

Abstract
Effective chemopreventive agents are needed against lung cancer, the leading cause of cancer death. Results from our previous work showed that dietary dihydromethysticin (DHM) effectively blocked initiation of lung tumorigenesis by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) in A/J mice, and it preferentially reduced 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL)-derived DNA adducts in lung. This study explored the mechanism(s) responsible for DHM’s differential effects on NNK/NNAL-derived DNA damage by quantifying their metabolites in A/J mice. The results showed that dietary DHM had no effect on NNK or NNAL abundance in vivo, indicating that DHM does not affect NNAL formation from NNK. DHM had a minimal effect on cytochrome P450 2A5 (CYP2A5, which catalyzes NNK and NNAL bioactivation in A/J mouse lung), suggesting that it does not inhibit NNAL bioactivation. Dietary DHM significantly increased O-glucuronidated NNAL (NNAL-O-gluc) in A/J mice. Lung and liver microsomes from dietary DHM-treated mice showed enhanced activities for NNAL O-glucuronidation. These results overall support the notion that dietary DHM treatment increases NNAL detoxification, potentially accounting for its chemopreventive efficacy against NNK-induced lung tumorigenesis in A/J mice. The ratio of urinary NNAL-O-gluc and free NNAL may serve as a biomarker to facilitate the clinical evaluation of DHM-based lung cancer chemopreventive agents.
Introduction
Lung cancer kills ∼160,000 people in the United States each year (Siegel et al., 2013). Although cigarette smoking cessation is the ideal strategy to reduce lung cancer risk, quitting is difficult because of the addictive nature of nicotine in tobacco products. An alternative strategy is to develop effective lung cancer chemopreventive agents that would be complementary to tobacco cessation to help prevent this deadly disease. Tobacco smoke contains various classes of carcinogens, including N-nitrosamines, polycyclic aromatic hydrocarbons, heterocyclic aromatic amines, and others. NNK [4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone] is a tobacco-specific N-nitrosamine with potent pulmonary carcinogenicity, inducing the formation of lung adenoma and adenocarcinoma in multiple species (Hecht, 1998). Substantial evidence suggests that NNK in cigarettes contributes to lung adenocarcinoma incidence among U.S. smokers (Hecht, 2014), which makes blocking NNK-induced lung tumorigenesis a plausible approach to decrease tobacco smoke-induced lung cancer.
We recently demonstrated that dihydromethysticin (DHM) (Fig. 1) effectively blocked NNK-induced lung adenoma formation in A/J mice when administered during the initiation phase (Narayanapillai et al., 2014), positioning DHM as a promising candidate for lung cancer prevention. Mechanistically, DHM dose-dependently reduced pulmonary DNA adducts derived from 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL), one major metabolite of NNK (Carmella et al., 1993) and a potent pulmonary carcinogen (Upadhyaya et al., 1999). Interestingly DHM had minimal effects on DNA adducts derived directly from NNK bioactivation. Dihydrokavain (DHK) (Fig. 1), despite its close structural similarity to DHM, was completely void of these properties and thus can serve as a control compound for mechanistic investigation of DHM.
Fig. 1.

Dihydromethysticin (DHM) and dihydrokavain (DHK).
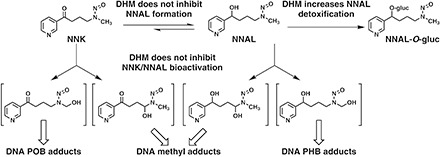
Based on the known pathways of NNK and NNAL metabolism (Hecht, 1998), we hypothesized that the preferential reduction in NNAL-derived DNA damage by DHM in the lung may be mediated via inhibiting NNAL formation from NNK, inhibiting cytochrome P450 enzyme-mediated NNAL bioactivation, or enhancing UDP-glucuronosyltransferase (UGT)–mediated NNAL detoxification (Fig. 2). We therefore quantified NNK, NNAL, and glucuronidated NNAL (NNAL-gluc) in serum and urine samples from mice exposed to dietary DHM in comparison with those from control mice or DHK-treated mice. We also evaluated the effect of DHM on the activity of cytochrome P450 2A5 (CYP2A5) in liver microsomes. The results indicate that dietary DHM treatment increases NNAL glucuronidation, potentially leading to enhanced detoxification and accounting for the preferential reduction in NNAL-derived DNA damage.
Fig. 2.

Potential mechanisms of DHM on NNAL metabolism, inhibiting NNAL formation, inhibiting NNAL bioactivation, or enhancing NNAL detoxification, which could account for its preferential reduction in NNAL-derived DNA damage.
