

Abstract
Background
Due to the high cost and low reproducibility of many microarray experiments, it is not surprising to find a limited number of patient samples in each study, and very few common identified marker genes among different studies involving patients with the same disease. Therefore, it is of great interest and challenge to merge data sets from multiple studies to increase the sample size, which may in turn increase the power of statistical inferences. In this study, we combined two lung cancer studies using micorarray GeneChip®, employed two gene shaving methods and a two-step survival test to identify genes with expression patterns that can distinguish diseased from normal samples, and to indicate patient survival, respectively.
Results
In addition to common data transformation and normalization procedures, we applied a distribution transformation method to integrate the two data sets. Gene shaving (GS) methods based on Random Forests (RF) and Fisher's Linear Discrimination (FLD) were then applied separately to the joint data set for cancer gene selection. The two methods discovered 13 and 10 marker genes (5 in common), respectively, with expression patterns differentiating diseased from normal samples. Among these marker genes, 8 and 7 were found to be cancer-related in other published reports. Furthermore, based on these marker genes, the classifiers we built from one data set predicted the other data set with more than 98% accuracy. Using the univariate Cox proportional hazard regression model, the expression patterns of 36 genes were found to be significantly correlated with patient survival (p < 0.05). Twenty-six of these 36 genes were reported as survival-related genes from the literature, including 7 known tumor-suppressor genes and 9 oncogenes. Additional principal component regression analysis further reduced the gene list from 36 to 16.
Conclusion
This study provided a valuable method of integrating microarray data sets with different origins, and new methods of selecting a minimum number of marker genes to aid in cancer diagnosis. After careful data integration, the classification method developed from one data set can be applied to the other with high prediction accuracy.
Background
Gene expression profiling is increasingly being used to aid adenocarcinomas (AD) identification, classification, and prognosis [1-3]. Recent studies suggested that primary solid tumors carrying an AD metastatic gene-expression signature were most likely to be associated with metastasis and poor clinical outcome, i.e. metastatic signature genes are encoded in primary AD tumors [4]. These results provide multiple data sets with similar diseased samples and also indicate the potential and effective use of gene expression profiling analysis in early cancer detection.
Microarray experiments are expensive and usually exhibit high noise within each experiment and low reproducibility among multiple data sets [5]. Thus, it is not surprising to find very few common marker genes among different studies with the same diseased samples [1,2]. In addition, since cancer patients for microarray experiments are usually limited, it is beneficial to combine data from different studies to increase the sample size, which may then increase the power of the statistics analysis. When combining different data sets, one has to consider at least the data scales, distributions, and sample similarity [4-6]. Therefore, valid mathematical methods to preprocess/transform data sets are necessary to obtain an integrated data set.
Beer et al [1] and Bhattacharjee et al [2] reported top 50 (and top100) genes and top 175 genes, respectively, in their studies to separate normal and different states of AD samples. Among their gene lists, at least 25 genes were used to differentiate normal from diseased samples. These gene lists may be appropriate for microarray-based AD diagnosis. However, the long gene list would add significant costs (time and labor) to PCR-based clinical tests. For the latter application, a statistically valid means of selecting fewer marker genes without compromising the prediction accuracy is of great importance.
The traditional method to select a set of marker genes is as follows: 1) rank the genes according to their significance in gene expression differences between diseased and normal samples using a statistical test (e.g. t-test); 2) use a classification method to evaluate the prediction error by using the top one gene, followed by the top two genes, the top three genes and so on until a pre-specified number of genes or a minimum prediction error is reached [1,2]. This method neglects the gene-gene interactions that may exert significant effects on the traits of interest. For example, assuming that gene 1 (g1) and gene 2 (g2) are the top two genes, when we consider two genes jointly, other two genes (not g1 or g2) may have more significant effect than g1 and g2 due to gene-gene interactions [7]. It is also impractical to search every possible gene combination (i.e. every one gene, every two genes, every three genes, and so on) due to the large number of genes in microarray data sets. We herein proposed a gene shaving method based on Random Forests (GSRF) and another based on Fisher's Linear Discrimination (GSFLD) to search for a set of genes whose expression levels can accurately classify diseased and normal samples. Fisher's Linear Discrimination (FLD) is a traditional classification method that has computational efficiency, while Random Forests (RF), as proposed recently by Breiman [8,9] is based on growing an ensemble of trees (classifiers) on bootstrapped samples, which improves the classification accuracy significantly. Identification of genes that are correlated to survival via gene expression analysis may lead to a better clinical prognosis. Proportional hazard regression model [10] is a very popular method to model survival-related data, and the principal component analysis (PCA) is an effective method to reduce data dimensions when dealing with multiple-variable data sets (e.g. microarray data).
In this analysis, we have 1) developed a statistically and biologically valid means of integrating two microarray gene expression data sets (Affymetrix's HumanFL and HumanGenome_U95Av2); 2) developed new methods to identify the minimum number of genes necessary for accurate prediction of disease status between healthy and AD patients, and to evaluate if the classifier built on one data set can be applied to the other; and 3) applied both a proportional hazard regression model and a PCA to identify a set of genes whose expression patterns are highly correlated with patient survival.
