werner and Mitchell (2013). The “electrical tape” method described in the previous section can also be used in live embryos as well as more traditional half-rose chambers or glass bottom Petri dishes (Wadsworth, 2007; Werner & Mitchell, 2013).
Figure 3.
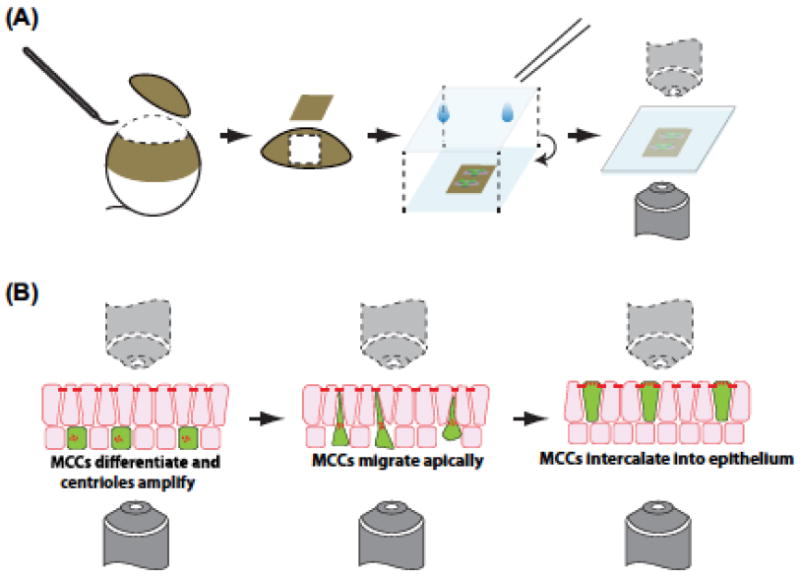
Ciliated epithelial explants.
(A) Illustration of dissecting off the animal cap of the embryo to generate an epithelial explant. (B) Process of visualizing multiciliated radial intercalation in which the cells migrate into the outer epithelium.
For imaging centriole biogenesis in MCCs, several special considerations should be made. As mentioned above, centriole biogenesis initiates around stage 16, prior to the cells undergoing radial intercalation and joining the outer epithelium. This means that the MCCs are approximately 10 mm below the surface when centrioles are amplifying. While this is within the range of confocal microscopy, it is challenging to get high-resolution images. Molecular solutions to this issue will be discussed below, but from an imaging perspective there are several helpful hints.
The use of skin explants provides two advantages: First, the sample is much flatter than whole embryos making deeper images more facile (Figure 3(A)). Second, the explants can be flipped upside down and imaged from underneath, which in some situations allows higher-resolution imaging (Figure 3(B)) (Stubbs et al., 2006). While other imaging techniques such as two-photon or light sheet illumination would also be beneficial, the detailed description of these methods is beyond the scope of this chapter. Additionally, other groups have successfully imaged MCCs from mouse using super-resolution techniques, and this will obviously be beneficial to translate into Xenopus (Al Jord et al., 2014; Zhao et al., 2013).
1.4 VISUALIZATION OF CENTRIOLES VIA ANTIBODY STAINING AND IMMUNOFLUORESCENCE
In addition to the fluorescent-tagged protein-based methods of visualizing centrioles as described above, it is also possible and practical to visualize centrioles and associated structures using immunofluorescence. For a detailed protocol in performing antibody staining and immunofluorescence in Xenopus epithelium, please refer to Ref. Werner and Mitchell (2013). In addition to the general protocol, several particular points should be noted when performing immunofluorescence in Xenopus embryos, as described below.
Fixatives need to be optimized for each individual antigen. Generally, it is advisable to try the following fixatives as a starting point: 3% paraformaldehyde (PFA) in phosphate-buffered saline (PBS), and MEMFA (one volume of 37% formaldehyde, one volume of MEM salt including 1 M MOPS, 20 mM EGTA, 10 mM MgSO4, pH 7.4, and eight volumes of ultra-pure H2O). High-purity, methanol-free formaldehyde, such as SigmaeAldrich F8775, is recommended since it will not interfere with phalloidin staining, or Dent’s fixative (80% MeOH, 20% DMSO, v/v).
Different antibodies may have their own preferences over blocking buffers. Five percent BSA in PBST (PBS þ0.1% Triton X-100) or 10% heat-inactivated goat serum in PBST are two good starting points for testing antibodies.
2. DRIVING SPECIFIC EXPRESSION IN XENOPUS MCCs
While targeted injection of mRNA into specific embryo blastomeres does allow for a certain level of spatial control over gene expression and protein synthesis in early Xenopus embryos, it is sometimes desirable to achieve targeted gene expression both temporally and spatially in MCCs alone. To achieve this goal, the a-tubulin promoter is placed upstream of the desired gene in a plasmid construct (Stubbs et al., 2006). The a-tubulin promoter is transcriptionally active specifically in MCCs starting at stage 13 and remains active throughout MCC development. Generally, injection of 10e50 pg of plasmid DNA containing the a-tubulin promoter is sufficient to drive the expression of the desired gene in MCCs. However, it should be taken into account that a certain level of mosaicism may arise from the injection as part of the inherited problems of DNA injection into early Xenopus embryos. It is possible to circumvent this problem by using transgenic methods to generate stable transgenic lines; however, significantly more effort and extended time are expected (minimum 18 months for each Xenopus laevis generation and 6 months for each Xenopus tropicalis generation) (Amaya & Kroll, 1999; Loeber, Pan, & Pieler, 2009). Alternatively, F0 transgenic animals can also be generated and analyzed. While a certain degree of mosaicism still presents under these conditions, one can typically identify samples that contain consistently strong expression. As will be discussed below, a certain level of mosaicism is often preferable, as it provides an internal control for cell biological analysis.
