

Abstract
Chronic kidney disease (CKD) has been associated with impaired host response and increased susceptibility to infections. Leukocyte recruitment during inflammation must be tightly regulated to protect the host against pathogens. FGF23 levels are increased in blood during CKD, and levels of this hormone have been associated with a variety of adverse effects in CKD patients. Here, we have shown that CKD impairs leukocyte recruitment into inflamed tissue and host defense in mice and humans. FGF23 neutralization during CKD in murine models restored leukocyte recruitment and host defense. Intravital microscopy of animals with chronic kidney failure showed that FGF23 inhibits chemokine-activated leukocyte arrest on the endothelium, and downregulation of FGF receptor 2 (FGFR2) on PMNs rescued host defense in these mice. In vitro, FGF23 inhibited PMN adhesion, arrest under flow, and transendothelial migration. Mechanistically, FGF23 binding to FGFR2 counteracted selectin- and chemokine-triggered β2 integrin activation on PMNs by activating protein kinase A (PKA) and inhibiting activation of the small GTPase Rap1. Moreover, knockdown of PKA abolished the inhibitory effect of FGF23 on integrin activation. Together, our data reveal that FGF23 acts directly on PMNs and dampens host defense by direct interference with chemokine signaling and integrin activation.
Introduction
Chronic kidney disease (CKD) is a common condition in developed countries, and the incidence can be high as 400 cases per 1,000,000 inhabitants (1). Loss of renal function is associated with a proinflammatory milieu and a concomitantly impaired immune system. In patients with CKD, the uremia-associated defect in the immune system has a broad clinical impact on morbidity and mortality. These patients are highly susceptible to infections and have an increased risk of atherosclerosis (2). In patients with CKD, the proinflammatory milieu is most likely caused by activation of immune cells and an increased oxidative stress, although the underlying condition may vary by severity and cause of CKD. Both phenomena are highly interrelated because proinflammatory cytokines might generate oxidative stress, whereas oxidative stress might modulate inflammation (3).
Little information is available about the mechanism of how CKD affects host defense. In a host with normal kidney function, leukocyte recruitment during inflammation and infection is initiated by selectin-mediated rolling on the vessel wall (4). Rolling leukocytes sense chemokines presented by activated endothelial cells, which bind their respective GPCRs on leukocytes (5, 6). Inside-out signaling induced by these receptors triggers CD18 (β2) integrin activation, which involves integrin clustering and a conformational change from a bent and inactive conformation to a fully extended conformation with high binding affinity for intercellular adhesion molecule-1 (ICAM-1) (7). The interaction of activated β2 integrins with ICAM-1 leads to leukocyte arrest on the endothelium and initiates transendothelial migration (8).
The family of FGFs consists of proteins that regulate cell proliferation, migration, differentiation, and survival (9). FGF23 is a recently discovered FGF and functions as an endocrine hormone that regulates phosphorus homeostasis through binding to FGF receptor (FGFR) and klotho, its coreceptor in the kidney and parathyroid glands (10–12). FGF23 levels are often 2- to 5-fold above the normal range during early and intermediate stages of CKD, but they can reach levels 1,000-fold above normal in advanced renal failure (13, 14). While compensatory increases in FGF23 levels help patients with CKD to maintain normal serum phosphate levels despite even severely reduced renal function, recent prospective studies of CKD and non-CKD patients demonstrated that elevated FGF23 levels during CKD are independently associated with infectious events and all-cause mortality, and they have a dose-dependent association between elevated FGF23 levels and greater risks of major cardiovascular events and mortality (13, 15–18). Although leukocytes express different high-affinity tyrosine kinase receptors (FGFR1, -2, and -4) — with FGFR2 being the only cell surface–bound FGFR — the effects of FGF23 on integrin activation and neutrophil activation and recruitment have not been investigated yet (19).
In this study, we analyzed the role of FGF23 in regulating the inflammatory response and host defense during CKD. We report that elevated FGF23 levels during CKD limit leukocyte recruitment and impair host defense by interfering with leukocyte integrin activation and recruitment.
Results
Neutrophil recruitment and host defense during pneumonia is decreased in mice with chronic kidney failure.
To investigate the effect of chronic kidney failure on host defense, we performed 5/6-nephrectomy in WT mice. This model system has been shown to reliably induce CKD in the murine system (20, 21). Ten days after surgery, pneumonia was induced in these mice by intratracheal injection of viable E. coli bacteria (22). In sham-operated animals, the induction of pneumonia led to a substantial recruitment of neutrophils into the bronchoalveolar lavage (BAL), and only a few bacterial CFUs could be detected in the BAL, lung, and spleen 24 hours after inducing pneumonia (Figure 1, A–D). The recruitment of neutrophils into the BAL was severely impaired in mice with chronic kidney failure, and consequently, these mice displayed an impaired bacterial clearance with high bacterial counts in the BAL, lung, and spleen after 24 hours (Figure 1, A–D).
Figure 1. Neutrophil recruitment and host defense during pneumonia is decreased in mice with chronic kidney failure.

CKD was achieved in mice by 5/6-nephrectomy, and pneumonia was induced by E. coli instillation after 10 days. (A–D) Twenty-four hours after inducing pneumonia, neutrophils were counted in the BAL (A), and CFUs were analyzed in the BAL (B), lung tissue (C), and spleen (D) of sham-operated and CKD mice (n = 4). (E–H) Pneumonia was induce in sham mice and CKD mice, and sham mice after pretreatment with FGF23 and neutrophils were counted in the BAL (F), lung tissue (G), and spleen (H) (n = 4). (I–P) Pneumonia was also induced in CKD mice after injection of a neutralizing FGF23 antibody (3 mg/kg body weight) or isotype control (I–L) and mice transplanted with control or FGFR2-KD BM (M–P) (n = 4). (Q and R) Survival curves of sham-operated animals and CKD animals (Q) and sham-operated animals and CKD animals treated with isotype or FGF23 neutralizing Ab (R). *P < 0.05, ANOVA plus Bonferroni testing in E–L or 2-tailed t test in A–D and M–P.
As plasma levels of FGF23 are elevated during chronic kidney failure (Supplemental Figure 1, A and B; supplemental material available online with this article; doi:10.1172/JCI83470DS1) and leukocytes express different FGFRs, we pretreated sham-operated animals with FGF23 and observed a dose-dependent inhibition of neutrophil recruitment into the BAL following pneumonia induction, which was associated with an impaired bacterial clearance in the BAL, lung, and spleen (Figure 1, E–H). In addition, inhibition of FGF23 by a neutralizing antibody (3 mg/kg) in mice with chronic kidney failure after inducing pneumonia improved host defense by restoring neutrophil recruitment and rescuing bacterial clearance in the BAL, lung, and spleen (Figure 1, I–L). The administration of an FGF23-neutralizing antibody in sham-operated animals did not significantly change neutrophil recruitment or bacterial burden (Figure 1, I–L).
As leukocytes express FGFR1, -2, and -4, we wanted to identify the FGFR responsible for transducing the effect of FGF23 in leukocytes. As only FGFR2 is expressed on the cell surface of neutrophils (19), we knocked down FGFR2 by using shRNA. FGFR2 knockdown (FGFR2-KD) on neutrophils was verified by Western blot (Supplemental Figure 5C) (23). FGFR2-KD in mice with chronic kidney failure rescued the host defense by restoring neutrophil recruitment, enabling efficient bacterial clearance in the BAL, lung, and spleen (Figure 1, M–P). In addition, when mice with chronic kidney failure were pretreated with the pan-FGFR inhibitor PD173074, neutrophil recruitment was restored and these mice were capable of efficient bacterial clearance, as indicated by low bacterial counts in the BAL, lung, and spleen (Supplemental Figure 2, A–D). In order to quantify kidney function after 5/6-nephrectomy, we measured serum creatinine levels and observed a significant increase in these animals compared with sham-operated mice (Supplemental Figure 3). Survival following induction of pneumonia was also significantly decreased in animals with chronic kidney failure compared with sham-operated animals (Figure 1Q). However, if these animals were treated with an FGF23 neutralizing antibody, survival was significantly improved (Figure 1R). Treatment of sham-operated animals with an FGF23 neutralizing antibody did not significantly modulate survival compared with animals with chronic kidney failure that received an FGF23 neutralizing antibody (Figure 1R).
CKD impairs neutrophil activation and recruitment by affecting β2 integrin–dependent functions.