Materials and Methods
Chemicals
NNK, [13C6]NNK, [13C6]NNAL, [CD3]NNAL-N-gluc, and [4-CD2, CD3]NNAL-O-gluc were purchased from Toronto Research Chemicals (Toronto, Ontario, Canada). [Caution: NNK and NNAL are carcinogenic. They should be handled in a well-ventilated hood with extreme care, and with proper personal protective equipment.] AIN-93 G powdered diet was purchased from Harlan Teklad (Madison, WI). β-Glucuronidase (recombinant, expressed in an Escherichia coli–overproducing strain, cat. no. G8295) was obtained from Sigma-Aldrich (St. Louis, MO). Glass vials (2 ml of silane-treated), with or without fused inserts, were obtained from Chromtech (Apple Valley, MN). Kava was acquired from Gaia Herbs (Brevard, NC) as an ethanol extract of the wild crafted lateral root from Vanuatu, which was standardized to 150 mg/ml total kavalactones. DHM and DHK were isolated from kava following an established procedure (Narayanapillai et al., 2014). All other chemicals and solvents were acquired from Sigma-Aldrich or Fisher Scientific (Fairlawn, NJ) and used without further purification.
Animal Studies
These studies were approved by the University of Minnesota Institutional Animal Care and Use Committee and performed in accordance with National Institutes of Health guidelines. Female A/J mice, 6 weeks of age, were purchased from Jackson Laboratories (Bar Harbor, ME) and housed in the specific pathogen-free animal facilities of the Research Animal Resources, University of Minnesota. Food intake was monitored twice a week, and body weight was measured once a week. None of the treatment caused detectable changes to the mice based on food consumption and body weight changes.
Study 1.
After 1 week of acclimation, 48 female mice were randomized into 16 groups of three. Four groups were maintained on AIN-G powdered diet, four groups on AIN-G powdered diet supplemented with kava at a dose of 5 mg/g of diet, four groups on AIN-G powdered diet supplemented with DHM at a dose of 1 mg/g of diet and the other four groups AIN-G powdered diet supplemented with DHK at a dose of 1 mg/g of diet. Kava, DHM, or DHK supplemented diets were prepared following our reported procedures (Narayanapillai et al., 2014). After 1 week of consuming the various diets, each mouse was treated with NNK via i.p. injection (100 mg/kg of body weight in 100 µl saline). One group of mice from each treatment regimen was euthanized at 0.5, 1.0, 2.0, or 4.0 hours after NNK exposure, and serum and urine samples were collected. Serum collection and storage followed established procedures (Narayanapillai et al., 2014). For urine collection, each individual mouse was placed on a piece of aluminum foil, and urine was passively released upon CO2 euthanasia. Typically 25 to 150 µl of urine was collected from each mouse. The urine samples were stored at −80°C until analysis.
Study 2.
Fifteen female mice were randomized into three groups of five. One group was maintained on AIN-G powdered diet, one group on AIN-G powdered diet supplemented with DHM at a dose of 1 mg/g of diet, and the other group on AIN-G powdered diet supplemented with DHK at a dose of 1 mg/g of diet. One week thereafter, the mice were euthanized with CO2; the lung and liver tissues were collected and stored at −80°C until NNAL glucuronidation analysis.
CYP2A5 Enzymatic Assays
Liver tissues used herein were from previous studies (Narayanapillai et al., 2014). Mouse liver microsomes from control mice (n = 4) and mice treated with NNK, NNK + DHM (1 mg/g of diet), or NNK + DHK (1 mg/g of diet) (n = 3 each) were prepared as previously described elsewhere (von Weymarn and Murphy, 2003).
The effect of DHM and DHK on CYP2A5 enzymatic activity was evaluated using liver microsomes pooled from the four control mice. Briefly, aliquots of the pooled liver microsomes from untreated mice (75 µg protein) were added to reaction mixtures containing coumarin (2 or 20 μM), DHM, or DHK dissolved in methanol (10 or 100 µM) or methanol control and an NADPH-generating system in 50 mM Tris buffer (pH 7.4). The final reaction volume was 200 μl. After 15 minutes of incubation at 37°C, the reaction was terminated with the addition of 20 µl of trifluoroacetic acid (15%). The formation of 7-hydroxycoumarin from coumarin was measured by high-pressure liquid chromatography with fluorescence detection as previously described elsewhere (Bock and Bock-Hennig, 2010).
All experiments were performed in duplicate on at least three separate occasions. The enzymatic activity of CYP2A5 in each liver microsome preparation (20–80 µg protein) from the mice treated with NNK, NNK + DHM, or NNK + DHK (n = 3 each) was individually evaluated.
Urine Processing to Convert NNAL-gluc into NNAL and Liquid Chromatography with Tandem Mass Spectrometry Quantification
Urine samples were diluted 105 times with saline. The diluted samples were divided into two or three portions (0.25 ml each for estimation of free NNAL, free NNAL + NNAL-N-gluc, or total NNAL [the sum of free NNAL, NNAL-N-gluc, and NNAL-O-gluc]). Each sample was mixed with [13C6]NNAL as an internal standard (4 ng/ml), processed, and analyzed by liquid chromatography with tandem mass spectrometry (LC-MS/MS) as previously described elsewhere (Carmella et al., 2013).