3. GENERATING MOSAIC XENOPUS EMBRYOS TO CIRCUMVENT EMBRYO LETHALITY
Either overexpression or knockdown of certain genes or proteins at the whole embryo level may result in developmental defects and/or poor embryo survival, since the desired genes may be crucial for proper cellular functioning. These risks are especially prone to happen during the investigation of centriolar proteins and present a challenge for analysis of MCCs, as these cells only emerge later in development. Hence, it is often desirable to circumvent such developmental defects by introducing a degree of mosaicism into the embryos to be investigated in order to partially compensate for the lethal effects. Generally, the injection of exogenous mRNAs or MOs can be restricted to one or more cells at the 4, 8, 16, or 32-cell stage, therefore restraining the overexpression or knockdown effects within a subset of cells in the developed embryos. A more targeted approach, which may even allow population-specific expressions within certain tissues or regions can be executed at the 16e32 cell stage with the reference from a fate map of individual blastomeres (Dale & Slack, 1987; Moody, 1987a, 1987b). In all cases, it is recommended to label the cells that have been injected using molecular tracers such as fluorescein isothiocyanate (FITC)-labeled dextran (SigmaeAldrich 46945) or rhodamine-labeled dextran (micro-ruby, Life Technologies D-7162). Strong fluorescence can be observed immediately after injection, and the fluorescence persists after fixation and remains strong during immunofluorescence. Injection of mRNAs encoding fluorescent proteins such as eGFP or mCherry as tracers can also be adopted in experiments, although such fluorescent proteins usually result in lower signal intensities compared to labeled dextrans and may not perform well after fixation and immunofluorescence. If MOs are to be used in the mosaic knockdowns, MOs can be prelabeled by either FITC or lissamine-rhodamine upon purchase for tracing the MO presence within embryos, and the fluorescence is maintained after fixation. Finally, it is often desirable to have centriolar markers (e.g., Centrin-RFP) in all cells and a specific MO in a subset of cells. To accomplish this, one set of injections can be performed at the 2-cell stage and a second set of injections can be performed at the 4, 8, 16, or 32-cell stage. This allows for a direct comparison of “wild-type” and mutant cells in the same embryos and can be a particularly powerful approach.
4. CILIATED EPITHELIAL SPHEROID CULTURE
During embryonic development, MCCs of Xenopus embryos undergo radial intercalation into the outer epidermal layer, and the numerous centrioles within are subsequently distributed to the apical surface (Figure 3(B)). This event provides a unique opportunity to study radial intercalation and the essential role that centrioles play during this process (Werner et al., 2014). However, performing time-lapse confocal imaging on intercalating cells presents a challenge, since intercalation generally occurs perpendicular to the coverglass surface in mounted embryos, which results in a severe decrease in resolution due to the absorption of both excitation lasers and emission light from the tissue itself. In addition, to obtain sufficient resolution of cell movements during the intercalation process, deep Z-stacks are required, which further increases the potential threat of photobleaching. Hence, it may be desired to mount the intercalating epidermis in a way that the plane of intercalation is parallel to the coverglass and, therefore, parallel to the XY axis. To achieve this, Xenopus epithelial spheres derived from ectodermal explants can be used, which are much smaller in size compared to the whole embryo and, more importantly, enable confocal imaging of intercalating cells from the side of the sphere, thus greatly reducing scan time and hence the risk of photobleaching (Werner & Mitchell, 2013).
Use agarose-coated dishes in all experiments. The surfaces of certain high-quality dishes for cell culture are specifically precoated with polymers and thus become extremely adhesive to any exposed tissues. In such cases, isolated animal cap explants will “cling” to the bottom of the dish and are difficult to manipulate and transfer. To make agarose-coated dishes, pour hot 1% agar dissolved in 0.1x MMR into the dishes to be used, let stand for 30 s, and aspirate out the remaining agar solution. A very thin layer of agar coating will remain on the inner surface of the dish and prevent any adhesion to explants.
Isolate animal cap explants around stage 10 (Figure 3(A)) (Sive, Grainger, & Harland, 2007a). Remove any residual white remnants that derive from the yolk-rich inner embryo mass from the inside of semispherical ectodermal animal cap explants. This is best done using an eyebrow knife to scrape off the white mass.
Cut the ectodermal explant into small pieces of approximately 20e30 mm in diameter using an eyebrow knife. This can be done in 0.1x MMR to circumvent high salt concentration-induced quick involution of the explants, especially when the operator is relatively inexperienced.
Transfer the small tissue explants obtained into another dish with 0.5x MMR plus gentamycin. Use a wide-bore glass pipette and avoid getting explants into contact with air or the surface tension will rupture the explants. Under normal conditions, explants will naturally form spheres in 30 mine1 h.