To investigate how FGF23 affects leukocyte recruitment in vivo, we performed intravital microscopy of the cremaster muscle. Mice with chronic kidney failure had an increased leukocyte rolling velocity in postcapillary venules of the cremaster muscle (Figure 2A), a decreased number of adherent cells (Figure 2B), and a decreased number of leukocytes that emigrated into inflamed tissue (Figure 2C) compared with sham-operated control mice. Blocking FGFRs by injecting PD173074 in these mice restored leukocyte recruitment (Figure 2, A–C), suggesting that FGF23 affects different steps of the leukocyte recruitment cascade.
Figure 2. CKD impairs neutrophil slow rolling and arrest.

Sham-operated mice and CKD mice, after injection of vehicle or PD173074, were intrascrotally injected with TNF-α. (A–C) After 2 hours, the rolling velocity (A), the number of adherent cells (B), and the number of emigrated cells (C) were analyzed by intravital microscopy of postcapillary venules of the cremaster muscle (n = 4). Adoptive transfer was performed by labeling BM-derived cells from sham-operated mice, CKD mice, or sham-operated mice pretreated with FGF23 with green or red cell trackers. The labeled cells were injected into WT recipient mice 30 minutes after intrascrotal TNF-α injection. (D–F) After 1.5 hours, the rolling velocity (D), the percentage of adherent fluorescent cells (E), and the percentage of transmigrated fluorescent cells (F) were analyzed by intravital microscopy of postcapillary venules of the cremaster muscle (n = 4). For autoperfused flow chamber assays, a catheter was placed in a common aortic artery and connected to coated glass capillaries by PE-50 tubing. (G and H) Leukocyte rolling velocities on E-selectin and E-selectin/ICAM-1 (G) or P-selectin and P-selectin/ICAM-1 (H) were analyzed by video microscopy (n = 4).*P < 0.05, ANOVA plus Bonferroni testing in A–F or 2-tailed t test in G and H.
To exclude that endothelium-related factors in animals with chronic kidney failure are responsible for the observed effects, adoptive transfer experiments were performed. A timeline for these experiments is shown in Supplemental Figure 4A. BM cells from sham-operated mice and mice with chronic kidney failure were labeled with cell trackers and then injected into WT recipient animals in equal amounts 30 minutes after TNF-α injection (competitive recruitment assay). Compared with neutrophils from sham-operated mice, neutrophils from mice with chronic kidney failure displayed an increased rolling velocity and reduced adhesion in inflamed postcapillary venules of the cremaster muscle (Figure 2, D and E). Additionally, the neutrophils from sham-operated mice transmigrated at a higher number than neutrophils from mice with chronic kidney failure (Figure 2F). Pretreatment of mice with chronic kidney failure with PD173074 rescued leukocyte rolling velocity, intravascular adhesion, and transmigration, whereas pretreatment of sham-operated mice with FGF23 led to impaired leukocyte slow rolling, adhesion, and transmigration (Figure 2, D–F).
To confirm our intravital microscopy data, we performed autoperfused flow chamber experiments in vitro. Leukocytes from sham-operated mice and mice with chronic kidney failure had similar rolling velocities on E-selectin and P-selectin (Figure 2, G and H). Leukocytes from sham-operated animals displayed reduced rolling velocities in flow chambers coated with either E-selectin/ICAM1 or P-selectin/ICAM-1 (Figure 2, G and H). However, slow leukocyte rolling in leukocytes from mice with chronic kidney failure was impaired, as these cells rolled faster on E-selectin/ICAM-1 and P-selectin/ICAM-1, respectively (Figure 2, G and H). To selectively investigate chemokine-induced arrest in vivo, sham-operated mice and mice with chronic kidney failure were injected with CXCL1 i.v., and arresting leukocytes in the postcapillary venules of the cremaster muscle were analyzed by intravital microscopy. Chemokine-induced leukocyte arrest was reduced in mice with chronic kidney failure compared with sham-operated animals but could be rescued in mice with chronic kidney failure pretreated with PD173074 (Figure 3A). We also confirmed these findings by using flow chamber experiments in vitro, where the presence of CXCL1 induced firm arrest of leukocytes from sham-operated mice. However, leukocytes from animals with chronic kidney failure failed to firmly arrest in flow chambers coated with P-selectin/ICAM-1/CXCL1 (Figure 3B).
Figure 3. CKD impairs neutrophil activation and recruitment by affecting β2 integrin–dependent functions.

(A) Chemokine-induced arrest was investigated in vivo by injecting CXCL1 through an intravascular catheter and recording leukocyte arrest in postcapillary cremasteric venules by intravital microscopy (n = 4–5). (B) Chemokine-induced arrest was investigated in vitro by connecting glass capillaries coated with P-selectin/ICAM-1 or P-selectin/ICAM-1/CXCL1 to intraarterial catheters inserted in sham-operated mice or CKD mice (n = 4). FOV, field of view.(C–E) Murine WT PMNs were pretreated with control or FGF23 (1–100 ng/ml), and static adhesion on ICAM-1 coated surface (C) (n = 4), arrest under flow conditions in glass capillaries coated with P-selectin/ICAM-1 or P-selectin/ICAM-1/CXCL1 (D) (n = 9–12), and ICAM-1 binding (E) were analyzed (n = 3). (F) Superoxide production from untreated and FGF23-treated WT PMNs (n = 3). (G) Phagocytosis of pHrodo E. coli particles were analyzed by flow cytometry (n = 6). Control group (only cells) lacked coincubation with pHrodo E. coli particles. (H) Transendothelial migration of WT PMNs pretreated with control or FGF23 (n = 6). *P < 0.05, ANOVA plus Bonferroni testing in B–H or 2-tailed t test in A.
We have demonstrated that FGF23 inhibits chemokine-induced integrin activation on leukocytes in vivo. The main proinflammatory chemokine that triggers integrin activation on neutrophil during vascular inflammation in the murine system is CXCL1. Thus, we used stimulation with CXCL1 to investigate the inhibitory effect of FGF23 on neutrophil integrin activation. To confirm the in vivo data, we performed neutrophil adhesion assays and could demonstrate that PMN adhesion to ICAM-1 was decreased after pretreatment with FGF23 under both static conditions (Figure 3C) and flow conditions (Figure 3D) in vitro. To directly investigate the effect of FGF23 on integrin activation, we analyzed ICAM-1 binding by flow cytometry and observed a decreased ICAM-1 binding of PMNs pretreated with FGF23 (Figure 3E). To investigate whether FGF23 also inhibited integrin-dependent outside-in signaling on PMNs, we analyzed superoxide production and showed that FGF23 pretreatment significantly decreased ROS production of PMNs in vitro (Figure 3F). Using increasing concentrations of FGF23, we demonstrated that FGF23 significantly decreases neutrophil adhesion (Figure 3D), ICAM-1 binding (Figure 3E), and superoxide production (Figure 3F) at concentrations of 50 ng/ml and above. In addition, FGF23 pretreatment also inhibited integrin-dependent phagocytosis (Figure 3G) and transendothelial migration (Figure 3H). These data suggest that FGF23 can selectively inhibit different neutrophil functions.
FGF23 inhibits integrin activation by signaling through FGFR2.
In order to investigate the effect of FGF23 on neutrophil β2 integrin activation, we used HL60 cells in a reporter-antibody–based flow chamber assay (24). Selectin-mediated slow rolling and chemokine-induced arrest of neutrophils is mediated by the conformational change of the β2 integrin LFA-1. To directly investigate the effect of FGF23 on selectin-mediated LFA-1 activation, we used the human reporter antibody Kim127, which specifically recognizes the intermediate conformation of LFA-1. In flow chambers coated with E-selectin and Kim127, FGF23 significantly reduced the number of adherent cells, indicating an inhibition of the transition of LFA-1 into the intermediate conformation (Figure 4A). To directly investigate the effect of FGF23 on chemokine-induced LFA-1 activation, we used the human reporter antibody mAb24, which specifically recognizes the high-affinity conformation of LFA-1. FGF23 significantly reduced the number of adherent cells in flow chambers coated with P-selectin/CXCL1 and mAb24, suggesting that FGF23 inhibits chemokine-induced LFA-1 activation (Figure 4B). Rap1 is known to be involved in β2 integrin activation at the distal end of the signaling pathway, and we demonstrated that the inhibitory effect of FGF23 was reversed by pretreating the cells with a constitutive active Tat-Rap1 fusion protein (Tat-Rap1-CA) (Figure 4, A and B).
Figure 4. FGF23 inhibits integrin activation by signaling through FGFR2.

