

Abstract
Objective
Nonketotic hyperglycinemia is a neurometabolic disorder characterized by intellectual disability, seizures, and spasticity. Patients with attenuated nonketotic hyperglycinemia make variable developmental progress. Predictive factors have not been systematically assessed.
Methods
We reviewed 124 patients stratified by developmental outcome for biochemical and molecular predictive factors. Missense mutations were expressed to quantify residual activity using a new assay.
Results
Patients with severe nonketotic hyperglycinemia required multiple anticonvulsants, whereas patients with developmental quotient (DQ) > 30 did not require anticonvulsants. Brain malformations occurred mainly in patients with severe nonketotic hyperglycinemia (71%) but rarely in patients with attenuated nonketotic hyperglycinemia (7.5%). Neonatal presentation did not correlate with outcome, but age at onset ≥ 4 months was associated with attenuated nonketotic hyperglycinemia. Cerebrospinal fluid (CSF) glycine levels and CSF:plasma glycine ratio correlated inversely with DQ; CSF glycine > 230 μM indicated severe outcome and CSF:plasma glycine ratio ≤ 0.08 predicted attenuated outcome. The glycine index correlated strongly with outcome. Molecular analysis identified 99% of mutant alleles, including 96 novel mutations. Mutations near the active cleft of the P‐protein maintained stable protein levels. Presence of 1 mutation with residual activity was necessary but not sufficient for attenuated outcome; 2 such mutations conferred best outcome. Divergent outcomes for the same genotype indicate a contribution of other genetic or nongenetic factors.
Interpretation
Accurate prediction of outcome is possible in most patients. A combination of 4 factors available neonatally predicted 78% of severe and 49% of attenuated patients, and a score based on mutation severity predicted outcome with 70% sensitivity and 97% specificity. Ann Neurol 2015;78:606–618
Nonketotic hyperglycinemia (NKH; glycine encephalopathy, Mendelian Inheritance in Man 605899) is an autosomal recessive neurometabolic condition caused by deficient activity of the glycine cleavage enzyme system. Patients usually present in the neonatal period with increasing lethargy, coma, seizures, and apnea, which spontaneously resolves after 1 to 3 weeks.1, 2, 3 They develop intellectual disability, seizures, persistent axial hypotonia, and spasticity. Some patients have congenital brain malformations, including agenesis of the corpus callosum or retrocerebellar cyst with hydrocephalus.4, 5, 6 Patients with attenuated NKH exhibit a variable degree of intellectual disability and seizures, hyperactivity, chorea, and intermittent episodes of lethargy and ataxia.2, 3, 7, 8 Up to 15% of neonatally presenting patients and 50% of infancy‐presenting patients show this attenuated phenotype,2, 3 but elements predicting the neurodevelopmental outcome in NKH have not been systematically studied.
The glycine cleavage enzyme complex is composed of 4 proteins. The P‐protein, encoded by the GLDC gene, is a pyridoxal phosphate‐dependent glycine decarboxylase; the H‐protein, encoded by the GCSH gene, is a lipoic acid containing hydrogen‐carrier protein; the T‐protein, encoded by the AMT gene, is a tetrahydrofolate‐dependent protein; and the L‐protein is a lipoamide dehydrogenase. Patients with classic NKH have mutations in GLDC or AMT, including exonic deletions in GLDC.9, 10, 11 The glycine cleavage enzyme is expressed in liver, brain, and placenta, and at low levels in Epstein–Barr virus (EBV)‐transformed lymphoblasts.12 Deficient enzyme activity causes elevated glycine levels in plasma and cerebrospinal fluid (CSF) with an elevated CSF:plasma glycine ratio.13 Lower CSF glycine levels and a lower ratio were reported for some patients with attenuated NKH.7, 14 Treatment most commonly involves sodium benzoate given to lower plasma glycine levels, and dextromethorphan or ketamine with the intent to reduce the excessive stimulating activity of glycine on N‐methyl‐D‐aspartate receptors, with unclear effect on outcome.
Predicting the prognosis of patients with NKH is important for families to make decisions on treatment following diagnosis, particularly in the neonatal intensive care setting, where withdrawal of life‐sustaining therapy may be considered.15 Differences in treatment response may exist between outcome categories, requiring early identification for appropriate treatment. In this study, we reviewed the outcome of patients with NKH in relation to biochemical, enzymatic, and molecular markers in addition to basic clinical and radiological findings.
Patients and Methods
Clinical Studies
The primary study was approved by the ethics committees of the Colorado Multiple Institutional Review Board and Catholic University Leuven, and written informed consent was obtained from all study subjects. A second limited exempt chart review study was approved by the Colorado Multiple Institutional Review Board. Some clinical details of 12 patients were previously described.2, 5, 16, 17, 18 All patients had classic NKH evidenced by elevated CSF glycine levels, elevated CSF:plasma glycine ratio, normal urine organic acids, and mutations in either GLDC or AMT excluding variant NKH.19, 20 Medical records were abstracted through a questionnaire with data listed in the Supplementary Table, and a clinical investigator (J.L.K.V.H. or J.B.H.) examined the vast majority of patients. Age at symptom onset was considered neonatal if symptoms occurred in the first week of life, with later onset specified. Glycine levels were recorded at diagnosis, excluding blood contaminated CSF samples.20 Brain magnetic resonance imaging (MRI) reports were reviewed for structural abnormalities. Because in NKH seizure propensity increases during the first year of life and improves on benzoate treatment, therapy resistance of the seizure disorder was estimated from the number of concurrent antiepileptic drugs (AEDs) used after age 1 year and while on benzoate therapy.2 The glycine index was calculated by subtracting the molar glycine intake in food from a dietary recall from the molar dose of sodium benzoate needed to normalize plasma glycine levels divided by body weight, which reflects a whole body balance of glycine metabolism.18 Enzyme assay results were recorded. Neurodevelopmental outcome was systematically assessed. Patients whose sole developmental milestone consisted of the ability to smile were given a developmental age of ≤6 weeks and categorized as severe NKH. Those making developmental progress beyond this stage were categorized as attenuated NKH, and formal developmental testing at an age > 2 years was used to derive a developmental quotient (DQ). Typical measures used were Bayley Scales of Infant Development, Mullen Scales of Early Learning, or Wechsler Scale of Intelligence. For each child, a DQ was calculated. The DQ is a measure of the rate of development. By using the mean or median age at which a milestone presents, a functional age equivalent for a child's development is given. It is the ratio of development age to chronological age. Subjects were subcategorized as poor attenuated NKH if DQ < 20, as intermediate attenuated NKH if DQ ≥ 20 and < 50, and as mild attenuated NKH if DQ ≥ 50. Patients without formal developmental assessment were categorized as attenuated NKH, not otherwise specified, if they could grasp and sit or walk independently, and as severe NKH if they could only smile.
