

Abstract
Mitochondria are a distinguishing feature of eukaryotic cells. Best known for their critical function in energy production via oxidative phosphorylation (OXPHOS), mitochondria are essential for nutrient and oxygen sensing and for the regulation of critical cellular processes, including cell death and inflammation. Such diverse functional roles for organelles that were once thought to be simple may be attributed to their distinct heteroplasmic genome, exclusive maternal lineage of inheritance, and ability to generate signals to communicate with other cellular organelles. Mitochondria are now thought of as one of the cell’s most sophisticated and dynamic responsive sensing systems. Specific signatures of mitochondrial dysfunction that are associated with disease pathogenesis and/or progression are becoming increasingly important. In particular, the centrality of mitochondria in the pathological processes and clinical phenotypes associated with a range of lung diseases is emerging. Understanding the molecular mechanisms regulating the mitochondrial processes of lung cells will help to better define phenotypes and clinical manifestations associated with respiratory disease and to identify potential diagnostic and therapeutic targets.
Introduction
Maternally inherited and thought to be of bacterial descent, mitochondria are iconic double-membrane structures full of convoluted cristae that are present in nearly all cells and possess their own genome, transcriptome, and proteome. Revolutionary studies of bioenergetics in the 1950s, 1960s, and 1970s deemed mitochondria the “powerhouses” of the cell; however, while mitochondria throughout our bodies produce the bulk of the ATP needed for cells to live, our understanding of mitochondrial biology has undergone major transformations since the mysteries of oxidative phosphorylation (OXPHOS) were unraveled decades ago (1). In the last decade, the arrival of state-of-the-art high-throughput genomics, metabolomics, and complementary discoveries in signal transduction pathways have helped rapidly expand the list of genes encoding mitochondrial proteins as well as identify metabolites that are linked to human disease. It is becoming increasingly clear that abnormal mitochondrial signatures and mitochondrial dysfunction underlie the pathological mechanisms behind a plethora of lung diseases, including but not limited to chronic obstructive pulmonary disease (COPD), asthma, and lung cancer. It is also becoming clear that alterations in the mitochondrial genome, proteome, and metabolome may act not only as independent pathological processes, but also synergistically with existing pathological mechanisms to predispose, promote, or exacerbate lung disease. Given that lung diseases are among the leading causes of death worldwide, with four respiratory disease categories appearing in the worldwide top ten causes of mortality, together accounting for one in six deaths globally, the need for novel diagnostic and therapeutic approaches is critical (2). This article will discuss the multiple regulatory facets of mitochondrial function in normal lung homeostasis and the prominence of mitochondrial dysfunction in acute and chronic lung injury and in lung disease. It will also highlight the concept that mitochondria are no longer thought of as simple, discrete, kidney bean–shaped energy factories, but are now believed to encompass a cell- and tissue-specific, dynamic organellar network that fuses, divides, and directs a vast array of functions central to cellular life, death, and differentiation (1).
Bioenergetics and nutrient sensing
The mammalian lung is composed of over forty different cell types, which are regionally and spatially localized throughout the organ and contain varying levels of mitochondria (3). Functionally, nearly every cell in the lung depends on mitochondrial metabolic activities, requiring a constant supply of energy from OXPHOS. Mitochondria are at the hub of cellular metabolism, regulating the continuous aerobic oxidation of fatty acids (FAs) and consuming the end products of glucose, glutamine, and amino acid degradation in order to aerobically produce ATP from oxygen and H2O (Figure 1). The oxygen consumption rate of the lung is comparable to the oxygen consumption rates of other organs, including the intestine, pancreas, and spleen, but it is considerably lower than that of the brain, heart, and kidney (4). Likewise, the ATP content of the lung is similar to that of other organs, such as the brain, liver, and kidney (5), and is mostly dependent on mitochondrial sources (4).
Figure 1. Mitochondrial OXPHOS occurs in the IMM and is responsible for the continuous aerobic oxidation of FAs and the end products of glucose, glutamine, and amino acid degradation in order to aerobically produce ATP from oxygen and H2O.

Lung mitochondria preferentially use glucose-derived substrates, such as pyruvate, for oxidative energy production; however, other energy sources, including FAs, Krebs cycle intermediates, glycerol-3-phosphate, and glutamate, are also used, with the highest O2 consumption rates achieved with succinate as a substrate (6). Lung mitochondria also have a unique advantageous metabolic adaptation to aerobic OXPHOS, owing to the fact that the lung possesses its own isoform of electron transport chain (ETC) complex IV, cytochrome c oxidase (COX subunit IV-2), which is present in all lung cells, is oxygen sensitive, and renders lung COX two-fold more active (oxygen-binding) compared with COX in other tissues (7).
Type II alveolar epithelial cells (AECs), which continuously release surfactant by exocytosis onto the epithelial cell surface, have approximately three times greater mitochondrial volume per cell than other lung cells, such as endothelial or type I AECs (8). Bronchial (ciliated) epithelial cells, which require mitochondria for cilia beating, as well as vascular smooth muscle cells (VSMCs) and alveolar macrophages (AMs) are also rich in mitochondria (9). During differentiation from type II to type I as a part of the normal physiological replacement or repair mechanism, type II AECs reduce the number and size of mitochondria (10), resulting in type I AECs having a lower energy demand with less COX expression (11). Type II AECs rely on mitochondria to produce acetyl-CoA for de novo FA synthesis, allowing for the generation of phospholipids that are needed to produce pulmonary surfactants. Irregularities in phospholipid production, such as an excessive increase in cardiolipin or abnormalities in the carnitine acyltransferase pathway, disrupt surfactant composition and, consequently, lung function (12). Under altered physiologic states, such as starvation, type II AECs rely on FAs as a source of energy (13). High rates of FA synthesis correlate with morphological transformations in lung mitochondria, and under conditions of cellular stress, mitochondria of type II AECs transform into lamellar bodies to facilitate more surfactant production (14). Such alterations in mitochondrial bioenergetic metabolism allow lung cells to adapt to cellular stresses; however, excessive or prolonged modifications to these processes may be pathogenic to normal lung function and have profound effects on the clinical indices of many lung diseases, including COPD, pulmonary hypertension (PH), asthma, cystic fibrosis (CF), and lung cancer, as discussed below.
COPD is a debilitating lung disease encompassing airway inflammation (chronic bronchitis), destruction of lung tissue (emphysema), and remodeling of the small airways (15). The pathogenesis of COPD involves aberrant inflammatory and dysregulated cellular responses of the lung to cigarette smoke (CS) exposure. In lung epithelial cells, cytotoxic exposure to CS reduces mitochondrial OXPHOS (15, 16), whereas treatment with nontoxic doses of CS increases mitochondrial metabolic activity (17, 18), inducing a metabolic shift from glucose (glycolysis) to palmitate (β-oxidation) metabolism (18). Loss of acetyl-CoA and the Krebs cycle intermediate succinate is observed in basal cells of smokers (19). Additionally, human airway smooth muscle (ASM) cells (20) and quadriceps, diaphragmatic, and external intercostal muscle of COPD (21) patients display altered OXPHOS with increased COX activity (22, 23) (Table 1).
Table 1. Mitochondrial bioenergetics in lung health and disease.