(A) Flow chambers were coated with E-selectin or E-selectin/Kim127 and perfused with HL60 cells pretreated with FGF23 suspended in human plasma, and the number of adherent cells per field of view (FOV) was analyzed by video microscopy (n = 4). (B) Flow chambers were coated with P-selectin/IL-8 or P-selectin/IL-8/mAb24 and perfused with HL60 cells with FGF23, and the number of adherent cells per FOV was analyzed (n = 4). (C) Flow chambers were coated with E-selectin or E-selectin/Kim127 and perfused with HL60 FGFR2-KD cells pretreated with or without FGF23 (100 ng/ml), and the number of adherent cells per FOV was analyzed by video microscopy (n = 4). (D) Flow chambers were coated with P-selectin/IL-8 or P-selectin/IL-8/mAb24 and perfused with HL60 FGFR2-KD cells pretreated with or without FGF23 (100 ng/ml), and the number of adherent cells per FOV was analyzed by video microscopy (n = 4). (E) LFA-1 clustering on the cell membrane of HL60 cells was analyzed by confocal microscopy (n = 5–8). (F) Exemplary micrographs. Scale bars: 5 μm. *P < 0.05, ANOVA plus Bonferroni testing in A–E.
In order to investigate the effect of FGF23 on neutrophil β2 integrin activation through its proposed receptor FGFR2, we generated an FGFR2-KD in the promyelocytic HL60 cell line by using shRNA (25). FGFR2-KD on the protein level was verified by Western blot (Supplemental Figure 5A). The FGFR2-KD abolished the inhibitory effect of FGF23 on selectin- and chemokine-induced conformational LFA-1 activation (Figure 4, C and D). Besides conformational changes, integrin activation also causes clustering of these molecules. To investigate whether FGF23 also inhibits clustering of LFA-1, we performed confocal microscopy and could observe a significant decrease of IL-8–stimulated HL60 cells with clustered LFA-1 on the cell surface (Figure 4, E and F). However, this effect was reversed in FGFR2-KD HL60 cells. These data indicate that FGF23 signals through FGFR2 to exert its inhibitory effect on the selectin-induced intermediate conformation as well as on the chemokine-induced high-affinity conformation of LFA-1 on leukocytes. Previous studies could show that elevated serum levels of FGF23 during CKD mediate left ventricular hypertrophy independent of α-klotho (17, 26). In order to investigate whether FGF23 also inhibits integrin activation on neutrophils independent of α-klotho, we pretreated isolated human neutrophils with or without FGF23 and/or the α-klotho inhibitor D-saccharid acid 1,4-lactone monohydrate (DLAS) and analyzed mAb24 binding after IL-8 stimulation. The addition of DLAS did not reverse the inhibitory effect of FGF23 on mAb24, thus indicating a klotho-independent mode of action for FGF23 (Supplemental Figure 6).
FGF23 deactivates integrins by inhibitory signaling through PKA.
Previous studies have indicated that the exogenous activation of the protein kinase A (PKA) in leukocytes is involved in integrin deactivation (27). In order to investigate whether FGF23 exerts its inhibitory effect through PKA activation, we analyzed the serine phosphorylation of PKA substrates. IL-8 is the human analogue to the murine chemokine CXCL-1, and it triggers integrin activation on neutrophils during vascular inflammation. IL-8 stimulation of isolated human neutrophils caused a significant increase in serine phosphorylation of PKA substrates, as did FGF23 pretreatment of isolated human neutrophils without IL-8 stimulation (Figure 5, A and B). However, the phosphorylation of PKA substrates, indicating its (inhibitory) activity, was significantly potentiated when FGF23 pretreated cells were consecutively stimulated with IL-8 (Figure 5, A and B). However, the effect of FGF23 pretreatment on PKA activation was absent in FGFR2-KD HL60 cells (Figure 5C), indicating that FGF23 signals through binding to FGFR2 (Figure 5C). PKA-KD in HL60 (Supplemental Figure 5B) significantly increased mAb24 binding at baseline and after IL-8 stimulation, indicating increased integrin activation (Figure 5D). In addition, PKA-KD abolished the inhibitory effect of FGF23 on integrin activation (Figure 5D). In order to show that FGF23 treatment leads to sustained PKA activation, we pretreated neutrophils with FGF23, washed the cells, and analyzed PKA substrate phosphorylation at different time points. We detected increased PKA substrate phosphorylation up to 2 hours after FGF23 had been washed away (Figure 5E). To investigate the effect of FGF23 on the activation of Rap1 as a downstream effector involved in integrin activation, we pretreated isolated human neutrophils with FGF23 and observed an abrogated Rap1 activation after IL-8 stimulation compared with stimulated control neutrophils (Figure 5F).
Figure 5. FGF23 deactivates integrins by inhibitory signaling through PKA.

PKA substrate serine phosphorylation in isolated human neutrophils pretreated with or without FGF23 and stimulated with IL-8. (A) Western blot from four experiments. (B) Quantification by densitometry. (C) PKA substrate serine phosphorylation in control and FGFR2-KD HL60 cells pretreated with or without FGF23 and stimulated with IL-8 (Western blot from four experiments). NS, nonsilencing. (D) Control and PKA- KD HL60 cells were stimulated with IL-8 (100 ng/ml) at 37°C for 5 minutes, and binding of the reporter antibody mAb24 was assessed by flow cytometry (n = 6). (E) PKA substrate phosphorylation in murine PMNs after FGF23 pretreatment (100 ng/ml) was analyzed at different time points after washing away FGF23 (Western blot from four experiments). (F) Rap1 activation in human neutrophils with or without FGF23 pretreatment and IL-8 stimulation (Western blot from four experiments). *P < 0.05, ANOVA plus Bonferroni testing in B and D.
Selectin-mediated slow rolling and chemokine-induced arrest is impaired in human patients with CKD.
To extend our findings to the human system, we conducted flow chamber experiments with whole blood samples from CKD patients (stages 3–5) and healthy volunteers. There was no significant difference in demographical data, except for estimated glomerular filtration rate (eGFR) and serum creatinine levels (Table 1). Leukocytes from all groups displayed similar rolling velocities on E-selectin or P-selectin alone (Figure 6, A and B). Leukocytes from healthy volunteers reduced their rolling velocity on E-selectin/ICAM-1 or P-selectin/ICAM-1, respectively (Figure 6, A and B), whereas this reduction in the rolling velocity was significantly less pronounced in leukocytes from CKD stage 3 patients and was virtually abolished in leukocytes from CKD stage 4–5 patients (Figure 6, A and B). To show that FGF23 is responsible for abolished slow leukocyte rolling, we pretreated whole blood samples from healthy volunteers with increasing doses of FGF23 and observed a complete abrogation of neutrophil slow rolling on E-selectin/ICAM-1 or P-selectin/ICAM-1 at FGF23 concentrations above 100 ng/ml (Figure 6, C and D). To test whether chemokine-induced arrest is also impaired in human CKD patients, we performed adhesion flow chamber experiments and observed a significantly reduced chemokine-induced arrest in neutrophils from CKD stage 3–5 patients compared with healthy controls (Figure 6E). Preincubating leukocytes from CKD patients with PD173074 rescued chemokine-induced arrest (Figure 6E). In contrast, pretreating blood samples from healthy volunteers with recombinant FGF23 reduced chemokine-induced neutrophil arrest on P-selectin/ICAM-1/IL-8 in a dose-dependent manner (Figure 6F).
Table 1. Patient data.