Molecular Studies
Exons and at least 50 nucleotides into the flanking intron sequences of AMT (GenBank NM_000481.2, NP_000472.2; Ensembl ENSG00000145020) and GLDC (GenBank NM_000170.2, NP_000161.2; Ensembl ENSG00000178445) were sequenced. Phase was established from parental samples. Deletions and duplications in GLDC were analyzed by either multiplex ligation‐dependent probe amplification,10 or by a targeted array‐based comparative genomic hybridization utilizing a customized 60k oligonucleotide microarray with a positive threshold of 3 consecutive probes allowing the detection of 300bp deletions, and verified by dual real‐time polymerase chain reaction (PCR) with Taqman probes. Sequence variants were compared with 6 other species, and conservation was recorded. Splice site mutations were analyzed in silico using the BDGP Splice Site Prediction by Neural Network program,21 and for select patients by analysis of mRNA in EBV transformed lymphoblasts. To evaluate the effect on splicing of a deep intronic mutation in AMT (Patient 75), the generated cDNA was sequenced in 2 overlapping fragments. To evaluate whether the maternal mutation c.2203‐2A>G; IVS18‐2A>G (Patient 310) created a leaky splicing error, we evaluated for the presence of normally spliced mRNA not containing the paternal mutation c.547delG in the mRNA of the patient and mother by digestion of the cDNA with NcoI followed by agarose electrophoresis, where the paternal mutation abolishes a restriction site.
Protein Expression and Modeling Studies
The impact of each mutation on residual enzyme activity was systematically evaluated. Exonic deletions, nonsense mutations, and frameshift mutations were categorized as having no residual activity, except for a frameshift mutation that affected the final 20 amino acids. Splice site mutations affecting the 2 conserved donor or acceptor nucleotides and predicted in silico as likely affecting splicing were considered to leave no residual activity.
For missense mutations in GLDC, the impact on residual P‐protein activity was assessed by expression in vitro in COS cells and measuring the residual enzyme activity on a glycine exchange assay modified by the use of purified human recombinant lipoylated H‐protein.22 Missense mutations were introduced by site‐directed mutagenesis into a full‐length GLDC cDNA in the pCMV6‐Entry vector (RC211292; Origine, Rockville, MD), and with mock and wild‐type GLDC included in each experiment, were transfected into COS7 cells using Lipofectamine 2000 (Life Technologies, Carlsbad, CA). Cells were harvested after 48 hours, and P‐protein activity was measured by the glycine exchange assay containing excess lipoylated, recombinant human H‐protein.22 Activities were normalized by protein concentration measured by the Bradford method23 and mutant enzyme activity expressed as percentage of concurrent wild‐type enzyme activity determined in the same experiment. Transfection efficiency was monitored by quantitative PCR for GLDC cDNA content. To generate recombinant, lipoylated human H‐protein, GCSH cDNA PCR amplified from a human liver cDNA library was cloned as a thioredoxin fusion protein into the pBAD‐DEST 49 vector (Life Technologies), expressed in bacteria, and purified from the cell lysate by boiling for 1 minute followed by chromatography on a diethylaminoethyl cellulose column. Lipoate‐protein ligase (LplA) from Escherichia coli was expressed, purified, and used to lipoylate purified human H‐protein as described using a molar ratio of 0.5:1.0 LplA:H‐protein.24 Lipoylation was confirmed by Western blotting (ab97625; Abcam, Cambridge, MA) and by mass spectrometry. The N‐terminal thioredoxin tag was removed from the lipoylated H‐protein by enterokinase digestion (New England Biolabs, Ipswich, MA), and reaction products were separated using a Q Sepharose Fast Flow (GE Healthcare Bio‐Sciences, Pittsburgh, PA).
Following transfection in COS cells, steady state P‐protein levels were identified by Western blotting following sodium dodecyl sulfate–polyacrylamide gel electrophoresis using an antibody against the P‐protein from Abcam (ab97625), with β‐actin as a loading control. Coexpression studies used cotransfection with vectors containing both mutants compared to transfection with a single mutation and an empty vector. A model of human P‐protein encoded by the GLDC gene was created using the homology modeling program ESyPred3D based on the crystal structure of holo glycine decarboxylase from Synechocystis (Protein Data Bank ID: 4LHC).25, 26 The relative locations of mutated residues in the 3‐dimensional model were viewed using the program RasTop 2.2 (http://www.geneinfinity.org/rastop/).
Mutation Score
A mutation score was constructed as the sum of both alleles with a mutation without residual activity scored −1 and with residual activity +2. For the presumed mutation score, the AMT mutations p.M1T and p.R320H were considered to have no residual activity and the mutation p.I106T to have residual activity based on the occurrence in multiple families. Thus, the presence of 2 mutations without residual activity will give a score of −2, whereas the presence of at least 1 mutation with known residual activity will give a score of ≥1.
Statistics
Descriptive statistics were used to summarize the data. The difference between severe and attenuated variables is evaluated by Student t test for variables that have normal distribution, and by Mann–Whitney U test for variables that are not normally distributed as evaluated with Wilk–Shapiro. Subcategories of attenuated NKH were evaluated by 1‐way analysis of variance (ANOVA). The significance of a difference in proportion is calculated by the chi‐square test. Pearson and Spearman correlation tests were conducted to identify the correlation between outcomes and predictors, and identify colinearity among the predictors according to the data distribution. Multiple linear regression was performed to model the DQ in relation to predictors. Multiple ordinal logistic regression was performed to model the relationship between predictors and disease severity or AEDs. SPSS Statistics package version 21 (IBM, Armonk, NY) and SAS version 9.3 (SAS, Cary, NC) were used to perform the analyses. A p value < 0.05 was considered statistically significant.
Results
Clinical Severity: Developmental Outcome, Seizures, and Brain Malformations
Of 124 patients, there were 26 patients (21%) who died in the neonatal or early infantile period. The 56 patients (45%) with severe outcome NKH represent a uniform very poor development limited to smiling, or rarely able to roll from side to prone. There were 42 patients (34%) with attenuated NKH: 6 patients with poor attenuated NKH (DQ < 20), 12 with intermediate attenuated NKH (DQ = 20–50), 15 with mild attenuated NKH (DQ ≥ 50), and 9 with attenuated NKH where the DQ was not available. The development presented a continuum from a DQ < 10 up to the normal range with intelligence quotient of 82 in a child who follows regular education curriculum with assistance. The number of AEDs used for seizure control related to the degree of developmental delay. All patients with severe NKH were treated with multiple (≥2) anticonvulsants and had persistent seizures, whereas patients with a DQ > 30 never required anticonvulsant therapy. The difference between severe NKH patients (mean 3.1 AEDs), attenuated poor and intermediate NKH combined (mean 1 AED), and mild NKH (mean 0 AEDs) was significant (p < 0.001; Fig 1A). In patients with severe NKH, 71% had brain malformations identified on MRI, whereas only 7.5% of patients with attenuated NKH had malformations, all hypoplastic corpus callosum (chi‐square, p < 0.001; Table 1). Severe brain malformations (corpus callosum agenesis or cerebellar cyst with hydrocephalus) only occurred in patients with severe NKH or in neonatal death. Thus, 3 features related to clinical severity—developmental delay, number of AED, and brain malformation—interrelated as a class. We next examined clinical predictors of these severity classes.