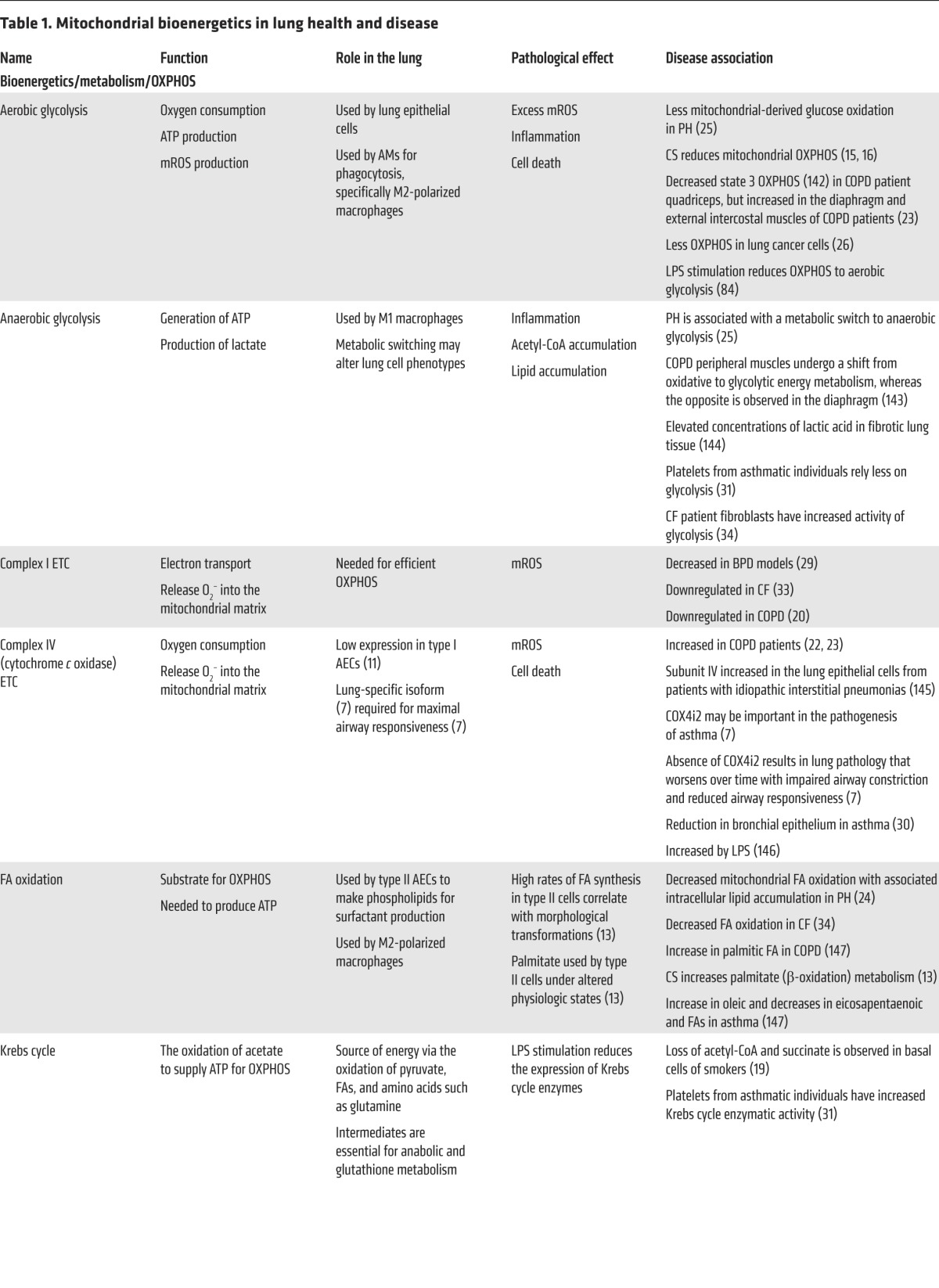
PH is characterized by obstruction of small pulmonary arteries, increased pulmonary arterial pressure, and lung vasculopathy. Vascular obstruction occurs as a consequence of excessive proliferation and apoptosis resistance of vascular cells. Chronic repression of mitochondrial metabolism, including decreased mitochondrial FA oxidation and transport (24), is associated with metabolic switching from mitochondrial-derived glucose oxidation to cytoplasmic-derived anaerobic glycolysis (25). The metabolic shift to anaerobic glycolysis contributes mechanistically to the apoptosis-resistant, proliferative phenotype of PH. Such metabolic switching, a phenomenon also referred to as the Warburg effect (26), is also observed in non–small cell lung cancer (NSCLC), a type of lung cancer that accounts for 85% of all lung cancers and originates from bronchial/epithelial cells (27). In NSCLC, the switch from production of ATP via OXPHOS to anaerobic glycolysis is thought to be a more efficient way to produce ATP and other metabolic precursors in a hypoxic environment.
Oxygen-associated alterations in complex II and I in response to hyperoxia are also observed in models of bronchopulmonary dysplasia (BPD), a chronic lung disorder of infants and children who receive prolonged mechanical ventilation to treat respiratory distress syndrome (RDS) (28, 29). Similarly, asthma, a heterogeneous chronic inflammatory disease characterized by variable airway obstruction, airway remodeling, and bronchial hyperresponsiveness, is also associated with a reduction in OXPHOS, specifically decreased expression and activity of COX in bronchial epithelium (30), and increased Krebs cycle enzymatic activity in platelets (31). Absence of the lung-specific isoform of COX, COX4i2, results in reduced airway responsiveness and a lung pathology that worsens over time, thus highlighting the potential importance of COX4i2 in the pathogenesis of asthma (7). Loss of the complex I OXPHOS proteins CISD1 and MT-ND4 (32), decreased complex I activity (33), and decreased FA oxidation (34) are also associated with CF, a lethal autosomal recessive disease associated with abnormal transport of chloride and sodium ions across the epithelium, leading to viscous airway secretions (Table 1).
Mitochondrial dynamics and biogenesis
Changes in bioenergetic processes, such as those observed in the lung diseases described above, may alter mitochondrial shape, movement, and cellular interactions. Mitochondria form a dynamic interconnected intracellular network, changing cellular location via cytoskeletal motors and altering size and shape in response to the metabolic needs of the cell. Mitochondria undergo membrane remodeling through cycles of fusion and division (35); the balance of these processes controls mitochondrial structure and metabolism as well as the cell cycle and results in the intermixing of the mitochondrial population in the cell both during normal mitochondrial turnover in homeostatic physiology and in response to mitochondrial or cellular stress (35). Increased fusion or reduced fission promotes the formation of elongated mitochondrial networks, whereas increased fission or reduced fusion causes mitochondrial fragmentation. Cells that primarily use OXPHOS metabolism, such as type II AECs, have more fusion and more elongated mitochondrial networks (15), whereas the mitochondria in cells that are more glycolytic and less reliant on OXPHOS, such as lung microvascular endothelial cells, appear more punctate (36).
Mitochondrial fusion is mediated by the dynamin-related GTPases mitofusin 1 and 2 (MFN1/2) at the outer mitochondrial membrane (OMM) and by the dynamin-related protein optic atrophy 1 (OPA1) at the inner mitochondrial membrane (IMM). Mitochondrial fission requires the recruitment of dynamin-related protein 1 (DRP1) from the cytosol to receptors (mitochondrial fission protein 1 [FIS1], mitochondrial fission factor [MFF], and mitochondrial elongation factors 1 and 2 [MID51 and MID49]) (ref. 35 and Figure 2). Hyperfusion has been documented in COPD and in lung cancer (17, 18, 37, 38), whereas loss of fusion, specifically loss of MFN2, has been associated with lung cancer (38) and PH (39). The role of mitochondrial fission and fusion in lung cancer may be microenvironment specific; increased fission may allow cancer cells to proliferate rapidly and invade the surrounding tissue, while increased fusion may allow for cell survival during times of stress or drug toxicity (27). Similarly, mitochondrial distribution within lung endothelial cells influences mitochondrial signaling in PH with the perinuclear clustering of mitochondria associated with the regulation of hypoxia-sensitive genes (40).
Figure 2. Mitochondrial fission, fusion, mitophagy, and cell death.
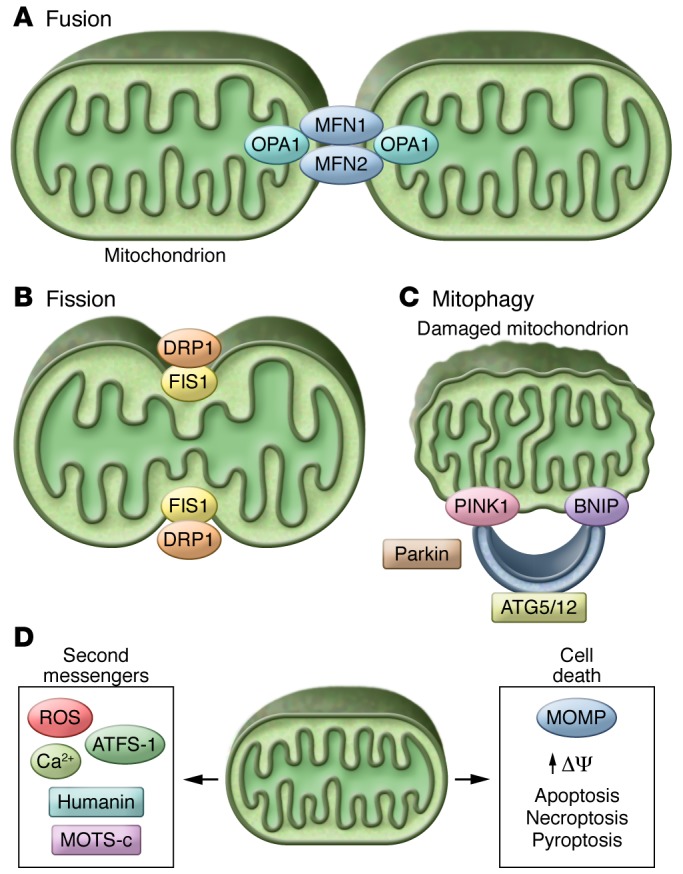
Mitochondrial biogenesis and mitophagy allow cells to quickly replace metabolically dysfunctional mitochondria with fresh, undamaged organelles. (A) Mitochondrial fusion is mediated by the dynamin-related GTPases MFN1 and MFN2 at the OMM and by OPA1 in the IMM. (B) Mitochondrial fission requires the recruitment of DRP1 from the cytosol to receptors on the OIMM (FIS1, MFF, MID49, and MID51), which causes constriction of the mitochondria and eventual division of the organelle in two. (C) Metabolically active cells, such as type II AECs, have developed robust programs to maintain mitochondrial quality. Damaged or defective mitochondria are removed via mitophagy, which is regulated by PINK1, BNIP, Parkin, and ATG5/12. (D)Mitochondrial-derived second messengers trigger a series of stress response pathways that provide both short-term and long-term benefits in increased stress resistance and longevity. However, excessive activation of these pathways may ultimately become detrimental to the cell, leading to the activation of programmed cell death pathways, including apoptosis, necroptosis, and pyroptosis.