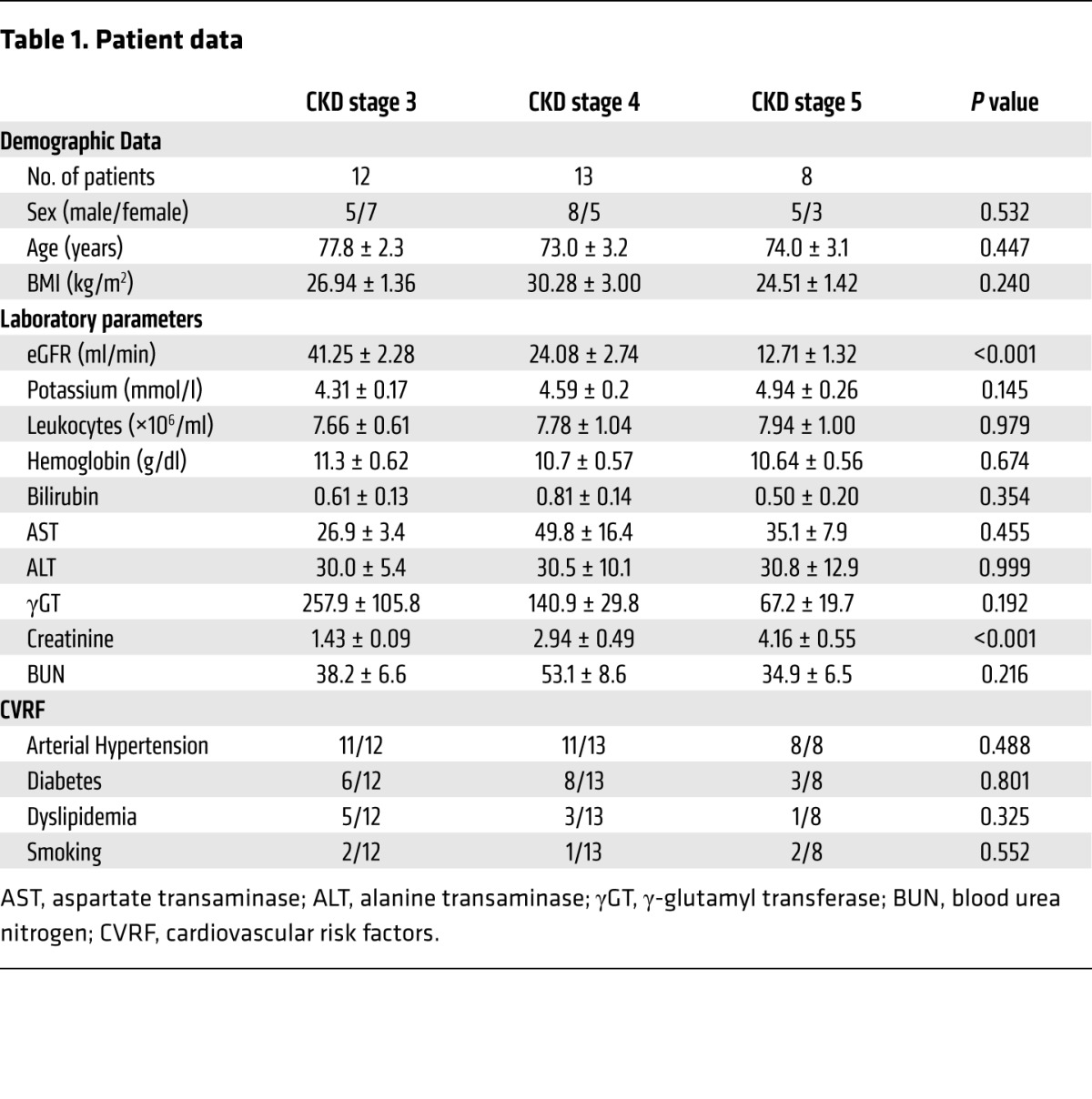
Figure 6. Selectin-mediated slow rolling and chemokine-induced arrest is impaired in human patients with CKD.

(A and B) Heparinised whole blood samples were obtained from healthy volunteers and CKD stage 3–5 patients and perfused through coated flow chambers, and the rolling velocity on E-selectin and E-selectin/ICAM-1 (A) or P-selectin and P-selectin/ICAM-1 (B) was analyzed by video microscopy (n = 8–13). (C and D) Whole blood samples from healthy volunteers were incubated with FGF23, and the rolling velocity in flow chambers with E-selectin and E-selectin/ICAM-1 (C) or P-selectin and P-selectin/ICAM-1 (D) was analyzed (n = 4). (E) To investigate chemokine-induced arrest, whole blood samples from healthy volunteers and CKD stage 3–5 patients were perfused through flow chambers coated with P-selectin/ICAM-1 or P-selectin/ICAM-1/IL-8 (n = 8–13). (F) Whole blood samples from healthy volunteers were incubated with FGF23, and the chemokine-induced arrest in flow chambers coated with P-selectin/ICAM-1 or P-selectin/ICAM-1/IL-8 was analyzed (n = 4). *P < 0.05, ANOVA plus Bonferroni testing in A, B, and E or 2-tailed t test in C, D, and F.
Discussion
This study indicates that CKD in the murine system and in human patients reduces neutrophil recruitment and host defense during inflammatory processes. We demonstrate that CKD affects different β2 integrin–dependent steps of the leukocyte recruitment cascade. FGF23 accumulates in the serum during the course of CKD (13, 14). We identify that FGF23 contributes to the defective leukocyte recruitment and host response. By using shRNA-mediated protein KD, we show that the FGFR2 on leukocytes is required for the inhibitory effect of FGF23 in β2 integrin activation. By using biochemical assays, we demonstrate that FGF23 activates PKA, and this results in deactivation of the downstream signaling intermediate Rap1. In a model of E. coli–induced pneumonia, we demonstrate the pathophysiological relevance of these observations.
While it is known that CKD patients have a higher incidence of recurrent bacterial infections (28), the underlying molecular mechanisms that are responsible for this phenomenon remain poorly defined. In addition, CKD may have both pro- and antistimulatory effects on immune cell recruitment. Interstitial leukocyte infiltration into the kidney itself may be initially increased during CKD, and CKD patients often present with progressive atherosclerotic diseases caused by a proinflammatory milieu, which may link these observations (2). However, it has also been shown that initial priming of neutrophils during CKD leads to unresponsiveness of neutrophils to further stimuli, accompanied by impaired migratory behavior and severely compromised phagocytic capacity, which contributes to vulnerability to exogenous pathogens (e.g., bacteria and fungi) and renders CKD patients prone to recurrent infections (29–31). This is in line with our findings that demonstrate decreased neutrophil recruitment and severely impaired host defense under inflammatory conditions in the murine system, as well as distinct impairments in several steps of the leukocyte recruitment cascade in samples from patients with CKD.
CKD causes accumulation of FGF23 in the plasma (17). Although various effects of increased FGF23 levels with respect to phosphate homeostasis regulation and myocardial hypertrophy have been described, the available evidence for a direct effect of FGF23 on the immune system is very scarce (17). It has been described that FGF23 may inhibit the extrarenal expression of vitamin D–activating enzyme 1α-hydroxylase in monocytes, thus regulating 1,25-dihydroxyvitamin D synthesis (32, 33). Moreover, a recently published study among chronic hemodialysis patients demonstrated that increased FGF23 levels were independently associated with a higher infection rate and higher all-cause mortality (18). However, this study included dialysis patients, and the mechanisms might differ from the data in our study. Yet, our findings are also in accordance with earlier evidence showing that high FGF23 levels are associated with higher mortality among CKD patients (15). However, FGF23 has not been shown to be directly linked to immune cell recruitment during inflammatory processes to this point. To our knowledge, this is the first report for the inhibitory effect of FGF23 on neutrophil activation and recruitment and, thus, the first report linking increased FGF23 levels during CKD to immune dysfunction. Shalhoub and colleagues observed an increased mortality of FGF23-neutralizing antibody–treated rats during chronic treatment over a time period of up to 6 weeks, which is possibly caused by increased aortic calcification (34). However, the short time (24 hours) of FGF23 neutralization in this study reversed neutrophil dysfunction in CKD mice and improved survival in an acute inflammatory model of bacterial pneumonia.
The innate immune response requires a tight regulation, as inadequate leukocyte recruitment may also harm the host itself (35). Several antiinflammatory factors have been identified to this date, including interleukins and their receptors such as an IL-R1 antagonist, IL-4, IL-6, IL-10, IL-11, IL-13, and TGF-β (36). Although these molecules act by diverse molecular signaling routes, they share a similarity in that they all act indirectly on the immune function, i.e., by affecting translational regulation. However, research over the last years could identify several antiinflammatory factors that modulate integrin activation directly by interacting with neutrophils. The first, GDF-15, has been shown to directly counteract chemokine-induced β2 integrin activation on neutrophils by activation of the inhibitory small GTPase Cdc42, resulting in deactivation of Rap1 (37). Additionally, we previously showed that GDF-15 also directly deactivates β3 integrins on platelets, resulting in both an antithrombotic effect as well as an antiinflammatory component (27). Several other endogenous regulators of neutrophil integrin activation have been identified, including Del-1, which inhibited neutrophil recruitment during periodontitis in an IL-17–dependent manner, and AnnexinA1, which limited arterial leukocyte recruitment during the pathogenesis of atherosclerosis (38, 39). With respect to our study, we observed that FGF23 modulated neutrophil integrin activation already after a short preincubation time, thus ruling out an indirect mode of action via translational regulation.