Incubate explants until control (nonexplanted) embryos reach stage 16e20.
Prepare agarose pads for mounting and imaging. Melt 2% agarose in 0.1x MMR to 90 oC in a heat block or microwave oven. Apply a drop of agarose to a glass microscopic slide that has been positioned between two other slides. The other two slides must be fixed to the bench surface using two layers of electrical tape.Place another glass slide on top of the agarose and perpendicular to all other three slides. A thin layer of agarose will form inside the buffer zone created by two layers of electrical tape between the two slides. For a graphical illustration of the procedure, see Ref. Werner and Mitchell (2013).
Carefully remove the top slide from the agarose pad by sliding it along the other ones. In most cases, some air bubbles will be trapped inside the buffer zone and leaving small holes on the agarose pad, which can be immediately used for mounting the epithelial spheres inside. Small holes can also be cut using fine forceps or punching the pad with pipette tips to sit the spheres within.
Carefully transfer the epithelial spheres into the holes on the agarose pad using a wide-bore glass pipette. Gently place a coverslip on the agarose pad and seal the rim with melted petroleum jelly or silicon grease. The spheroid cultures are now ready to be used for live imaging.
5. MANIPULATING MCC FORMATION
The MCCs that form on the surface of Xenopus embryos exhibit a stereotypical spacing pattern that reflects the requirement for cell vertices during the process of radial intercalation (Figure 4). For example, one can greatly increase the number of MCC precursors, but only a fraction of these will intercalate due to the limitations of the number of proper vertices (Stubbs et al., 2006). The transcriptional program that leads to MCC development has been the focus of numerous studies and several factors are known to modulate MCC formation. Here, we will describe several of these factors and how they can be exploited to study centriole formation and function.
Figure 4.

Modulation of multiciliated cell fate.
Illustration of multiciliated cell phenotypes obtained with the manipulation of various transcriptional regulators (as labeled).
Notch signaling appears to provide one of the first levels of regulation for MCCs at around stage 12. Activation of the Notch pathway using mRNA injections to over-express the intracellular domain of Notch (pCS2-NICD) leads to a severe decrease in the number of MCCs, while conversely the inhibition of the Notch pathway via overexpression of a dominant negative Mastermind (pCS2-dnMM) construct leads to an overproduction of MCCs (Figure 4 and Table 2) (Deblandre, Wettstein, Koyano-Nakagawa, & Kintner, 1999). However, the effect of each of these constructs is restricted to the initial cell lineage in that ectopic expression within outer epithelial cells fails to convert these cells to the MCC’s lineage. In stark contrast, the loss of multicilin (MCIDAS), which is a member of the gemini family of proteins, results in a loss of MCCs. More importantly, ectopic expression of MCIDAS using a dexamethasone inducible construct (pCS2-hGR-MCIN) leads to the conversion of outer epithelial cells into MCCs, such that the entire epithelium is now covered with MCCs (Figure 4 and Table 2) (Stubbs, Vladar, Axelrod, & Kintner, 2012). Two downstream targets of MCIDAS are the transcription factors MYB and FoxJ1. MYB controls centriole duplication and FoxJ1 controls ciliogenesis. Ectopic expression of MYB leads to more MCCs but fails to convert outer cells into MCCs (similar to dnMM) (Tan et al., 2013). In contrast, ectopic expression of FoxJ1 does not create more MCCs due to a lack of centriole amplification but rather lead to the formation of ectopic cilia such that outer cells will use their existing centrioles to nucleate one to two cilia per cell (Stubbs, Oishi, Izpisua Belmonte, & Kintner, 2008).
Table 2.
Tools for Manipulating Multiciliated Cell Formation
| Regulator | Dose | Effects |
|---|---|---|
|
| ||
| Notch-ICD (intracellular domain of Notch) (Deblandre et al., 1999) | 175 pg mRNA | Overexpression in early embryos activates Notch signaling, thus reducing or preventing MCC formation |
| dnHMM (dominant-negative version of mastermind) (Fryer, Lamar, Turbachova, Kintner, & Jones, 2002) | 1 ng mRNA | Overexpression in early embryos inhibits Notch signaling, thus promoting MCC formation |
| FoxJ1 (Stubbs et al., 2008) | 1e5 ng mRNA | Overexpression induces nodelike ciliated cells; |
| 75 ng Morpholino oligo | knockdown inhibits formation of both node and epidermal ciliated cells | |
| MYB (Tan et al., 2013) | 500 pg mRNA | Overexpression promotes MCC formation in epidermis |
| hGR-MCI (inducible version of multicilin) (Stubbs et al., 2012) | 40 pg mRNA; 2 mM of dexamethasone for induction (Ryan et al., 2004); | Overexpression will convert all epidermal cells into MCCs; |
| 50 ng MO | knockdown prevents MCC formation in epidermis | |
While each of these factors can provide useful tools for the study of centrioles, MCIDAS is particularly useful. The formation of ectopic MCCs (with temporal control using human glucocorticoid receptor-fused version of MCIDAS, hGR-MCI together with dexamethasone induction) eliminates the issues of imaging MCCs undergoing centriole amplification deep in the tissue (see above). When outer cells are converted to MCCs, one can easily visualize the formation of nascent centrioles budding off of the deuterosome. Additionally, it has been observed that deuterosomes are formed in MCCs ectopically expressing both MCIDAS and MYB, suggesting that these MCCs undergo typical deuterosome-mediated centriole amplification. Table 2 outlines the optimal dose and experimental use of these constructs as well as the expected outcomes.