Fission and fusion are intimately linked to the formation of new mitochondria and allow the cell to maintain a healthy pool of mitochondria. Mitochondria are not formed de novo, but mitochondrial biogenesis results from the growth and division of preexisting mitochondria (41). Mitochondrial biogenesis is regulated mostly at the transcriptional level and requires the coordinated expression of both nuclear-encoded and mitochondrial-encoded proteins, including mitochondrial transcription factor A (TFAM), PPAR coactivator-1α (PGC-1α), AMPK, and nuclear respiratory factors 1 and 2 (NRF-1/2) (41). Mitochondrial biogenesis can occur in distal lung cells, including type II AECs (42), in small blood vessel SMCs, and in inflammatory cells of the alveolar region (9) and is thought to arise during growth, conditions of high-energy demand, or cellular stress. In the parenchyma, type II AECs initiate mitochondrial biogenesis during acute lung injury (ALI), pneumonia, hyperoxic lung injury (43), and Staphylococcus aureus–associated sepsis (44). ALI, acute respiratory distress syndrome (ARDS), and sepsis remain significant sources of morbidity and mortality in the critically ill patient population (45). ALI and ARDS result from the inflammatory response of the lung to both direct and indirect insults and are characterized by severe hypoxemia, hypercapnia, diffuse infiltration visible in the chest x-ray, and a substantial reduction in pulmonary compliance (45). Mitochondrial biogenesis is increased in bronchial smooth muscle remodeling in asthma (46) and in lung cancer; however, in lung cancer, it is unclear whether these alterations contribute to tumorigenesis or are a consequence of carcinogenesis (47). In contrast, loss of mitochondrial biogenesis is associated with COPD, which may be associated with a significantly lower body mass index and lower muscle mass (ref. 48 and Table 2).
Table 2. Mitochondrial dynamics in lung health and disease.
Mitophagy
Metabolically active cells, such as type II AECs, have developed robust programs of mitochondrial quality control consisting of mitochondrial biogenesis and mitochondrial removal. Damaged or defective mitochondria are removed by selective encapsulation into double-membraned autophagosomes that are delivered to the lysosome for degradation, a process called mitophagy (15). To date, the best-documented mitophagy regulators are PTEN-induced kinase 1 (PINK1), which is expressed at low levels in healthy mitochondria (mitochondria with normal mitochondrial membrane potential [Δψm]), the BH-3 only BCL2 protein BNIP3, and the E3 ubiquitin ligase Parkin. When the Δψm is low (i.e., under conditions of stress), damaged, depolarized mitochondria stabilize PINK1 or BNIP3, which accumulates on the OMM and recruits Parkin or the autophagy protein LC3B, respectively (15). Parkin ubiquitinates various OMM proteins, including MFN1/2, and recruits autophagosomes (Figure 2 and ref. 49). Mitochondrial biogenesis and mitophagy allow cells to quickly replace metabolically dysfunctional mitochondria before energy failure (43).
The role of mitophagy in lung disease is complex. In some cases, mitophagy-related processes appear to be pathogenic, whereas in others, these processes are protective. Specifically, COPD patients have increased mitochondrial fission and increased mitophagy (15). CS also induces PINK1/Parkin-associated mitophagy, which regulates mitochondrial ROS (mROS) production and cellular senescence in primary human bronchial epithelial cells (50), and CS-induced mitophagy regulates necroptosis in lung epithelial cells and in experimental COPD murine models (15). PINK1-induced mitophagy triggers pulmonary vascular remodeling and PH (51), while S. aureus infection increases PINK1 to induce ALI (52). Conversely, defective mitophagy leads to CS-induced cellular senescence in human lung fibroblasts and small AECs (53), and loss of PINK1 and defective mitophagy promote pulmonary fibrosis (PF) in animal models and in human idiopathic pulmonary fibrosis (IPF) (54, 55). PF is characterized by irreversible destruction of lung architecture, abnormal wound healing, and deposition of extracellular matrix (ECM) proteins, leading to disruption of gas exchange and death from respiratory failure. Lung fibrosis is either idiopathic (54) or arises from exposure to environmental toxins, such as fibers, asbestos, metals, pesticides, chemotherapeutic drugs, viruses, or radiotherapy. While the pathogenic role of mitophagy in lung disease is perplexing, the differential role of mitophagy in specific cell types in the lung and in diseases such as COPD and IPF may help to explain the differences in obvious clinical, radiological, and pathologic features and may offer novel routes for therapeutic intervention or biomarker development.
Genetic regulation
Mitochondrial biogenesis and mitophagy also allow cells to quickly replace or segregate mitochondria with pathogenic damaged mitochondrial DNA (mtDNA) away from the rest of the cell. Mammalian mtDNA encodes 37 genes, 24 of which are dedicated to processing 13 key genes essential to OXPHOS and energy production, while others encode transfer RNAs (tRNAs) and rRNAs essential for expression of these genes (35). Until a few years ago, it was assumed that mtDNA was homoplasmic for a single mtDNA genotype. However, with the arrival of deep sequencing, it has become clear that low-level heteroplasmy in mtDNA is present in most tissues, including the lung (56), where a portion is maternally inherited and another portion is presumed to arise from de novo acquired mutations (35). mtDNA is 3 to 10 times more susceptible to oxidative DNA damage than nuclear DNA (43). Chronic oxidation of over 50% of mtDNA without prompt repair results in a reduction of mtDNA copy number, loss of OXPHOS, and altered mitochondrial dynamics (43). Such persistent mtDNA damage is lethal to some cell populations, whereas oxidant-induced alterations in mtDNA/protein stability may acutely influence the behavior of other cell populations, thereby facilitating cellular adaptation (43). Pathogenic defects in or loss of mtDNA is associated with a number of lung diseases, including COPD (57), PH (58), lung cancer (59), asthma (60), and IPF (ref. 61 and Table 3). Asthma is not considered a mitochondrial syndrome per se; however, maternal inheritance is a risk factor for asthma and other atopic diseases (62), and mitochondrial haplogroups are associated with increased serum IgE levels (63, 64). Similarly, inherited mtDNA haplotypes may also predispose or confer susceptibility to COPD (ref. 65 and Table 2).
Table 3. mtDAMPs in the lung.
Second messenger signaling
Recent technological advances have identified hundreds of mitochondrial proteins that vary in a cell-specific and tissue-dependent manner; however, the biochemical functions of the majority of these proteins remain unknown (1). With the other 1,500 or so proteins that make up the mitochondrion encoded by the nucleus, mitochondria have developed a symbiotic codependency on the nuclear genome and must signal to it in a retrograde fashion to ensure survival and adaptation. The best example of such signaling is the generation of mROS where, under normal physiological conditions, basal ROS released from the mitochondria acts as a second messenger to maintain cellular homeostasis (66). mROS production is tightly regulated by the ETC and antioxidant systems in mitochondria and is generated by a one-electron reduction of molecular O2 to yield superoxide (O2−) (ref. 43 and Figure 1). mROS production leads to translocation of NRF2, resulting in expression of antioxidant and antiinflammatory proteins, such as the mitochondrial sirtuins (SIRT3, -4, and -5), which directly regulate expression of antioxidant genes. Genetic ablation of Nrf2 enhances susceptibility to CS-induced emphysema (67) and bleomycin-induced PF (68) in mice. SIRT5 is upregulated by CS in lung epithelial cells (69), SIRT3 regulates cell proliferation and apoptosis in NSCLC cells (70), and Sirt4 knockout mice spontaneously develop lung tumors (71), suggesting a key role for mitochondrial antioxidant systems in lung disease.
Mitochondria may also actively regulate homeostasis at the cellular and organismal level via peptides encoded within their genome (72, 73) or via the mitochondrial unfolded protein response (UPR) pathway (74); however, little is known about the function of these processes in the lung. Mitochondria are key regulators of the second messengers calcium (Ca2+) and iron (Fe), which control a diverse range of cellular processes, including mROS production. The formation of mitochondrial iron-sulfur (Fe-S) clusters is essential for many of the ETC complexes (complexes I and II) and other enzymes important for mitochondrial metabolism (75). Similarly, Ca2+ mobilization and the activation of Ca2+-binding proteins control a diverse range of cellular processes, including mitochondrial biogenesis (76). Loss or overload of mitochondrial Fe or Ca2+ can lead to mitochondrial dysfunction (77) and is associated with PH (76, 78), NSCLC (77), asthma (78, 79), and CF (33).