Direct LC-MS/MS Detection and Quantification of NNAL-O-gluc, NNK, and NNAL in Urine and Serum Samples
Urine samples were diluted 105 times with saline. The diluted samples (0.1 ml each) were mixed with [4-CD2, CD3]NNAL-O-gluc and/or [CD3]NNAL-N-gluc at a final concentration of 5 ng/ml. For NNK and NNAL analysis, [13C6]NNK and [13C6]NNAL at a final concentration of 10 ng/ml each were added as the internal standards. The serum samples were diluted 104 times with saline, and each of the diluted samples (0.1 ml) was mixed with [4-CD2, CD3]NNAL-O-gluc, [13C6]NNK, and [13C6]NNAL at a final concentration of 10 ng/ml each.
LC-MS/MS analysis was performed using an Agilent 1100 series capillary high-pressure liquid chromatography system (Agilent Technologies, Palo Alto, CA) interfaced to a TSQ Quantum Discovery Max triple quadrupole mass spectrometer (Thermo Electron, San Jose, CA) operated in selective reaction monitoring mode and tuned to NNAL-N-gluc.
NNK, NNAL, NNAL-N-gluc, and NNAL-O-gluc were analyzed simultaneously on a Phenomenex Luna C18 (150 × 0.5 mm, 3 microns) capillary column, eluted under the following conditions: 95% A (10 mM ammonium acetate): 5% B (acetonitrile) held for 5 minutes, followed by a linear gradient to 30% B in 2 minutes, a 3-minute hold at 30% B, a linear gradient to 50% B in 1 minute, a 2-minute hold at 50% B, a linear gradient to 5% B in 1 minute, and a 11-minute hold at 5% B. The flow rate was 10 µl/min.
For NNAL-N-gluc, the mass transitions monitored were as follows: NNAL-N-gluc (m/z 386 → 210, collision energy (CE) = 14; and m/z 386 → 180, CE = 25) and [CD3]NNAL-N-gluc (m/z 389 → 213, CE = 14; and m/z 389 → 183, CE = 25). For NNAL-O-gluc, the mass transitions monitored were as follows: NNAL-O-gluc (m/z 386 → 210, CE = 14; and m/z 386 → 162, CE = 20) and [4-CD2,CD3]NNAL-O-gluc (m/z 391 → 215, CE = 14; and m/z 391 → 167, CE = 20). For NNK, the mass transitions monitored were as follows: [12C6]NNK (m/z 208 → 122, CE = 12) and [13C6] NNK (m/z 214 → 128, CE = 12). For NNAL, the mass transitions monitored were as follows: [12C6]NNAL (m/z 210 → 92, CE = 12) and [13C6]NNAL (m/z 216 → 98, CE = 12).
NNAL Glucuronidation Activity of Mouse Lung and Liver Microsomes.
Lung and liver microsomes were prepared from the respective tissues (100 mg) in study 2 as described previously elsewhere (Wong et al., 2003). The microsomes were resuspended in 10 mM Tris buffer (pH 7.4, 1 mM EDTA, and 20% glycerol, 0.2 ml) and stored at −80°C. Protein concentrations were determined by the standard methods.
NNAL glucuronidating activity was evaluated via a reported procedure (Crampsie et al., 2011). Briefly, liver or lung microsomes (0.1 mg of protein) were preincubated with 1.1 mg/mL alamethicin, 8.5 mM saccharolactone, 10 mM MgCl2, and 50 mM KH2PO4 on ice for 10 minutes and then at 37°C for 3 minutes. After preincubation, NNAL and UDPGA were added to achieve final concentrations of 5 mM and 2 mM, respectively, in a total reaction volume of 100 µl. The reaction mixture was incubated for 1 hour at 37°C. The samples were then mixed with [4-CD2, CD3]NNAL-O-gluc (5 pmol). The reactions were terminated by the addition of 0.1 volumes of 0.3 M Ba(OH)2 and ZnSO4 each, while being cooled on ice. The precipitate was removed by centrifugation, and NNAL-O-gluc in the supernatant was purified using a Resprep C18 SPE column (cat. no. 26030; Restek, Bellefonte, PA), which was preactivated with methanol and equilibrated with H2O. The sample retained on the column was washed with doubly distilled H2O (1 ml) and eluted with 5% methanol (0.5 ml). The eluted samples were dried using a SpeedVac (Thermo Fisher Scientific, Waltham, MA), reconstituted in 100 μl of H2O, and analyzed for NNAL-O-gluc by LC-MS/MS by the method described earlier.
Results
Effects of DHM, DHK, or Kava on NNK and NNAL Abundance in A/J Mouse Urines and Sera.