6. FULLY-AUTOMATIC, PROGRAM-ASSISTED COUNTING OF CENTRIOLES IN CONFOCAL IMAGES USING ImageJ
When investigating the molecular mechanisms related to centriole proliferation and/ or development in ciliated cells, it is often necessary to make statistical analysis on the number of centrioles within each ciliated cell in control versus experimental group(s). In most cases, manual counting is applied to collect centriole counts from samples, which is both time-consuming and labor-intensive. Several commercially available software packages, such as Nikon NIS-Documentation™, have built-in functions that allow automatic cell border tracking, image segmentation, and particle counting within each of the region of interests (ROI) that greatly reduces both experiment time and human labor. However, one drawback is that commercial software is often instrument-bound and/or expensive to buy, which restricts their widespread application. Here, we introduce a method using the built-in macro function of the GNU Freeware ImageJ for both fully automatic and program-assisted centriole counting. Essentially, two critical steps define the core conception of automatic centriole counting. First, maximum-projected Z-stack confocal images need to be segmented into single cells that contribute to multiple ROIs. Secondly, centrioles within each ROI (i.e., cell) need to be counted separately without interference from neighboring cells.
Prepare embryos or animal cap explants for confocal microscopy. Generally, at least two channels are required in samples that will be imaged. The first channel (C0) labels centrioles using centriole-specific markers such as GFP-Centrin4, while the second channel (C1) labels cell boundaries using membrane-specific markers such as memRFP, LifeAct-RFP, or fluorescently labeled phalloidin (recommended).
Prepare confocal images for analysis. An image size of at least 512 × 512 pixels (1024 × 1024 or higher is ideal) is required for successful image analysis.Instrument and scanning parameters: 40e60x oil lens, 2 mm thick Z-stack in total, 10 optical sections with 0.2 mm distance in between. Final output images should be multichannel (not RGB) images that have been superimposed using maximum projection along the Z-axis with the centriole channel in the C0 position.
Program requirements for imaging analysis: all the codes used within the section have been tested using ImageJ 1.49h 64-bit version with adaptiveThr plugin installed (downloadable at https://sites.google.com/site/qingzongtseng/adaptivethreshold).
If using machine-assisted counting (which is more useful when the signals from cell boundaries channel is not very strong), cell boundaries are manually drawn using the built-in ROI function of ImageJ and added into the ROI manager on the cell boundaries channel. Images need to be presplit into two independent images before the ROIs are drawn (Image > Color > Split Channels). After all the ROIs have been set up, transfer all ROIs to the centrioles channel (Edit > Selection > Restore Selection or Ctrl þ Shift þ E).
Load the macro scripts below (Box 1; this only needs to be done once and the code will remain open for all subsequent analyses) using Plugins > Macros > Edit. Make sure the centriole window has the focus and press Ctrl þ R to run the script. The script will attempt to count the number of centrioles within each ROI in an exhaustive pattern until all ROIs have been counted. After counting, the centriole counts within each cell will be displayed in the summary window and can be subsequently exported (column 2). If desired, the area size of each ROI (i.e., cell) can be calculated by dividing the Total Area of the centrioles (column 3) over the %Area (column 5). The threshold level in line 2 and centriole size in line 9 need to be determined empirically.
The fully automatic script has the ability to batch-process whole folders containing unlimited number of images, make analysis, and save the results into designated folders as tab-separated text files. Several parameters, as displayed in the program initiation page, need to be empirically determined and may differ on images obtained from separate sets of experiments. Hence, it is suggested to use images from the same set of experiments during each batch run. The code itself is extensively annotated and minor adjustments can be performed by referring to the in-line comments when editing the macro.
BOX 1. CODE FOR AUTOMATED QUANTIFICATION OF CENTRIOLE NUMBER.
//----------------------Set global variables var
minimum_cell_size;
var maximum_cell_size;
var adaptiveThr_pixel_block_size_cell; var
adaptiveThr_subtract_value_cell;
var gaussian_radius;
var minimum_centriole_size; var
maximum_centriole_size;
var adaptiveThr_pixel_block_size_centriole; var
adaptiveThr_subtract_value_centriole;
var edge_count;
var min_count_threshold; var output_2_file;
var watershed_count;
get_all_parameters(); //set all parameters for counting
dir_Input = getDirectory(“Choose Input Directory ”); // Get Input directory
dir_Output = getDirectory(“Choose Output Directory ”); // Get Output directory
list = getFileList(dir_Input); //Get file name
output_2_file = File.open(dir_Output + “output.txt”); //set output summary file
for (i=0; i<list.length; i++)
{
//load image for processing
showProgress(i+1, list.length); open(dir_Input + list[i]);
Current_File_title = list[i];
//--------------------------------------Beginning of image processing
// Note: the 1st Channel is Centrin while the 2nd Channel is Phalloidin.
