

Abstract
Bacteroides spp. have been proposed as indicators of fecal contamination in microbial source tracking (MST) methodologies. The aim of this study was to develop new qPCR assays that target host‐specific Bacteroidal 16S ribosomal RNA genes, to determine the source of fecal contamination in water. Denaturing gradient gel electrophoresis (DGGE) was used to select for host‐specific bands of Bacteroides associated with a fecal pollution source and later to design four qPCR host‐specific assays. A set of common primers for Bacteroides spp., four different Bacteroides spp. host‐associated hydrolysis probes (human, cattle, pig, and poultry), and one hydrolysis probe for the Bacteroides genus were designed. This set of qPCR assays together with other previously developed Bacteroidetes MST targets were used to analyze water samples with fecal contamination from the four sources studied. The host‐specific Bacteroides qPCRs designed for human (HMprobeBac), pig (PGprobeBac), and poultry (PLprobeBac) were highly specific for its sources (1.0, 0.97, and 1.0, respectively) although its sensitivity was lower (0.45, 0.50, and 0.73, respectively). The cattle‐specific qPCR was totally unspecific and was discarded for future experiments. When compared to previously designed assays, the human and pig qPCRs showed better accuracies (0.86 and 0.84) than their counterparts HF183 and Pig‐2‐Bac (0.38 and 0.65). Thus, the newly designed human, pig, and poultry qPCR assays outperform other methods developed until date and may be useful for source tracking purposes.
Keywords: Bacteroides, DGGE, microbial source tracking, quantitative PCR
Introduction
Fecal pollution of water is one the main causes of disease worldwide, particularly in developing countries (WHO 2008), and thus it is a global public health concern. In addition, it has a negative impact on many economic activities and leads to environmental deterioration. As a result, it is necessary to devise methods to monitor the levels of fecal pollution and determine its source(s) (Hagedorn et al. 2003). The identification and tracking of fecal source(s) may contribute to improving the management of the pollution at its source, thereby reducing economic losses and resolving legal issues (Jagals and Grabow 1996; Blanch et al. 2004).
Several microbiological methods such as those involving fecal coliforms, Escherichia coli, enterococci, and sulfite‐reducing clostridia are routinely used as microbial indicators to detect the presence of fecal pollution in water (Scott et al. 2002; Blanch et al. 2006). Unfortunately, such methods do not discriminate between human and animal fecal sources. In contrast, some Bacteroides spp. have been reported to be highly host specific (Kreader 1995) and consequently some species or markers have been proposed as new indicators of fecal pollution in water through microbial source tracking (MST) (Fiksdal et al. 1985; US EPA 2005). Bacteroides spp. are one of the most abundant genera of bacteria in the gastrointestinal tract of humans and other warm‐blooded animals. They represent up to a third of total gut microbiota (Eckburg et al. 2005; Ley et al. 2008; Wei et al. 2013; Ziemer 2014). Since Bacteroides spp. are anaerobic bacteria, cultivable Bacteroides can only survive 6 days under oxygen stress conditions and 2 or 3 days in the summer, with high temperatures (Avelar et al. 1998; Ballesté and Blanch 2010). This short environmental persistence hampers detection of Bacteroides by culture methods; however, their nucleic acids may persist longer periods (Kreader 1998).
For this reason, different MST molecular markers that target the order Bacteroidales have been proposed in recent years (Harwood et al. 2014). Among these, human‐associated HF183 (Bernhard and Field 2000a), belonging to Bacteroides dorei (Haugland et al. 2010), has been widely evaluated. However, it has been shown that HF183 is not entirely specific to human fecal sources (Green et al. 2014).
The aim of this study is to develop new host‐specific fecal pollution markers based on Bacteroides spp. To this end, the 16S rRNA genes of populations of Bacteroides occurring in samples with a unique and known fecal pollution source (cattle, pig, poultry, or human) were analyzed by denaturing gradient gel electrophoresis (DGGE). Host‐specific 16S rRNA gene sequences were selected to establish a set of common primers and various host‐specific hydrolysis probes that could be used in qPCR assays. The new qPCRs were then tested in point‐source water samples and compared with previously proposed MST molecular markers based on Bacteroidales.
Materials and Methods
Bacterial strains and growth conditions
Bacteriodes coprophilus DSM 18228T, B. coprosuis DSM 18011T, B. eggerthii DSM 20697T, B. finegoldi DSM 17565T, B. fragilis DSM 2151T, B. gallinarum DSM 18171T, B. intestinalis DSM 17393T, B. ovatus DSM 1896T, B. pyogenes DSM 20611T, B. salanitronis DSM 18170T, B. suis DSM 20612T, B. thetaiotaomicron DSM 2079T, B. uniformis DSM 6597T, B. vulgatus DSM 1447T, and Parabacteroides distasonis DSM 20701T were used as reference strains to establish the optimal DGGE gradient and conditions.
Type strains were grown using BPRM (Bacteroides Phage Recovery Media) broth (Tartera et al. 1992) and BPRM agar (Pronadisa, Laboratorios Conda, Madrid, Spain) without antibiotics at 37°C in anaerobic conditions (Anaerocult®; Merck Millipore, Darmstadt, Germany).
Host‐specific wastewater and slurry samples
To devise the qPCR assays, a total of 114 wastewater samples were collected from various sources and the DGGE assay was performed on them. Thirty‐four human sewage samples were obtained from five urban wastewater treatment plants located in the metropolitan area of Barcelona, Catalonia (NE Spain). Two of the wastewater treatment plants served populations of 384,000 inhabitants (28 samples); two plants served populations of between 100,000 and 200,000 inhabitants (four samples); and the other plant served a population of more than 2,000,000 inhabitants (two samples). All sewage samples were collected from the influent sewer entering the plants right after the bar screen and before the primary treatment.
A total of 25 samples of poultry slurry were collected from two poultry slaughterhouses that weekly process 60,000 chickens each. A total of 30 samples of pig slurry were obtained: 19 from four pig slaughterhouses that process 12,500, 15,000, 15,000, and 5000 pigs each week, respectively; and 11 from a fattening farm containing 14,000 pigs. Finally, 25 samples of cattle slurry were collected from six slaughterhouses processing between 250 and 2000 calves weekly (15 samples); and from two farms: one with 50 cows and the other with 200 calves (10 samples). All slaughterhouses were located in Catalonia (NE Spain) and had separate pipes for animal slurry and human wastewater from workers’ lavatories and showers.
In all samplings, 1 L of water or slurry were collected in sterile polyethylene containers and refrigerated at 4°C for up to 6 h before being processed in accordance with standardized protocols (Anonymous 1994).
Once the qPCR assays were designed and in order to test them, a total of 44 additional wastewater samples (11 human samples, 11 poultry samples, 12 pig samples, and 10 cattle samples) were freshly collected from the same locations described above.