We first determined the ratio of NNK and total NNAL in the urine samples from A/J mice with different dietary treatments, including control, DHM, DHK, and kava. Ratios instead of absolute quantities were reported because we did not control for urine excretion during the experimental period, and some mice might have urinated between NNK treatment and euthanasia. In addition, the relative amounts of urine loss during collection might be different among the mice. However, these variables were not critical to the ratio. As shown in Fig. 3A, there were no significant differences in the ratios of NNK to total NNAL among different treatment regimens, suggesting that DHM and kava treatment had no effect on NNAL formation from NNK. To confirm this, the absolute amount of NNK and NNAL in the serum samples from control and DHM-treated mice were quantified (Fig. 3B). The results also showed no significant differences between DHM-treated mice and controls.
Fig. 3.

The effect of dietary DHM, DHK, or kava on the abundance of NNK and NNAL in A/J mouse urine (A) and serum (B). Mice maintained on the specified diet were given NNK and euthanized at different time points thereafter (n = 3 per group except for the urine samples with DHM treatment at 0.5 hours; n = 2 because one mouse urinated upon NNK administration, so urine collection at euthanasia was not successful). Statistical analysis was performed with one-way analysis of variance for urine samples and with two-tailed Student’s t test for serum samples. None of the differences were statistically significant. Cont: control diet.
Effect of Dietary DHM on CYP2A5 Activity.
CYP2A5 is the major catalyst of NNK and NNAL bioactivation in A/J mice (Hollander et al., 2011; Zhou et al., 2012; Megaraj et al., 2014). Mouse liver microsomes were prepared and analyzed for coumarin 7-hydroxylation activity [a CYP2A5-specific reaction (von Weymarn and Murphy, 2003)] in the presence of DHM or DHK. Coumarin 7-hydroxylation was inhibited by 62% with 10 μM DHK whereas DHM exhibited minimal inhibition at 10 µM and a concentration of 100 μM was required to achieve similar inhibition as DHK at 10 µM (Table 1). The concentration of DHM and DHK (10 µM) was based on our preliminary pharmacokinetic quantification of DHM in the mouse lung tissues (data not shown). The CYP2A5 activity in liver microsomes from mice consuming dietary DHM or DHK also did not differ from that in control mice (Table 2). A DHM-enriched kava fraction also showed no inhibition on CYP2A13 (data not shown), the major human pulmonary CYP responsible for NNK/NNAL bioactivation (Megaraj et al., 2014). Overall these results suggest that inhibiting CYP2A5-mediated NNAL bioactivation is unlikely to be the primary mode of action of DHM.
TABLE 1.
Effect of DHM and DHK on coumarin 7-hydroxylation (CYP2A5) activity in mouse liver microsomes
Mouse liver microsomes were incubated with 2 µM coumarin and NADPH for 15 minutes at 37°C in the presence or absence of DHM or DHK, which were dissolved in methanol. Hence, 0.4% methanol was added to the control reactions.
| Concentration (µM) | Percentage of Inhibitiona | |
|---|---|---|
| DHK | 10 | 62 ± 6 |
| DHM | 10 | 19 ± 9 |
| DHM | 100 | 71 ± 10 |
Average of three independent experiments performed in duplicate.
TABLE 2.
Effect of dietary DHM and DHK on coumarin 7-hydroxylation (CYP2A5) activity in mouse liver microsomes (n = 3)
Mouse liver microsomes (20–80 µg protein) were incubated with 20 µM coumarin and NADPH for 15 minutes at 37°C.
| 7-Hydroxycoumarin Formationa |
|
|---|---|
| pmol/min/mg | |
| Control | 12 ± 3 |
| DHK (1 mg/g of diet) | 16 ± 4 |
| DHM (1 mg/g of diet) | 17 ± 4 |
Statistical analysis was performed with one-way analysis of variance. None of the differences were statistically significant.
Effect of Dietary DHM and Kava on the Urinary Ratio of NNAL-O-gluc to Free NNAL in A/J Mice.
The best characterized pathway for NNAL detoxification is UGT-catalyzed glucuronidation. Both N- and O-glucuronides of NNAL (NNAL-N-gluc and NNAL-O-gluc) have been detected in smokers’ urine (Carmella et al., 2002). The ratio of urinary NNAL-gluc (the sum of NNAL-N-gluc and NNAL-O-gluc) and free NNAL is a convenient parameter to evaluate glucuronidation-mediated NNAL detoxification (Carmella et al., 2013). The urinary ratios of NNAL-gluc to free NNAL for DHM or kava-treated mice were about twice those of NNK control mice at all four times of urine collection (Fig. 4). The differences were significantly different at 0.5, 2, and 4-hour time points (P < 0.05). DHK, the inactive analog, had no effect.
Fig. 4.

Effects of dietary DHM, DHK, and kava on the ratio of NNAL-gluc and free NNAL in A/J mouse urine. Mice maintained on the specified diet were given NNK and euthanized at different time points thereafter (n = 3 per group except for the urine samples with DHM treatment at 0.5 hours; n = 2). Statistical analysis was performed with one-way analysis of variance followed by Dunnett’s test relative to the corresponding NNK treatment group; *P < 0.05; **P < 0.01. Cont: control diet.