selectWindow(Current_File_title); //Get image
run(“8-bit”); //adaptiveThr only works on 8-bit images
run(“Split Channels”); // separate Phalloidn and centrin channels
selectWindow(“C2-” + Current_File_title); // process Phalloidin to identify cell boundaries
run(“adaptiveThr ”, “using=Mean
from=adaptiveThr_pixel_block_size_cell
then=adaptiveThr_subtract_value_cell”); // Threshold
selectWindow(“C2-” + Current_File_title); // Reconfirm window
run(“Gaussian Blur.”, “sigma=gaussian_radius”); // Apply Gaussian Blur
wait(1000); //System wait time for medium-slow computers, necessary, otherwise the current window may lose focus.
run(“Make Binary”); // Transform to binary
run(“Invert LUT”); //Need BLACK cells over WHITE cell boundaries
//Determine whether edge counts should be included if (edge_count == true)
{
run(“Analyze Particles.”, “size=minimum_cell_size-maximum_cell_size circularity=0.20-1.00 clear add”); //Identify single cells, at least 500 in size
}
else
{
run(“Analyze Particles.”, “size=minimum_cell_size-maximum_cell_ size circularity=0.20-1.00 clear exclude add”); //Identify single cells, at least 500 in size
}
selectWindow(“C1-” + Current_File_title); //Switch to basal bodies window
run(“adaptiveThr ”, “using=[Weighted mean] from
=adaptiveThr_pixel_block_size_centriole then
=adaptiveThr_subtract_value_centriole ”); // Apply threshold
run(“Invert LUT”); // Invert LUT, need to be BLACK centrin signals over WHITE background
run(“Make Binary”); // Transform to binary, need to be BLACK cells over
WHITE cell boundaries
if (watershed_count == true)
run(“Watershed”); // Try to separate touching particles
selectWindow(“C2-” + Current_File_title);
run(“Select All”); //Get Selection from Phalloidin channel
selectWindow(“C1-” + Current_File_title);
run(“Restore Selection”); //Transfer selection to Centrin channel
n = roiManager(“count”); // Count centrin signals (i.e.: basal bodies) for (j=0; j<n; j++)
{
roiManager(“select”, j);
run(“Analyze Particles.”, “size=min_centriole_size-maximum_centriole_size clear summarize”);
}
// Save Summary window
selectWindow(“Summary”);
saveAs(“text”, dir_Output + Current_File_title + “.txt”);
lines = split(getInfo(), “yn”); //save output to one large summary file
print(output_2_file, lines[0] + ’ yt’ + “Mother Cell Area”); //Write form titles
for (m=1; m<lengthOf(lines);m++)
{
values = split(lines[m], “yt”); //False counts filter centriole_count = values[1];
if (centriole_count > min_count_threshold)
{
centriole_area = parseFloat(values[2]);
centriole_area_percentage = parseFloat(values[4]);
cell_area = centriole_area / centriole_area_percentage * 100;
//Count the total area of mother cell
print(output_2_file, lines[m] + ' yt' + cell_area);//Write mother cell area
}
}
//--------------------------------------End of Image processing
// Cleanup
close(); //Close Channel 1d close(); //Close Channel 2
// close(dir_Input+list[i]);
//setBatchMode(true);
}
//------------------File selection function function
get_all_parameters()
{
Dialog.create(“Please set program parameters”);
Dialog.addMessage(“Parameters for detecting cell boundaries”);
Dialog.addSlider(“Minimum Cell Size”, 2500, 10000, 5000);
Dialog.addSlider(“Maximum Cell Size”, 3000, 100000, 50000);
Dialog.addSlider(“Pixel Block Size for Cell Boundary”, 3, 1000, 437);
Dialog.addSlider(“Subtraction Value for Cell Boundary”, -100, 100, -12);
Dialog.addSlider(“Gaussian Blur Radius”, 0, 30, 3);
Dialog.addMessage(“Parameters for detecting basal bodies”)
Dialog.addSlider(“Minimum Basal Body Size”, 10, 100, 10);
Dialog.addSlider(“Maximum Basal Body Size”, 10, 100, 40);
Dialog.addSlider(“Pixel Block Size for Basal Bodies”, 3, 100, 60);
Dialog.addSlider(“Subtraction Value for Basal Bodies”, -100, 100, -11);
Dialog.addMessage(“Misc”);
Dialog.addCheckbox(“Including Edges?”, false);
Dialog.addCheckbox(“Watershed?”, false);
Dialog.addSlider(”Minimum Basal Bodies Count Threshold Per Cell?”, 0, 50, 10);
Dialog.addMessage(“Basal Body Counter v0.1 by Siwei Zhang, 16 Oct 2014”);
Dialog.show();
minimum_cell_size = Dialog.getNumber(); maximum_cell_size = Dialog.getNumber();
adaptiveThr_pixel_block_size_cell = Dialog.getNumber();
adaptiveThr_subtract_value_cell = Dialog.getNumber();
gaussian_radius = Dialog.getNumber();
minimum_centriole_size = Dialog.getNumber();
maximum_centriole_size = Dialog.getNumber();
adaptiveThr_pixel_block_size_centriole = Dialog.getNumber();
adaptiveThr_subtract_value_centriole = Dialog.getNumber();
edge_count = Dialog.getCheckbox();
watershed_count = Dialog.getCheckbox();