Figure 1.
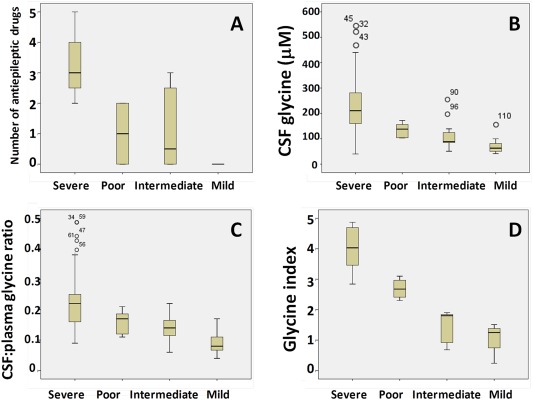
Differences in antiepileptic drugs, glycine levels, and glycine index by disease category. (A) The differences between severe nonketotic hyperglycinemia (NKH), poor attenuated NKH, intermediate attenuated NKH, and mild attenuated NKH are shown for the number of antiepileptic drugs (A), the cerebrospinal fluid (CSF) glycine levels (B), the CSF:plasma glycine ratios (C), and the glycine index (D). [Color figure can be viewed in the online issue, which is available at www.annalsofneurology.org.]
Table 1.
Clinical Predictors: Age at Onset, Brain Malformations, Glycine Levels, and Ratio
| Category | Severe | Attenuated Alla | Attenuated Poor | Attenuated Intermediate | Attenuated Mild |
|---|---|---|---|---|---|
| Age at onset | |||||
| No. | 56 | 42 | 6 | 12 | 15 |
| 1st week | 49 | 20 | 5 | 5 | 7 |
| >1 week | 7 | 22 | 1 | 7 | 8 (>4 months in 7) |
| Oldest | 2 months | 3 year | 2 months | 4 months | 3 year |
| Brain malformation | |||||
| No. (with brain imaging data) | 45 | 40 | 6 | 12 | 15 |
| Corpus callosum agenesis | 9 | 0 | 0 | 0 | 0 |
| Corpus callosum hypoplasia | 18 | 3 | 1 | 0 | 0 |
| Cerebellar cyst/hydrocephalus | 4 | 0 | 0 | 0 | 0 |
| Brain atrophy | 1 | 0 | 0 | 0 | 0 |
| No malformations | 13 | 34 | 5 | 12 | 15 |
| Glycine levels | |||||
| Plasma glycine levels, μM | |||||
| Median, No. | 1,055, n = 48 | 827, n = 37b | 844, n = 6 | 760, n = 12 | 829, n = 15 |
| Range | 324–2,048 | 342–1,590 | 552–1,433 | 509–1,120 | 470–1,196 |
| CSF glycine levels, μM | |||||
| Median, No. | 213, n = 46 | 89, n = 31c | 138, n = 6 | 89, n = 11 | 63, n = 11 |
| Range | 40–510 | 41–230 | 103–172 | 51–230 | 41.4–154 |
| CSF:plasma glycine ratio | |||||
| Median, No. | 0.22, n = 45 | 0.12, n = 29c | 0.17, n = 5 | 0.14, n = 11 | 0.08, n = 9 |
| Range | 0.09–0.45 | 0.04–0.22 | 0.11–0.21 | 0.06–0.22 | 0.04–0.17 |
The category Attenuated All also included patients with attenuated nonketotic hyperglycinemia not otherwise specified where the developmental quotient is not known.
p < 0.01, c p < 0.001, Mann–Whitney U test.
CSF = cerebrospinal fluid.
Basic Predictors: Age at Onset and Gender
Age at onset was commonly neonatal in all categories (see Table 1). Onset delayed beyond the first week was present in 12% of patients with severe NKH, and in 52% of patients with attenuated NKH. Delayed onset in severe NKH was between 2 weeks and 2 months, and ≤4 months of age in poor or intermediate attenuated NKH. In mild attenuated NKH, 53% of patients had onset beyond the first week of life, 7 patients at >4 months of age, which predicted good outcome. There was no significant gender difference in age at onset, DQ, or number of AEDs used, but mean CSF glycine was higher in females than in males (219 vs 155 μM, p = 0.02).
Glycine Biochemistry
The plasma glycine levels, the CSF glycine levels, and the CSF:plasma glycine ratio were higher in severe NKH than in attenuated NKH (see Table 1). Despite substantial overlap in the ranges, the following findings were predictive: a CSF glycine level >230 μM predicted severe NKH with a sensitivity of 43%, and a CSF:plasma glycine ratio ≤ 0.08 predicted attenuated NKH with a sensitivity of 28%. Within the group of attenuated NKH, there was a significant difference in CSF glycine and in the CSF:plasma ratio between subclasses (ANOVA, both p < 0.02) but not in plasma glycine levels (see Fig 1B, C). Attenuated mild NKH had significantly lower CSF glycine levels (good 73 vs intermediate/poor 121 μM, p < 0.01) and ratios (good 0.095 vs intermediate/poor 0.15, p < 0.01).
In attenuated NKH patients, the continuous outcome variable of DQ correlated significantly with age at onset, number of AEDs, CSF glycine level, CSF:plasma glycine ratio, and glycine index (Table 2 and Fig 2). Multiple linear regression showed a best model of outcome in DQ when related to both age at onset = 1.06 ± 0.42 and CSF glycine level = −0.19 ± 0.075 μM with constant 53 ± 9.1, p = 0.01. Multiple logistic regression identified only 2 independent predictors for disease severity: age at onset (point estimate = 1.526, 95% confidence interval [CI] = 1.183–1.968, p = 0.001) and CSF glycine level (point estimate = 0.993, 95% CI = 0.989–0.997, p < 0.001).
Table 2.