The activation of mitochondrial-derived second messengers may trigger a protective or hormetic response that provides both short-term benefits and the potential for long-term benefits in increased stress resistance and longevity (80). However, excessive activation of these pathways may ultimately become detrimental to the cell. Mitochondria consistently play a vital role in stress responses and programmed cell death pathways. The decline of Δψm, constitutive opening of mitochondrial pores, arrest of OXPHOS, interruption of mitochondrial protein import, and leakage of cytochrome c into the cytoplasm have all been associated with cell death pathways. Mitochondria regulate four forms of cell death, including (a) extrinsic apoptosis, (b) intrinsic apoptosis, (c) necrosis/necroptosis (15), and (d) pyroptosis, all of which have been documented in lung cells in various models of lung disease (15, 81–83).
mtDAMPs
A wide variety of mitochondrial-derived molecules, which at normal physiological concentrations act as second messengers in the lung, can also behave as mitochondrial damage–associated molecular patterns (mtDAMPs) when produced in excess or in an alternative cellular compartment. DAMPs arise from endogenous molecules secreted or released from intracellular or extracellular sources as a result of tissue injury and primarily activate pathogen recognition receptors (PRRs), including retinoic acid inducible gene–like (RIG-1–like) receptors (RLRs), TLRs, and nuclear oligomerization domain–like (NOD-like) receptors (NLRs), resulting in the induction of inflammatory cascades (84). Oxidized, fragmented mtDNA released from damaged mitochondria in response to stress or injury (85, 86) is one of the most important mtDAMPs needed to regulate innate immunity (Figure 3). Oxidized mtDNA is thought to act as a sentinel molecule in the cell, such that before an externally applied oxidant stress rises to a level that threatens the nuclear genome with mutation, oxidative mtDNA damage triggers death of the affected cell and promotes the propagation of signals to alert neighboring and roaming cells (43). We have previously shown that mtDNA released from dysfunctional mitochondria in response to stress and/or infection activates the NLRP3 inflammasome (86) and that circulating extracellular mtDNA is associated with mortality in medical intensive care unit (ICU) patients (45). Others have replicated these findings (87, 88) and shown that mtDNA also activates the DNA sensor cyclic GMP–AMP synthase (cGAS) (89) intracellularly and activates neutrophils (87), vascular endothelial cells (90), and AMs extracellularly (91). Injection of mitochondrial lysates in the rat causes lung inflammation (87), and mtDNA released as a result of injury in murine models of PF stimulates TGF-β1 release from AECs (92). These findings suggest that mtDNA is a fundamental signaling molecule in the lung for the regulation and initiation of inflammation (Table 3).
Figure 3. mtDAMPs.

A wide variety of mitochondrial-derived molecules, which at normal physiological concentrations act as second messengers in the lung, can also behave as mtDAMPs when produced in excess or in an alternative cellular compartment. DAMPs primarily activate PRRs, including RLRs, TLRs, and NLRs, resulting in the induction of inflammatory cascades.
ATP can also act as a mtDAMP in the lung. Normal lung function requires ATP release by AECs to optimize airway surface layer hydration, mucus composition, and mucociliary clearance (93). However, excessive ATP release from dying or damaged cells acts as a DAMP and is recognized by the purinergic receptor P2X7, which is expressed specifically on cells of the immune system and is involved in the release of IL-1β (94). ATP levels are increased in bronchoalveolar lavage fluid (BALF) of COPD (95) and asthma patients as well as in murine asthma (96) and PF (bleomycin) models (97), all of which are associated with increased inflammation. TFAM is an integral regulator of mtDNA integrity that, when released from mitochondria, acts as a mtDAMP to regulate inflammatory responses (98). Similarly, N-formyl peptides (NFPs) originating from mitochondrial proteins act as chemoattractants for neutrophils and may link trauma, systemic inflammatory response syndrome (SIRS), and cardiovascular collapse (99). SIRS, a leading cause of death in ARDS patients, is a nonspecific inflammatory state caused by ischemia, inflammation, trauma, infection, or a combination of insults that triggers the release of inflammatory mediators from damaged lung tissue. Cardiolipin (Figure 1), which tethers members of the ETC to the IMM, directly activates inflammasome-mediated immune responses (100) and generates lipid mediators during ALI (12), when it is released into the cytosol or extracellular space. Cardiolipin concentrations are also elevated in BALF of individuals with pneumonia (12) and in smoking COPD patients (101), and mice given intratracheal injection of cardiolipin display lower lung compliance with greater elasticity and resistance (12). Finally, lysocardiolipin acyltransferase (LYCAT), a cardiolipin-remodeling enzyme, is significantly altered in lung tissues from patients with IPF (ref. 102 and Table 3).
mROS is the most universal and best-documented mtDAMP. The exact source of mROS may change with the mitochondrial stressor and particular disease state, but investigations into the source of mROS in many lung diseases is limited by the lack of highly specific in vivo mROS-sensing agents (103). That said, there is compelling evidence for a role of mROS in the pathogenesis of a number of lung diseases, including PF (54), COPD (20, 50), asthma (104), CF (105), lung cancer (106), BPD (107), and PH (108).
Mitochondria and inflammation
Lung inflammation is caused by pathogens or by exposure to toxins, pollutants, irritants, and allergens. The innate immune response of the lung relies on resident AMs for the detection of infectious agents, cellular stresses, or tissue damage. Bacterial or viral constituents as well as DAMPs secreted by lung epithelial cells ligate PRRs on AMs, causing AMs to secrete proinflammatory cytokines that activate alveolar epithelial receptors, which leads to recruitment of activated neutrophils. Mitochondria play a key role in the correct functioning of immune cells in the lung, including AMs and CD4+ Th2 and CD8+ (T cytotoxic) cells. Proinflammatory M1 macrophages exhibit robust glycolysis, and profibrotic/antiinflammatory M2–polarized macrophages increase oxygen consumption through the induction of mitochondrial biogenesis and FA oxidation (109). Mitochondria regulate mROS and AM phagocytosis in response to bacteria (110). Mitochondrial proteins and metabolites also interact and regulate TLR signaling (84), directly linking the activation of the downstream proinflammatory cytokines IL-1β, IL-6, and TNF-α to mitochondrial function.
The IMM ETC uncoupling protein 2 (UCP2) regulates NLR-mediated caspase-1 activation through the stimulation of lipid synthesis in macrophages with implications for sepsis-induced lung injury (111). Activation of NLRP3 signaling, a key regulator of IL-1β and IL-18 secretion, is regulated by mitochondrial pathways (84, 112), and the NLRP3 inflammasome adaptor (ASC) and UCP2 regulate hypoxia-induced PH in mice (51, 113, 114). NLRP3 inflammasome activation by mROS in bronchial epithelial cells is required for allergic inflammation (104) and in AMs contributes to mechanical stretch-induced lung inflammation and injury (115). Caspase-1–dependent IL-1β secretion is critical for host defence against Chlamydia pneumoniae lung infection (116), and mitochondrial Ca2+-dependent NLRP3 activation exacerbates the Pseudomonas aeruginosa–driven inflammatory response in CF (117).
NLRP3 signaling is also regulated by the mitochondrial RLR signaling protein MAVS. RLR signaling resulting in the production of type I IFNs and other proinflammatory cytokines that promote adaptive antiviral immunity is regulated by mtDNA and a number of mitochondrial proteins (84, 118). AMs detect respiratory syncytial virus (RSV) via MAVS, and loss of MAVS may underlie the development of RSV-induced severe lung inflammation (118). MAVS also regulates the response of lung mast cells to influenza A virus (IAV) (119) and may be responsible for CS enhancement of virus-induced pulmonary innate immune and remodeling responses in mice (120).
Mitochondria also play a role in the adaptive immune response of lymphocytes in the airway and lung parenchyma. Specifically, mitochondria regulate antigen processing and presentation and localize to the immune synapse during T cell activation (121). Mitochondrial metabolism also maintains the memory T cell phenotype (122) and dictates the different inflammatory and suppressive CD4+ Th cells (109). A balanced Th1 and Th2 response is suited to the immune challenge, and a dysregulated response is linked to a variety of chronic inflammatory lung conditions such as asthma and chronic bronchitis (123). Nuclear factor of activated T cells (NFAT) regulates mitochondrial remodeling and contributes to apoptosis resistance in PH and cancer (124), and mitochondrial-derived proteins modulate BALF eosinophilia by regulating both eosinophil apoptosis and Th2-type cytokine production (125).
Reciprocally, inflammatory pathways also regulate mitochondrial function (84). Bacterial pathogens hijack the mitochondrial cell death machinery of host cells, and viral infection alters the mitochondrial proteome, increases mROS and mitochondrial biogenesis, and attenuates mitochondrial lipid β-oxidation (84). Examples of such modulation in the lung include the following: influenza infection increasing long-chain acylcarnitine secretion from mitochondria, which in turn inhibit the surface adsorption of pulmonary surfactant, thereby increasing the risk for lung injury (126); LPS activating MAPK kinase 3 (MKK3), which regulates mitochondrial biogenesis and mitophagy in sepsis-induced lung injury (127); S. aureus decreasing cardiolipin availability in pneumonia models (52); and RSV infection increasing lung mitochondrial bioenergetics (128).
Therapeutic targeting of mitochondria in lung disease
Given the central role of mitochondria and mROS in human disease, several natural antioxidants, such as vitamin C, vitamin E, and curcumin, have been investigated both in vitro and in vivo; however, most of these were found to be ineffective in attenuating mROS production in response to an environmental stimulus or in patients with lung disease (129–132). While there has been considerable advancement in the development of mitochondria-targeted small molecule antioxidants (3) and alternative approaches to targeting mROS (133–137) appear promising, such mROS-targeted therapeutic approaches must be used with caution. mROS behave as a cytoprotective agent that leaves cells less susceptible to subsequent perturbations. This response, termed mitohormesis, is being rapidly dissected in many model systems and must be considered in the design of all mitochondrial-targeted therapeutics for the treatment of lung disease (66). Other mitochondrial-targeted therapeutic strategies that could potentially be used to treat lung diseases include the use of metabolic modulating compounds such as dicholoroacetate (138), histone deacetylase inhibitors (139, 140), and fission inhibitors. Stimulating adaptive mitochondrial biogenesis and mitophagy may be a useful adjuvant therapy for ALI in sepsis (44), and transfer of mitochondria from bone marrow–derived mesenchymal stem cells to injured alveolar epithelium may be beneficial in ARDS, asthma, or COPD (141).