Results
We obtained two data sets (referred to as data set 1 and data set 2 in the following discussion) of Affymetrix's GeneChip® ".CEL" files from Beer et al. [1] and Bhattacharjee et al. [2]. The two data sets were produced from 2 chip genereations, HumanFL and HumanGenome_U95Av2, respectively. Only normal (NL) and adenocarcinomas (AD) samples were used in this article because 1) data set 1 only has NL and AD samples; 2) AD is the predominant histological subtype representing ~30% of all Lung Cancer (LC), and its progressive course and resultant patient survival are difficult to predict [1,3].
The original CEL files were processed using dChip software [11]. Twelve chips (L100, L102, L107, L27, L37, L54, L81, L88, L89, L90, L92, and L96) from data set 1 and 3 chips (AD382, AD315, and NL1698) from data set 2 were discarded during the quality control step (see Methods). Two chips (L111 and L24) in data set 1 were also removed due to the extremely low survival times (1.5 and 1.6 months, respectively). To improve the consistency of the tumor samples, only 84 AD samples with greater than 40% of tumor cells were used from data set 2 [1]. Thus, a total of 82 (72AD and 10 NL) and 99 (83 AD and 16NLs) samples from data set 1 and data set 2, respectively, and 6,124 common probe sets (the list is available upon request) were used for data analysis. Because the replicate samples (chips) in data set 2 were clustered together by Hierarchical Clustering Analysis (HCA), and had high correlation coefficients (R2 > 0.95, except one 0.84), the average intensities from replicate samples were used for further analysis.
It is not uncommon to find different data scales and distributions among microarray data sets from different studies that used the same platform (e.g. Affymetrix's GeneChip®) [5]. The two data sets used here were from two generations of Affymetrix's GeneChip® and were found to have different gene expression intensities. For example, the median gene expression intensities of AD/NL samples in the two data sets were 914/966 and 141/149, respectively. The maximum intensities were also quite different (40,898 and 12,190, respectively). In addition, the scatter plot and Quantile-Quantile (Q-Q) plot showed that the distributions of the two data sets were dissimilar (data not shown). HCA clustered the samples into two distinct groups according to the data sources rather than disease status (Figure 1A).
Figure 1.
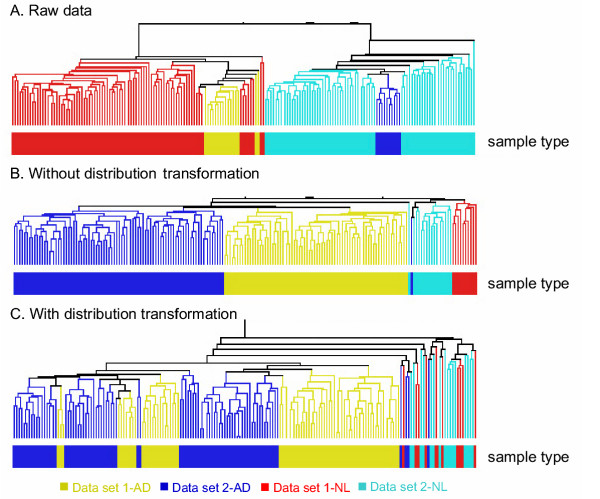
Hierarchical Clustering Analysis of two data sets. A: raw data without any normalization, B: partial normalized data without distribution transformation, and C: partial normalized data with distribution transformation. AD and NL refer to adenocarcinomas (AD) patients and normal (NL) samples, respectively.
Effects of data preprocessing
A total of 6,124 common probe sets were found after gene mapping. Among these, a total of 1,567 probe sets were removed by the Student's t-Test because they had inconsistent gene expression patterns between the two data sets. Therefore, a total of 4,557 common probe sets remained for further analysis.
In addition to common data transformation and normalization procedures, we added a distribution transformation (disTran, or simply transformation) step before gene normalization (see Methods). Data processed with disTran showed a greatly improved consistency in gene expression patterns between the two data sets (see scatter plot in Figure 2), i.e. the expression levels of corresponding genes are plotted closely to the theoretical diagonal line. The Q-Q plot (Figure 3) with disTran also showed that the distributions of the two data sets are nearly identical with almost all data points falling on the theoretical line. Without disTran, HCA could not merge the two data sets as seen by the two distinct clusters reflecting the two data sources in Figure 1A. However, with disTran, similar patient samples (AD or NL) from the two data sets were clustered together, and diseased and normal samples were more distinctly separated (Figure 1B and 1C). Improvement of data uniformity was also supported by PCA, with the first two components explaining 10% more variance in the transformed data than that in the non-transformed data (data not shown). In the following analysis, we used the distribution transformed combined data set.
Figure 2.
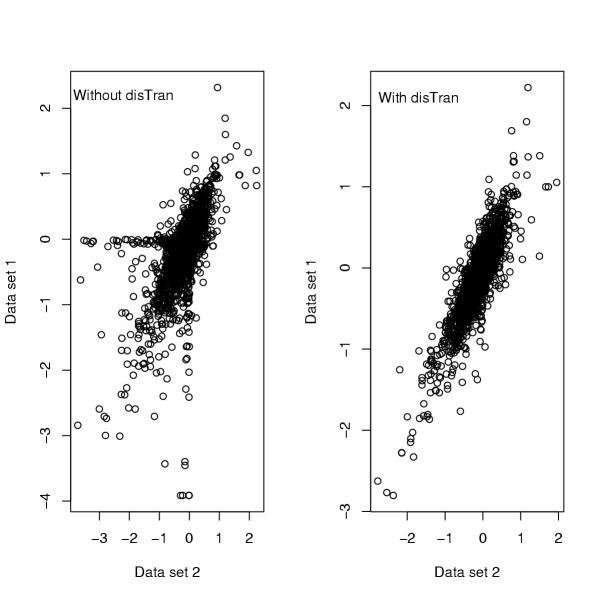
Scatter plot comparing data distributions with and without distribution transformation (disTran).