Relation of Developmental Quotient with Outcome and Predictive Variables
| Predictive Factor | No. | Pearson R 2 | p | Spearman Rho | p |
|---|---|---|---|---|---|
| Attenuated NKH only | |||||
| Age at onset | 33 | 0.247 | 0.003 | 0.384 | 0.03 |
| Antiepileptic drugs | 33 | −0.260 | 0.002 | −0.582 | <0.001 |
| CSF glycine | 28 | −0.315 | 0.002 | −0.664 | <0.001 |
| CSF:plasma glycine | 24 | −0.328 | 0.003 | −0.544 | 0.005 |
| Glycine index | 12 | −0.424 | 0.02 | −0.765 | 0.004 |
| Attenuated and severe | |||||
| Age at onset | 88 | 0.311 | <0.001 | 0.433 | <0.001 |
| Antiepileptic drugs | 56 | −0.599 | <0.001 | −0.827 | <0.001 |
| CSF glycine | 74 | −0.296 | <0.001 | −0.681 | <0.001 |
| CSF:plasma glycine | 70 | −0.289 | <0.001 | −0.622 | <0.001 |
| Plasma glycine | 81 | −0.081 | 0.01 | −0.266 | 0.02 |
| Glycine index | 20 | 0.645 | <0.001 | −0.904 | <0.001 |
| Mutation score | 81 | −0.479 | <0.001 | 0.674 | <0.001 |
Correlation between developmental quotient and each of the predictive factors.
CSF = cerebrospinal fluid; NKH = nonketotic hyperglycinemia.
Figure 2.
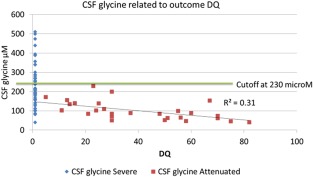
Relation between cerebrospinal fluid (CSF) glycine levels and developmental quotient (DQ). There is a direct linear correlation between CSF glycine levels and DQ. The cutoff at 230 μM is shown.
Protein, Enzyme Activity, Glycine Index
Patients affected with AMT mutations (15%) are less common than patients with GLDC mutations (85%), with no GCSH mutations identified. The proportion of T‐protein–deficient patients was not significantly different in severe NKH (12 of 56) versus attenuated NKH (5 of 42; chi‐square, p = 0.25). Also, plasma and CSF glycine levels, CSF:plasma glycine ratio, age at onset, and DQ did not differ significantly between patients deficient in P‐ and T‐protein. The enzyme activity in liver depended on the affected protein (P‐protein 0–10%, n = 4; T‐protein 10–31%, n = 4), as previously reported.27 Residual activity in EBV‐transformed lymphoblasts (n = 10) ranged from 0 to 83% of normal activity, and as previously noted proved unreliable, with a patient with a severe outcome who had 83% residual activity.21
In contrast, the glycine index had a strong relation with outcome (see Fig 1D). It was highest in the severely affected patients (average = 4.01, range = 2.84–4.87, n = 8), between 2 and 3 in poor attenuated patients (average = 2.69, range = 2.30–3.10, n = 4), and <2 in all intermediate or mild attenuated patients (average = 1.27, range = 0.24–1.90, n = 8; ANOVA, p < 0.001). As the glycine index reflects residual glycine catabolism on a whole body level, this indicated that patients with intermediate and mild attenuated NKH had more residual activity than patients with poor attenuated NKH and severe NKH, suggesting a contributing genetic factor.
Genetic Studies
The mutation spectrum showed extensive intragenic molecular heterogeneity in classic NKH, including 78 novel mutations in GLDC and 18 novel mutations in AMT. All mutations identified are listed in the Supplementary Table at the cDNA and protein level and were submitted to the Leiden Open Variant Databases (http://databases.lovd.nl/shared/genes) of GLDC and AMT (Figs 3 and 4). Of the 209 alleles in GLDC, there were 50.2% missense mutations, 8.1% nonsense mutations, 5.7% frameshift mutations, 13.4% splice site mutations, 20.6% exonic deletions, 1 exonic duplication, and 1 in‐frame 3bp deletion resulting in a single amino acid deletion. Of the 114 mutations, 72 mutations were only identified in a single allele. Recurring mutations (≥5×) included p.R515S (13×), del exon 1–2 (12×), IVS22+1G>C (10×), p.A389V (8×), del exon 3‐21 (7×), p.A802V (6×), p.A202V (5×), p.E167X (5×), and IVS19‐1G>A (5×). The exonic deletions were heterogeneous, ranging from a single exon to entire gene deletions, and were as frequent on the maternal (n = 21) as the paternal (n = 22) allele. Some patients had 2 small nonoverlapping deletions not detectable by sequencing, making deletion analysis diagnostically imperative. A mutation was not identified in 2 alleles, giving current analysis methods 99% sensitivity in GLDC. The AMT gene with 38 alleles had a different mutation spectrum, with 76.3% missense mutations, 7.9% splice site mutations, 13.2% frameshift mutations, and 1 nonsense mutation, but no exonic deletions or duplications. All recurring mutations were missense: p.R320H (7×), p.M1T (4×), and p.I106T (4×). In silico analysis confirmed the expected pathogenicity of mutations of the 2 nucleotides of each consensus splice site (score < 0.46), except for IVS18‐2A>G in GLDC (score = 0.64), where RNA analysis confirmed the presence of normally spliced mRNA, indicating a leaky splicing mutation (data not shown). The in silico prediction of pathogenicity of the deep splice site mutation IVS6+5G>C in AMT was confirmed by RNA analysis showing the presence of sequence compatible with the insertion of intron 6 sequence. The in silico predicted pathogenicity of the deep splice site mutation IVS23+5G>A in GLDC could not be further verified, as lymphoblasts were unavailable. An ethnic concentration was present for AMT p.I106T in the Southern Netherlands and GLDC p.S132L in New Zealand, reflecting a founder effect. Most parents were carriers, but a de novo mutation not present in the blood of the parents was identified in 3 patients (1% of alleles), although germline mosaicism could not be excluded.
Figure 3.

Mutations in GLDC. Missense mutations are shown above the diagram of the exonic structure of the GLDC gene. Missense mutations with residual activity are shown in green, missense mutations without residual activity are shown in red, and mutations not expressed are shown in yellow. Frameshift mutations (gray) and splice site mutations (orange) are shown below the exonic structure. The length of observed exonic deletions is shown in red and duplications in blue. The frequency of occurrence is provided for recurring mutations.
Figure 4.
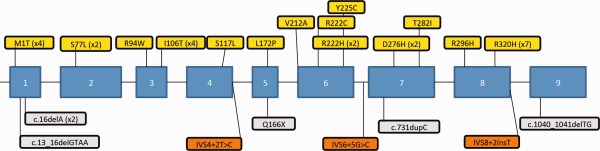
Mutations in AMT. Missense mutations (yellow) are shown above the diagram of the exonic structure of the AMT gene. Frameshift mutations (gray) and splice site mutations (orange) are shown below the exonic structure.