Conclusion
Lung diseases are among the leading causes of death worldwide, with lung infections, lung cancer, and COPD together accounting for over 9.5 million deaths in 2008 (2). In the next two decades, it is predicted that the proportion of deaths and disability arising from chronic lung diseases will rise significantly, yet few advances have been made to effectively treat the majority of lung diseases, with suboptimal therapeutic options eliciting only modest improvements in disease symptoms. Mitochondrial dysfunction is rapidly advancing as a key pathological feature that appears early and consistently in the development of lung disease. It is clear that abnormal mitochondrial signatures, including metabolic switching, altered mitochondrial biogenesis and mitophagy, increased occurrence of mtDNA mutations, abnormalities in mitochondrial-derived signaling, and the activation of mtDAMPs, play a substantial role in a number of lung diseases. While the pathogenic role of each of these mitochondrial processes in the lung remains complex, precise differential mitochondrial signatures in specific lung cell types may help to elucidate the clinical, radiological, and pathological alterations associated with each disease. To be able to identify such disease-associated mitochondrial signatures, a rigorous mapping of mitochondrial turnover and dynamics, mitochondrial metabolic activity, mtDNA sequence heterogeneity, and profiling of lung-specific mitochondrial proteins represent potential approaches that may bear dividends for a better understanding of normal physiologic and pathologic processes in the lung. Mitochondria therefore offer promising potential targets in the search for novel diagnostics and therapeutics in lung diseases.
Acknowledgments
This work was supported by NIH grants P01-HL114501, R01-HL055330, and R01-HL079904 (to A.M.K. Choi), K99HL125899 (to S.M. Cloonan), and P0-HL105339, and by the American Lung Association (Biomedical Research Grant, RG-348928 to S.M. Cloonan).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2016;126(3):809–820. doi:10.1172/JCI81113.
References
- 1.Pagliarini DJ, Rutter J. Hallmarks of a new era in mitochondrial biochemistry. Genes Dev. 2013;27(24):2615–2627. doi: 10.1101/gad.229724.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.European Respiratory Society . European Lung White Book. Yorkshire, United Kingdom: European Respiratory Society; 2013. [Google Scholar]
- 3.Natarajan V, Parinandi NL. Mitochondrial Function In Lung Health And Disease. New York, New York, USA: Humana Press; 2014. [Google Scholar]
- 4.Fisher AB, Scarpa A, LaNoue KF, Bassett D, Williamson JR. Respiration of rat lung mitochondria and the influence of Ca2+ on substrate utilization. Biochemistry. 1973;12(7):1438–1445. doi: 10.1021/bi00731a026. [DOI] [PubMed] [Google Scholar]
- 5.Williamson JR, Corkey BE. Assay of citric acid cycle intermediates and related compounds — update with tissue metabolite levels and intracellular distribution. Methods Enzymol. 1979;55:200–222. doi: 10.1016/0076-6879(79)55025-3. [DOI] [PubMed] [Google Scholar]
- 6.Mustafa MG, Cross CE. Effects of short-term ozone exposure on lung mitochondrial oxidative and energy metabolism. Arch Biochem Biophys. 1974;162(2):585–594. doi: 10.1016/0003-9861(74)90219-7. [DOI] [PubMed] [Google Scholar]
- 7.Huttemann M, et al. Cytochrome c oxidase subunit 4 isoform 2-knockout mice show reduced enzyme activity, airway hyporeactivity, and lung pathology. FASEB J. 2012;26(9):3916–3930. doi: 10.1096/fj.11-203273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Massaro GD, Gail DB, Massaro D. Lung oxygen consumption and mitochondria of alveolar epithelial and endothelial cells. J Appl Physiol. 1975;38(4):588–592. doi: 10.1152/jappl.1975.38.4.588. [DOI] [PubMed] [Google Scholar]
- 9.Carraway MS, Suliman HB, Kliment C, Welty-Wolf KE, Oury TD, Piantadosi CA. Mitochondrial biogenesis in the pulmonary vasculature during inhalational lung injury and fibrosis. Antioxid Redox Signal. 2008;10(2):269–275. doi: 10.1089/ars.2007.1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moriguchi K, et al. Differentiation of human pulmonary alveolar epithelial cells revealed by peroxisome changes in pulmonary proteinosis. Exp Mol Pathol. 1984;40(2):262–270. doi: 10.1016/0014-4800(84)90082-0. [DOI] [PubMed] [Google Scholar]
- 11.Hirai K, Ogawa K. Cytochemical quantitation of cytochrome oxidase activity in rat pulmonary alveolar epithelial cells and possible defect in type I cells. J Electron Microsc (Tokyo) 1986;35(1):19–28. [PubMed] [Google Scholar]
- 12.Ray NB, et al. Dynamic regulation of cardiolipin by the lipid pump Atp8b1 determines the severity of lung injury in experimental pneumonia. Nat Med. 2010;16(10):1120–1127. doi: 10.1038/nm.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Agarwal AR, Yin F, Cadenas E. Short-term cigarette smoke exposure leads to metabolic alterations in lung alveolar cells. Am J Respir Cell Mol Biol. 2014;51(2):284–293. doi: 10.1165/rcmb.2013-0523OC. [DOI] [PubMed] [Google Scholar]
- 14.Pattle RE, Schock C, Dirnhuber P, Creasey JM. Lamellar transformation of lung mitochondria under conditions of stress. Nature. 1972;240(5382):468–469. doi: 10.1038/240468a0. [DOI] [PubMed] [Google Scholar]
- 15.Mizumura K, et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest. 2014;124(9):3987–4003. doi: 10.1172/JCI74985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van der Toorn M, et al. Cigarette smoke-induced blockade of the mitochondrial respiratory chain switches lung epithelial cell apoptosis into necrosis. Am J Physiol Lung Cell Mol Physiol. 2007;292(5):L1211–L1218. doi: 10.1152/ajplung.00291.2006. [DOI] [PubMed] [Google Scholar]
- 17.Ballweg K, Mutze K, Konigshoff M, Eickelberg O, Meiners S. Cigarette smoke extract affects mitochondrial function in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2014;307(11):L895–L907. doi: 10.1152/ajplung.00180.2014. [DOI] [PubMed] [Google Scholar]
- 18.Hoffmann RF, et al. Prolonged cigarette smoke exposure alters mitochondrial structure and function in airway epithelial cells. Respir Res. 2013;14: doi: 10.1186/1465-9921-14-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deeb RS, Walters MS, Strulovici-Barel Y, Chen Q, Gross SS, Crystal RG. Smoking-associated disordering of the airway basal stem/progenitor cell metabotype. Am J Respir Cell Mol Biol. doi: 10.1165/rcmb.2015-0055OC. [published online ahead of print July 10, 2015]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wiegman CH, et al. Oxidative stress-induced mitochondrial dysfunction drives inflammation airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2015;136(3):769–780. doi: 10.1016/j.jaci.2015.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rabinovich RA, et al. Mitochondrial dysfunction in COPD patients with low body mass index. Eur Respir J. 2007;29(4):643–650. doi: 10.1183/09031936.00086306. [DOI] [PubMed] [Google Scholar]
- 22.Sauleda J, et al. Cytochrome oxidase activity and mitochondrial gene expression in skeletal muscle of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998;157(5 pt 1):1413–1417. doi: 10.1164/ajrccm.157.5.9710039. [DOI] [PubMed] [Google Scholar]
- 23.Ribera F, et al. Mitochondrial electron transport chain function is enhanced in inspiratory muscles of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2003;167(6):873–879. doi: 10.1164/rccm.200206-519OC. [DOI] [PubMed] [Google Scholar]
- 24.Talati M, Hemnes A. Fatty acid metabolism in pulmonary arterial hypertension: role in right ventricular dysfunction and hypertrophy. Pulm Circ. 2015;5(2):269–278. doi: 10.1086/681227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu W, et al. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc Natl Acad Sci U S A. 2007;104(4):1342–1347. doi: 10.1073/pnas.0605080104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 27.Lennon FE, Salgia R. Mitochondrial dynamics: biology and therapy in lung cancer. Expert Opin Investig Drugs. 2014;23(5):675–692. doi: 10.1517/13543784.2014.899350. [DOI] [PubMed] [Google Scholar]
- 28.Sureshbabu A, Bhandari V. Targeting mitochondrial dysfunction in lung diseases: emphasis on mitophagy. Front Physiol. 2013;4: doi: 10.3389/fphys.2013.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Das KC. Hyperoxia decreases glycolytic capacity, glycolytic reserve and oxidative phosphorylation in MLE-12 cells and inhibits complex I and II function, but not complex IV in isolated mouse lung mitochondria. PLoS One. 2013;8(9): doi: 10.1371/journal.pone.0073358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mabalirajan U, et al. Mitochondrial structural changes and dysfunction are associated with experimental allergic asthma. J Immunol. 2008;181(5):3540–3548. doi: 10.4049/jimmunol.181.5.3540. [DOI] [PubMed] [Google Scholar]