Figure 3.
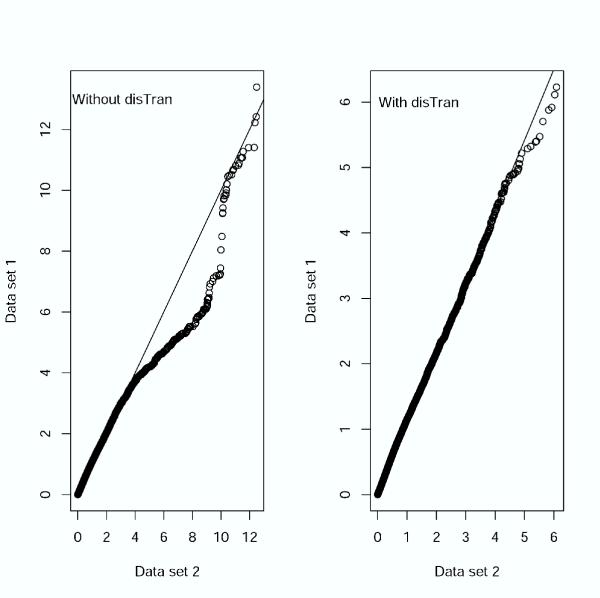
Quantile-Quantile (Q-Q) plot comparing data distributions with and without distribution transformation (disTran).
Signature genes to predict AD and normal samples
Since healthy normal samples were distinctively separated from AD samples by PCA and HCA (Figure 1C), we then used two different methods, GSRF and GSFLD, to select marker genes that can predict NL and AD samples. Using the GSRF approach to analyze the combined data set, the out-of-bag errors and prediction errors were all zero when using more than 15 probe sets. Errors started to occur when the probe sets were reduced to less than 15. The out-of-bag errors, prediction errors, and the probe set IDs are listed in Table 1. Our results show that the GSRF classifiers based on a minimum of 6 probe sets can predict the AD and normal samples with no error. Since there was one sample misclassified when we used 13 probe sets, we believed that the use of 15 probe sets for GSRF prediction may be more reliable. The GSRF method does not appear to be over-fit since the prediction accuracy was essentially unchanged as the number of genes used as the identifier increased (Table 1). Table 1 also shows that the classifiers built from one data set can predict the samples in the other with 100% accuracy, suggesting the usefulness of the GSRF technique for analysis of microarray data derived from independent yet related studies.
Table 1.
The out-of-bag and prediction errors1 of the last 15 nested probe sets2 using GSRF.
| No. of probe sets | Out-of-bag error | Prediction error | Probe set ID (HG_U95Av2) | |||
| D13 | D23 | C3 | D1→D23 | D2→D13 | ||
| 1 | 1 | 1 | 2 | 1 | 2 | 268_at |
| 2 | 1 | 1 | 1 | 0 | 1 | + 35868_at |
| 3 | 1 | 0 | 1 | 0 | 1 | + 1596_g_at |
| 4 | 1 | 0 | 1 | 0 | 0 | + 38430_at |
| 5 | 0 | 0 | 1 | 0 | 0 | + 32542_at |
| 6 | 0 | 0 | 0 | 0 | 0 | + 40282_s_at |
| 7–12 | 0 | 0 | 0 | 0 | 0 | +198_at, 32184_at, 39031_at, 36627_at, 1815_g_at, 39631_at |
| 13 | 0 | 1 | 0 | 0 | 0 | + 40419_at |
| 15 | 0 | 0 | 0 | 0 | 0 | +1814_at, 39350_at |
1: The number of misclassified samples. 2: Sets of genes (probe sets) produced by iteratively removing 10% of the least significant genes at a time. 3: D1: Data set 1; D2: Data set 2; C: Combined data set; D1→D2: use D1 to predict D2; D2→D1: use D2 to predict D1. +: Additional probe set ID plus those identified from lower number probe-set list(s). For example, the 3 probe sets contains 1596_g_at in addition to those (35868_at and 268_at) identified from the 2 probe sets prediction.
A similar analysis was conducted using the GSFLD method. In contrast to the GSRF results, when more than 10 probe sets were used, the error rate became larger as more genes were involved (data not shown). When using identifiers with 10 probe sets or less, both the leave-one-out cross-validation error and the prediction error from one data set to the other were considerably low (<3%) as shown in Table 2. Considering the cross-validation and prediction errors, it seems that the number of probe sets between 5 and 10 is the best. Since fewer genes are more sensitive to experimental errors, we focus our discussion on the GSFLD results based on 10 probe sets in the following paragraph. Selected marker genes and their functional classifications from the two gene shaving analysis methods are listed in Table 3 [see Additional file 1].
Table 2.