Residual Activity of Mutations in Expression Analysis
In many metabolic conditions, residual enzyme activity determines clinical outcome. We analyzed the impact of mutations on residual activity and in relation to neurodevelopmental outcome. There were differences in the mutation classes between severe NKH and attenuated NKH. Nonsense mutations, frameshift mutations, exonic deletions, and duplications, which have no residual activity, were more frequent in severe than in attenuated NKH patients (38 % vs 11% of alleles, p < 0.001), and adding mutations affecting consensus donor and acceptor splice sites, which usually result in absent activity, accentuated this difference (severe NKH 57 of 109, attenuated NKH 18 of 84 alleles, p < 0.001). Patients with both alleles from these classes had severe NKH, whereas each attenuated NKH patient had at least 1 missense mutation. Two exceptional mutations were classified as mild: a frameshift mutation that affects only the penultimate 20 amino acids, and a leaky splice site mutation IVS18‐2A>G.
We hypothesized that missense mutations that recurred in attenuated NKH (p.A202V, p.T269M, p.A389V, p.A802V in GLDC and p.I106T in AMT) conferred residual enzyme activity, whereas others that occurred in homozygosity in severe NKH patients (p.S132L and p.R515S in GLDC and p.R320H and p.M1T in AMT) had no residual activity. We expressed selected missense mutations and measured residual activity of the P‐protein using the glycine exchange reaction with recombinant human H‐protein, which after the lipoyl–ligase reaction was >80% lipoylated by mass spectrometry, and was no longer rate limiting at 4 μM. The mutations p.S132L, p.P304L, p.C382Y, p.T437I, p.R515S, p.A569T, p.G618R, p.S657R, p.G761R, p.S808R, p.A833V, p.P893L, and p.H950R had no measurable residual activity. Mutations with residual activity were classified as very mild with >10% activity (p.L82S, p.A202V, p.I381T, p.A389V, p.L548V, p.A802V, p.A802E), mild with 1 to 10% activity (p.T269M, p.C291Y, p.Q366R, p.G607S, p.R630P), and intermediate with measurable activity 0.5 to 1% of wild‐type activity (p.A283P, p.R461Q, p.A678del; see Supplementary Table).
To relate residual activity to outcome, we analyzed 60 patients who survived the neonatal period with known neurodevelopment outcome, and for whom both mutations in GLDC were identified and the residual activity determined (Fig 5). Both developmental outcome as DQ and number of AEDs used differed significantly with the number of alleles with residual activity (Fisher exact test, both p < 0.001). Patients with 2 severe mutations used multiple (2–4) AEDs; patients with 2 residual activity–conferring alleles used no or rarely 1 AED; whereas patients with a single residual activity–conferring allele used no (in 47%) or 1 to 3 AEDs. All 29 patients with 2 severe mutations had severe NKH; all 8 patients with 2 mutations with measurable residual activity had attenuated NKH, 6 of them mild, whereas 91% of patients with a single allele with residual activity had attenuated NKH, the majority poor or intermediate. Even patients with a single intermediate mutation still had attenuated outcome (Patients 18, 61, 79), indicating that very little in vitro residual activity is required. Exceptions exist where severe outcome occurred even in the presence of a single very mild allele (eg, p.A802V [Patient 39] and p.A389V [Patient 35]). This indicates that an allele with residual activity is necessary but not sufficient for attenuated outcome, and that factors in addition to genotype influence outcome. Further illustrating this, the average DQ was higher in patients with 2 residual activity–conferring mutations than in those with a single such mutation (53 vs 28, p = 0.01), but within the group of attenuated patients there was no correlation between DQ and the summed residual activity of both alleles (r 2 < 0.001). Substantial phenotypic differences existed within the same genotype; for instance, p.A389V/p.R515S was seen in a severe (Patient 35) and an attenuated intermediate phenotype (Patient 148).
Figure 5.
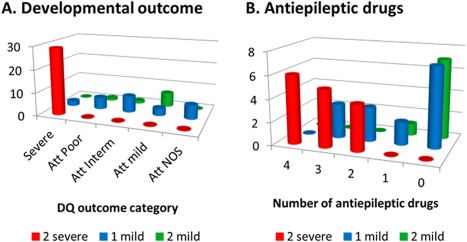
Number of residual activity–conferring mutations and disease severity. (A) The frequency of developmental outcome in categories of severe, attenuated (Att) poor, attenuated intermediate, attenuated mild, or attenuated not otherwise specified (NOS; without known developmental quotient [DQ]) is shown in relation to the number of alleles with a mutation conferring no residual activity (severe mutation) or alleles with a mutation conferring residual activity (mild mutation). (B) The frequency of patients requiring a number of antiepileptic medications in relation to the number of severe or mild alleles.
Expanding this analysis to all patients showed a significant difference in the mutation score between outcome categories (ANOVA, p < 0.001; post hoc Tukey difference only significant between severe 1.38 ± 1.41 and attenuated −1.43 ± 0.83, p < 0.001; Fig 6). A score of −2 predicted severe outcome with sensitivity of 62% and specificity of 100%, and a score ≥1 predicted attenuated outcome with a sensitivity of 81% and a specificity of 94% (Table 3). Combined, this mutation score had a predictive sensitivity of 70% and specificity of 97%.
Figure 6.
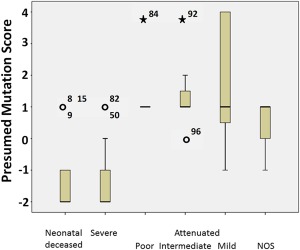
Mutation score by disease category. The mutation score is compared between neonatal deceased, severe nonketotic hyperglycinemia (NKH), poor attenuated NKH, intermediate attenuated NKH, mild attenuated NKH, and attenuated NKH not otherwise specified (NOS). The mutation score is significantly higher in attenuated NKH than in severe or neonatal decreased NKH. [Color figure can be viewed in the online issue, which is available at www.annalsofneurology.org.]
Table 3.
Presumed Mutation Score as a Predictor
| Score | Severe | Attenuated |
|---|---|---|
| −2 | 34 | 0 |
| ≥1 | 2 | 34 |
| Other | 19 | 10 |
| Total | 55 | 42 |
Number of severe or attenuated nonketotic hyperglycinemia patients with a specific mutation score.
Molecular Modeling and Protein Stability
The relative locations of the expressed mutations are shown on the tertiary structure of human P‐protein modeled on the homologous α2 homodimer structure of Synechocystis species glycine decarboxylase (Fig 7).26 Mutated amino acids located near the active site pocket (A833, L548, H950, A389, S808, I381, Q366, C382, G607, P893) tended to have stable protein levels but highly variable residual activity. For instance, mild mutations Q366R, I381T, and G607S affect amino acids located in a loop or domain that is mobile upon pyridoxal–phosphate binding in Thermus thermophilus, and Q366 resides in the lipoamide channel.28 In contrast, mutated amino acids located on the outer side of protein away from the active pocket (A283, A202, C291, T269, R461, P304, R515, S132, A569, A678, T437, A802) tended to have decreased protein levels of variable residual activity, likely due to protein instability. For instance, mild mutations p.T269M and p.A202V located in the 9th α‐helix, and severe mutations p.R515S and p.G761R affected the structural interaction of the N‐ and C‐terminal halves of the protein.28 Cotransfection studies were done to review if observed combination of 2 mutations would affect the residual activity given the dimeric structure of P‐protein. The severe but stable mutant p.S657R did not significantly reduce the residual activity or protein levels of p.A802V (5.9%; Patient 39) compared to sham (7.2%, p = 0.28; Patient 52), but the unstable p.R515S decreased the activity and the protein amount of p.A389V (4.2% activity; Patients 35, 148) compared to sham (11.2%, p < 0.001; Patient 278), indicating a dominant negative effect for unstable mutants at the protein level (Fig 8).