- 31.Xu W, Cardenes N, Corey C, Erzurum SC, Shiva S. Platelets from Asthmatic Individuals Show Less Reliance on Glycolysis. PLoS One. 2015;10(7): doi: 10.1371/journal.pone.0132007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Valdivieso AG, Santa-Coloma TA. CFTR activity and mitochondrial function. Redox Biol. 2013;1:190–202. doi: 10.1016/j.redox.2012.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shapiro BL, Lam LF. Calcium and age in fibroblasts from control subjects and patients with cystic fibrosis. Science. 1982;216(4544):417–419. doi: 10.1126/science.7071590. [DOI] [PubMed] [Google Scholar]
- 34.Joseloff E, et al. Serum metabolomics indicate altered cellular energy metabolism in children with cystic fibrosis. Pediatr Pulmonol. 2014;49(5):463–472. doi: 10.1002/ppul.22859. [DOI] [PubMed] [Google Scholar]
- 35.Mishra P, Chan DC. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat Rev Mol Cell Biol. 2014;15(10):634–646. doi: 10.1038/nrm3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mishra P, Carelli V, Manfredi G, Chan DC. Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 2014;19(4):630–641. doi: 10.1016/j.cmet.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aravamudan B, et al. Cigarette smoke-induced mitochondrial fragmentation and dysfunction in human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2014;306(9):L840–L854. doi: 10.1152/ajplung.00155.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rehman J, et al. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J. 2012;26(5):2175–2186. doi: 10.1096/fj.11-196543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ryan JJ, et al. PGC1α-mediated mitofusin-2 deficiency in female rats and humans with pulmonary arterial hypertension. Am J Respir Crit Care Med. 2013;187(8):865–878. doi: 10.1164/rccm.201209-1687OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Al-Mehdi AB, et al. Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci Signal. 2012;5(231): doi: 10.1126/scisignal.2002712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jornayvaz FR, Shulman GI. Regulation of mitochondrial biogenesis. Essays Biochem. 2010;47:69–84. doi: 10.1042/bse0470069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Athale J, et al. Nrf2 promotes alveolar mitochondrial biogenesis and resolution of lung injury in Staphylococcus aureus pneumonia in mice. Free Radic Biol Med. 2012;53(8):1584–1594. doi: 10.1016/j.freeradbiomed.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schumacker PT, et al. Mitochondria in lung biology and pathology: more than just a powerhouse. Am J Physiol Lung Cell Mol Physiol. 2014;306(11):L962–L974. doi: 10.1152/ajplung.00073.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chang AL, Ulrich A, Suliman HB, Piantadosi CA. Redox regulation of mitophagy in the lung during murine Staphylococcus aureus sepsis. Free Radic Biol Med. 2015;78:179–189. doi: 10.1016/j.freeradbiomed.2014.10.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakahira K, et al. Circulating mitochondrial DNA in patients in the ICU as a marker of mortality: derivation and validation. PLoS Med. 2013;10(12): doi: 10.1371/journal.pmed.1001577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trian T, et al. Bronchial smooth muscle remodeling involves calcium-dependent enhanced mitochondrial biogenesis in asthma. J Exp Med. 2007;204(13):3173–3181. doi: 10.1084/jem.20070956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roberts ER, Thomas KJ. The role of mitochondria in the development and progression of lung cancer. Comput Struct Biotechnol J. 2013;6: doi: 10.5936/csbj.201303019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Remels AH, et al. Peroxisome proliferator-activated receptor expression is reduced in skeletal muscle in COPD. Eur Respir J. 2007;30(2):245–252. doi: 10.1183/09031936.00144106. [DOI] [PubMed] [Google Scholar]
- 49.Chu CT, Ji J, Dagda RK. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol. 2013;15(10):1197–205. doi: 10.1038/ncb2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ito S, et al. PARK2-mediated mitophagy is involved in regulation of HBEC senescence in COPD pathogenesis. Autophagy. 2015;11(3):547–559. doi: 10.1080/15548627.2015.1017190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Haslip M, et al. Endothelial uncoupling protein 2 regulates mitophagy and pulmonary hypertension during intermittent hypoxia. Arterioscler Thromb Vasc Biol. 2015;35(5):1166–1178. doi: 10.1161/ATVBAHA.114.304865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen BB, et al. E3 ligase subunit Fbxo15 and PINK1 kinase regulate cardiolipin synthase 1 stability and mitochondrial function in pneumonia. Cell Rep. 2014;7(2):476–487. doi: 10.1016/j.celrep.2014.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ahmad T, et al. Impaired mitophagy leads to cigarette smoke stress-induced cellular senescence: Implications for chronic obstructive pulmonary disease. FASEB J. 2015;29(7):2912–2929. doi: 10.1096/fj.14-268276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Patel AS, et al. Epithelial cell mitochondrial dysfunction and PINK1 are induced by transforming growth factor-β1 in pulmonary fibrosis. PLoS One. 2015;10(3): doi: 10.1371/journal.pone.0121246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bueno M, et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J Clin Invest. 2015;125(2):521–538. doi: 10.1172/JCI74942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Burgstaller JP, et al. MtDNA segregation in heteroplasmic tissues is common in vivo and modulated by haplotype differences and developmental stage. Cell Rep. 2014;7(6):2031–2041. doi: 10.1016/j.celrep.2014.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ning W, et al. Comprehensive analysis of gene expression on GOLD-2 Versus GOLD-0 smokers reveals novel genes important in the pathogenesis of COPD. Proc Am Thorac Soc. 2006;3(6): doi: 10.1513/pats.200603-031MS. [DOI] [PubMed] [Google Scholar]
- 58.Belostotsky R, et al. Mutations in the mitochondrial seryl-tRNA synthetase cause hyperuricemia, pulmonary hypertension, renal failure in infancy and alkalosis, HUPRA syndrome. Am J Hum Genet. 2011;88(2):193–200. doi: 10.1016/j.ajhg.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fliss MS, et al. Facile detection of mitochondrial DNA mutations in tumors and bodily fluids. Science. 2000;287(5460):2017–2019. doi: 10.1126/science.287.5460.2017. [DOI] [PubMed] [Google Scholar]
- 60.Liang L, et al. An epigenome-wide association study of total serum immunoglobulin E concentration. Nature. 2015;520(7549):670–674. doi: 10.1038/nature14125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gazdhar A, et al. Time-dependent and somatically acquired mitochondrial DNA mutagenesis and respiratory chain dysfunction in a scleroderma model of lung fibrosis. Sci Rep. 2014;4: doi: 10.1038/srep05336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Litonjua AA, Carey VJ, Burge HA, Weiss ST, Gold DR. Parental history and the risk for childhood asthma. Does mother confer more risk than father? Am J Respir Crit Care Med. 1998;158(1):176–181. doi: 10.1164/ajrccm.158.1.9710014. [DOI] [PubMed] [Google Scholar]
- 63.Zifa E, et al. Mitochondrial genetic background plays a role in increasing risk to asthma. Mol Biol Rep. 2012;39(4):4697–4708. doi: 10.1007/s11033-011-1262-8. [DOI] [PubMed] [Google Scholar]
- 64.Raby BA, et al. A common mitochondrial haplogroup is associated with elevated total serum IgE levels. J Allergy Clin Immunol. 2007;120(2):351–358. doi: 10.1016/j.jaci.2007.05.029. [DOI] [PubMed] [Google Scholar]
- 65.Zheng S, et al. Role of mtDNA haplogroups in COPD susceptibility in a southwestern Han Chinese population. Free Radic Biol Med. 2012;53(3):473–481. doi: 10.1016/j.freeradbiomed.2012.05.019. [DOI] [PubMed] [Google Scholar]
- 66.Yun J, Finkel T. Mitohormesis. Cell Metab. 2014;19(5):757–766. doi: 10.1016/j.cmet.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rangasamy T, et al. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest. 2004;114(9):1248–1259. doi: 10.1172/JCI21146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cho HY, Reddy SP, Yamamoto M, Kleeberger SR. The transcription factor NRF2 protects against pulmonary fibrosis. FASEB J. 2004;18(11):1258–1260. doi: 10.1096/fj.03-1127fje. [DOI] [PubMed] [Google Scholar]