min_count_threshold = Dialog.getNumber();
return;
}
CONCLUSION
Centriole biogenesis has been the focus of research in many labs. The vast majority of studies have been performed in cell culture or the early Caenorhabditis elegans embryo. This is due to the fact that many deletions of proteins involved in centriole formation are embryonic lethal. In addition to the role that centrioles play in organizing microtubules during interphase (MTOC) and mitosis (spindle pole), they also have the capacity to act as basal bodies nucleating the microtubule-based cilium. While the regulation of cell cycle-dependent centriole duplication has been widely studied, the massive amplification of centrioles that occurs within MCCs is only starting to be addressed at a molecular level. MCCs are found in a variety of tissues including the mammalian lung, oviduct, and ependyma. These tissues, as well as primary cultures, have proven to be extremely useful for the study of MCC formation and function. Here, we have described the use of the skin of Xenopus embryos as a source for addressing the molecular regulation of centriole amplification. Xenopus has the advantage that the ciliated tissue develops on the external surface of the animal, thus eliminating the need for tissue dissection or long-term tissue culture. The ease of molecular manipulations and the speed of development have also made Xenopus an appealing system with which to address the biology of MCCs and, specifically, the fascinating question of how these cells uncouple centriole biogenesis from the cell cycle and how they utilize the poorly characterized structure the deuterosome to nucleate centrioles de novo. Xenopus has been instrumental in recent advances in understanding both the transcriptional program leading to MCCS as well as the initial molecular characterization of the deuterosome. However, clearly there is much to be learned about these unique cells and we hope that the techniques provided here will be a spring board for future work in this area.
References
- Al Jord A, Lemaitre AI, Delgehyr N, Faucourt M, Spassky N, Meunier A. Centriole amplification by mother and daughter centrioles differs in multiciliated cells. Nature. 2014;516(7529):104–107. doi: 10.1038/nature13770. http://dx.doi.org/10.1038/nature13770. [DOI] [PubMed] [Google Scholar]
- Amaya E, Kroll KL. A method for generating transgenic frog embryos. Methods in Molecular Biology. 1999;97:393–414. doi: 10.1385/1-59259-270-8:393. http://dx.doi.org/10.1385/1-59259-270-8:393. [DOI] [PubMed] [Google Scholar]
- Antoniades I, Stylianou P, Skourides PA. Making the connection: ciliary adhesion complexes anchor basal bodies to the actin cytoskeleton. Developmental Cell. 2014;28(1):70–80. doi: 10.1016/j.devcel.2013.12.003. http://dx.doi.org/10.1016/j.devcel.2013.12.003. [DOI] [PubMed] [Google Scholar]
- Brownlee CW, Rogers GC. Show me your license, please: deregulation of centriole duplication mechanisms that promote amplification. Cellular and Molecular Life Sciences. 2013;70(6):1021–e1034. doi: 10.1007/s00018-012-1102-6. http://dx.doi.org/10.1007/s00018-012-1102-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien YH, Werner ME, Stubbs J, Joens MS, Li J, Chien S, et al. Bbof1 is required to maintain cilia orientation. Development. 2013;140(16):3468–3477. doi: 10.1242/dev.096727. http://dx.doi.org/10.1242/dev.096727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale L, Slack JM. Fate map for the 32-cell stage of Xenopus laevis. Development. 1987;99(4):527–551. doi: 10.1242/dev.99.4.527. [DOI] [PubMed] [Google Scholar]
- Dammermann A, Pemble H, Mitchell BJ, McLeod I, Yates JR, 3rd, Kintner C, et al. The hydrolethalus syndrome protein HYLS-1 links core centriole structure to cilia formation. Genes & Development. 2009;23(17):2046–e2059. doi: 10.1101/gad.1810409. http://dx.doi.org/10.1101/gad.1810409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deblandre GA, Wettstein DA, Koyano-Nakagawa N, Kintner C. A two-step mechanism generates the spacing pattern of the ciliated cells in the skin of Xenopus embryos. Development. 1999;126(21):4715–4728. doi: 10.1242/dev.126.21.4715. [DOI] [PubMed] [Google Scholar]
- Fryer CJ, Lamar E, Turbachova I, Kintner C, Jones KA. Mastermind mediates chromatin-specific transcription and turnover of the Notch enhancer complex. Genes & Development. 2002;16(11):1397–1411. doi: 10.1101/gad.991602. http://dx.doi.org/10.1101/gad.991602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalnins VI, Porter KR. Centriole replication during ciliogenesis in the chick tracheal epithelium. Zeitschrift fur Zellforschung und mikroskopische Anatomie. 1969;100(1):1–30. doi: 10.1007/BF00343818. [DOI] [PubMed] [Google Scholar]