Figure 7.
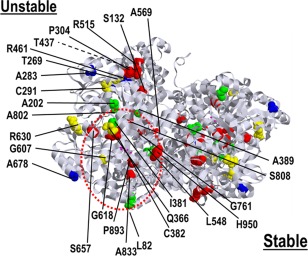
Molecular modeling of mutated amino acids on the structure of the P‐protein. The homology structure of the human P‐protein dimer modeled from Synechocystis species is shown with the amino acids of the expressed missense mutations highlighted. Amino acids involved in missense mutations without residual activity on expression study are shown in red, with <1% residual activity in blue, with 1 to 10% residual activity in yellow, and with >10% residual activity in green. The active site lysine 754 is shown in pink. Amino acids that when mutated result in a stable protein (identified in the lower part) tend to cluster around the active site fold (red circle), whereas amino acids that when mutated result in unstable protein (identified in the upper part) tend to be located away from the active site fold.
Figure 8.
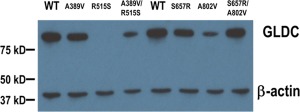
Western blot analysis of expressed mutations in GLDC. The missense mutations present in 2 patients were expressed in COS cells, and the level of P‐protein identified by Western blot. The amount of β‐actin protein is shown as a loading control. WT = wild type. [Color figure can be viewed in the online issue, which is available at www.annalsofneurology.org.]
Neonatal and Early Infantile Deceased Patients
As a group, neonatally deceased patients were similar to severe patients. They had more severe mutations (65% vs 54% of alleles, p < 0.001) and fewer brain malformations (48% vs 71%, p < 0.001), but similar CSF glycine levels and CSF:plasma glycine ratio. On close observation, we identified a few patients who had residual activity–conferring mutations, without brain malformations, and with low CSF glycine level and CSF:plasma glycine ratio (eg, Patients 33a and 33b with p.T269M, and Patient 19 with p.G607S). Because >50% of attenuated NKH patients presented neonatally, we presume that these patients at least had the potential for attenuated outcome.
Discussion
At the outset of this study, we encountered patients who presented neonatally and had been given a dismal prognosis, but who made substantial developmental progress. This inaccurate prognostic information was frustrating for the families. The majority of patients with attenuated NKH, even mild, presented neonatally, and predicting prognosis based on age at onset alone was inaccurate. Moreover, single case studies of a new treatment, such as dextromethorphan, exhibited the same prognostic pitfall attributing the attenuated outcome to the drug treatment when, because of the neonatal presentation, the patient was expected to have a severe outcome.29 A more accurate prognostic stratification is required for adequate clinical trial evaluation. A reliable measure to predict outcome in NKH was not yet available,30 and past small studies had contradictory conclusions on the predictive value of glycine levels.7, 14 In this study, we categorized patients with NKH according to outcome, rather than age at onset, using developmental attainment as the primary outcome measure, which correlated well with neurological indicators such as the presence of brain malformations and the number of AEDs used to treat epilepsy. Attenuated NKH was distinguished from severe NKH by the presence of a variable degree of development,2 and despite its continuous nature was subdivided in categories to facilitate statistical analysis.
This study provides strong evidence that genotype is a primary determinant of outcome, and presence of residual activity relates to outcome, as had previously been hypothesized from individual cases.7, 8, 11, 22 Two mutations without residual enzyme activity are associated with severe outcome, and attenuated outcome requires at least 1 mutation providing residual enzyme activity. Lack of residual enzyme activity in severe patients results in a larger glycine pool, which requires more sodium benzoate to normalize glycine levels as reflected in a greater glycine index,18 which showed the best discrimination between outcome categories. This larger glycine pool results in increased brain and CSF glycine levels. CSF glycine correlated significantly although not strongly with outcome measures, but at the extremes CSF glycine >230 μM predicted severe outcome and low CSF:plasma glycine ratio ≤ 0.08 predicted attenuated outcome. Of the clinically useful predictors, which are listed with sensitivity and specificity in Table 4, age at onset, brain imaging, CSF glycine, and CSF:plasma glycine ratio are available in the immediate newborn period to enable counseling of parents. Using these 4 predictors available at diagnosis (age at onset ≥4 months, CSF glycine >230 μM, CSF:plasma glycine ratio ≤0.08, and brain malformations on MRI), 78% of severe patients and 49% of attenuated patients would be accurately identified. A simplified panel of 7 mutations (p.A202V, p.A389V, p.A802V/E, p.T269M, p.L548V, p.G607S in GLDC and p.I106T in AMT) identified 55% of attenuated patients, and could be implemented for rapid prediction in the neonatal period.
Table 4.
Clinically Useful Predictors of Outcome
| Parameter | Outcome | Sensitivity | Specificity |
|---|---|---|---|
| Onset > 4 months | Attenuated mild | 47% | 100% |
| Onset 1st week | Severe | 87% | 70% |
| CSF glycine > 230 μM | Severe | 43% | 100% |
| CSF:plasma glycine ratio ≤ 0.08 | Attenuated | 28% | 100% |
| Brain malformation severe | Severe | 29% | 100% |
| Brain malformation all (including HCC) | Severe | 71% | 92% |
| No epilepsy (no AEDs) | Attenuated | 70% | 100% |
| Glycine index > 3a | Severe | 87% | 100% |
| 2 nonmissense mutations | Severe | 36% | 95% |
| Mutation scoreb = −2 | Severe | 59% | 97% |
| Mutation scoreb ≥ 1 | Attenuated | 76% | 94% |
A glycine index of 3 mmol/kg/day for a child on breast milk or regular infant formula with intake of 150 ml/kg/day calculates to a sodium benzoate dose of 510–540 mg/kg/day to bring glycine levels within therapeutic range (120–300 μM).
The mutation score provides a score of −1 for a mutation expected to leave no residual activity and +2 for a mutation that is expected to have residual activity. The sum of both alleles scored as is currently known is used.
AED = antiepileptic drug; CSF = cerebrospinal fluid; HCC = hypoplastic corpus callosum.