Enumeration of Escherichia coli and somatic coliphages
Escherichia coli and somatic coliphages were used as indicators of bacterial fecal pollution and viral fecal pollution, respectively. Escherichia coli was enumerated by membrane filtration, following previously standardized methods (American Public Health Association, American Water Works Association & Water Environment Federation 1999) using Chromocult® Coliform agar (Merck, Darmstadt, Germany) incubated at 44.5°C for 24 h. Somatic coliphages were counted by the ISO 10705‐2 double agar layer method (Anonymous 2000) using E. coli strain WG5 (ATCC 700078) as the host. Each enumeration was performed in duplicate.
Nucleic acid isolation
Genomic DNA was obtained from reference strains by centrifuging 1.5 mL of a 48 h culture at 12,000g for 5 min. The pellet was washed twice in Tris‐EDTA buffer (Tris‐HCl 10 mmol/L, pH = 8.0 and EDTA 1 mmol/L) and the cells were then incubated at 100°C for 10 min. Finally, the mixture was centrifuged at 12,000g for 5 min and the supernatant was subjected to DNA amplification.
DNA extraction from the samples was performed with the QIAamp DNA blood minikit (Qiagen GmbH, Hilden, Germany), following the manufacturer's instructions. The DNA was suspended in a final volume of 50 μL of elution buffer. The integrity of the genomic DNA extracted was evaluated by 0.8% agarose gel electrophoresis and ethidium bromide staining.
Primers and PCR amplification
The 32F and 708R primer pair (Table 1) was used to amplify a 670‐bp fragment of the 16S rRNA gene using the DNA extracted from each wastewater sample or from 2‐day‐old cultures of the reference strains. Each 25 μL of PCR mixture contained 12.5 μL 2X DreamTaq Green DNA Polymerase (Fermentas, Madrid, Spain), 400 nmol/L of each primer, and 2.5 μL of DNA. PCR amplification was performed using a GeneAmp PCR system 2700 (Applied Biosystems, Barcelona, Spain) with the following conditions: initial denaturation step at 94°C for 5 min; then 35 cycles consisting of 94°C for 30 sec, 65°C for 1 min (this temperature was decreased by 1°C every cycle until the touchdown temperature of 61°C was reached), and 72°C for 1 min and 30 sec; and finally a 6 min extension at 72°C. Bacteriodes fragilis DSM 2151T was used as a positive control; a negative control containing no template was also included in each experiment.
Table 1.
Primers and probes used in this study
| Name | Sequence (5′ to 3′) | Use | Targeta | Reference |
|---|---|---|---|---|
| 32F | AACGCTAGCTACAGGCTT | PCR and sequencing | Bacteroides genus specific | Bernhard and Field (2000b,b) |
| 708R | CAATCGGAGTTCTTCGTG | PCR | Bacteroides genus specific | Bernhard and Field (2000b,b) |
| 32F‐GCb | CGCCCGGGGCGCGCCCCGGGCGGGGCGGGGGCACGGGGGGAACGCTAGCTACAGGCTT | PCR‐DGGE | Bacteroides genus specific | Myers et al. (1985a,b) |
| 580R | CGCTCCCTTTAAACCCAAT | DGGE‐PCR and sequencing | Bacteroides genus specific | This study |
| pGEMup | tgtaatacgactcactat | PCR and sequencing | pGEM‐T Easy plasmid | Serra‐Moreno et al. (2008) |
| Bac‐FW | GGCGCACGGGTGAGTAAC | qPCR | Uncultured Bacteroides | This study |
| Bac‐Rev | TGTGGGGGACCTTCCTCTC | qPCR | Uncultured Bacteroides | This study |
| HMprobeBac | FAM‐GTGAGGGCATCTAATCA | qPCR probe humans | Uncultured Bacteroides | This study |
| PLprobeBac | FAM‐TCCGCATGAAGGACTT | qPCR probe poultry | Uncultured Bacteroides | This study |
| PGprobeBac | FAM‐TATGATAGCATTAGAGTGTGACGAA | qPCR probe pigs | Uncultured Bacteroides | This study |
| CWprobeBac | FAM‐CTATGGGATGGGGATGC | qPCR probe cattle | Uncultured Bacteroides | This study |
| Bacprobe | FAM‐CGGGGTAACGGCCCA | qPCR common probe | Bacteroides genus specific | This study |
| HF183f | ATCATGAGTTCACATGTCCG | qPCR primers | Human Bacteroides | Seurinck et al. (2005) |
| HF183r | TACCCCGCCTACTATCTAATG | qPCR primers | Human Bacteroides | Seurinck et al. (2005) |
| Pig‐2‐Bac | FAM‐TCCACGGGATAGCC | qPCR probe pigs | Pig Bacteroides | Mieszkin et al. (2009) |
| Pig‐2‐Bac41F | GCATGAATTTAGCTTGCTAAATTTGAT | qPCR primers pigs | Pig Bacteroides | Mieszkin et al. (2009) |
| Pig‐2‐Bac163Rm | ACCTCATACGGTATTAATCCGC | qPCR primers pigs | Pig Bacteroides | Mieszkin et al. (2009) |
| Rum‐2‐Bac | FAM‐ATGAGGTGGATGGAATT | qPCR probe cattle | Cattle Bacteroides | Mieszkin et al. (2010) |
| BacB2‐590F | ACAGCCCGCGATTGATACTGGTAA | qPCR primers cattle | Cattle Bacteroides | Mieszkin et al. (2010) |
| Bac708Rm | CAATCGGAGTTCTTCGTGAT | qPCR primers cattle | Cattle Bacteroides | Mieszkin et al. (2010) |
Species that show the highest similarity to the probes according to BLAST analysis (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
The GC clamp attached to the 5′ end is denoted in italics.
The positive samples were reamplified by nested PCR using the pair of primers 32F‐GC and 580R (Table 1), and the PCR mixture prepared as described above. Conditions for the nested PCR were an initial denaturation step at 94°C for 5 min; then 35 cycles consisting of 94°C for 30 sec, 65°C for 1 min (this temperature was decreased by 1°C every cycle until the touchdown temperature of 63°C was reached), and 72°C for 1 min and 30 sec; and finally a 6 min extension at 72°C.
An aliquot of 5 μL of each PCR or nested‐PCR product was analyzed by 1.5% agarose (w/v) gel electrophoresis. DNA bands were stained with ethidium bromide and visualized by fluorescing when exposed to UV light.