- 69.Wang Y, Zhu Y, Xing S, Ma P, Lin D. SIRT5 prevents cigarette smoke extract-induced apoptosis in lung epithelial cells via deacetylation of FOXO. Cell Stress Chaperones. 2015;20(5):805–810. doi: 10.1007/s12192-015-0599-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li H, Feng Z, Wu W, Li J, Zhang J, Xia T. SIRT3 regulates cell proliferation and apoptosis related to energy metabolism in non-small cell lung cancer cells through deacetylation of NMNAT2. Int J Oncol. 2013;43(5):1420–1430. doi: 10.3892/ijo.2013.2103. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 71.Jeong SM, et al. SIRT4 has tumor-suppressive activity and regulates the cellular metabolic response to DNA damage by inhibiting mitochondrial glutamine metabolism. Cancer Cell. 2013;23(4):450–463. doi: 10.1016/j.ccr.2013.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hashimoto Y, et al. A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer’s disease genes and Abeta. Proc Natl Acad Sci U S A. 2001;98(11):6336–6341. doi: 10.1073/pnas.101133498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lee C, et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab. 2015;21(3):443–454. doi: 10.1016/j.cmet.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Haynes CM, Petrova K, Benedetti C, Yang Y, Ron D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev Cell. 2007;13(4):467–480. doi: 10.1016/j.devcel.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 75.White K, et al. Genetic and hypoxic alterations of the microRNA-210-ISCU1/2 axis promote iron-sulfur deficiency and pulmonary hypertension. EMBO Mol Med. 2015;7(6):695–713. doi: 10.15252/emmm.201404511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Navarro-Sastre A, et al. A fatal mitochondrial disease is associated with defective NFU1 function in the maturation of a subset of mitochondrial Fe-S proteins. Am J Hum Genet. 2011;89(5):656–667. doi: 10.1016/j.ajhg.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huang H, Shah K, Bradbury NA, Li C, White C. Mcl-1 promotes lung cancer cell migration by directly interacting with VDAC to increase mitochondrial Ca2+ uptake and reactive oxygen species generation. Cell Death Dis. 2014;5: doi: 10.1038/cddis.2014.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Waypa GB, et al. Increases in mitochondrial reactive oxygen species trigger hypoxia-induced calcium responses in pulmonary artery smooth muscle cells. Circ Res. 2006;99(9):970–978. doi: 10.1161/01.RES.0000247068.75808.3f. [DOI] [PubMed] [Google Scholar]
- 79.Girodet PO, et al. Calcium channel blocker reduces airway remodeling in severe asthma. A proof-of-concept study. Am J Respir Crit Care Med. 2015;191(8):876–883. doi: 10.1164/rccm.201410-1874OC. [DOI] [PubMed] [Google Scholar]
- 80.Finkel T, et al. The ins and outs of mitochondrial calcium. Circ Res. 2015;116(11):1810–1819. doi: 10.1161/CIRCRESAHA.116.305484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kovarova M, et al. NLRP1-dependent pyroptosis leads to acute lung injury and morbidity in mice. J Immunol. 2012;189(4):2006–2016. doi: 10.4049/jimmunol.1201065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chen ZH, et al. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS One. 2008;3(10): doi: 10.1371/journal.pone.0003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen ZH, et al. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc Natl Acad Sci U S A. 2010;107(44):18880–18885. doi: 10.1073/pnas.1005574107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cloonan SM, Choi AM. Mitochondria: commanders of innate immunity and disease? Curr Opin Immunol. 2012;24(1):32–40. doi: 10.1016/j.coi.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 85.Garcia N, Chavez E. Mitochondrial DNA fragments released through the permeability transition pore correspond to specific gene size. Life Sci. 2007;81(14):1160–1166. doi: 10.1016/j.lfs.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 86.Nakahira K, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12(3):222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang Q, Itagaki K, Hauser CJ. Mitochondrial DNA is released by shock and activates neutrophils via p38 map kinase. Shock. 2010;34(1):55–59. doi: 10.1097/SHK.0b013e3181cd8c08. [DOI] [PubMed] [Google Scholar]
- 88.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 89.West AP, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015;520(7548):553–557. doi: 10.1038/nature14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sun S, et al. Mitochondrial DAMPs increase endothelial permeability through neutrophil dependent and independent pathways. PLoS One. 2013;8(3): doi: 10.1371/journal.pone.0059989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang JZ, Liu Z, Liu J, Ren JX, Sun TS. Mitochondrial DNA induces inflammation and increases TLR9/NF-κB expression in lung tissue. Int J Mol Med. 2014;33(4):817–824. doi: 10.3892/ijmm.2014.1650. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 92.Li G, et al. DNaseI protects against Paraquat-induced acute lung injury and pulmonary fibrosis mediated by mitochondrial DNA. BioMed Res Int. 2015;2015: doi: 10.1155/2015/386952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Button B, Okada SF, Frederick CB, Thelin WR, Boucher RC. Mechanosensitive ATP release maintains proper mucus hydration of airways. Sci Signal. 2013;6(279): doi: 10.1126/scisignal.2003755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.McDonald B, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330(6002):362–366. doi: 10.1126/science.1195491. [DOI] [PubMed] [Google Scholar]
- 95.Lommatzsch M, et al. Extracellular adenosine triphosphate and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;181(9):928–934. doi: 10.1164/rccm.200910-1506OC. [DOI] [PubMed] [Google Scholar]
- 96.Idzko M, et al. Extracellular ATP triggers and maintains asthmatic airway inflammation by activating dendritic cells. Nat Med. 2007;13(8):913–919. doi: 10.1038/nm1617. [DOI] [PubMed] [Google Scholar]
- 97.Riteau N, et al. Extracellular ATP is a danger signal activating P2X7 receptor in lung inflammation and fibrosis. Am J Respir Crit Care Med. 2010;182(6):774–783. doi: 10.1164/rccm.201003-0359OC. [DOI] [PubMed] [Google Scholar]
- 98.Julian MW, Shao G, Vangundy ZC, Papenfuss TL, Crouser ED. Mitochondrial transcription factor A, an endogenous danger signal, promotes TNFα release via RAGE- and TLR9-responsive plasmacytoid dendritic cells. PLoS One. 2013;8(8): doi: 10.1371/journal.pone.0072354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wenceslau CF, McCarthy CG, Szasz T, Goulopoulou S, Webb RC. Mitochondrial N-formyl peptides induce cardiovascular collapse and sepsis-like syndrome. Am J Physiol Heart Circ Physiol. 2015;308(7):H768–H777. doi: 10.1152/ajpheart.00779.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Iyer SS, et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity. 2013;39(2):311–323. doi: 10.1016/j.immuni.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lusuardi M, Capelli A, Carli S, Tacconi MT, Salmona M, Donner CF. Role of surfactant in chronic obstructive pulmonary disease: therapeutic implications. Respiration. 1992;59(suppl 1):28–32. doi: 10.1159/000196100. [DOI] [PubMed] [Google Scholar]
- 102.Huang LS, et al. The mitochondrial cardiolipin remodeling enzyme lysocardiolipin acyltransferase is a novel target in pulmonary fibrosis. Am J Respir Crit Care Med. 2014;189(11):1402–1415. doi: 10.1164/rccm.201310-1917OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Logan A, et al. Using exomarkers to assess mitochondrial reactive species in vivo. Biochim Biophys Acta. 2014;1840(2):923–930. doi: 10.1016/j.bbagen.2013.05.026. [DOI] [PubMed] [Google Scholar]
- 104.Kim SR, et al. NLRP3 inflammasome activation by mitochondrial ROS in bronchial epithelial cells is required for allergic inflammation. Cell Death Dis. 2014;5: doi: 10.1038/cddis.2014.460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Velsor LW, Kariya C, Kachadourian R, Day BJ. Mitochondrial oxidative stress in the lungs of cystic fibrosis transmembrane conductance regulator protein mutant mice. Am J Respir Cell Mol Biol. 2006;35(5):579–586. doi: 10.1165/rcmb.2005-0473OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Galli F, et al. Oxidative stress and antioxidant therapy in cystic fibrosis. Biochim Biophys Acta. 2012;1822(5):690–713. doi: 10.1016/j.bbadis.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 107.Datta A, et al. Mouse lung development and NOX1 induction during hyperoxia are developmentally regulated and mitochondrial ROS dependent. Am J Physiol Lung Cell Mol Physiol. 2015;309(4):L369–L377. doi: 10.1152/ajplung.00176.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Adesina SE, et al. Targeting mitochondrial reactive oxygen species to modulate hypoxia-induced pulmonary hypertension. Free Radic Biol Med. 2015;87:36–47. doi: 10.1016/j.freeradbiomed.2015.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Berod L. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med. 2014;20(11):1327–1333. doi: 10.1038/nm.3704. [DOI] [PubMed] [Google Scholar]
- 110.West AP, et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature. 2011;472(7344):476–480. doi: 10.1038/nature09973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Moon JS, et al. UCP2-induced fatty acid synthase promotes NLRP3 inflammasome activation during sepsis. J Clin Invest. 2015;125(2):665–680. doi: 10.1172/JCI78253. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 112.Ichinohe T, Yamazaki T, Koshiba T, Yanagi Y. Mitochondrial protein mitofusin 2 is required for NLRP3 inflammasome activation after RNA virus infection. Proc Natl Acad Sci U S A. 2013;110(44):17963–17968. doi: 10.1073/pnas.1312571110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cero FT, et al. Absence of the inflammasome adaptor ASC reduces hypoxia-induced pulmonary hypertension in mice. Am J Physiol Lung Cell Mol Physiol. 2015;309(4):L378–L387. doi: 10.1152/ajplung.00342.2014. [DOI] [PubMed] [Google Scholar]