Although both NNAL-N-gluc and NNAL-O-gluc have been detected in human urine, such information is not available for mice. We therefore analyzed their relative abundance in A/J mouse urine.
NNAL-N-gluc and NNAL-O-gluc were first quantified as the released aglycons upon NaOH or β-glucuronidase treatment (Carmella et al., 2013). No NNAL-N-gluc was detected in mouse urine, irrespective of treatment regimens (data not shown). To confirm the absence of NNAL-N-gluc in mouse urine, representative mouse urine samples (two from NNK-treated mice, two from NNK + DHM-treated mice, and two from NNK + DHK-treated mice, all at the 2-hour time point) were directly analyzed for NNAL-N-gluc and NNAL-O-gluc via a modified LC-MS/MS method with the corresponding deuterium-labeled compounds as internal standards. NNAL-N-gluc (RT = 4.9 minutes) and NNAL-O-gluc (RT = 12.8 minutes) were readily separated (Fig. 5). No appreciable amount of NNAL-N-gluc was detected (<0.1% of NNAL-O-gluc), but NNAL-O-gluc was readily detectable in micromolar abundance (Panel A). Similar results were observed in the other samples.
Fig. 5.

LC-MS/MS chromatograms of urinary NNAL-N-gluc and NNAL-O-gluc from a mouse exposed to NNK and dietary DHM with urine collection 2 hours after NNK. (A) Mass transition monitoring m/z 386 → 210 of NNAL-N-gluc (RT = 4.9 minutes) and NNAL-O-gluc (RT = 12.8 minutes). (B) Mass transition monitoring of the internal standards m/z 389 → 213 of [CD3]NNAL-N-gluc and m/z 391 → 215 of [4-CD2, CD3]NNAL-O-gluc.
These data demonstrated that the majority of NNAL-gluc in A/J mouse urine is NNAL-O-gluc. Using this method, the urine samples were analyzed again to directly quantify NNAL-O-gluc. The ratios of NNAL-O-gluc and free NNAL from this direct quantification were in good agreement with the ratios from the indirect quantification (data not shown).
To confirm the increase in NNAL-O-gluc abundance upon DHM treatment, we quantified NNAL-O-gluc in serum samples from A/J mice with and without dietary DHM treatment via the direct LC-MS/MS method. Higher amounts of NNAL-O-gluc were detected in all samples from DHM-treated mice in comparison with those from control mice (Fig. 6A), which was also reflected in the ratio of NNAL-O-gluc to free NNAL (Fig. 6B).
Fig. 6.

Quantities of NNAL-O-gluc (A) and ratio of NNAL-O-gluc to free NNAL (B) in serum samples from NNK-exposed A/J mice upon dietary DHM in comparison with NNK-exposed mice on control diet (n = 3 per group). Statistical analysis was performed with two-tailed Student’s t test between DHM treatment and control group; *P < 0.05 and **P < 0.01. Cont: control diet.
Effect of Dietary DHM on UGT Enzymatic Activity in A/J Mouse Lungs and Livers.
Upon establishing the increase in NNAL-O-gluc with DHM treatment, we evaluated whether such an increase may be mediated via enhanced NNAL glucuronidation activity in lung and liver tissues upon dietary DHM treatment. In both tissues the extent of NNAL glucuronidation increased with dietary DHM treatment (Fig. 7); the increase was significant in liver whereas DHK treatment had no such effects. We next explored whether DHM could biochemically enhance NNAL glucuronidation; we incubated DHM (10 µM) with liver microsomes from the control mice and quantified the NNAL glucuronidation. DHK was evaluated as well. Under such conditions, DHM and DHK had no significant effect on NNAL glucuronidation (data not shown).
Fig. 7.

NNAL-O-gluc formation by (A) lung microsomes and (B) liver microsomes from A/J mice exposed to dietary DHM or DHK in comparison with control (n = 5). We incubated 5 mM NNAL for 30 minutes with NNAL-O-gluc quantified by LC-MS/MS. Statistical analysis was performed with one-way analysis of variance followed by Dunnett’s test relative to the control group; *P < 0.05.
Discussion
Dietary DHM has demonstrated potent efficacy in blocking NNK-induced lung tumorigenesis in A/J mice by a unique mechanism—preferential reduction in DNA damage derived from NNAL in the target lung tissues—while having minimal effect on DNA damage derived directly from NNK (Narayanapillai et al., 2014). Based on the well-characterized metabolism of NNK and NNAL (Hecht, 1998), we investigated the impact of dietary DHM treatment on NNAL formation from NNK, on CYP2A5 activity (the enzyme likely responsible for NNAL bioactivation), and on NNAL glucuronidation, one detoxification pathway for NNAL (Fig. 2) that could contribute to DHM-induced preferential reduction in NNAL-derived DNA damage.