DGGE analysis of PCR products
DGGE was performed with a DCode system (Bio‐Rad, Hercules, CA) as previously described (Ballesté and Blanch 2011). Electrophoresis was performed with 1‐mm thick 8% (w/v) polyacrylamide gels (30% acrylamide–bisacrylamide [37.5:1]) submerged in 1× Tris–acetate acid–EDTA (40 mmol/L Tris, 20 mmol/L sodium acetate, 1 mmol/L EDTA; pH = 7.4) at 60°C. About 800–1000 ng of nested‐PCR product from the environmental samples and 100 ng (NanoDrop ND‐1000 spectrophotometer; Thermo Scientific, Wilmington, DE) of nested‐PCR product from the reference strains were loaded into individual lanes in the gel. The following electrophoresis conditions were selected in accordance with the one of the perpendicular DGGE and time of travel experiments (data not shown): 17 h at 85 V and 60°C in a linear 40–65% denaturing gradient (100% denaturant agent was defined as 7 mol/L urea and 40% [v/v] formamide). The gels were stained for 45 min in 1X sodium chloride–Tris–EDTA buffer (100 mmol/L NaCl, 10 mmol/L Tris, 1 mmol/L EDTA; pH = 7.4) with SYBRGold nucleic acid stain (Molecular Probes Inc., Eugene, OR) and visualized under UV radiation using a ChemiDoc™ MP Imaging System (BioRad). The gels were scanned and analyzed using the Quantity One 4.6.7 program (Bio‐Rad).
Extraction of DGGE bands and sequencing
DGGE bands that were present in all the samples with a particular fecal pollution source and absent in the rest were selected and excised with a sterile razor blade. They were introduced into the wells of 1.5% agarose gel, sealed with melted 1.5% agarose, and analyzed by electrophoresis. The bands were visualized via ethidium bromide staining. DNA was extracted and purified using a QIAquick® gel extraction kit (Qiagen GmbH) following the manufacturer's instructions. The product was reamplified with the primers 32F and 580R (Table 1), and analyzed again by gel electrophoresis, and DNA was extracted and purified as explained above. The sequencing reaction was performed using a BigDye Terminator cycle‐sequencing ready‐reaction kit (Applied Biosystems) and by adding 5 μL of DNA. The reaction was performed under the following conditions: 25 cycles of 96°C for 30 sec, 50°C for 5 sec, and 60°C for 4 min. The product was analyzed using an automated DNA sequencer (ABI Prism 3700; PerkinElmer, Thermo Fisher Scientific, Waltham, MA USA [service provided by the Serveis Cientificotècnics of the University of Barcelona]).
The 16S rRNA gene sequences were edited and aligned using version 7.0.1 of the BioEdit program (Hall 1999). BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) was used to search for homology of each sequence with Bacteroides spp. and, when the information was available, to look for coincidences with the corresponding fecal source.
qPCR procedures
Clone construction
To generate standards for the qPCR assays, the four host‐specific 16S rRNA gene fragments were cloned in pGEM‐T Easy vectors following the manufacturer's instructions (Promega Biotech Ibérica, Barcelona, Spain). Each construction was transformed by electroporation (2.5 kV, 25 F capacitance, and 200 V resistance) into E. coli DH5α electrocompetent cells. The ampicillin‐resistant colonies that contained the vector with the insert were selected, verified by PCR, and plasmids were purified using a Qiagen Plasmid Midi purification kit (Qiagen Inc., Valencia, CA). A NanoDrop ND‐1000 spectrophotometer (Thermo Scientific) was used to evaluate the concentration and purity of the constructs containing each band.
To calculate the number of gene copies (GC) in the stock prepared for each gene, the following equation was used: [concentration of pGEM‐T Easy::insert (ng/μL)/molecular mass (ng/mol)] × 6.022 × 1023 molecules/mol = number of molecules of pGEM‐T Easy::insert/μL. Ten‐fold serial dilutions of the stock were performed with double‐distilled water and stored at −20°C until used. The stocks were amplified in triplicate in five independent experiments, and the average of the threshold cycle (Ct) results was used to elaborate standard curves.
Bac‐Fw and Bac‐Rev primers and probe sets
The 16S rRNA gene sequences were edited and aligned using version 7.0.1 of the BioEdit program (Hall 1999). Primers and probes were selected in the sequence of each DGGE fragment of 16S rRNA genes using the software tool Primer Express 3.0 (Applied Biosystems) to be used in a standardized amplification protocol. A set of common forward and reverse primers and specific hydrolysis probes for each fecal source (human, pig, poultry, and cattle) were designed. In addition, a probe common to all the origins was also designed. All primers and hydrolysis probes were commercially synthesized by Applied Biosystems. HMprobeBac, PGprobeBac, PLprobeBac, CWprobeBac, and Bacprobe were MGB probes with an FAM reporter and a nonfluorescent quencher (NFQ). The amplification conditions were used as described previously (Gómez‐Doñate et al. 2012). Briefly, amplification was performed in a 20‐μL reaction mixture with TaqMan Environmental Real‐Time PCR Master Mix 2.0 (Applied Biosystems). The mixture contained 900 nmol/L each primer, 250 nmol/L the corresponding probe, and 7 μL of the DNA sample or quantified plasmid DNA. The thermal‐cycler conditions were as follows: an initial setup of 2 min at 50°C, followed by 10 min at 95°C, 40 cycles of 15 sec of denaturation at 95°C, and 1 min of annealing/extension at 60°C. All the samples, standards, and positive and negative controls were run in triplicate. The threshold cycle (Ct) obtained was defined as the average of the triplicate data obtained. Ct data were expressed as the number of GC according to the values obtained with the standard for each qPCR reaction. In order to confirm the specificity of the primers and probes for their target genomes, NCBI (National Center for Biotechnology Information) data entries for Bacteroides spp. were used. Their specificity was also tested by cross reactions using DNA isolated from the other sources.
Use of host‐specific MST markers based on Bacteroidetes qPCR methods
Three previously described qPCR assays used for MST studies were also tested for comparison with those developed in this study: HF183, specific for humans (Seurinck et al. 2005); Pig‐2‐Bac, specific for pigs (Mieszkin et al. 2009); and Rum‐2‐Bac, specific for ruminants (Mieszkin et al. 2010). The assays and conditions used were as previously reported. Briefly, HF183 is a real‐time polymerase chain reaction (PCR) assay using SybrR Green I (Table 1), while Pig‐2‐Bac and Rum‐2‐Bac are real‐time qPCR assays using FAM‐labeled TaqMan probes (Table 1). Wastewater samples from known and single fecal sources were used to test their specificities. Traditional indicators (E. coli and somatic coliphages) were enumerated as detailed in the section “Enumeration of E. coli and somatic coliphages” and used as references of the fecal load of the wastewater and slurry samples.