FGF23 exerts its inhibitory effect on integrin activation by a molecular pathway, including the small GTPase Rap1, which is a well-known signaling intermediate involved in integrin activation in various cell types (40, 41). In this study, we identified FGFR2 on neutrophils as the receptor for FGF23, thus providing a direct molecular pathway responsible for the observed inhibitory effects of FGF23. Importantly, FGFR3 is not expressed by neutrophils, and FGFR1 and FGFR4 are localized in the cytosol but not on the cell surface of neutrophils (19). This adds to our finding that the inhibitory effect of FGF23 on neutrophil integrin activation is mediated by binding and signaling via FGFR2.
By using biochemical analysis, we also demonstrated that the inhibitory molecule PKA gets activated upon FGF23 binding to FGFR2. FGF23 levels and PKA activation have also been associated with nonhematopoietic cells lines, e.g., in osteoclasts (42). Furthermore, low levels of FGF23 have been demonstrated to activate PKA in proximal tubule-like cells in vitro, although this required the presence of parathyroid hormone (PTH) (43). Further functional assays with HL60 cells strengthened the concept that PKA is indeed necessary to mediate the inhibitory effect of FGF23 on the intracellular signaling cascade and, thus, necessary to deactivate integrins. This is in accordance with previous studies indicating that the activation of PKA is linked to decreased neutrophil recruitment and reduced integrin-dependent neutrophil functions, e.g., migration, ROS production, polarization, and chemotaxis (44–48). In addition, we could previously demonstrate that the inhibitory effect of GDF-15 on β3 integrin activation in platelets is mediated by PKA activation (27). To our surprise, PKA was not only activated after FGF23 pretreatment, but was even more pronounced after IL-8 stimulation. Thus, it appears that the chemokine-triggered activating signaling processes are simultaneously accompanied by initiation of counterregulatory mechanisms, in this case PKA, that aid in balancing integrin activation and avoiding an overshooting response. This concept is also supported by our findings that chemokine-induced integrin activation is significantly increased in the absence of PKA in HL60, which drives the balance toward the integrin-activating pathways in the system. FGF23 levels have been reported to be elevated up to 1,000-fold in patients with CKD (13, 14, 49, 50). However, FGF23 levels vary among different publications and may be based on different methods used for measuring FGF23 levels (radioactive assays or ELISA-based methods). It is possible that some methods measure only active FGF23, whereas other methods determine total FGF23, which includes active as well as inactive FGF23. We measured FGF23 using a commercially available ELISA assay. Analysis of FGF23 levels in patients and mice revealed up to 1,000-fold elevated FGF23 levels in CKD mice compared with sham-operated control mice, as well as in CKD stage 5 patients compared with healthy control subjects (Supplemental Figure 1). These measurements justified the use of FGF23 concentration of up to 100 ng/ml for in vivo and in vitro experiments. Additionally, experiments with lower FGF23 concentrations demonstrated a dose-response kinetic for the inhibitory effect of FGF23 on integrin deactivation.
In conclusion, our data indicate a previously unknown role for FGF23 in neutrophil integrin deactivation, which inhibits neutrophil activation, adhesion, and transendothelial migration. We have demonstrated the inhibitory effect of FGF23 in several in vivo models as well as in vitro; thus, it is reasonable to consider FGF23-mediated inhibition of neutrophil activation and recruitment during CKD as a general principle. These findings may be of great interest to further elucidate the therapeutic potential of FGF23 for the control of inflammatory disorders in future studies.
Methods
Animals.
We used 8- to 12-week-old male C57BL/6 mice (Janvier Labs). The mice were kept in a barrier facility under specific pathogen–free (SPF) conditions.
Reagents.
Unless otherwise stated, all reagents were obtained from Sigma-Aldrich.
Murine model of chronic kidney failure.
Chronic kidney failure in mice was induced by 5/6-nephrectomy. Mice were anesthetized by i.p. injection of ketamine (125 μg/g body weight; Pfizer) and xylazine (12.5 μg/g body weight; Bayer). After epidermal hair removal and disinfection, a skin incision was performed and the kidneys in the retroperitoneal space were exposed. The right kidney was ligated and removed. The lower renal artery supplying the caudal half of the left kidney was ligated, and the cranial pole of the left kidney was thermocoagulated so that only 1/6 of the global renal tissue remained functional. After skin closure, animals were injected with 200 μl saline i.p. for volume resuscitation and kept on a heating pad until the animals had fully recovered from the procedure. Animals were used for further experiments 10 days after 5/6-nephrectomy.
E. coli–induced pneumonia.
E. coli–induced pneumonia was performed in mice as previously reported (22). Mice were anesthetized by i.p. injection of ketamine/xylazine and injected intratracheally with 6 × 106 viable E. coli (8 × 106 for survival analysis). After 24 hours, the mice were sacrificed and the lungs were lavaged 5× with 0.7 ml physiologic saline solution. The number of neutrophils in the BAL was counted using Kimura staining. E. coli CFUs in the lung, blood, and spleen were counted by serial plating on trypticase soy agar plates (TSA) agar plates (22). The number of detected CFUs was used to determine the actual E. coli bacterial burden after 24 hours. Some mice were injected with PD173074 (1 mg/kg body weight/day), recombinant FGF23 (carrier free, R&D Systems, catalog 2604-FG/CF) or an FGF23-neutralizing antibody (clone 58-5, Amgen; 3 mg/kg body weight) (34) 15 minutes after induction of pneumonia. To calculate the FGF23 dose for in vivo experiments, mice were weighed before the experiment and the blood volume was calculated by assuming a mean murine blood volume of 80 ml/kg bodyweight. In order to achieve an FGF23 concentration of 100 ng/ml, a mouse weighting 25 g was injected with 200 ng FGF23 via the tail vein 15 minutes after induction of pneumonia. In order to demonstrate effective FGF23 neutralization by the antibody, we injected the FGF23-neutralizing antibody in CKD mice and measured FGF23 serum levels before and after the administration of the neutralizing antibody. The administration of the FGF23-neutralizing antibody sharply decreased the detectable, circulating FGF23 levels, indicating effective neutralization (Supplemental Figure 7). In order to show this neutralization effect, we used the same antibody against FGF23 in the in vivo experiments and in ELISA. The rationale behind this approach is that, once the neutralization antibody binds to its epitope on FGF23, the same epitope cannot be detected by the same antibody that was used in the ELISA.
Hematopoietic stem cell isolation and retroviral transduction.
Retroviral supernatants were generated by transfecting a packaging cell line with the respective constructs using Lipofectamine 3000 (Thermo Fischer Scientific) as described previously (23). Supernatants were collected and used for infection of hematopoietic stem and progenitor cells as described previously (51). Briefly, BM cells were isolated from WT mice and were subjected to lineage depletion by using a lineage cell depletion kit (Miltenyi Biotec) according to the manufacturer’s protocol. Purified hematopoietic stem and progenitor cells were preincubated overnight in BM stimulation medium (DME, Invitrogen), 20% FCS supplemented with 10 ng/ml of murine IL-3, 5 ng/ml of murine IL-6, and 50 ng/ml of murine stem cell factor (all cytokines from PeproTech). RetroNectin-coated culture dishes (50 μg/ml; Takara Bio Inc.) were used to concentrate the viral particles. The prestimulated BM was seeded for overnight incubation on these plates with a density of 2 × 106/plate in 3 ml BM stimulation medium. After 2 days of transduction, efficiency was measured by flow cytometry. BM transplantation was performed as described previously (52). Recipient animals were lethally irradiated with 950 cGy to destroy the native BM, and only transduced donor stem cells were transplanted into recipient mice by tail-vein injection. The degree of BM reconstitution with donor stem cells was verified by flow cytometry (>95%), and the animals were used for experiments 6 weeks after transplantation. In order to show that FGFR2 shRNA effectively knocked down FGFR2 on neutrophils after transplantation, we injected lethally irradiated WT mice with BM stem cells that had been retrovirally transduced with FGFR2 or scrambled control shRNA. Flow cytometry of circulating neutrophils showed a strong reduction of FGFR2 expression in FGFR2-KD mice compared with control mice (Supplemental Figure 8).
Intravital microscopy of the cremaster muscle.