We found that dietary DHM treatment had no effect on the abundance of NNK and NNAL in serum or urine samples (Fig. 3), demonstrating that DHM does not inhibit the reductive formation of NNAL from NNK. Results from numerous studies suggest that kavalactones can directly inhibit cytochrome 450 enzymes or indirectly regulate cytochrome 450 enzymes via transcriptional induction (Ma et al., 2004; Zou et al., 2004; Lim et al., 2007; Guo et al., 2010; Li et al., 2011). DHM, one of the major kavalactones, had minimal effect on CYP2A5 enzymatic activity in the mouse liver microsomes at 10 µM. Although it inhibited CYP2A5 by 71% at 100 µM, such a high concentration is likely physiologically irrelevant. DHK indeed was a better CYP2A5 inhibitor in this assay, but it failed to block NNK-induced lung tumorigenesis (Narayanapillai et al., 2014). CYP2A5 activity in liver microsomes from mice exposed to dietary DHM treatment was similar to that from control mice, suggesting that DHM did not affect CYP2A5 activity in vivo. These data overall suggest that it is unlikely that dietary DHM treatment inhibits the bioactivation of NNAL.
NNAL can be detoxified via several mechanisms, such as N-oxidation and glucuronidation. NNAL glucuronidation has been characterized in several species, including humans (Carmella et al., 2002; Wiener et al., 2004; Chen et al., 2008; Balliet et al., 2010). Although both NNAL-N-gluc and NNAL-O-gluc have been detected in humans (Carmella et al., 2002), we only detected NNAL-O-gluc in A/J mice, indicating a species difference between mice and human in NNAL detoxification. We observed a clear increase in NNAL-O-gluc formation in A/J mice exposed to dietary DHM but not DHK. In addition, both lung and liver microsomes from DHM-treated mice had enhanced NNAL glucuronidating activity. This again was not observed in DHK-treated mice. These results overall support the mechanism that dietary DHM treatment enhances NNAL glucuronidation both in the lung and liver tissues, which may lead to increased NNAL detoxification and reduction in NNAL-derived DNA damage in A/J mice. Dietary DHM may up-regulate UGT enzyme(s) via transcriptional activation of AHR as DHM is a potential AHR agonist (Li et al., 2011) and AHR can transcriptionally activate UGTs (Bock and Bock-Hennig, 2010).
Interestingly the concentration of free NNAL was not reduced in A/J mouse sera upon dietary DHM treatment despite the significant increase in the level of NNAL-O-gluc. The concentration of free NNAL in the lung tissues also did not appear to be reduced upon dietary DHM treatment (data not shown). The lack of reduction in free NNAL in the sera and lung tissues raises the question whether the increase in NNAL glucuronidation by DHM is critical for its reduction in NNAL-induced DNA damage.
It should be pointed out that the amount of free NNAL we quantified was for the whole lung tissues, which contain different type of cells, including type II cells, Clara cells, and small cells. As demonstrated by Devereux et al., different cells in the lung have varied sensitivity to NNK/NNAL-induced DNA damage; the amount of O6-mG adduct was 2 to 8 times higher in Clara cells and type II cells [potentially the target cells for tumorigenesis (Belinsky et al., 1991)] in comparison with small cells or the whole lung tissues (Devereux et al., 1993). These different types of cells also respond differently at the molecular level upon NNK treatment (Belinsky et al., 1988, 1996). It remains to be determined whether DHM treatment may have different effects on these cells. It is possible that DHM may selectively enhance the UGT activity in a subpopulation of the lung cells, which significantly reduces free NNAL in the target cells and prevents DNA adduct formation and tumorigenesis, even though the levels of free NNAL in the whole lung and sera are not affected. Work is ongoing to test these hypotheses.
Because DHM-induced increase in NNAL-O-gluc formation may account for its lung cancer chemopreventive activity, the urinary ratio of NNAL-gluc to free NNAL has the potential to facilitate the translational development of DHM. Results from several human studies demonstrate that such a ratio varies among individuals (Carmella et al., 1995; Park et al., 2015). It is possible that individuals with a low basal urinary ratio of NNAL-gluc to free NNAL may more likely benefit from DHM in reducing their lung cancer risk via enhancing/restoring their detoxification capacity. Such a biomarker could also be used to monitor the chemopreventive effect of DHM; if DHM increases the urinary ratio of NNAL-gluc to free NNAL in a specific individual, DHM may likely reduce the lung cancer risk via enhancing NNAL detoxification. Glucuronidation is also involved in the detoxification of other carcinogens (Bock, 1991; Dellinger et al., 2007). DHM may induce UGT(s) to help detoxify carcinogens besides NNAL, which would have a broader impact to reduce human cancer risk. On the other hand, glucuronidation is critical in the metabolism of various therapeutic drugs (Burchell, 2003) and DHM usage may change their pharmacokinetics and pharmacodynamics. Future studies are needed to investigate these aspects.