The leave-one-out cross-validation and prediction errors1 of the last 10 nested probe sets2 using GSFLD.
| No. of probe sets | Leave-one-out cross-validation error | Predict error | Probe set ID (HG_U95Av2) | |||
| D13 | D23 | C3 | D1→D23 | D2→D13 | ||
| 1 | 2 | 1 | 4 | 1 | 2 | 35868_at |
| 2 | 0 | 0 | 2 | 0 | 1 | + 38430_at |
| 3 | 0 | 0 | 1 | 0 | 1 | + 36247_f_at |
| 4 | 0 | 0 | 1 | 1 | 1 | + 32542_at |
| 5 | 0 | 0 | 1 | 0 | 1 | + 39220_at |
| 6 | 1 | 0 | 1 | 1 | 1 | + 38299_at |
| 7 | 1 | 0 | 1 | 1 | 1 | + 1596_g_at |
| 8 | 1 | 0 | 1 | 1 | 1 | + 39350_at |
| 9 | 1 | 0 | 1 | 1 | 1 | + 31525_s_at |
| 10 | 0 | 0 | 0 | 1 | 1 | + 37407_s_at |
1: The number of misclassified samples. 2: Sets of genes (probe sets) produced by iteratively removing 10% of the least significant genes at a time. 3: D1: Data set 1; D2: Data set 2; C: Combined data set; D1→D2: use D1 to predict D2; D2→D1: use D2 to predict D1. +: Additional probe set ID plus those identified from lower number probe-set list(s). For example, the 3 probe sets contains 36247_f_at in addition to those (38430_at and 35868_at) identified from the 2 probe sets prediction.
GSRF selected 15 probe sets (representing 13 genes), 6 (TGFBR2, FHL1, AGER, COX7A1, DF, STOM) of which were also identified by Bhattacharjee et al. [2] in their NL cluster. Another 6 genes or their family members (EDNRB, EMP2, FABP4, GPC3, LMO2, TEK) were found in the CancerGene Database, an integrated database of cellular genes (mostly experimentally validated) involved in different cancers [12]. In terms of the gene function, 2 (TGFBR2 and GPC3) are tumor suppressor genes, 3 (AGER, LMO2, and TEK) are oncogenes, and 3 are cancer-related genes or belong to gene families that are cancer-related. In comparison, GSFLD selected 10 probe sets (representing 10 genes), among which two (FHL1 and AGER) were found in the top 25 genes of the NL cluster by Bhattacharjee et al. [2]. Another 6 genes or their family members are cancer related (MYH11, SCGB1A1, FABP4), oncogenic (IL6, TEK), or tumor-suppressor genes (GPC3) based on the CancerGene Database. The two gene shaving methods together identified 5 common marker genes (AGER, DF, FHL1, GPC3, TEK) that should be more reliable to use for AD diagnosis.
Marker genes to predict AD patient survival
Applying the univariate Cox proportional hazard regression model separately to the two preprocessed data sets, we found 44 common probe sets (representing 36 genes) whose expression patterns are significantly (p < 0.05) correlated with patient survival (Table 4 [see Additional file 2]). Because the expression of these genes may be coordinately regulated, we applied PCA, a data reduction method, to further reduce the probe set list. Using the above procedure with an α = 0.8, we further reduced the list of probe sets to 20 (representing 16 genes). The prediction survival curves using these 16 genes were compared with the original Kaplan-Meier survival curves (Figure 4). It is noted that the tumor stages have different survival rates, and the predicted curves are very similar to the observed survival curves. The results also suggest that these16 genes are able to predict patient survival time.
Figure 4.
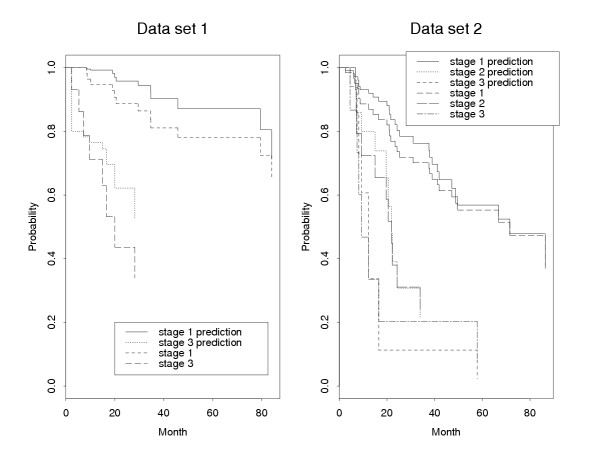
Comparison between the original Kaplan-Meier survival curves and the predicted survival curves using the selected 16 genes. Data set 1 has 72 patients with tumor stages 1 and 3, while data set 2 has 83 patients with tumor stages 1, 2, and 3.
Among the top 36 survival significant genes, 11 of them were identified by Beer et al. [1] and Bhattacharjee et al. [2], and additional 15 genes or related family members are present in the CancerGene Database. Among these 26 genes, 7 are tumor-suppressor genes (BBC3, BMP2, CDS1, HOXA4, NME2, RRM1, TPM2), 9 oncogenes (ARHC, CCR7, GAPD, GPI, LTBR, CKAP4 (P63), PLD3, RALA, TMP21), and 2 cancer-related genes (RGS7, SLC7A6) [12]. The expression of gene GPI (survival rank #2) was correlated with more tumor aggressiveness and inferior prognosis in pulmonary adenocarcinomas [12]. We classified selected marker genes into seven functional classes based on Gene Ontology information (Tables 3, 4 [see Additional files 1,2]).
Discussion
As more and more microarray data are released to the public domain (e.g. Gene Expression Omnibus at NCBI), it is becoming increasingly recognized that more information may be derived if multiple, independently generated data sets targeting the same biological question can be integrated. This is especially important given the large variation, both biological (e.g. differences between patients or disease states) and non-biological (e.g., variations in microarray production or hybridization), associated with microarray analysis.