Intravital microscopy of anesthetized mice was performed as described (23, 25, 52–56). To induce inflammation, mice received an intrascrotal injection of 500 ng TNF-α (R&D Systems) 2 hours before the exteriorization of the cremaster muscle. The cremaster muscle was prepared for intravital imaging as previously described (23, 25, 52–56). For the investigation of chemokine-induced arrest, mice were injected i.v. with 500 ng CXCL1. Intravital microscopy was carried out on an upright microscope (Axioskop, Zeiss) with a 40×0.75 NA saline immersion objective. Leukocyte rolling velocity and leukocyte arrest were determined by transillumination intravital microscopy, whereas leukocyte extravasation was investigated by near-infrared reflected-light oblique transillumination microscopy. Recorded images were analyzed using ImageJ and AxioVision (Zeiss) software. Emigrated cells were determined in an area reaching out 75 μm to each side of a vessel for a vessel length distance of 100 μm (representing 1.5 × 104 μm2 tissue area). The microcirculation was recorded using a digital camera (Sensicam QE, Cooke Corporation). Blood flow centerline velocity was measured using a dual photodiode sensor system (Circusoft Instrumentation). Centerline velocities were converted to mean blood flow velocities as previously described. To perform the transfer of sham-operated leukocytes and CKD leukocytes, BM from sham-operated control animals and CKD mice was isolated and consecutively labeled with 2 mM green or orange cell tracker (Invitrogen) according to the manufacturer’s instructions. The cells were injected i.v. in a 1:1 ratio 30 minutes after intrascrotal injection of WT mice with TNF-α (57).
Static adhesion assay.
Isolated neutrophils were plated on ICAM-1–coated 24-well plates and incubated at 37°C and 5% CO2 for 1 hour. After removal of nonadherent cells by washing with PBS, adherent neutrophils per field of view were analyzed by microscopy (PrimoVert, Zeiss).
ICAM-1 binding assay.
ICAM-1 binding was analyzed as described previously (25, 57).
ROS production.
ROS production was analyzed as described previously (57). Briefly, isolated neutrophils were plated on poly-RGD–coated (20 mg/ml) 96-well plates with CaCl2 (1 mM), MgCl2 (1 mM), and cytochrome c (0.1 mM) in the presence of TNF-α (0.5 mg/ml) and/or SOD (~45 U), and plates were analyzed on a plate reader.
Phagocytosis.
pHrodo E. coli bioparticles (Invitrogen) were opsonized with 50% mouse serum at 37°C for 30 minutes. Isolated neutrophils were incubated with opsonized bioparticles at 37°C and 5% CO2 for 60 minutes. The phagocytosis of bioparticles was analyzed by flow cytometry (BD FacsCanto, BD Biosciences).
Transendothelial migration.
Transendothelial migration of isolated murine neutrophils though a bEnd.5 endothelial cell monolayer was analyzed as described before (58).
LFA-1 clustering.
LFA-1 clustering was analyzed as described before (25). Briefly, HL60 cells were stimulated with 100 ng/ml IL-8 (PeproTech) for 5 minutes. After fixation with 1% PFA/PBS, cells were blocked with 3% BSA/PBS for 30 minutes and incubated with anti–LFA-1 antibody (clone TS2/4, BioLegend) at 4°C overnight, followed by incubation with Alexa Fluor 488–coupled anti-mouse secondary antibody (Invitrogen) for 1 hour. Images were obtained using a spinning disc confocal microscope (Cell Observer SD, Zeiss).
Autoperfused murine flow chamber assay.
Autoperfused flow chamber experiments were performed as previously described (23, 25, 52–54). Briefly, rectangular glass capillaries were coated with E-selectin (2.5 μg/ml) alone or in combination with ICAM-1 (2 μg/ml; R&D Systems) for 2 hours and then blocked for 1 hour using casein (Thermo Fisher Scientific). To control the wall shear stress in the capillary, a PE-50 tubing (BD Biosciences) was connected to one side of the capillary. The other side of the chamber was connected to a PE-10 tubing (BD Biosciences) and inserted into a mouse carotid artery. Leukocyte rolling was recorded for 1 minute using an SW40/0.75 objective and a digital camera (Sensicam QE, Cooke Corporation).
Human flow chamber assay.
Blood-perfused flowchamber assays with human whole blood were performed as described previously (24, 31). Glass capillaries were coated with E-selectin (3.5 μg/ml), P-selectin (20 μg/ml), E-selectin/ICAM-1 (3.5/3.5 μg/ml), or P-selectin/ICAM-1 (20/5 μg/ml) for 2 hours. Chambers were blocked with casein 1% (Fisher Scientific) for 1 hour and, afterward, perfused with heparinised whole blood samples at a constant shear stress of 5–6 dynes/cm2. It has been demonstrated before that >90% of rolling cells in this system are polymorphonuclear neutrophils (24). For the analysis of chemokine-induced arrest, glass capillaries were coated with P-selectin (50 μg/ml) and ICAM-1 (5 μg/ml) or P-selectin (50 μg/ml), ICAM-1 (5 μg/ml), and IL-8 (5 μg/ml) for 2 hours. Rolling and adhering cells per field of view were counted after 2 minutes of perfusion with whole blood at 5–6 dynes/cm2, and the ratio of adhering to rolling cells was calculated.
Cell lines and constructs.
Stable KD of FGFR2 and PKA in promyelocytic HL60 (ATCC) cells was achieved by lentiviral transduction of shRNA as described previously (FGFR2 shRNA: 5′-CCGGATATGGATCAGAGGAGTAAATCTCGAGATTTACTCCTCTGA TCCATATTTTTTG-3′, PKA shRNA: 5′-CCGGTAGATCTCACCAAGCGCTTTGCTCGA GCAAAGCGCTTGGTGAGATCTATTTTTG-3′, or nonsense control shRNA: 5′-CCGGCAACAAGATGAAGAGCACCAACTCGAGTTGGTGCTCTTCATCTTGTTGTTTTTG-3′) (25). Lentiviral particles were produced in HEK293 cells cotransfected with the shRNA-expressing pLKO1.puro vector, pVSV-G, pMDLg, and p–RSV-REV. Downregulation of FGFR2 (FGFR2 antibody, clone D4H9, Santa Cruz Biotechnology Inc.) and PKA (PKA Cα antibody, catalog 4782, Cell Signaling Technology) in transduced HL60 was confirmed by Western blot and maintained by puromycin selection.
Reporter antibody adhesion flow chamber assay.
Adhesion flow chamber experiments were carried out as previously described (24, 25). Briefly, protein-G precoated glass capillaries were coated with E-selectin (6.6 μg/ml) and IgG1 (25 μg/ml) or Kim127 (25 μg/ml, purified from hybridoma) for 1 hour and blocked with casein (Thermo Fisher Scientific). In other experiments, capillaries were coated with P-selectin (20 μg/ml), IL-8 (50 μg/ml, PeproTech), and IgG1 (5 μg/ml) or mAb24 (5 μg/ml, gift from N. Hogg). HL60 cells were resuspended in human plasma with a density of 5 × 106/ml living cells. The flow chamber was perfused with the cell suspension for 2 minutes and washed with phosphate-buffered saline (1 mM MgCl2/CaCl2) for 1 minute. In representative images, the number of cells per field of view was determined.
Reporter antibody flow cytometry assay.
HL60 cells pretreated with or without FGF23 (100 ng/ml) were stimulated with IL-8 (100 ng/ml) for 5 minutes at 37°C in the presence of mAb24 (5 μg/ml). After centrifugation, cells were fixed with 2% paraformaldehyde and stained with secondary goat anti-mouse Alexa Fluor 488–coupled antibody (Invitrogen). mAb24 binding was analyzed by flow cytometry (BD FacsCanto, BD Biosciences).
PKA activation assay.
Isolated human neutrophils or HL60 cells were resuspended in PBS (1 mM CaCl2/MgCl2), incubated with FGF23 at 37 °C for 30 minutes, and stimulated with IL-8 (100 ng/ml) at 37°C for 3 minutes. Cells were lysed with RIPA buffer. Lysates were boiled with sample buffer, run on 10% SDS-PAGE, and immunoblotted using antibodies against phosphorylated PKA substrates or PKA Cα subunit (Cell Signaling Technology, catalogs 4782 and 9624). Immunoblots were developed using an ECL system (GE Healthcare).
Rap1 activation assay.