Molecular analysis provides an important predictor. This report greatly expands the number of mutations beyond those previously reported. Combined sequencing and exonic deletion–duplication analysis identified 99% of causative alleles in GLDC and all in AMT, and constitutes the preferred confirmatory diagnostic testing. Due to the documented 1% de novo mutation rate, carrier status of parents should be verified and not assumed in the context of recurrence risk counseling and prenatal diagnosis. The large genetic heterogeneity complicates the analysis of genotype to phenotype relation. Caution is necessary when using generic rules in predicting whether a mutation would retain residual activity. Phylogenetic conservation was unhelpful, with most mutations affecting conserved amino acids regardless of residual activity, yet p.A569T affects a poorly conserved amino acid but had no residual activity. Mutations affecting the 2 essential splice donor/acceptor nucleotides severely disrupted splicing, but IVS18‐2A>G was a notable exception. To evaluate missense mutations, we developed a new effective expression study, with results agreeing with the few previous publications.27, 31 The amount of residual activity associated with attenuated outcome was very low. Mutations that result in unstable protein tend to be located away from the active cleft, and even reduce the protein amount of the other allele likely due to dimer, formation as was shown for the p.R515S mutation. In this study, 69 of 124 patients carried at least 1 GLDC missense mutation, and 13 of 27 GLDC missense mutants created unstable proteins, including common mutations such as p.R515S. The impact of current therapy with benzoate and dextromethorphan has been limited. A theoretically new therapeutic option of a chaperone or a proteostasis regulator that increases protein levels could increase enzyme activity and positively affect outcome in at least 27% of patients.32 The relation between residual activity and outcome is complex. The number of alleles with residual activity relates to outcome, but there was not a direct correlation between DQ and the amount of residual activity in this eukaryotic expression system. Patients with a similar mild mutation sometimes had a very different outcome in this study, and substantial intrafamilial variability in large families has been noted.14 This suggests that factors additional to genotype contribute to outcome.11, 31 For patients with attenuated NKH, the nature and timing of early treatment has been suggested as a potential factor.30
Although this study is the largest ever in NKH, it does have limitations. Patients were evaluated from a very wide geographic region, allowing for generalization in this rare condition, and therefore encountered a variety of methods to evaluate developmental outcome. Analysis of developmental and behavioral outcome subcategories was not possible and will require a systematic analysis at a few centers using standardized measures. A wide spectrum of behavioral and developmental problems was noted in preliminary analysis of a few patients with attenuated NKH. The MRI review was limited to brain malformations as described in the report of the reading radiologist, and detailed analysis, such as the distribution of diffusion restriction, requires further study.33, 34 A detailed description of the seizure type, electroencephalogram, or response to specific antiepileptic medications could not be systematically captured, and requires a separate study. The number of AEDs used, which could be accurately recorded, is significant because the need for >2 AEDs, as seen in severe NKH, relates to the presence of therapy‐refractory epilepsy.35
This study documented several factors predicting prognosis, including the presence of brain malformations, the CSF glycine level, the CSF:plasma glycine ratio, and the type of genetic mutation. This information is valuable to parents and clinicians to allow better prediction of outcome. Prediction of severe outcome can help families in considering withdrawal of life‐sustaining therapy in the neonatal apneic phase, given the current lack of effective therapies in this setting.15, 36 It is also important in the stratification of patients in treatment studies. Different treatment responses may exist between severe and attenuated NKH.30, 36 The relation between residual activity and outcome suggests that novel strategies to improve residual activity such as chaperones may have a beneficial clinical effect and should be researched. The complex relationship between genotype and phenotype indicates that additional factors will need to be identified to make such a strategy fully successful.
Authorship
Conception, design, and coordination of the study was by J.L.K.V.H. Acquisition and analysis of clinical data were by J.L.K.V.H., J.B.H., C.R.C., K.J.B., and G.H.S., and of laboratory data were by M.A.S., G.H.S., H.J.S., E.B.S., G.C.‐S., V.M., G.M., D.A.A., J.R.T., and K.W. Statistical analysis was done by J.L.K.V.H. and S.T. The manuscript was written by J.L.K.V.H., M.A.S., and C.R.C.
Potential Conflicts of Interest
Nothing to report.
Supporting information
Supplementary Information Table 1.
Supplementary Information Table 1.
Acknowledgment
This study was supported by the NKH Crusaders Fund, Hope for NKH Fund, CU Nonketotic Hyperglycinemia Fund, Joseph's Goal Fund, Brodyn's Friends Fund, Smiles for Miles NKH Research Fund, Lucy's BEElievers Fund, Madi's Mission to Find a Cure for NKH Fund, Les Petits Bourdons, Children's Clinical Research Organization at Children's Hospital Colorado, and vector core of the Rocky Mountain Neurological Disorders Core Center Grant NIH/NS048154. Sequencing was supported by the University of Colorado DNA Sequencing and Analysis Core.
We thank the families who participated in this study; the numerous physicians and genetic counselors who referred patients and provided clinical information; the nutritionists who calculated glycine intake, S. Gaughan, C. Burns, and K. Vande Kerckhove; the genetic counselors who helped with patient enrollment, M. Raymond and L. Sremba; and Drs T. Benke and A. Brooks‐Kayal for helpful discussions.