Nimgaonkar et al. [5] conducted a series of experiments and compared the reproducibility of gene expressions across two generations of the Affymetrix Genechip, the same as we compared in this study: HumanFL and HumanGenome_U95Av2. They reported 2,200 (27%) of the total Affymetrix mapped 8,044 probe sets had negative correlations, i.e. the gene expression patterns changed in opposite directions between the two generations. Similarly, we found the discrepancy (1,567 probe sets in 6,124 common probe sets, ~26%) in gene expression patterns between the two data sets. Therefore, removing 'outlier' genes (i.e. genes having significantly different expression patterns between the two data sets) during data preprocessing is an important step for joint analysis in order to minimize the discrepancies between data sets, especially when the causes for such variations are not known. We acknowledge that we may have lost some relevant genes by the outlier screening. For instance, only ~50% of the genes in data set 2 were used in the final analysis. However, this drawback is unavoidable when combining different data sets originating from different types of microarray chips/platforms that differ in the number of genes/probe sets. A standardized microarray platform for each genome may be a good way to solve the problem.
Another significant source of variations can be ascribed to the hybridization signal intensities that differed greatly between these two chip generations/data sets. Data set 1 had higher overall intensity and lower variance than data set 2. These differences may be due to a ten-fold decrease in photo-multiplier tube (PMT) settings used for data acquisition associated with data set 2 [5], or altered probe design (e.g. probe numbers for each gene and the probe sequences). As a result, data distributions varied greatly between the two raw and even normalized (without disTrans) data sets. Several techniques have been reported to unify these data sets using various rescaling, filtering, and normalization methods [4,6]. We have also tested several methods, and the one we report here, unique distribution transformation based on weighted averages of normal and diseased samples, produced the best data integration results in terms of data scale, data distribution and sample (e.g. diseased vs. normal) clustering (Figure 1,2,3).
Commonly used microarray analysis methods, such as HCA and neural network, are effective for pattern classification and prediction, but do not provide gene ranking for identification of marker genes. To aid marker gene identification, we have developed GSRF and GSFLD methods by combining the idea of gene shaving, an unsupervised classification [13], with the supervised classification techniques, RF [8,9] and FLD [14]. Both methods use backward stepwise procedures to shave off a proportion of the least important genes at a time, forming nested gene sets that are subjected to marker gene set selection. Both methods produce ranked gene lists in the order of their significance/importance, thereby permitting a minimum number of genes to be selected without compromising the prediction accuracy. Comparing the two methods, GSFLD is easy to compute but could be over-fit, while GSRF is not over-fit but computationally more extensive. Considering the prediction accuracy, the GSRF method performs better than the GSFLD method. Although there are 5 and 59 common probe sets found in the top 10 and top 100 marker probe sets, respectively, by the two methods, the gene lists are not identical. The reason for this may be due to different algorithms or the fact that some genes are highly correlated in gene expression patterns. Nevertheless, different sets of genes may be equally useful to predict AD and NL samples.
The Cox proportional hazard regression model is often used when there is incomplete information (censoring) concerning the patient survival time. Based on the model, a forward selection method is applied to the two data sets separately, and the common marker genes are considered to be significantly correlated with patient survival. Since the expression patterns of these genes may be correlated, it is possible to further reduce the gene list to a smaller subset by PCA. The number of components is chosen so that at least 80% of the variation in the data can be explained. Based on these criteria, we are able to identify a smaller set of genes that are significantly correlated with survival. The method we used is especially efficient when there are many correlated covariates.
Although some of our selected marker genes/probe sets were not found in other similar studies [1,2,6,12], their association with AD can not be excluded and warrant further investigations. For example, PGK1 was recently experimentally validated to be significantly associated with lung cancer patient survival by 2D PAGE, immunohistochemistry of tissue arrays, and ELISA analysis [15].
Conclusions
This paper presents a unique and statistically efficient technique of integrating different microarray data sets and two new methods (GSRF and GSFLD) of selecting a minimal number of cancer-related marker genes. Two generations of Affymetrix lung cancer data sets were used to test the validity and efficiency of the methods. After careful data integration, a handful of marker genes were selected and the classification method developed from one data set can be applied to the other with high prediction accuracy. A two-step survival test selected a minimal set of 16 genes highly correlated with patient survival. These techniques would be of practical value in reducing PCR-based clinical cost in adenocarcinomas diagnosis and prognosis.
Methods
Sources of experimental data
The original 96 and 254 CEL files in data set 1 and data set 2 were processed using dChip software [11]. Any chip with a probe set outlier percentage no less than 5% is discarded for quality insurance.
Data processing
Gene mapping (common probe set identification)
Based on probe selection methods and sequence information, the Affymetrix company mapped 6,623 probe sets from the HumanFL chip to 7,094 probe sets from the HumanGenome_U95Av2 chip (spread-sheet file of PN600444HumanFLComp.zip is downloadable from http://www.affymetrix.com/support/technical/comparison_spreadsheets.affx). Since multiple probe sets having different target sequences could be mapped to the same UniGene clusters, mapping common genes using probe sets' sequence comparison is more reliable and conservative than using UniGene clusters. In order to obtain a more stringent subset of matched genes/probe sets, we conducted the following procedures:
1) Selected those probe sets with overlap sequence identity ≥65% and overlap sequence ≥50 bases in the column of 'old → new Target Seq Relationship'. After this step, a total of 6,616 probe sets remained.