Statistical analysis
Sensitivity (r), specificity (s), and accuracy (a) were calculated according to the following formulas: r = [TP/(TP + FN)]; s = [TN/(TN + FP)]; and a = [(TP + TN)/all samples], where TP is the number of samples that were positive for the qPCR marker of their own species (true positive), TN is the number of samples that were negative for a qPCR marker of another species (true negative), FP is the number of samples that were positive for a qPCR marker of another species (false positive), and FN is the number of samples that were negative for a qPCR marker of their own species (false negative).
Theory
Host‐specific bacterial identification by DGGE pattern‐band technique was successfully applied in previous studies for identifying host‐specific Bifidobacterium (Ballesté and Blanch 2011). These strains were used to discriminate between human, cow, poultry, and pig fecal pollution from high and moderate polluted samples (Gómez‐Doñate et al. 2012). As Bacteroides spp. is one of the most abundant genera of bacteria in the gastrointestinal tract of humans and other warm‐blooded animals, the design of Bacteroides host‐specific qPCRs could become a robust and reliable technique for MST studies.
Results
DGGE analysis of PCR products
The DGGE profiles of the fragments corresponding to Bacteroides 16S rRNA genes varied depending on the source of the fecal pollution in the water sample. The profiles of the human and poultry wastewater samples showed fewer bands than the cow and pig samples (Fig. 1), which had more heterogeneous profiles. The broadest bands common to all the samples from the same source and differential from those of other fecal sources were selected for further analysis. The selected bands were sequenced and compared with the BLAST and NCBI (National Center for Biotechnology Information) databases. Finally, only one band for each fecal origin was selected (Fig. 1). The bands selected for each source were all identified by BLAST as Bacteroides spp.
Figure 1.
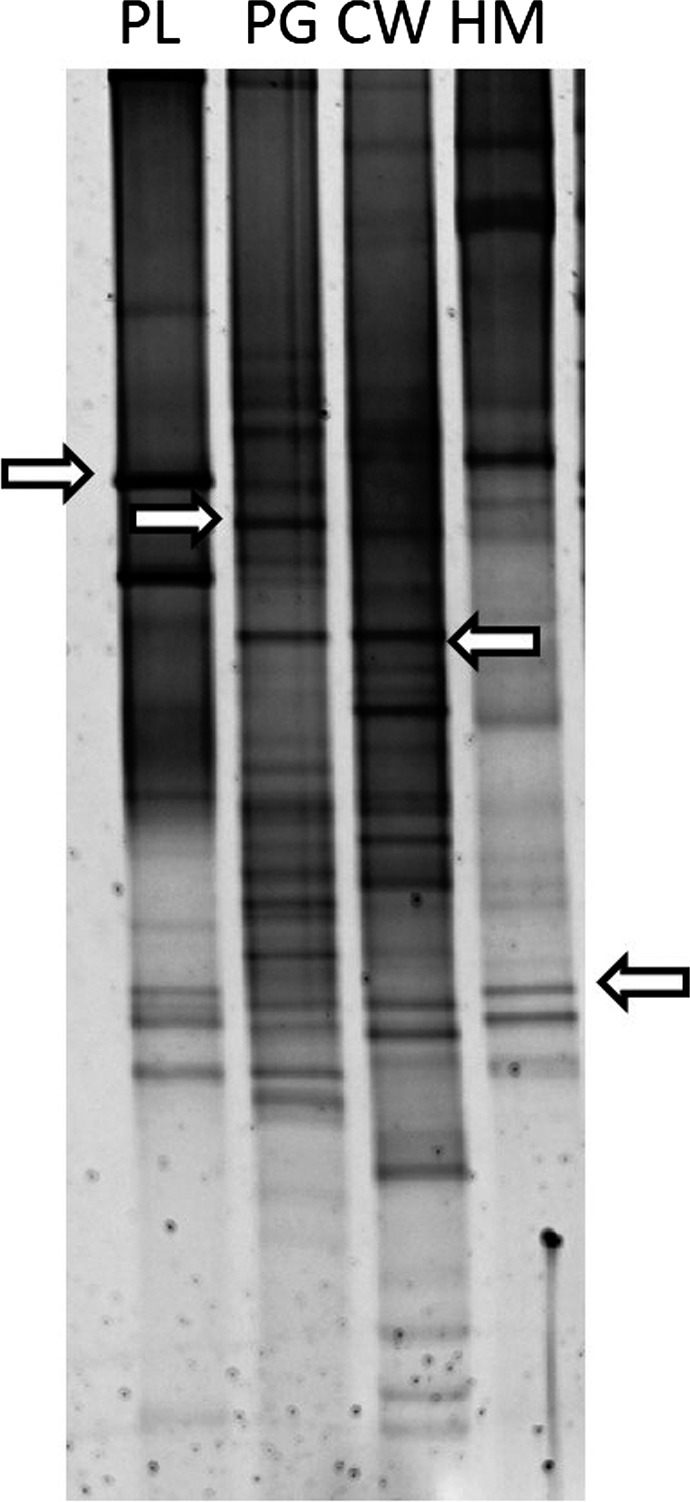
Representative denaturing gradient gel electrophoresis profiles of 16S rRNA of Bacteroides from wastewater samples of known fecal origin. Selected bands for each source are indicated with arrows (PL, poultry; PG, pig; CW, cattle; HM, human).
qPCR standards of host‐specific Bacteroides spp.
A region of the 16S rRNA gene sequence which had a variable zone between two conserved areas for each of the four hosts was selected to design the forward and reverse primers, and the Bacprobe common to the four origins. The specific probes for each source of fecal pollution (human, cattle, poultry, and pig) and a probe common to the Bacteroides genus were designed in the variable zone of each respective sequence (Fig. 2).
Figure 2.

Alignment of the sequences of the 16S rRNA genes from isolated denaturing gradient gel electrophoresis bands of human (HM), pig (PG), cattle (CW), and poultry (PL) wastewater, and general for Bacteroides (Bac). The locations of the primers Bac‐FW and Bac‐Rev, and the set of probes (HMprobeBac, PGprobeBac, CWprobeBac, PLprobeBac and Bacprobe) are indicated.