Rap1 activation in isolated human neutrophils following chemokine stimulation was investigated as described previously, with some modifications (37). Briefly, isolated human neutrophils or HL60 cells were incubated with FGF23 at 37°C for 30 minutes and consecutively stimulated with IL-8 (100 ng/ml) at 37°C for 3 minutes. After lysis with ice-cold lysis buffer (50 mm TrisHCl [pH 7.4]; 500 mM NaCl; 1% NP40; 10% glycerol; 2.5 mM MgCl2; 10 μg/ml PEFA-Block; 25 mM NaF; 1 mM vanadate; and 1 mM PMSF [1% Roche HALT protease inhibitor cocktail]), GTP-bound Rap1 (Rap1-GTP) was precipitated from whole cell lysates using GST-Ral beads (made in-house). Precipitated proteins were separated using SDS-PAGE, transferred to a PVDF membrane, and immunoblotted against Rap1. To determine the level of total Rap1, a small portion of the whole cell lysate was mixed with SDS sample buffer and separated by SDS-PAGE. Rap1 was detected using a rabbit polyclonal antibody (clone sc-65, Santa Cruz Biotechnology Inc.).
Measuring FGF23 levels.
FGF23 concentrations were analyzed using an ELISA assay as described in the manufacturer’s protocol (R&D Systems, catalog DY2604-05).
Statistics.
Statistical analysis was performed with SPSS (version 22.0) using a Wilcoxon test or 2-tailed t test as appropriate. More than two groups were compared using 1-way ANOVA followed by Bonferroni testing. Data distribution was assessed using a Kolmogorov-Smirnov test or Shapiro-Wilks test. Survival analysis was performed using a log-rank test. All data are represented as mean ± SEM. P values less than 0.05 were considered statistically significant.
Study approval.
All animal experiments were approved by the Landesamt für Natur, Umwelt und Verbraucherschutz Northrhine-Westfalia (LANUV, Düsseldorf, Germany). Approval to obtain whole blood samples from healthy volunteers and CKD patients was obtained from the Institutional Review Board (Ethik-Kommission der Ärztekammer Westfalen-Lippe und der medizinischen Fakultät der Westfälischen Wilhelms Universität Münster, approval number 2012-319-f-S). Written informed consent was received from participants prior to inclusion in the study. The study was registered in the German Clinical Trial Registry (study ID: DRKS00005794).
Author contributions
JR performed the experiments, analyzed the data, and wrote the manuscript. JO performed experiments. HVA, MM, SR, HJP, and MU contributed to writing the manuscript. AZ conceived of the study and wrote the manuscript.
Supplementary Material
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (ZA428/11-1 and SFB1009_A5 to A. Zarbock) and Cells-in-Motion Cluster of Excellence EXC 1003–CiM (University of Münster, Germany; to A. Zarbock). The authors would like to thank Nadja Giesbrecht and Charlotte Sohlbach for expert technical support.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2016;126(3):962–974. doi:10.1172/JCI83470.
Contributor Information
Jan Rossaint, Email: rossaint@uni-muenster.de.
Jessica Oehmichen, Email: jessica.oehmichen@web.de.
Hugo Van Aken, Email: hva@uni-muenster.de.
Stefan Reuter, Email: Stefan.Reuter@ukmuenster.de.
Melanie Meersch, Email: meersch@googlemail.com.
Mark Unruh, Email: MLUnruh@salud.unm.edu.
Alexander Zarbock, Email: zarbock@uni-muenster.de.
References
- 1.Levey AS, Coresh J. Chronic kidney disease. Lancet. 2012;379(9811):165–180. doi: 10.1016/S0140-6736(11)60178-5. [DOI] [PubMed] [Google Scholar]
- 2.Recio-Mayoral A, Banerjee D, Streather C, Kaski JC. Endothelial dysfunction, inflammation and atherosclerosis in chronic kidney disease — a cross-sectional study of predialysis, dialysis and kidney-transplantation patients. Atherosclerosis. 2011;216(2):446–451. doi: 10.1016/j.atherosclerosis.2011.02.017. [DOI] [PubMed] [Google Scholar]
- 3.Vaziri ND. Oxidative stress in uremia: nature, mechanisms, and potential consequences. Semin Nephrol. 2004;24(5):469–473. doi: 10.1016/j.semnephrol.2004.06.026. [DOI] [PubMed] [Google Scholar]
- 4.Phillipson M, Kubes P. The neutrophil in vascular inflammation. Nat Med. 2011;17(11):1381–1390. doi: 10.1038/nm.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ley K. Arrest chemokines. Microcirculation. 2003;10(3–4):289–295. doi: 10.1038/sj.mn.7800194. [DOI] [PubMed] [Google Scholar]
- 6.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7(9):678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 7.Luo BH, Carman CV, Springer TA. Structural basis of integrin regulation and signaling. Annu Rev Immunol. 2007;25:619–647. doi: 10.1146/annurev.immunol.25.022106.141618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zarbock A, Deem TL, Burcin TL, Ley K. Galphai2 is required for chemokine-induced neutrophil arrest. Blood. 2007;110(10):3773–3779. doi: 10.1182/blood-2007-06-094565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005;16(2):139–149. doi: 10.1016/j.cytogfr.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 10.Shimada T, et al. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci U S A. 2001;98(11):6500–6505. doi: 10.1073/pnas.101545198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kurosu H, et al. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem. 2006;281(10):6120–6123. doi: 10.1074/jbc.C500457200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Urakawa I, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444(7120):770–774. doi: 10.1038/nature05315. [DOI] [PubMed] [Google Scholar]
- 13.Isakova T, et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011;79(12):1370–1378. doi: 10.1038/ki.2011.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shimada T, et al. Circulating fibroblast growth factor 23 in patients with end-stage renal disease treated by peritoneal dialysis is intact and biologically active. J Clin Endocrinol Metab. 2010;95(2):578–585. doi: 10.1210/jc.2009-1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gutierrez OM, et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med. 2008;359(6):584–592. doi: 10.1056/NEJMoa0706130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Isakova T, et al. Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA. 2011;305(23):2432–2439. doi: 10.1001/jama.2011.826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Faul C, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest. 2011;121(11):4393–4408. doi: 10.1172/JCI46122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chonchol M, Greene T, Zhang Y, Hoofnagle AN, Cheung AK. Low vitamin D and high fibroblast growth factor 23 serum levels associate with infectious and cardiac deaths in the HEMO study. J Am Soc Nephrol. 2016;27(1):227–237. doi: 10.1681/ASN.2014101009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haddad LE, Khzam LB, Hajjar F, Merhi Y, Sirois MG. Characterization of FGF receptor expression in human neutrophils and their contribution to chemotaxis. Am J Physiol Cell Physiol. 2011;301(5):C1036–C1045. doi: 10.1152/ajpcell.00215.2011. [DOI] [PubMed] [Google Scholar]
- 20.Hayakawa S, et al. Cardiac myocyte-derived follistatin-like 1 prevents renal injury in a subtotal nephrectomy model. J Am Soc Nephrol. 2015;26(3):636–646. doi: 10.1681/ASN.2014020210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leelahavanichkul A, et al. Chronic kidney disease worsens sepsis and sepsis-induced acute kidney injury by releasing High Mobility Group Box Protein-1. Kidney Int. 2011;80(11):1198–1211. doi: 10.1038/ki.2011.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ittner A, et al. Regulation of PTEN activity by p38Δ-PKD1 signaling in neutrophils confers inflammatory responses in the lung. J Exp Med. 2012;209(12):2229–2246. doi: 10.1084/jem.20120677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Block H, et al. Crucial role of SLP-76 and ADAP for neutrophil recruitment in mouse kidney ischemia-reperfusion injury. J Exp Med. 2012;209(2):407–421. doi: 10.1084/jem.20111493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuwano Y, Spelten O, Zhang H, Ley K, Zarbock A. Rolling on E- or P-selectin induces the extended but not high-affinity conformation of LFA-1 in neutrophils. Blood. 2010;116(4):617–624. doi: 10.1182/blood-2010-01-266122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Herter JM, Rossaint J, Block H, Welch H, Zarbock A. Integrin activation by P-Rex1 is required for selectin-mediated slow leukocyte rolling and intravascular crawling. Blood. 2013;121(12):2301–2310. doi: 10.1182/blood-2012-09-457085. [DOI] [PubMed] [Google Scholar]