2) If one 'Old_Probe_Set' matched multiple 'New_Probe_Set' names, selected those that have identity ≥80% and sequence ≥100 bases. After this step, there are a total of 6,333 probe sets left. There were multiple old probe sets (as well as their matching new probe sets) corresponding to the same gene in this list.
3) Selected only one 'Old_Probe_Set' per 'New_Probe_Set' from the above output with the following priorities: select those that are 'identical' first, then select those with the highest percentage, and finally, if the percentage was the same, chose the one with the longest sequence. However, if the percentages and the sequence lengths were the same, all probe sets were retained. After this step, a total of 6,073 probe sets remained.
We also removed 7 out of these 6,073 probe sets because they were absent in data set 2. In addition, we added 58 common probe sets (with prefix of 'AFFX') because they were not listed in Affymetrix's spread-sheet but instead were listed in both raw data sets. The total number of common probe sets used in the analysis was 6,124.
Data transformation and normalization
We conducted the following data transformation and normalization steps using GeneSpring (v6.1) [16] and in-house C program [see Additional file 3]. Unique procedures in this study are in bold:
1) Assigned 0.01 to those probe sets with processed expression intensities less then 0.01;
2) Per chip normalization: the expression of each probe set in each chip was divided by the median of the chip;
3) Gene filtering: Student's t-Test was used to filter out genes that had significantly different expression patterns between the two data sets. AD and NL samples were calculated separately without assuming equal variances. Since there was a multiple test problem, Bonferroni correction is applied. The p-value cut-off was set to be 0.00001.
4) Distribution transformation (disTran): After the above normalizations, the scales and distributions of the two data sets were still different (Figure 2 and 3). Because of this, we proposed a distribution transformation method to transform the two data sets to a similar distribution. For two random variables X and Y with Cumulative Distribution Function (CDF) FX(x) and FY(y), respectively, let
Z = F-1X (FY(Y)) (1)
Z and X then have the same distribution. Since the CDF of a random variable is usually unknown, we used the empirical distribution as an estimate of the CDF. Specifically, we conducted the following transformation: 1) Using data set 1, we constructed a reference sample by using an equation of ((mean expression of AD samples)/2 + (mean expression of NL samples)/2). The gene expression data of the reference sample was then denoted by G = (g1, g2,...,gn), where g1 ≤ g2 ≤ ... ≤ gn and n is the number of genes; 2) Considering the gene expression data of the reference sample as X and the gene expression data of each sample in data set 1 or data set 2 as Y, we transformed each of the samples in the two data sets using formula (1) such that the gene expression data of each sample had the same distribution as that of the constructed sample. For each of the samples with gene expression data Q = (q1, q2,...,qn), this transformation was equivalent to the transformation of qi to gj if the rank of qi in Q is j.
5) Per gene normalization: each gene is divided by the mean of corresponding NL samples.
6) Natural log transformation of the above processed data.
Except for the above distribution transformation, three other similar transformations were tested for step 4 above: i) Transformed data set 2 to a reference sample, which is the average of all samples in data set 1; ii) Transformed both data sets according to a reference sample, which is the average of 10 normal samples in data set 1; iii) The same as ii) except using the average of all 82 samples as the reference sample in data set 1. We also tested three slightly different gene normalization methods for step 5 above, i.e. divide each gene's intensity by either i) its median, ii) the median of the normal samples only, or iii) the means of the normal samples. Since some genes had notably different data scales (~10 fold) with a similar number of samples, we chose to use iii) for this paper in order to keep a better representation of the raw data. For a similar reason, we did a distribution transformation after partial normalization (i.e. after steps 1 and 2 above) within each data set because it minimized the errors from different chips.
Clustering and statistical tests
HCA on patient samples was carried out in GeneSpring® (version 6.1) [16]. Pearson correlation was used as the similarity measure, and no further branching if the distance is less than 0.01. The plotting of data distribution (scatter plot and Q-Q plot) and Kaplan-Meier survival curves were implemented using functions (e.g. qqplot(), coxph(), survfit()) in R and S-PLUS® (version 6.1) [17], respectively.
Marker (signature) gene selection
GSRF
Random Forests is based on construction of classification trees using bootstrapped samples of the original data set. After a large number of trees are generated, they vote for the most popular class. The predicted class of each sample is determined by the votes of the tree classifiers for which the given sample is "out-of-the bag", i.e. not included in the bootstrapped samples used to build the tree. This prediction is called out-of-bag prediction and the corresponding error is called out-of-bag error, which is an unbiased estimate of the true error rate. For a variable (gene), its margin is the proportion of votes for its true class minus the maximum proportion of votes for each of the other classes. The importance of a variable is quantified by average lowering of the margin across all samples when this variable is randomly permutated. In summary, when we applied RF to a data set, we obtained an out-of-bag error rate as an 'importance measure' for each variable.
In principle, we can choose the marker genes based on the 'importance measure'. However, it is difficult to set a cut-off value when there are many variables and most of them have very similar 'importance measures'. We proposed a gene shaving method to choose marker genes based on RF. The gene shaving method was originally proposed by Hastie et al. [13] based on PCA. Our gene shaving process was as following:
1) Let S1 denote all the variables in the original data set. Run the RF using the variables in S1 and get an 'importance measure' for each of the variables.