Primers and probes were selected according to the requirements of the Primer Express 3.0 software (Applied Biosystems). Using the five qPCR assays, five standard curves were plotted: one for the assay targeting the Bacteroides genus regardless the fecal origin, and one assay specific to each of the four pollution sources. The fragment of the 16S rRNA gene amplified from each DGGE band and cloned to generate the standard curves had 500 bp for the human source, 541 bp for poultry, 478 bp for pig, 543 bp for cattle, and 543 bp for the common Bacteroides genus assay. By using the primers Bac‐FW and Bac‐Rev, the amplicon size for each fecal source was 215 bp for human, cattle, and the common Bacteroides genus probe; and 214 bp for poultry and pig. The standard curves were reproducible, and the average slopes of the three replicates of the five independent amplifications were −3.3725 with a standard deviation (SD) of 0.033 for humans; −3.5061 (SD, 0.076) for poultry; −3.4589 (SD, 0.079) for pig; −3.4900 (SD, 0.064) for cattle; and −3.2805 (SD, 0.023) for the Bacteroides genus. Accordingly, the amplification efficiencies (E) for the assays were as follows: 97.7% (range, 96.4–100.1%) for human origin; 92.9% (range, 90.1–98.9%) for poultry origin; 94.1% (range, 90.7–97.9%) for pig origin; 93.3% (range, 90.8–96.3%) for cattle origin; and 101.8% (range, 100.7–102.9%) for the Bacteroides genus. Since the number of copies of the 16S rRNA gene of different Bacteroides spp. are not yet well defined (Hong et al. 2008), the quantitative expression of the qPCR calculations was based on GC and not on the number of bacterial cells. Thus, the calculated limits of detection in the qPCR assay are: 21.1 GC/μL for human; 13.9 GC/μL for pig; 16.4 GC/μL for poultry; 33.9 GC/μL for cattle; and 33.7 GC/μL for the Bacteroides genus.
Performance of host‐specific qPCR assays with wastewater samples of known fecal origin
The new host‐specific Bacteroides‐based probes showed high specificity for their respective hosts (Tables 2, 3) in the 44 wastewater samples from the four origins, except for the cattle‐specific probe. Since the assay for cattle was not specific, it was only used to analyze five samples and dropped from further studies. The HMprobeBac and PLprobeBac qPCR assays (for human and poultry, respectively) did not show any false positives with the samples from other fecal sources, and the PGprobeBac qPCR assay (pig specific) showed false‐positive reaction with a cattle sample (Table 2).
Table 2.
Microbial indicators and qPCR results for wastewater samples. Host‐specific Bacteroides qPCRs developed in this study: HMprobeBac (human), PLprobeBac (poultry), PGprobeBac (pig), CWprobeBac (cow), and Bacprobe (total Bacteroides). Host‐specific Bacteroides qPCRs developed by other authors and assayed in this study: HF183 (human), Pig‐2‐Bac (pig), and Rum‐2‐Bac (ruminants). SOMCPH: somatic coliphages. Relative abundance of Bacteroides cells in the samples were calculated with Bacprobe results considering five copies of 16S rRNA gene in Bacteroides thetaiotaomicron (Pei et al. 2010)
| Source | Host‐specific Bacteroides qPCRs in this study (GC/mL) | Other proposed host‐specific targets (GC/mL) | B. thetaiotaomicroncells/mL | Escherichia coli (cfu/mL) | SOMCPH (pfu/mL) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| HMprobeBac | PLprobeBac | PGprobeBac | CWprobeBac | Bacprobe | HF183 | Pig‐2‐Bac | Rum‐2‐Bac | ||||
| Human | |||||||||||
| Positive (%) | 5/11 (45.5) | 0/11 (0) | 0/11 (0) | 5/5 (100) | 11/11 (100) | 6/6 (100) | 2/9 (22.2) | 0/11 (0) | 11/11 (100) | 11/11 (100) | |
| Max value | 2.05 × 109 | 0 | 0 | 4.59 × 106 | 1.80 × 108 | 1.14 × 107 | 2.12 × 106 | 0 | 3.6 × 107 | 9.15 × 106 | 9.77 × 105 |
| Min value | 0 | 0 | 0 | 4.44 × 104 | 4.56 × 104 | 4.42 × 106 | 0 | 0 | 9.1 × 103 | 2.01 × 104 | 8.80 × 103 |
| Meana | 7.95 × 107 | 1.48 × 106 | 1.28 × 107 | 7.56 × 106 | 2.12 × 105 | 2.6 × 106 | 2.96 × 105 | 4.20 × 104 | |||
| Poultry | |||||||||||
| Positive (%) | 0/11 (0) | 8/11 (72.7) | 0/11 (0) | 5/5 (100) | 11/11 (100) | 6/6 (100) | 5/9 (55.6) | 0/11 (0) | 11/11 (100) | 11/11 (100) | |
| Max value | 0 | 2.84 × 109 | 0 | 2.52 × 106 | 1.28 × 109 | 3.61 × 106 | 2.99 × 106 | 0 | 2.6 × 108 | 1.72 × 105 | 9.25 × 106 |
| Min value | 0 | 0 | 0 | 5.74 × 103 | 6.25 × 105 | 2.03 × 106 | 0 | 0 | 1.2 × 105 | 6.31 × 104 | 2.70 × 102 |
| Meana | 5.42 × 107 | 1.61 × 105 | 3.93 × 107 | 2.82 × 106 | 6.71 × 105 | 7.9 × 106 | 2.70 × 105 | 4.20 × 104 | |||
| Pig | |||||||||||
| Positive (%) | 0/12 (0) | 0/12 (0) | 6/12 (50) | 5/5 (100) | 12/12 (100) | 5/9 (55.6) | 12/12 (100) | 0/12 (0) | 12/12 (100) | 12/12 (100) | |
| Max value | 0 | 0 | 5.68 × 107 | 5.52 × 108 | 5.24 × 108 | 9.42 × 105 | 3.03 × 108 | 0 | 1.1 × 108 | 6.00 × 105 | 2.95 × 105 |
| Min value | 0 | 0 | 0 | 5.98 × 105 | 2.39 × 105 | 0 | 1.10 × 107 | 0 | 4.8 × 104 | 1.35 × 104 | 1.57 × 104 |
| Meana | 5.40 × 106 | 2.80 × 107 | 5.61 × 107 | 1.23 × 105 | 4.15 × 107 | 1.1 × 107 | 7.80 × 104 | 5.65 × 104 | |||
| Cattle | |||||||||||
| Positive (%) | 0/10 (0) | 0/10 (0) | 1/10 (10) | 5/5 (100) | 10/10 (100) | 5/5 (100) | 7/10 (70) | 7/10 (70) | 10/10 (100) | 10/10 (100) | |
| Max value | 0 | 0 | 4.86 × 102 | 1.16 × 107 | 1.80 × 109 | 3.32 × 106 | 3.45 × 106 | 2.70 × 103 | 3.6 × 108 | 3.54 × 106 | 3.25 × 106 |
| Min value | 0 | 0 | 0 | 2.97 × 105 | 5.79 × 106 | 1.04 × 105 | 0 | 0 | 1.2 × 106 | 2.51 × 104 | 1.00 |
| Meana | 4.86 × 102 | 2.28 × 106 | 1.46 × 108 | 4.12 × 105 | 2.02 × 104 | 2.06 × 102 | 2.9 × 107 | 2.14 × 105 | 4.73 × 103 | ||
Geometric mean calculated from positive results.