Abbreviations
- AHR
aryl hydrocarbon receptor
- CE
collision energy
- CYP2A5
cytochrome P450 2A5
- DHK
dihydrokavain
- DHM
dihydromethysticin
- LC-MS/MS
liquid chromatography with tandem mass spectrometry
- NNAL
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol
- NNAL-gluc
glucuronidated NNAL
- NNK
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone
- O6-mG
O6-methylguanine
- RT
retention time
- UGT
UDP-glucuronosyltransferase
Authorship Contributions
Participated in research design: Narayanapillai, Upadhyaya, Hecht, Murphy, Xing.
Conducted experiments: Narayanapillai, von Weymarn, Carmella, Leitzman, Paladino, Upadhyaya.
Contributed new reagents or analytic tools: von Weymarn, Narayanapillai.
Performed data analysis: Narayanapillai, von Weymarn, Carmella, Hecht, Murphy, Xing.
Wrote or contributed to the writing of the manuscript: Narayanapillai, Hecht, Murphy, Xing.
Footnotes
This work was supported by a grant from the National Institutes of Health National Cancer Institute [Grant R01 CA193278].
References
- Balliet RM, Chen G, Dellinger RW, Lazarus P. (2010) UDP-glucuronosyltransferase 1A10: activity against the tobacco-specific nitrosamine, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol, and a potential role for a novel UGT1A10 promoter deletion polymorphism in cancer susceptibility. Drug Metab Dispos 38:484–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belinsky SA, Devereux TR, White CM, Foley JF, Maronpot RR, Anderson MW. (1991) Role of Clara cells and type II cells in the development of pulmonary tumors in rats and mice following exposure to a tobacco-specific nitrosamine. Exp Lung Res 17:263–278. [DOI] [PubMed] [Google Scholar]
- Belinsky SA, Dolan ME, White CM, Maronpot RR, Pegg AE, Anderson MW. (1988) Cell specific differences in O6-methylguanine-DNA methyltransferase activity and removal of O6-methylguanine in rat pulmonary cells. Carcinogenesis 9:2053–2058. [DOI] [PubMed] [Google Scholar]
- Belinsky SA, Nikula KJ, Baylin SB, Issa JP. (1996) Increased cytosine DNA-methyltransferase activity is target-cell-specific and an early event in lung cancer. Proc Natl Acad Sci USA 93:4045–4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock KW. (1991) Roles of UDP-glucuronosyltransferases in chemical carcinogenesis. Crit Rev Biochem Mol Biol 26:129–150. [DOI] [PubMed] [Google Scholar]
- Bock KW, Bock-Hennig BS. (2010) UDP-glucuronosyltransferases (UGTs): from purification of Ah-receptor-inducible UGT1A6 to coordinate regulation of subsets of CYPs, UGTs, and ABC transporters by nuclear receptors. Drug Metab Rev 42:6–13. [DOI] [PubMed] [Google Scholar]
- Burchell B. (2003) Genetic variation of human UDP-glucuronosyltransferase: implications in disease and drug glucuronidation. Am J Pharmacogenomics 3:37–52. [DOI] [PubMed] [Google Scholar]
- Carmella SG, Akerkar S, Hecht SS. (1993) Metabolites of the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in smokers’ urine. Cancer Res 53:721–724. [PubMed] [Google Scholar]
- Carmella SG, Akerkar SA, Richie JP, Jr, Hecht SS. (1995) Intraindividual and interindividual differences in metabolites of the tobacco-specific lung carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) in smokers’ urine. Cancer Epidemiol Biomarkers Prev 4:635–642. [PubMed] [Google Scholar]
- Carmella SG, Le Ka KA, Upadhyaya P, Hecht SS. (2002) Analysis of N- and O-glucuronides of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL) in human urine. Chem Res Toxicol 15:545–550. [DOI] [PubMed] [Google Scholar]
- Carmella SG, Ming X, Olvera N, Brookmeyer C, Yoder A, Hecht SS. (2013) High throughput liquid and gas chromatography-tandem mass spectrometry assays for tobacco-specific nitrosamine and polycyclic aromatic hydrocarbon metabolites associated with lung cancer in smokers. Chem Res Toxicol 26:1209–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Dellinger RW, Sun D, Spratt TE, Lazarus P. (2008) Glucuronidation of tobacco-specific nitrosamines by UGT2B10. Drug Metab Dispos 36:824–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crampsie MA, Jones N, Das A, Aliaga C, Desai D, Lazarus P, Amin S, Sharma AK. (2011) Phenylbutyl isoselenocyanate modulates phase I and II enzymes and inhibits 4-(methylnitrosamino)-1-(3-pyridyl)- 1-butanone-induced DNA adducts in mice. Cancer Prev Res (Phila) 4:1884–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellinger RW, Chen G, Blevins-Primeau AS, Krzeminski J, Amin S, Lazarus P. (2007) Glucuronidation of PhIP and N-OH-PhIP by UDP-glucuronosyltransferase 1A10. Carcinogenesis 28:2412–2418. [DOI] [PubMed] [Google Scholar]