2) Delete a proportion (10% in the present study) of variables with the smallest 'importance measure'. Let S2 denote all remaining variables. Run RF using the variables in S2 and get an 'importance measure' for each of the variables in S2.
Continue this procedure until Sk contains only one variable. In this way, we get a group of nested variable sets S1 ⊃ S2 ⊃ S3 ... ⊃ Sk. For each variable set, we have an out-of-bag error and may use it to choose the best variable set. In the present study, we used the combination of out-of-bag errors and prediction errors between the two data sets to choose the best gene sets. We first applied the above mentioned stepwise procedure of GSRF to the combined data set. In each step, we generated 10,000 trees to calculate the 'importance measure' of each gene and then shaved off 10% of the probe sets according to their 'importance measures'. Next, we did the following prediction steps for each set of the probe sets beginning from the smallest one: 1) applied RF to data set 1 to build 10,000 trees (classifiers); 2) applied the 10,000 classifiers to classify the samples in data set 1, and obtained an out-of-bag error for data set 1; 3) applied the 10,000 classifiers from data set 1 to classify the samples in data set 2 and obtained a prediction error, i.e. the prediction error using data set 1 to predict data set 2; 4) repeated steps 1–3 for data set 2 to obtain an out-of-bag error of data set 2 and a prediction error for data set 1.
GSFLD
One sample with multivariate gene expression data is denoted as G = (g1, g2,...,gn), where n is the number of genes. Fisher's method is to transform the multivariate data G to one dimension by the linear combination of α1g1 + α2g2 + ... + αngn. Choose αi (i = 1,..,n) such that the two classes are separated as much as possible [14]. After this transformation, each sample has one value (this value is called the 'super-gene expression data' in the following discussion). Let 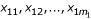 and
and 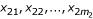 denote the super-gene expression data for the samples of the two classes (AD and NL), respectively. Let
denote the super-gene expression data for the samples of the two classes (AD and NL), respectively. Let 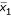 and
and 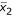 denote the sample mean of the two classes, respectively, and
denote the sample mean of the two classes, respectively, and 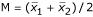 denotes the middle point. For a new sample with gene expression data G = (g1, g2,...,gn), the super-gene expression data of this sample is x0 = α1g1 + α2g2 + ... + αngn. The Fisher's discriminant function is that if x0 ≥ M, this sample is predicted to be class one; otherwise it is predicted to be class two. For each gene, we used the correlation coefficient between this gene and the super-gene as the 'importance measure'. We used the 'importance measure' and the gene shaving method discussed above to get nested gene sets, and used the leave-one-out cross-validation prediction error to choose the best gene sets. [see Additional file 4].
denotes the middle point. For a new sample with gene expression data G = (g1, g2,...,gn), the super-gene expression data of this sample is x0 = α1g1 + α2g2 + ... + αngn. The Fisher's discriminant function is that if x0 ≥ M, this sample is predicted to be class one; otherwise it is predicted to be class two. For each gene, we used the correlation coefficient between this gene and the super-gene as the 'importance measure'. We used the 'importance measure' and the gene shaving method discussed above to get nested gene sets, and used the leave-one-out cross-validation prediction error to choose the best gene sets. [see Additional file 4].
Survival-related genes selection
We considered 72 patients from data set 1 and 83 patients from data set 2. The clinical data between the two studies were similar in terms of age, gender, smoking status, and the percentage of cancer stage. However, survival (patient survival time in month from the operation date to death or last follow-up as of the study) distributions between the two data sets are quite different (Figure 4).
To select survival-related genes, we used the forward selection of the Cox regression model. We selected a number of probe sets whose gene expression patterns are significantly correlated (p < 0.05) to survival time in both data sets. PCA was used to further reduce the probe set list. Suppose we select m probe sets in the above step, then we do a principal component analysis on the combined samples with m probe sets each. For each sample, let xi denotes ith principal component, corresponding to eigenvalue λi and eigenvector ei, where λ1 ≥ ... ≥ λm. The goal is to determine the first L principal components such that 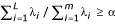 , here α is the proportion of variance explained (80% in this study) by the first L principal components.
, here α is the proportion of variance explained (80% in this study) by the first L principal components.
Using the first L principal components as the new variables, we applied the multivariate Cox proportional hazard regression model with different baseline hazard functions to the two data sets. For each gene k, this gene is selected if 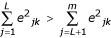 , where ejk is the kth element of ej. In other words, a gene is selected if it contributes more to the first L principal components than to the other principal components.
, where ejk is the kth element of ej. In other words, a gene is selected if it contributes more to the first L principal components than to the other principal components.
Author's contributions
HJ and YD initiated and designed the study. HJ evaluated different algorithms of data processing and clustering, conducted the distribution test, HCA and PCA, annotated the cancer gene functions, and drafted the manuscript. HSC performed the survival test and initial distribution test, and helped with the manuscript. LT conducted the GSFLR test. QS and SZ conducted GSRF. CJT helped with gene mapping algorithm development and the manuscript. YD and JC obtained gene expression intensity using the dChip program, tested several classification methods, and analyzed/classified gene functions. YD checked the quality of the preprocessed data. SZ developed the distribution transformation programs, guided the overall study, and helped with the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Selected marker genes to separate AD from NL samples using GSRF and GSFLD.
Selected survival-related genes using univariate Cox proportional hazard regression model and PCA.
The C program to integrate 2 data sets. It contains 5 files: integrate.exe, README4integration.txt, pparameter1.txt, data1.txt, and data2.txt (the last two are example data sets).