Table 3.
Sensitivity, specificity, and accuracy of the qPCR assays analyzed. Host‐specific Bacteroides qPCRs developed in this study: HMprobeBac (human), PLprobeBac (poultry), PGprobeBac (pig), CWprobeBac (cow), and Bacprobe (total Bacteroides). Host‐specific Bacteroides qPCRs developed by other authors and assayed in this study: HF183 (human), Pig‐2‐Bac (pig), and Rum‐2‐Bac (ruminants)
| Value | Host‐specific Bacteroides qPCRs in this study | Other proposed host‐specific targets | ||||||
|---|---|---|---|---|---|---|---|---|
| HMprobeBac | PLprobeBac | PGprobeBac | CWprobeBac | Bacprobe | HF183 | Pig‐2‐Bac | Rum‐2‐Bac | |
| Sensitivity | 0.45 | 0.73 | 0.50 | 1.00 | 1.00 | 1.00 | 1.00 | 0.70 |
| Specificity | 1.00 | 1.00 | 0.97 | 0.00 | 1.00 | 0.20 | 0.50 | 1.00 |
| Accuracy | 0.86 | 0.93 | 0.84 | 0.25 | 1.00 | 0.38 | 0.65 | 0.93 |
The common qPCR assay for the Bacteroides genus showed amplification in all the samples tested. The concentration detected was equal to or greater than that of the corresponding host‐specific Bacteroides qPCR assay (Table 2). This observation suggests that the qPCR assay for the Bacteroides genus not only detects the studied fecal sources present in the sample, but also other Bacteroides strains that are part of host's gastrointestinal microbiota present in the samples.
Host‐specific Bacteroides qPCRs assays previously described in other studies were incorporated to our study with the same samples. Because these assays were later incorporated to the study, with some assays less samples were analyzed (Table 2). The human specific qPCR HF183 (Seurinck et al. 2005) showed 100% detection with human samples but 88% of false positives with samples of different fecal origins. The pig‐specific qPCR assay Pig‐2‐Bac (Mieszkin et al. 2009) showed 100% amplification of pig samples, but 50% false positives, especially with poultry and cattle samples. Finally, the cattle qPCR Rum‐2‐Bac assay (Mieszkin et al. 2010) was 100% specific (Table 3), without any cross reactivity, with 70% of cattle samples showing positive amplification, although the GC detected was lower than for the other host‐specific qPCR assays.
Thus, the host‐specific Bacteroides qPCRs designed in this study for human and pig showed a higher specificity in samples from Northeastern Spain than the previously designed ones (HF183 and Pig‐2‐Bac) for the Bacteroidetes group. In contrast, the Rum‐2‐Bac outperformed our cattle‐specific assay (CWprobeBac) (Table 3).
Discussion
Bacteroides spp. have been proposed as indicators of fecal pollution in water because they fulfill most of the requirements of a fecal indicator (Fiksdal et al. 1985; USEPA 2005; Hurst 2007). Moreover, it has been reported that there are some species of Bacteroides which are highly host‐specific, and which can be used to discriminate the sources of fecal pollution in water (Kreader 1995; Harwood et al. 2014). Bacteroides is an strictly anaerobic bacteria and hence not able to survive under oxygen stress conditions (Avelar et al. 1998). The reduced environmental persistence of this genus makes its detection by cultivable methods difficult. It is therefore advisable to target the corresponding 16S rRNA gene using culture‐independent molecular methods (Kreader 1998; Ballesté and Blanch 2010).
In this study, 16S rRNA gene amplification with specific primers for Bacteroides genus, followed by DGGE analysis, allowed us to differentiate bands with different sequences belonging to different Bacteroides spp. for each fecal origin. It has been considered that, based on the 16S rRNA gene sequences, DGGE is sufficiently sensitive to allow detection of bacteria that constitute up to 1% of the total bacteria community (Muyzer and Smalla 1998). Thus, in our study, only the most predominant Bacteroides spp. should have been detected. In some cases, however, different DNA sequences present the same melting domains (Ballesté and Blanch 2011), as observed with bands selected for pig and cow (Fig. 1).
Using this combined 16S rRNA gene amplification and DGGE technique, it was possible to observe that human and poultry samples showed less diversity in the 16S rRNA fragments within their Bacteroides populations than the pig and cattle samples. The most useful band for each fecal origin was selected on the basis of the most dominant band coincident in various samples analyzed of the same origin and its absence in samples of other origins. There is a delicate equilibrium between the required specificity and the need to cover most Bacteroides spp. within the same fecal source. The problem of samples showing negative results within their own source could be due to the presence of an abundant Bacteroides spp. population with a 16S rRNA gene showing a different sequence than that of the band selected in our study. Despite 114 samples being used for the selection of the most useful band, the analysis of more samples or samples from different sites would allow the evaluation of the extent of the false‐negative results, considering the high diversity observed. For example, some poultry samples were collected from abattoirs that sacrifice different animal species. Since each organism has specific Bacteroides microbiota, which has coevolved with it, it is possible that the band selected belonged, for instance, to the Bacteroides species corresponding to hens and therefore there would be no amplification if a sample contained fecal pollution from turkey. The variability within the Bacteroidales group has previously been reported and it has been suggested that multiple targets could be necessary to accurately assess fecal pollution (Lamendella et al. 2013).
Meanwhile, the lack of specificity observed in the assay intended to be specific for cattle could be attributable to the opposite effect. In addition to strains in cattle, the sequence could also be present (albeit in minimal proportions, hence not visible by DGGE) in other sources. This limitation may be overcome by increasing the number of samples analyzed by DGGE and the selection of a new band; although our attempts have resulted unsuccessful so far (data not shown).
Concerning the common Bacteroides genus assay, it showed detection in all the fecal samples, showing GC equal to or higher than the respective specific assay. This observation could be explained by this assay not only detecting a specific target linked with a fecal origin, as the specific assays do, but also most of the Bacteroides strains present in the gastrointestinal tract regardless the host.
The problems encountered with the qPCR assays designed in this study appear to be shared with other assays. When using the host‐specific Bacteroidetes assay (Seurinck et al. 2005; Mieszkin et al. 2009), it was observed that the human HF183 and pig Pig‐2‐Bac assays showed good sensitivity but a lower specificity than the host‐specific Bacteroides qPCR assays developed in this study. It should be considered that these assays were designed from samples from different geographical regions. Differences in the Bacteroides strains, as well as in the bacteriophages infecting Bacteroides, have been observed in samples with different geographical origins (Puig et al. 1999; Ishikawa et al. 2013; Lin et al. 2013). The reason for these differences is not clear, but they could be due to different selection of gut microbiota in different ethnic groups or the influence of different feeding habits (Yatsunenko et al. 2012; Kelder et al. 2014), and because Bacteroides spp. have established close mutualism with their hosts (Bäckhed et al. 2005). Finally, the cow‐specific assay, Rum‐2‐Bac, showed a high specificity for its fecal source. Unfortunately, in our samples, we detected a low GC concentration (2 logarithmic units) in highly polluted samples. These low concentrations may be limiting the use of Rum‐2‐Bac assay in environmental samples with a low fecal pollution level. Overall, the host‐specific Bacteroides sp. qPCRs designed in this study for human (HMprobeBac) and pig (PGprobeBac) presented a higher accuracy than the HF183 and Pig‐2‐Bac qPCRs, respectively.