- Khokha MK, Chung C, Bustamante EL, Gaw LW, Trott KA, Yeh J, et al. Techniques and probes for the study of Xenopus tropicalis development. Developmental Dynamics. 2002;225(4):499–510. doi: 10.1002/dvdy.10184. http://dx.doi.org/10.1002/dvdy.10184. [DOI] [PubMed] [Google Scholar]
- Kieserman EK, Lee C, Gray RS, Park TJ, Wallingford JB. High magnification in vivo imaging of Xenopus embryos for cell and developmental biology. Cold Spring Harbor Protocols. 2010;2010(5) doi: 10.1101/pdb.prot5427. http://dx.doi.org/10.1101/pdb.prot5427 pdb prot5427. [DOI] [PubMed] [Google Scholar]
- Klos Dehring DA, Vladar EK, Werner ME, Mitchell JW, Hwang P, Mitchell BJ. Deuterosome-mediated centriole biogenesis. Developmental Cell. 2013;27(1):103–112. doi: 10.1016/j.devcel.2013.08.021. http://dx.doi.org/10.1016/j.devcel.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Kieserman E, Gray RS, Park TJ, Wallingford J. Whole-mount fluorescence immunocytochemistry on Xenopus embryos. Cold Spring Harbor Protocols. 2008;2008 doi: 10.1101/pdb.prot4957. http://dx.doi.org/10.1101/pdb.prot4957 pdb prot4957. [DOI] [PubMed] [Google Scholar]
- Loeber J, Pan FC, Pieler T. Generation of transgenic frogs. Methods in Molecular Biology. 2009;561:65–72. doi: 10.1007/978-1-60327-019-9_4. http://dx.doi.org/10.1007/978-1-60327-019-9_4. [DOI] [PubMed] [Google Scholar]
- Mimoto MS, Christian JL. Manipulation of gene function in Xenopus laevis. Methods in Molecular Biology. 2011;770:55–75. doi: 10.1007/978-1-61779-210-6_3. http://dx.doi.org/10.1007/978-1-61779-210-6_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell B, Jacobs R, Li J, Chien S, Kintner C. A positive feedback mechanism governs the polarity and motion of motile cilia. Nature. 2007;447(7140):97–101. doi: 10.1038/nature05771. http://dx.doi.org/10.1038/nature05771. [DOI] [PubMed] [Google Scholar]
- Mitchell B, Stubbs JL, Huisman F, Taborek P, Yu C, Kintner C. The PCP pathway instructs the planar orientation of ciliated cells in the Xenopus larval skin. Current Biology. 2009;19(11):924–929. doi: 10.1016/j.cub.2009.04.018. http://dx.doi.org/10.1016/j.cub.2009.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody SA. Fates of the blastomeres of the 16-cell stage Xenopus embryo. Developmental Biology. 1987a;119(2):560–578. doi: 10.1016/0012-1606(87)90059-5. [DOI] [PubMed] [Google Scholar]
- Moody SA. Fates of the blastomeres of the 32-cell-stage Xenopus embryo. Developmental Biology. 1987b;122(2):300–319. doi: 10.1016/0012-1606(87)90296-x. [DOI] [PubMed] [Google Scholar]
- Nigg EA, Cajanek L, Arquint C. The centrosome duplication cycle in health and disease. FEBS Letters. 2014;588(15):2366–2372. doi: 10.1016/j.febslet.2014.06.030. http://dx.doi.org/10.1016/j.febslet.2014.06.030. [DOI] [PubMed] [Google Scholar]
- Park TJ, Mitchell BJ, Abitua PB, Kintner C, Wallingford JB. Dishevelled controls apical docking and planar polarization of basal bodies in ciliated epithelial cells. Nature Genetics. 2008;40(7):871–879. doi: 10.1038/ng.104. http://dx.doi.org/10.1038/ng.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park AY, Shen TL, Chien S, Guan JL. Role of focal adhesion kinase Ser-732 phosphorylation in centrosome function during mitosis. Journal of Biological Chemistry. 2009;284(14):9418–9425. doi: 10.1074/jbc.M809040200. http://dx.doi.org/10.1074/jbc.M809040200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan K, Russ AP, Levy RJ, Wehr DJ, You J, Easterday MC. Modulation of eomes activity alters the size of the developing heart: implications for in utero cardiac gene therapy. Human Gene Therapy. 2004;15(9):842–855. doi: 10.1089/hum.2004.15.842. http://dx.doi.org/10.1089/hum.2004.15.842. [DOI] [PubMed] [Google Scholar]
- Sive HL, Grainger RM, Harland RM. The early development of Xenopus laevis: A laboratory manual. Plainview, New York: Cold Spring Harbor Laboratories; 1998. [Google Scholar]
- Sive HL, Grainger RM, Harland RM. Animal cap isolation from Xenopus laevis. Cold Spring Harbor Protocols. 2007a;2007 doi: 10.1101/pdb.prot4744. http://dx.doi.org/10.1101/pdb.prot4744 pdb prot4744. [DOI] [PubMed] [Google Scholar]
- Sive HL, Grainger RM, Harland RM. Isolating Xenopus laevis testes. Cold Spring Harbor Protocols. 2007b;2007 doi: 10.1101/pdb.prot4735. http://dx.doi.org/10.1101/pdb.prot4735 pdb prot4735. [DOI] [PubMed] [Google Scholar]