- 114.Dromparis P, Paulin R, Sutendra G, Qi AC, Bonnet S, Michelakis ED. Uncoupling protein 2 deficiency mimics the effects of hypoxia and endoplasmic reticulum stress on mitochondria and triggers pseudohypoxic pulmonary vascular remodeling and pulmonary hypertension. Circ Res. 2013;113(2):126–136. doi: 10.1161/CIRCRESAHA.112.300699. [DOI] [PubMed] [Google Scholar]
- 115.Wu J, Yan Z, Schwartz DE, Yu J, Malik AB, Hu G. Activation of NLRP3 inflammasome in alveolar macrophages contributes to mechanical stretch-induced lung inflammation and injury. J Immunol. 2013;190(7):3590–3599. doi: 10.4049/jimmunol.1200860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Shimada K, et al. Caspase-1 dependent IL-1beta secretion is critical for host defense in a mouse model of Chlamydia pneumoniae lung infection. PLoS One. 2011;6(6): doi: 10.1371/journal.pone.0021477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rimessi A, Bezzerri V, Patergnani S, Marchi S, Cabrini G, Pinton P. Mitochondrial Ca2+-dependent NLRP3 activation exacerbates the Pseudomonas aeruginosa-driven inflammatory response in cystic fibrosis. Nat Commun. 2015;6: doi: 10.1038/ncomms7201. [DOI] [PubMed] [Google Scholar]
- 118.Goritzka M, et al. Alveolar macrophage-derived type I interferons orchestrate innate immunity to RSV through recruitment of antiviral monocytes. J Exp Med. 2015;212(5):699–714. doi: 10.1084/jem.20140825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Graham AC, Hilmer KM, Zickovich JM, Obar JJ. Inflammatory response of mast cells during influenza A virus infection is mediated by active infection and RIG-I signaling. J Immunol. 2013;190(9):4676–4684. doi: 10.4049/jimmunol.1202096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kang MJ, et al. Cigarette smoke selectively enhances viral PAMP- and virus-induced pulmonary innate immune and remodeling responses in mice. J Clin Invest. 2008;118(8):2771–2784. doi: 10.1172/JCI32709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sena LA, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity. 2013;38(2):225–236. doi: 10.1016/j.immuni.2012.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Okoye I, et al. T cell metabolism. The protein LEM promotes CD8(+) T cell immunity through effects on mitochondrial respiration. Science. 2015;348(6238):995–1001. doi: 10.1126/science.aaa7516. [DOI] [PubMed] [Google Scholar]
- 123.Chodaczek G, Bacsi A, Dharajiya N, Sur S, Hazra TK, Boldogh I. Ragweed pollen-mediated IgE-independent release of biogenic amines from mast cells via induction of mitochondrial dysfunction. Mol Immunol. 2009;46(13):2505–2514. doi: 10.1016/j.molimm.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bonnet S, et al. The nuclear factor of activated T cells in pulmonary arterial hypertension can be therapeutically targeted. Proc Natl Acad Sci U S A. 2007;104(27):11418–11423. doi: 10.1073/pnas.0610467104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Maret M, et al. A role for Bid in eosinophil apoptosis and in allergic airway reaction. J Immunol. 2009;182(9):5740–5747. doi: 10.4049/jimmunol.0800864. [DOI] [PubMed] [Google Scholar]
- 126.Otsubo C, et al. Long-chain acylcarnitines reduce lung function by inhibiting pulmonary surfactant. J Biol Chem. 2015;290(39):23897–23904. doi: 10.1074/jbc.M115.655837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mannam P, et al. MKK3 regulates mitochondrial biogenesis and mitophagy in sepsis-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2014;306(7):L604–L619. doi: 10.1152/ajplung.00272.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Alsuwaidi AR, et al. Respiratory syncytial virus increases lung cellular bioenergetics in neonatal C57BL/6 mice. Virology. 2014;454–455:263–269. doi: 10.1016/j.virol.2014.02.028. [DOI] [PubMed] [Google Scholar]
- 129.Ciofu O, Lykkesfeldt J. Antioxidant supplementation for lung disease in cystic fibrosis. Cochrane Database Syst Rev. 2014;8: doi: 10.1002/14651858.CD007020.pub3. [DOI] [PubMed] [Google Scholar]
- 130.Wu TC, Huang YC, Hsu SY, Wang YC, Yeh SL. Vitamin E and vitamin C supplementation in patients with chronic obstructive pulmonary disease. Int J Vitam Nutr Res. 2007;77(4):272–279. doi: 10.1024/0300-9831.77.4.272. [DOI] [PubMed] [Google Scholar]
- 131.Aghdassi E, Royall D, Allard JP. Oxidative stress in smokers supplemented with vitamin C. Int J Vitam Nutr Res. 1999;69(1):45–51. doi: 10.1024/0300-9831.69.1.45. [DOI] [PubMed] [Google Scholar]
- 132.Black PN, Morgan-Day A, McMillan TE, Poole PJ, Young RP. Randomised, controlled trial of N-acetylcysteine for treatment of acute exacerbations of chronic obstructive pulmonary disease [ISRCTN21676344] BMC Pulm Med. 2004;4: doi: 10.1186/1471-2466-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zmijewski JW, et al. Mitochondrial respiratory complex I regulates neutrophil activation and severity of lung injury. Am J Respir Crit Care Med. 2008;178(2):168–179. doi: 10.1164/rccm.200710-1602OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jung SS, et al. Carbon monoxide negatively regulates NLRP3 inflammasome activation in macrophages. Am J Physiol Lung Cell Mol Physiol. 2015;308(10):L1058–L1067. doi: 10.1152/ajplung.00400.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Waypa GB, et al. Superoxide generated at mitochondrial complex III triggers acute responses to hypoxia in the pulmonary circulation. Am J Respir Crit Care Med. 2013;187(4):424–432. doi: 10.1164/rccm.201207-1294OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Jain M, et al. Mitochondrial reactive oxygen species regulate transforming growth factor-beta signaling. J Biol Chem. 2013;288(2):770–777. doi: 10.1074/jbc.M112.431973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Jaffer OA, et al. Mitochondrial-targeted antioxidant therapy decreases transforming growth factor-β-mediated collagen production in a murine asthma model. Am J Respir Cell Mol Biol. 2015;52(1):106–115. doi: 10.1165/rcmb.2013-0519OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mercken EM, Calvert LD, Singh SJ, Hageman GJ, Schols AM, Steiner MC. Dichloroacetate modulates the oxidative stress and inflammatory response to exercise in COPD. Chest. 2009;136(3):744–751. doi: 10.1378/chest.08-2890. [DOI] [PubMed] [Google Scholar]
- 139.Lam HC, et al. Histone deacetylase 6-mediated selective autophagy regulates COPD-associated cilia dysfunction. J Clin Invest. 2013;123(12):5212–5230. doi: 10.1172/JCI69636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ota C, et al. Histone deacetylase inhibitor restores surfactant protein-C expression in alveolar-epithelial type II cells attenuates bleomycin-induced pulmonary fibrosis in vivo. Exp Lung Res. 2015;41(8):422–434. doi: 10.3109/01902148.2015.1060275. [DOI] [PubMed] [Google Scholar]
- 141.Islam MN, et al. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat Med. 2012;18(5):759–765. doi: 10.1038/nm.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Jakobsson P, Jorfeldt L, Henriksson J. Metabolic enzyme activity in the quadriceps femoris muscle in patients with severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1995;151(2 pt 1):374–377. doi: 10.1164/ajrccm.151.2.7842194. [DOI] [PubMed] [Google Scholar]
- 143.Gosker HR, Wouters EF, van der Vusse GJ, Schols AM. Skeletal muscle dysfunction in chronic obstructive pulmonary disease and chronic heart failure: underlying mechanisms and therapy perspectives. Am J Clin Nutr. 2000;71(5):1033–1047. doi: 10.1093/ajcn/71.5.1033. [DOI] [PubMed] [Google Scholar]
- 144.Kottmann RM, et al. Lactic acid is elevated in idiopathic pulmonary fibrosis and induces myofibroblast differentiation via pH-dependent activation of transforming growth factor-β. Am J Respir Crit Care Med. 2012;186(8):740–751. doi: 10.1164/rccm.201201-0084OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kuwano K, et al. Oxidative stress in lung epithelial cells from patients with idiopathic interstitial pneumonias. Eur Respir J. 2003;21(2):232–240. doi: 10.1183/09031936.03.00063203. [DOI] [PubMed] [Google Scholar]
- 146.Bauerfeld CP, et al. TLR4-mediated AKT activation is MyD88/TRIF dependent and critical for induction of oxidative phosphorylation and mitochondrial transcription factor A in murine macrophages. J Immunol. 2012;188(6):2847–2857. doi: 10.4049/jimmunol.1102157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.De Castro J, Hernandez-Hernandez A, Rodriguez MC, Sardina JL, Llanillo M, Sanchez-Yague J. Comparison of changes in erythrocyte and platelet phospholipid and fatty acid composition and protein oxidation in chronic obstructive pulmonary disease and asthma. Platelets. 2007;18(1):43–51. doi: 10.1080/09537100600800776. [DOI] [PubMed] [Google Scholar]
- 148.Marsboom G, et al. Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ Res. 2012;110(11):1484–1497. doi: 10.1161/CIRCRESAHA.111.263848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Chaung WW, Wu R, Ji Y, Dong W, Wang P. Mitochondrial transcription factor A is a proinflammatory mediator in hemorrhagic shock. Int J Mol Med. 2012;30(1):199–203. doi: 10.3892/ijmm.2012.959. [DOI] [PMC free article] [PubMed] [Google Scholar]