Certain limitations of all Bacteroides qPCR methods should be considered when selecting the specific assay. First, the sensitivity of the assay if it is intended to apply it in MST in environments that present dilute or aged fecal pollution. Second, the selection of the method should take into account the area where it is to be applied. Validation of a given assay with local samples could reveal different outcomes from the results reported with samples from the location where the method was developed.
Conclusion
DGGE is a useful technique for identifying host‐specific bacteria from single fecal polluted samples. MST molecular targets based on these host‐specific Bacteroides allowed the design of host‐specific qPCR methods, with the exception of cow‐specific one. Although qPCR molecular targets showed moderate sensitivity (Jofre and Blanch 2010), these particular MST molecular indicators were rapid, straightforward, and accurate. These methods could be applied, together with traditional microbial indicators, for the development of predictive models, as previously reported (Ballesté and Blanch 2010; Sánchez et al. 2011).
Conflict of Interest
None declared.
Acknowledgments
This work was supported by the Spanish Government (research project CGL2011‐25401), also by the FP7 KBBE EU project AQUAVALENS, as well as by the research programs of the Xarxa de Referència en Biotecnologia (XRB) and the Comissionat per a Universitats i Recerca del DIUE of the regional authorities of Catalonia. Marta Gómez‐Doñate is the recipient of an FI grant from the regional authorities of Catalonia.
MicrobiologyOpen 2016; 5(1): 83–94
References
- American Public Health Association, American Water Works Association & Water Environment Federation . 1999. Standard methods for the examination of water and wastewater. Standard Methods, 541.
- Anonymous , 1994. Guidance on the preservation and handling of samples ‐ ISO 5667/3:1994. International Organization for Standardization, Geneva, Switzerland. [Google Scholar]
- Anonymous , 2000. ISO 10705‐2: Water quality. Detection and enumeration of bacteriophages ‐part 2: Enumeration of somatic coliphages. International Organization for Standardization, Geneva, Switzerland. [Google Scholar]
- Avelar, K. E. , Moraes S. R., Pinto L. J., Souza W. G., Domingues R., and Ferreira M. C.. 1998. Influence of stress conditions on Bacteroides fragilis survival and protein profiles. Zentralblatt für Bakteriologie 287:399–409. [PubMed] [Google Scholar]
- Bäckhed, F. , and Gordon J. I.. 2005. Host‐bacterial mutualism in the human intestine. Science 307:1915–1920. [DOI] [PubMed] [Google Scholar]
- Ballesté, E. , and Blanch A. R.. 2010. Persistence of Bacteroides species populations in a river as measured by molecular and culture techniques. Appl. Environ. Microbiol. 76:7608–7616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesté, E. , and Blanch A. R.. 2011. Bifidobacterial diversity and the development of new microbial source tracking indicators. Appl. Environ. Microbiol. 77:3518–3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhard, A. E. , and Field K. G.. 2000a. A PCR assay to discriminate human and ruminant feces on the basis of host differences in Bacteroides‐Prevotella genes encoding 16S rRNA. Appl. Environ. Microbiol. 66:4571–4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhard, A. E. , and Field K. G.. 2000b. Identification of nonpoint sources of fecal pollution in coastal waters by using host‐specific 16S ribosomal DNA genetic markers from fecal anaerobes. Appl. Environ. Microbiol. 66:1587–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanch, A. R. , Belanche‐Munoz L., Bonjoch X., Ebdon J., Gantzer C., Lucena F., et al. 2004. Tracking the origin of faecal pollution in surface water: an ongoing project within the European Union research programme. J. Water. Health 2:249–260. [PubMed] [Google Scholar]
- Blanch, A. R. , Belanche‐Munoz L., Bonjoch X., Ebdon J., Gantzer C., Lucena F., et al. 2006. Integrated analysis of established and novel microbial and chemical methods for microbial source tracking. Appl. Environ. Microbiol. 72:5915–5926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckburg, P. B. , Bik E. M., Bernstein C. N., Purdom E., Dethlefsen L., and Sargent M., et al. 2005. Diversity of the human intestinal microbial flora. Science 308:1635–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiksdal, L. , Maki J. S., LaCroix S. J., and Staley J. T.. 1985. Survival and detection of Bacteroides spp., prospective indicator bacteria. Appl. Environ. Microbiol. 49:148–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez‐Doñate, M. , Balleste E., Muniesa M., Blanch A. R.. 2012. New molecular quantitative PCR assay for detection of host‐specific Bifidobacteriaceae suitable for microbial source tracking. Appl. Environ. Microbiol. 78:5788–5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green, H. C. , Haugland R. A., Varma M., Millen H. T., Borchardt M. A., Field K. G., et al. 2014. Improved HF183 quantitative real‐time PCR assay for characterization of human fecal pollution in ambient surface water samples. Appl. Environ. Microbiol. 80:3086–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagedorn, C. , Crozier J. B., Mentz K. A., Booth A. M., Graves A. K., and Nelson N. J., et al. 2003. Carbon source utilization profiles as a method to identify sources of faecal pollution in water. J. Appl. Microbiol. 94:792–799. [DOI] [PubMed] [Google Scholar]
- Hall, T. A. 1999. BioEdit: a user‐friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41:95–98. [Google Scholar]
- Harwood, V. J. , Staley C., Badgley B. D., Borges K., and Korajkic A.. 2014. Microbial source tracking markers for detection of fecal contamination in environmental waters: relationships between pathogens and human health outcomes. FEMS Microbiol. Rev. 38:1–40. [DOI] [PubMed] [Google Scholar]
- Haugland, R. A. , Varma M., Sivaganesan M., Kelty C. A., Peed L., and Shanks O. C.. 2010. Evaluation of genetic markers from the 16S rRNA gene V2 region for use in quantitative detection of selected Bacteroidales species and human fecal waste by qPCR. Syst. Appl. Microbiol. 33:348–357. [DOI] [PubMed] [Google Scholar]
- Hong, P. Y. , Wu J.‐H., and Liu W.‐T.. 2008. Relative abundance of Bacteroides spp. in stools and wastewaters as determined by hierarchical oligonucleotide primer extension. Appl. Environ. Microbiol. 74:2882–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst, C. J. , Crawford R. L., Garland J. L., Lipson D. A., Mills A. L., and Stetzenbach L. D.. 2007. Manual of environmental microbiology, 3rd ed ASM Press, Washington, DC. [Google Scholar]