- Sive HL, Grainger RM, Harland RM. Synthesis and purification of digoxigenin-labeled RNA probes for in situ hybridization. Cold Spring Harbor Protocols. 2007c;2007 doi: 10.1101/pdb.prot4778. http://dx.doi.org/10.1101/pdb.prot4778 pdb prot4778. [DOI] [PubMed] [Google Scholar]
- Sive HL, Grainger RM, Harland RM. Xenopus laevis egg collection. Cold Spring Harbor Protocols. 2007d;2007 doi: 10.1101/pdb.prot4736. http://dx.doi.org/10.1101/pdb.prot4736 pdb prot4736. [DOI] [PubMed] [Google Scholar]
- Sive HL, Grainger RM, Harland RM. Xenopus laevis in vitro fertilization and natural mating methods. Cold Spring Harbor Protocols. 2007e;2007 doi: 10.1101/pdb.prot4737. http://dx.doi.org/10.1101/pdb.prot4737 pdb prot4737. [DOI] [PubMed] [Google Scholar]
- Sive HL, Grainger RM, Harland RM. Microinjection of RNA and preparation of secreted proteins from Xenopus oocytes. Cold Spring Harbor Protocols. 2010a;2010(12) doi: 10.1101/pdb.prot5538. http://dx.doi.org/10.1101/pdb.prot5538 pdb prot5538. [DOI] [PubMed] [Google Scholar]
- Sive HL, Grainger RM, Harland RM. Microinjection of Xenopus embryos. Cold Spring Harbor Protocols. 2010b;2010(12) doi: 10.1101/pdb.ip81. http://dx.doi.org/10.1101/pdb.ip81 pdb ip81. [DOI] [PubMed] [Google Scholar]
- Sorokin SP. Reconstructions of centriole formation and ciliogenesis in mammalian lungs. Journal of Cell Science. 1968;3(2):207–230. doi: 10.1242/jcs.3.2.207. [DOI] [PubMed] [Google Scholar]
- Steinman RM. An electron microscopic study of ciliogenesis in developing epidermis and trachea in the embryo of Xenopus laevis. American Journal of Anatomy. 1968;122(1):19–55. doi: 10.1002/aja.1001220103. http://dx.doi.org/10.1002/aja.1001220103. [DOI] [PubMed] [Google Scholar]
- Stubbs JL, Davidson L, Keller R, Kintner C. Radial intercalation of ciliated cells during Xenopus skin development. Development. 2006;133(13):2507–2515. doi: 10.1242/dev.02417. http://dx.doi.org/10.1242/dev.02417. [DOI] [PubMed] [Google Scholar]
- Stubbs JL, Oishi I, Izpisua Belmonte JC, Kintner C. The forkhead protein Foxj1 specifies node-like cilia in Xenopus and zebrafish embryos. Nature Genetics. 2008;40(12):1454–1460. doi: 10.1038/ng.267. http://dx.doi.org/10.1038/ng.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbs JL, Vladar EK, Axelrod JD, Kintner C. Multicilin promotes centriole assembly and ciliogenesis during multiciliate cell differentiation. Nature Cell Biology. 2012;14(2):140–147. doi: 10.1038/ncb2406. http://dx.doi.org/10.1038/ncb2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan FE, Vladar EK, Ma L, Fuentealba LC, Hoh R, Espinoza FH, et al. Myb promotes centriole amplification and later steps of the multiciliogenesis program. Development. 2013;140(20):4277–4286. doi: 10.1242/dev.094102. http://dx.doi.org/10.1242/dev.094102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitre BD, Cleveland DW. Centrosomes, chromosome instability (CIN) and aneuploidy. Current Opinion in Cell Biology. 2012;24(6):809–815. doi: 10.1016/j.ceb.2012.10.006. http://dx.doi.org/10.1016/j.ceb.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadsworth P. Studying mitosis in cultured mammalian cells. Cold Spring Harbor Protocols. 2007;2007 doi: 10.1101/pdb.prot4674. http://dx.doi.org/10.1101/pdb.prot4674 pdb prot4674. [DOI] [PubMed] [Google Scholar]
- Werner ME, Hwang P, Huisman F, Taborek P, Yu CC, Mitchell BJ. Actin and microtubules drive differential aspects of planar cell polarity in multiciliated cells. Journal of Cell Biology. 2011;195(1):19–26. doi: 10.1083/jcb.201106110. http://dx.doi.org/10.1083/jcb.201106110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner ME, Mitchell BJ. Using Xenopus skin to study cilia development and function. In: Marshall WF, editor. Methods in Enzymology, volume 525: Cilia, Part B. Vol. 525. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner ME, Mitchell JW, Putzbach W, Bacon E, Kim SK, Mitchell BJ. Radial intercalation is regulated by the Par complex and the microtubule-stabilizing protein CLAMP/Spef1. Journal of Cell Biology. 2014;206(3):367–376. doi: 10.1083/jcb.201312045. http://dx.doi.org/10.1083/jcb.201312045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Zhu L, Zhu Y, Cao J, Li S, Huang Q, et al. The Cep63 paralogue Deup1 enables massive de novo centriole biogenesis for vertebrate multiciliogenesis. Nature Cell Biology. 2013;15(12):1434–1444. doi: 10.1038/ncb2880. http://dx.doi.org/10.1038/ncb2880. [DOI] [PubMed] [Google Scholar]