References
- 1. Carson NAJ. Non‐ketotic hyperglycinaemia—a review of 70 patients. J Inherit Metab Dis 1982;5:126–128.. [Google Scholar]
- 2. Hennermann JB, Berger J‐M, Grieben U, et al. Prediction of long‐term outcome in glycine encephalopathy: a clinical survey. J Inherit Metab Dis 2012;35:253–261. [DOI] [PubMed] [Google Scholar]
- 3. Hoover‐Fong JE, Shah S, Van Hove JLK, et al. Natural history of nonketotic hyperglycinemia in 65 patients. Neurology 2004;63:1847–1853. [DOI] [PubMed] [Google Scholar]
- 4. Dobyns WB. Agenesis of the corpus callosum and gyral malformations are frequent manifestations of nonketotic hyperglycinemia. Neurology 1989;39:817–820. [DOI] [PubMed] [Google Scholar]
- 5. Van Hove JLK, Kishnani PS, Demaerel P, et al. Acute hydrocephalus in nonketotic hyperglycinemia. Neurology 2000;54:754–756. [DOI] [PubMed] [Google Scholar]
- 6. Press GA, Barshop BA, Haas RH, et al. Abnormalities of the brain in nonketotic hyperglycinemia: MR manifestations. AJNR Am J Neuroradiol 1989;10:315–321. [PMC free article] [PubMed] [Google Scholar]
- 7. Dinopoulos A, Matsubara Y, Kure S. Atypical variants of nonketotic hyperglycinemia. Mol Genet Metab 2005;86:61–69. [DOI] [PubMed] [Google Scholar]
- 8. Steiner RD, Sweetser DA, Rohrbaugh JR, et al. Nonketotic hyperglycinemia: atypical clinical and biochemical manifestations. J Pediatr 1996;128:243–246. [DOI] [PubMed] [Google Scholar]
- 9. Conter C, Rolland MO, Cheillan D, et al. Genetic heterogeneity of the GLDC gene in 28 unrelated patients with glycine encephalopathy. J Inherit Metab Dis 2006;29:135–142. [DOI] [PubMed] [Google Scholar]
- 10. Kanno J, Hutchin T, Kamada F, et al. Genomic deletion within GLDC is a major cause of non‐ketotic hyperglycinaemia. J Med Genet 2007;44:e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kure S, Kato K, Dinopoulos A, et al. Comprehensive mutation analysis of GLDC, AMT, and GCSH in nonketotic hyperglycinemia. Hum Mutat 2006;27:343–352. [DOI] [PubMed] [Google Scholar]
- 12. Kure S, Narisawa K, Tada K. Enzymatic diagnosis of nonketotic hyperglycinemia with lymphoblasts. J Pediatr 1992;120:95–98. [DOI] [PubMed] [Google Scholar]
- 13. Perry TL, Urquhart N, MacLean J, et al. Nonketotic hyperglycinemia. Glycine accumulation due to absence of glycine cleavage in brain. N Engl J Med 1975;292:1269–1273. [DOI] [PubMed] [Google Scholar]
- 14. Boneh A, Degani Y, Harari M. Prognostic clues and outcome of early treatment of nonketotic hyperglycinemia. Pediatr Neurol 1996;15:137–141. [DOI] [PubMed] [Google Scholar]
- 15. Boneh A, Allan S, Mendelson D, et al. Clinical, ethical and legal considerations in the treatment of newborns with non‐ketotic hyperglycinaemia. Mol Genet Metab 2008;94:143–147. [DOI] [PubMed] [Google Scholar]
- 16. Brunel‐Guitton C, Casey B, Coulter‐Mackie M, et al. Late‐onset nonketotic hyperglycinemia caused by a novel homozygous missense mutation in the GLDC gene. Mol Genet Metab 2011;103:193–196. [DOI] [PubMed] [Google Scholar]
- 17. Van Hove JLK, Kishnani P, Muenzer J, et al. Benzoate therapy and carnitine deficiency in non‐ketotic hyperglycinemia. Am J Med Genet 1995;59:444–453. [DOI] [PubMed] [Google Scholar]
- 18. Van Hove JLK, Vande Kerckhove K, Hennermann JB, et al. Benzoate treatment and the glycine index in nonketotic hyperglycinaemia. J Inherit Metab Dis 2005;28:651–663. [DOI] [PubMed] [Google Scholar]
- 19. Baker PR, Friederich MW, Swanson MA, et al. Variant non‐ketotic hyperglycinemia is caused by mutations in LIAS, BOLA3 and the novel gene GLRX5. Brain 2014;137(pt 2):366–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Korman SH, Gutman A. Pitfalls in the diagnosis of glycine encephalopathy (non‐ketotic hyperglycinemia). Dev Med Child Neurol 2002;44:712–720. [DOI] [PubMed] [Google Scholar]
- 21. Reese MG, Eeckman FH, Kulp D, et al. Improved splice site detection in Genie. J Comput Biol 1997;4:311–323. [DOI] [PubMed] [Google Scholar]
- 22. Kure S, Ichnohe A, Kojima K, et al. Mild variant of nonketotic hyperglycinemia with typical neonatal presentations: mutational and in vitro expression analyses in two patients. J Pediatr 2004;144:827–829. [DOI] [PubMed] [Google Scholar]
- 23. Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein‐dye binding. Anal Biochem 1976;72:248–254. [DOI] [PubMed] [Google Scholar]
- 24. Jiang Y, Cronan JE. Expression cloning and demonstration of Enterococcus faecalis lipoamidase (pyruvate dehydrogenase inactivase) as a Ser‐Ser‐Lys triad amidohydrolase. J Biol Chem 2005;280:2244–2256. [DOI] [PubMed] [Google Scholar]
- 25. Lambert C, Léonard N, De Bolle X, et al. ESyPred3D: prediction of proteins 3D structures. Bioinformatics 2002;18:1250–1256. [DOI] [PubMed] [Google Scholar]
- 26. Hasse D, Andersson E, Carlsson G, et al. Structure of the homodimeric glycine decarboxylase P‐protein from Synechocystis sp. PCC 6803 suggests a mechanism for redox regulation. J Biol Chem 2013;288:35333–35345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Applegarth DA, Toone JR. Nonketotic hyperglycinemia (glycine encephalopathy): laboratory diagnosis. Mol Genet Metab 2001;74:139–146. [DOI] [PubMed] [Google Scholar]
- 28. Nakai T, Nakagawa N, Maoka N, et al. Structure of P‐protein of the glycine cleavage system: implications for nonketotic hyperglycinemia. EMBO J 2005;24:1523–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hamosh A, McDonald JW, Valle D, et al. Dextromethorphan and high‐dose benzoate therapy for nonketotic hyperglycinemia in an infant. J Pediatr 1992;121:131–135. [DOI] [PubMed] [Google Scholar]
- 30. Korman SH, Boneh A, Ichnohe A, et al. Persistent NKH with transient or absent symptoms and a homozygous GLDC mutation. Ann Neurol 2004;56:139–143. [DOI] [PubMed] [Google Scholar]
- 31. Dinopoulos A, Kure S, Chuck G, et al. Glycine decarboxylase mutations: a distinctive phenotype of nonketotic hyperglycinemia in adults. Neurology 2005;64:1255–1257. [DOI] [PubMed] [Google Scholar]
- 32. Muntau AC, Leandro J, Staudigl M, et al. Innovative strategies to treat protein misfolding in inborn errors of metabolism: pharmacological chaperones and proteostasis regulators. J Inherit Metab Dis 2014;37:505–523. [DOI] [PubMed] [Google Scholar]
- 33. Khong P‐L, Lam BCC, Chung BHY, et al. Diffusion‐weighted MR imaging in neonatal nonketotic hyperglycinemia. AJNR Am J Neuroradiol 2003;24:1181–1183. [PMC free article] [PubMed] [Google Scholar]
- 34. Sener RN. Nonketotic hyperglycinemia: diffusion magnetic resonance imaging findings. J Comput Assist Tomogr 2003;27:538–540. [DOI] [PubMed] [Google Scholar]
- 35. Kwan P, Brodie MJ. Early identification of refractory epilepsy. N Engl J Med 2000;342:314–319. [DOI] [PubMed] [Google Scholar]
- 36. Korman SH, Wexler ID, Gutman A, et al. Treatment from birth of nonketotic hyperglycinemia due to a novel GLDC mutation. Ann Neurol 2006;59:411–415. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information Table 1.
Supplementary Information Table 1.