- Devereux TR, Belinsky SA, Maronpot RR, White CM, Hegi ME, Patel AC, Foley JF, Greenwell A, Anderson MW. (1993) Comparison of pulmonary O6-methylguanine DNA adduct levels and Ki-ras activation in lung tumors from resistant and susceptible mouse strains. Mol Carcinog 8:177–185. [DOI] [PubMed] [Google Scholar]
- Guo L, Shi Q, Dial S, Xia Q, Mei N, Li QZ, Chan PC, Fu P. (2010) Gene expression profiling in male B6C3F1 mouse livers exposed to kava identifies—changes in drug metabolizing genes and potential mechanisms linked to kava toxicity. Food Chem Toxicol 48:686–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht SS. (1998) Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines. Chem Res Toxicol 11:559–603. [DOI] [PubMed] [Google Scholar]
- Hecht SS. (2014) It is time to regulate carcinogenic tobacco-specific nitrosamines in cigarette tobacco. Cancer Prev Res (Phila) 7:639–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollander MC, Zhou X, Maier CR, Patterson AD, Ding X, Dennis PA. (2011) A Cyp2a polymorphism predicts susceptibility to NNK-induced lung tumorigenesis in mice. Carcinogenesis 32:1279–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Mei H, Wu Q, Zhang S, Fang JL, Shi L, Guo L. (2011) Methysticin and 7,8-dihydromethysticin are two major kavalactones in kava extract to induce CYP1A1. Toxicol Sci 124:388–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim ST, Dragull K, Tang CS, Bittenbender HC, Efird JT, Nerurkar PV. (2007) Effects of kava alkaloid, pipermethystine, and kavalactones on oxidative stress and cytochrome P450 in F-344 rats. Toxicol Sci 97:214–221. [DOI] [PubMed] [Google Scholar]
- Ma Y, Sachdeva K, Liu J, Ford M, Yang D, Khan IA, Chichester CO, Yan B. (2004) Desmethoxyyangonin and dihydromethysticin are two major pharmacological kavalactones with marked activity on the induction of CYP3A23. Drug Metab Dispos 32:1317–1324. [DOI] [PubMed] [Google Scholar]
- Megaraj V, Zhou X, Xie F, Liu Z, Yang W, Ding X. (2014) Role of CYP2A13 in the bioactivation and lung tumorigenicity of the tobacco-specific lung procarcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone: in vivo studies using a CYP2A13-humanized mouse model. Carcinogenesis 35:131–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanapillai SC, Balbo S, Leitzman P, Grill AE, Upadhyaya P, Shaik AA, Zhou B, O’Sullivan MG, Peterson LA, Lu J, et al. (2014) Dihydromethysticin from kava blocks tobacco carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced lung tumorigenesis and differentially reduces DNA damage in A/J mice. Carcinogenesis 35:2365–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SL, Carmella SG, Ming X, Vielguth E, Stram DO, Le Marchand L, Hecht SS. (2015) Variation in levels of the lung carcinogen NNAL and its glucuronides in the urine of cigarette smokers from five ethnic groups with differing risks for lung cancer. Cancer Epidemiol Biomarkers Prev 24:561–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R, Naishadham D, Jemal A. (2013) Cancer statistics, 2013. CA Cancer J Clin 63:11–30. [DOI] [PubMed] [Google Scholar]
- Upadhyaya P, Kenney PM, Hochalter JB, Wang M, Hecht SS. (1999) Tumorigenicity and metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol enantiomers and metabolites in the A/J mouse. Carcinogenesis 20:1577–1582. [DOI] [PubMed] [Google Scholar]
- von Weymarn LB, Murphy SE. (2003) CYP2A13-catalysed coumarin metabolism: comparison with CYP2A5 and CYP2A6. Xenobiotica 33:73–81. [DOI] [PubMed] [Google Scholar]
- Wiener D, Doerge DR, Fang JL, Upadhyaya P, Lazarus P. (2004) Characterization of N-glucuronidation of the lung carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL) in human liver: importance of UDP-glucuronosyltransferase 1A4. Drug Metab Dispos 32:72–79. [DOI] [PubMed] [Google Scholar]
- Wong HL, Murphy SE, Wang M, Hecht SS. (2003) Comparative metabolism of N-nitrosopiperidine and N-nitrosopyrrolidine by rat liver and esophageal microsomes and cytochrome P450 2A3. Carcinogenesis 24:291–300. [DOI] [PubMed] [Google Scholar]
- Zhou X, D’Agostino J, Xie F, Ding X. (2012) Role of CYP2A5 in the bioactivation of the lung carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in mice. J Pharmacol Exp Ther 341:233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, Henderson GL, Harkey MR, Sakai Y, Li A. (2004) Effects of kava (Kava-kava, 'Awa, Yaqona, Piper methysticum) on c-DNA-expressed cytochrome P450 enzymes and human cryopreserved hepatocytes. Phytomedicine 11:285–294. [DOI] [PubMed] [Google Scholar]