- Ishikawa, E. , Matsuki T., Kubota H., Makino H., Sakai T., Oishi K., et al. 2013. Ethnic diversity of gut microbiota: species characterization of Bacteroides fragilis group and genus Bifidobacterium in healthy Belgian adults, and comparison with data from Japanese subjects. J. Biosci. Bioeng. 116:265–270. [DOI] [PubMed] [Google Scholar]
- Jagals, P. , and Grabow W. O. K. 1996. An evaluation of sorbitol‐fermenting bifidobacteria as specific indicators of human faecal pollution of environmental water. Water SA, 22:235. [Google Scholar]
- Jofre, J. , and Blanch A. R.. 2010. Feasibility of methods based on nucleic acid amplification techniques to fulfil the requirements for microbiological analysis of water quality. J. Appl. Microbiol. 109:1853–1867. [DOI] [PubMed] [Google Scholar]
- Kelder, T. , Stroeve J. H. M., Bijlsma S., Radonjic M., and Roeselers G.. 2014. Correlation network analysis reveals relationships between diet‐induced changes in human gut microbiota and metabolic health. Nutr. Diabetes 4:e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreader, C. A. 1995. Design and evaluation of Bacteroides DNA probes for the specific detection of human fecal pollution. Appl. Environ. Microbiol. 61:1171–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreader, C. A. 1998. Persistence of PCR‐detectable Bacteroides distasonis from human feces in river water. Appl. Environ. Microbiol. 64:4103–4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamendella, R. , Li K. C., Oerther D., and Santo Domingo J. W.. 2013. Molecular diversity of Bacteroidales in fecal and environmental samples and swine‐associated subpopulations. Appl. Environ. Microbiol. 79:816–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley, R. E. , Lozupone C. A., Hamady M., Knight R., and Gordon J. I.. 2008. Worlds within worlds: evolution of the vertebrate gut microbiota. Nat. Rev. Microbiol. 6:776–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, A. , Bik E. M., Costello E. K., Dethlefsen L., Haque R., and Relman D. A.. 2013. Distinct distal gut microbiome diversity and composition in healthy children from Bangladesh and the United States. PLoS ONE 8:e53838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mieszkin, S. , Furet J. P., Corthier G., and Gourmelon M.. 2009. Estimation of pig fecal contamination in a river catchment by real‐time PCR using two pig‐specific Bacteroidales 16S rRNA genetic markers. Appl. Environ. Microbiol. 75:3045–3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mieszkin, S. , Yala J. F., Joubrel R., and Gourmelon M.. 2010. Phylogenetic analysis of Bacteroidales 16S rRNA gene sequences from human and animal effluents and assessment of ruminant faecal pollution by real‐time PCR. J. Appl. Microbiol. 108:974–984. [DOI] [PubMed] [Google Scholar]
- Muyzer, G. , and Smalla K.. 1998. Application of denaturing gradient gel electrophoresis (DGGE) and temperature gradient gel electrophoresis (TGGE) in microbial ecology. Antonie Van Leeuwenhoek 73:127–141. [DOI] [PubMed] [Google Scholar]
- Myers, R. M. , Fischer S. G., Maniatis T., and Lerman L. S.. 1985a. Modification of the melting properties of duplex DNA by attachment of a GC‐rich DNA sequence as determined by denaturing gradient gel electrophoresis. Nucleic Acids Res. 13:3111–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers, R. M. , Fischer S. G., Lerman L. S., and Maniatis T.. 1985b. Nearly all single base substitutions in DNA fragments joined to a GC‐clamp can be detected by denaturing gradient gel electrophoresis. Nucleic Acids Res. 13:3131–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei, A. Y. , Oberdorf W. E., Nossa C. W., Agarwal A., Chokshi P., Gerz E. A., et al. 2010. Diversity of 16S rRNA genes within individual prokaryotic genomes. Appl. Environ. Microbiol. 76:3886–3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig, A. , Queralt N., Jofre J., and Araujo R.. 1999. Diversity of Bacteroides fragilis strains in their capacity to recover phages from human and animal wastes and from fecally polluted wastewater. Appl. Environ. Microbiol. 65:1772–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez, D. , Belanche L. A., and Blanch A. R.. 2011. A software for the microbial source tracking problem. J. Mach. Learn. Res 17:56–62. [Google Scholar]
- Scott, T. M. , Rose J. B., Jenkins T. M., Farrah S. R., and Lukasik J.. 2002. Microbial source tracking: current methodology and future directions. Appl. Environ. Microbiol. 68:5796–5803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra‐Moreno, R. , Jofre J., and Muniesa M.. 2008. The CI repressors of Shiga toxin‐converting prophages are involved in coinfection of Escherichia coli strains, which causes a down regulation in the production of Shiga toxin 2. J. Bacteriol. 190:4722–4735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seurinck, S. , Defoirdt T., Verstraete W., and Siciliano S. D.. 2005. Detection and quantification of the human‐specific HF183 Bacteroides 16S rRNA genetic marker with real‐time PCR for assessment of human faecal pollution in freshwater. Environ. Microbiol. 7:249–259. [DOI] [PubMed] [Google Scholar]
- Tartera, C. , Araujo R., Michel T., and Jofre J.. 1992. Culture and decontamination methods affecting enumeration of phages infecting Bacteroides fragilis in sewage. Appl. Environ. Microbiol. 58:2670–2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- US Environmental Protection Agency . 2005. Microbial source tracking guide. Document EPA/600/R‐05/064. U. S. Environmental Protection Agency, Washington, DC. [Google Scholar]
- Wei, S. , Morrison M., and Yu Z.. 2013. Bacterial census of poultry intestinal microbiome. Poult. Sci. 92:671–683. [DOI] [PubMed] [Google Scholar]
- World health organization , 2008. The global burden disease. 2004 Update. Available at http://www.who.int/healthinfo/global_burden_disease/en/ (accessed 02 November 2015).
- Yatsunenko, T. , Rey F. E., Manary M. J., Trehan I., Dominguez‐Bello M. G., Contreras M., et al. 2012. Human gut microbiome viewed across age and geography. Nature 486:222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziemer, C. J. 2014. Newly cultured bacteria with broad diversity isolated from eight‐week continuous culture enrichments of cow feces on complex polysaccharides. Appl. Environ. Microbiol. 80:574–585. [DOI] [PMC free article] [PubMed] [Google Scholar]