The C program to run GSFLD test. It contains 3 files: gsfld.exe, README4gsfld.txt, and inputdata.txt (a small example data set). Click here to download the above files: http://aspendb.mtu.edu/project/ad/paper1_download.html
Acknowledgments
Acknowledgements
The authors thank Sandra Hubscher for proof-reading the manuscript. This work was partially supported by the Michigan Life Sciences Corridor Funds of the Michigan Economic Development Corporation (grant no. 085P1000477) to CJT.
Contributor Information
Hongying Jiang, Email: hojiang@mtu.edu.
Youping Deng, Email: ydeng@ksu.edu.
Huann-Sheng Chen, Email: hschen@mtu.edu.
Lin Tao, Email: ltao@mtu.edu.
Qiuying Sha, Email: qsha@mtu.edu.
Jun Chen, Email: jch5353@ksu.edu.
Chung-Jui Tsai, Email: chtsai@mtu.edu.
Shuanglin Zhang, Email: shuzhang@mtu.edu.
References
- Beer DG, Kardia SL, Huang CC, Giordano TJ, Levin AM, Misek DE, Lin L, Chen G, Gharib TG, Thomas DG, Lizyness ML, Kuick R, Hayasaka S, Taylor JM, Iannettoni MD, Orringer MB, Hanash S. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med. 2002;8:816–824. doi: 10.1038/nm733. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee A, Richards WG, Staunton J, Li C, Monti S, Vasa P, Ladd C, Beheshti J, Bueno R, Gillette M, Loda M, Weber G, Mark EJ, Lander ES, Wong W, Johnson BE, Golub TR, Sugarbaker DJ, Meyerson M. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci U S A. 2001;98:13790–13795. doi: 10.1073/pnas.191502998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber ME, Troyanskaya OG, Schluens K, Petersen S, Thaesler Z, Pacyna-Gengelbach M, van de Rijn M, Rosen GD, Perou CM, Whyte RI, Altman RB, Brown PO, Botstein D, Petersen I. Diversity of gene expression in adenocarcinoma of the lung. Proc Natl Acad Sci U S A. 2001;98:13784–13789. doi: 10.1073/pnas.241500798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet. 2003;33:49–54. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- Nimgaonkar A, Sanoudou D, Butte AJ, Haslett JN, Kunkel LM, Beggs AH, Kohane IS. Reproducibility of gene expression across generations of Affymetrix microarrays. BMC Bioinformatics. 2003;4:27. doi: 10.1186/1471-2105-4-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Critical Assessment of Microarray Data Analysis (CAMDA) 2003 Conference Submitted Abstracts (CAMDA 2003) http://www.camda.duke.edu/camda03/papers/
- Xiong M, Li W, Zhao J, Jin L, Boerwinkle E. Feature (gene) selection in gene expression-based tumor classification. Mol Genet Metab. 2001;73:239–247. doi: 10.1006/mgme.2001.3193. [DOI] [PubMed] [Google Scholar]
- Breiman L. Random forests. Machine Learning. 2001;45:5–32. doi: 10.1023/A:1010933404324. [DOI] [Google Scholar]
- Breiman L, Cutler A. Random Forests. Version 40. http://www.stat.berkeley.edu/users/breiman/RandomForests/cc_home.htm
- Cox DR. Regression models in life tables (with discussion) J Roy Sta Soc Ser B. 1972;34:187–220. [Google Scholar]
- Li C, Wong WH. Model-based analysis of oligonucleotide arrays: Expression index computation and outliers detection. Genome Biol. 2001;2:research0032.1–0032.11. doi: 10.1186/gb-2001-2-8-research0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CancerGene Database http://caroll.vjf.cnrs.fr/cancergene/
- Hastie T, Tibshirani R, Eisen MB, Alizadeh A, Levy R, Staudt L, Chan WC, Botstein D, Brown P. 'Gene shaving' as a method for identifying distinct sets of genes with similar expression patterns. Genome Biol. 2000;1:research0003.1–0003.21. doi: 10.1186/gb-2000-1-2-research0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson R, Wichern DW. Applied multivariate statistical analysis. New Jersey: Prentice-Hall; 1998. [Google Scholar]
- Chen G, Gharib TG, Wang H, Huang CC, Kuick R, Thomas DG, Shedden KA, Misek DE, Taylor JM, Giordano TJ, Kardia SL, Iannettoni MD, Yee J, Hogg PJ, Orringer MB, Hanash SM, Beer DG. Protein profiles associated with survival in lung adenocarcinoma. Proc Natl Acad Sci U S A. 2003;100:13537–13542. doi: 10.1073/pnas.2233850100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway AR. GeneSpring (version 61), Silicon Genetics, Redwood City, CA. 2003.
- S-PLUS (version 61), Insightful Corporation, Seattle, WA. 2003. http://www.insightful.com
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Selected marker genes to separate AD from NL samples using GSRF and GSFLD.
Selected survival-related genes using univariate Cox proportional hazard regression model and PCA.
The C program to integrate 2 data sets. It contains 5 files: integrate.exe, README4integration.txt, pparameter1.txt, data1.txt, and data2.txt (the last two are example data sets).
The C program to run GSFLD test. It contains 3 files: gsfld.exe, README4gsfld.txt, and inputdata.txt (a small example data set). Click here to download the above files: http://aspendb.mtu.edu/project/ad/paper1_download.html