- 26.Grabner A, et al. Activation of cardiac fibroblast growth factor receptor 4 causes left ventricular hypertrophy. Cell Metab. 2015;22(6):1020–1032. doi: 10.1016/j.cmet.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rossaint J, Vestweber D, Zarbock A. GDF-15 prevents platelet integrin activation and thrombus formation. J Thromb Haemost. 2012;11(2):335–344. doi: 10.1111/jth.12100. [DOI] [PubMed] [Google Scholar]
- 28.Thompson S, et al. Cause of death in patients with reduced kidney function. J Am Soc Nephrol. 2015;26(10):2504–2511. doi: 10.1681/ASN.2014070714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anders HJ, Ninichuk V, Schlondorff D. Progression of kidney disease: blocking leukocyte recruitment with chemokine receptor CCR1 antagonists. Kidney Int. 2006;69(1):29–32. doi: 10.1038/sj.ki.5000053. [DOI] [PubMed] [Google Scholar]
- 30.Anding K, Gross P, Rost JM, Allgaier D, Jacobs E. The influence of uraemia and haemodialysis on neutrophil phagocytosis and antimicrobial killing. Nephrol Dial Transplant. 2003;18(10):2067–2073. doi: 10.1093/ndt/gfg330. [DOI] [PubMed] [Google Scholar]
- 31.Rossaint J, et al. Acute loss of renal function attenuates slow leukocyte rolling and transmigration by interfering with intracellular signaling. Kidney Int. 2011;80(5):493–503. doi: 10.1038/ki.2011.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bacchetta J, et al. Fibroblast growth factor 23 inhibits extrarenal synthesis of 1,25-dihydroxyvitamin D in human monocytes. J Bone Miner Res. 2013;28(1):46–55. doi: 10.1002/jbmr.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Viaene L, Evenepoel P, Meijers B, Vanderschueren D, Overbergh L, Mathieu C. Uremia suppresses immune signal-induced CYP27B1 expression in human monocytes. Am J Nephrol. 2012;36(6):497–508. doi: 10.1159/000345146. [DOI] [PubMed] [Google Scholar]
- 34.Shalhoub V, et al. FGF23 neutralization improves chronic kidney disease-associated hyperparathyroidism yet increases mortality. J Clin Invest. 2012;122(7):2543–2553. doi: 10.1172/JCI61405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zarbock A, Kempf T, Wollert KC, Vestweber D. Leukocyte integrin activation and deactivation: novel mechanisms of balancing inflammation. J Mol Med. 2012;90(4):353–359. doi: 10.1007/s00109-011-0835-2. [DOI] [PubMed] [Google Scholar]
- 36.Opal SM, DePalo VA. Anti-inflammatory cytokines. Chest. 2000;117(4):1162–1172. doi: 10.1378/chest.117.4.1162. [DOI] [PubMed] [Google Scholar]
- 37.Kempf T, et al. GDF-15 is an inhibitor of leukocyte integrin activation required for survival after myocardial infarction in mice. Nat Med. 2011;17(5):581–588. doi: 10.1038/nm.2354. [DOI] [PubMed] [Google Scholar]
- 38.Drechsler M, et al. Annexin A1 counteracts chemokine-induced arterial myeloid cell recruitment. Circ Res. 2015;116(5):827–835. doi: 10.1161/CIRCRESAHA.116.305825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eskan MA, et al. The leukocyte integrin antagonist Del-1 inhibits IL-17-mediated inflammatory bone loss. Nat Immunol. 2012;13(5):465–473. doi: 10.1038/ni.2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kinashi T, Katagiri K. Regulation of lymphocyte adhesion and migration by the small GTPase Rap1 and its effector molecule, RAPL. Immunol Lett. 2004;93(1):1–5. doi: 10.1016/j.imlet.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 41.Kinashi T, et al. LAD-III, a leukocyte adhesion deficiency syndrome associated with defective Rap1 activation and impaired stabilization of integrin bonds. Blood. 2004;103(3):1033–1036. doi: 10.1182/blood-2003-07-2499. [DOI] [PubMed] [Google Scholar]
- 42.Lavi-Moshayoff V, Wasserman G, Meir T, Silver J, Naveh-Many T. PTH increases FGF23 gene expression and mediates the high-FGF23 levels of experimental kidney failure: a bone parathyroid feedback loop. Am J Physiol Renal Physiol. 2010;299(4):F882–F889. doi: 10.1152/ajprenal.00360.2010. [DOI] [PubMed] [Google Scholar]
- 43.Weinman EJ, Steplock D, Shenolikar S, Biswas R. Fibroblast growth factor-23-mediated inhibition of renal phosphate transport in mice requires sodium-hydrogen exchanger regulatory factor-1 (NHERF-1) and synergizes with parathyroid hormone. J Biol Chem. 2011;286(43):37216–37221. doi: 10.1074/jbc.M111.288357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mizuno R, et al. In vivo imaging reveals PKA regulation of ERK activity during neutrophil recruitment to inflamed intestines. J Exp Med. 2014;211(6):1123–1136. doi: 10.1084/jem.20132112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li J, Zhao L, He X, Zeng YJ, Dai SS. Sinomenine protects against lipopolysaccharide-induced acute lung injury in mice via adenosine A(2A) receptor signaling. PLoS One. 2013;8(3): doi: 10.1371/journal.pone.0059257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reber LL, Daubeuf F, Nemska S, Frossard N. The AGC kinase inhibitor H89 attenuates airway inflammation in mouse models of asthma. PLoS One. 2012;7(11): doi: 10.1371/journal.pone.0049512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu HP, Hsieh PW, Chang YJ, Chung PJ, Kuo LM, Hwang TL. 2-(2-Fluorobenzamido)benzoate ethyl ester (EFB-1) inhibits superoxide production by human neutrophils and attenuates hemorrhagic shock-induced organ dysfunction in rats. Free Radic Biol Med. 2011;50(12):1737–1748. doi: 10.1016/j.freeradbiomed.2011.03.026. [DOI] [PubMed] [Google Scholar]
- 48.Neel NF, et al. VASP is a CXCR2-interacting protein that regulates CXCR2-mediated polarization and chemotaxis. J Cell Sci. 2009;122(pt 11):1882–1894. doi: 10.1242/jcs.039057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Isakova T, et al. Associations of FGF23 with change in bone mineral density and fracture risk in older individuals. J Bone Miner Res. doi: 10.1002/jbmr.2750. [published online ahead of print November 21, 2015]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wolf M. Update on fibroblast growth factor 23 in chronic kidney disease. Kidney Int. 2012;82(7):737–747. doi: 10.1038/ki.2012.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mocsai A, Abram CL, Jakus Z, Hu Y, Lanier LL, Lowell CA. Integrin signaling in neutrophils and macrophages uses adaptors containing immunoreceptor tyrosine-based activation motifs. Nat Immunol. 2006;7(12):1326–1333. doi: 10.1038/ni1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zarbock A, Abram CL, Hundt M, Altman A, Lowell CA, Ley K. PSGL-1 engagement by E-selectin signals through Src kinase Fgr and ITAM adapters DAP12 and FcR gamma to induce slow leukocyte rolling. J Exp Med. 2008;205(10):2339–2347. doi: 10.1084/jem.20072660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zarbock A, Lowell CA, Ley K. Spleen tyrosine kinase Syk is necessary for E-selectin-induced α(L)β(2) integrin-mediated rolling on intercellular adhesion molecule-1. Immunity. 2007;26(6):773–783. doi: 10.1016/j.immuni.2007.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mueller H, et al. Tyrosine kinase Btk regulates E-selectin-mediated integrin activation and neutrophil recruitment by controlling phospholipase C (PLC) gamma2 and PI3Kgamma pathways. Blood. 2010;115(15):3118–3127. doi: 10.1182/blood-2009-11-254185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stadtmann A, et al. Rap1a activation by CalDAG-GEFI and p38 MAPK is involved in E-selectin-dependent slow leukocyte rolling. Eur J Immunol. 2011;41(7):2074–2085. doi: 10.1002/eji.201041196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stadtmann A, et al. The PSGL-1-L-selectin signaling complex regulates neutrophil adhesion under flow. J Exp Med. 2013;210(11):2171–2180. doi: 10.1084/jem.20130664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Germena G, Volmering S, Sohlbach C, Zarbock A. Mutation in the CD45 inhibitory wedge modulates integrin activation and leukocyte recruitment during inflammation. J Immunol. 2015;194(2):728–738. doi: 10.4049/jimmunol.1401646. [DOI] [PubMed] [Google Scholar]
- 58.Schnoor M, et al. Cortactin deficiency is associated with reduced neutrophil recruitment but increased vascular permeability in vivo. J Exp Med. 2011;208(8):1721–1735. doi: 10.1084/jem.20101920. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.