

Abstract
Response of cancer cells to chemotherapy-induced DNA damage is regulated by the ATM-Chk2 and ATR-Chk1 pathways. We investigated the association between phosphorylated H2AX (γ-H2AX), a marker of DNA double-strand breaks that trigger the ATM-Chk2 cascade, and phosphorylated Chk1 (pChk1), with pathological complete response (pCR) in triple-negative breast cancer (TNBC) patients treated with neoadjuvant chemotherapy. γ-H2AX and pChk1 were retrospectively assessed by immunohistochemistry in a series of pretreatment biopsies related to 66 patients. In fifty-three tumors hormone receptor status was negative in both the diagnostic biopsies and residual cancers, whereas in 13 cases there was a slight hormone receptor expression that changed after chemotherapy. Internal validation was carried out. In the entire cohort elevated levels of γ-H2AX, but not pChk1, were associated with reduced pCR rate (p = 0.009). The association tested significant in both uni- and multivariate logistic regression models (OR 4.51, 95% CI: 1.39–14.66, p = 0.012, and OR 5.07, 95% CI: 1.28–20.09, p = 0.021, respectively). Internal validation supported the predictive value of the model. The predictive ability of γ-H2AX was further confirmed in the multivariate model after exclusion of tumors that underwent changes in hormone receptor status during chemotherapy (OR 7.07, 95% CI: 1.39–36.02, p = 0.018). Finally, in residual diseases a significant decrease of γ-H2AX levels was observed (p < 0.001). Overall, γ-H2AX showed ability to predict pCR in TNBC and deserves larger, prospective studies.
Keywords: DNA damage and repair, triple-negative breast cancer, pathological complete response
INTRODUCTION
Triple-negative breast cancer (TNBC) accounts for approximately 20% of all breast cancer (BC) cases, and represents the most aggressive BC subtype [1]. Neoadjuvant chemotherapy (NACT) was historically delivered with the aim to shrink unresectable tumors or increase the rate of breast-conserving surgery for operable tumors [2]. Due to evidence linking pathological complete response (pCR) to improved survival outcomes [3], the neoadjuvant setting is increasingly exploited as a platform in the search for predictive biomarkers [4].
Escape from chemotherapy-induced death stimuli is a multifaceted phenomenon mediated by both cancer cell-intrinsic and -extrinsic factors [5]. Cancer cells “hijack” physiological mechanisms to endure perturbations arising or induced in their eco-system, such as exposure to chemotherapy. The pronounced ability to protect the genome is one of the best preclinically described way through which cancer cells survive chemotherapy [6]. Safeguarding genome integrity and preventing the accumulation of harmful mutations is a complex task, whose accomplishment requires a tight cooperation between a number of pathways. This intricate network, overall defined as the DNA damage repair (DDR), schematically operates through the coordinated activity of three major signals [7]: cell cycle checkpoints that halt the progression of the cell cycle when DNA damage is sensed, DNA repair mechanisms that remove DNA lesions, and apoptotic pathways that eliminate cells whose genetic lesions cannot be repaired [7]. Over the past decade, the complexity of the DDR has been the focus of intense preclinical investigations, and nowadays we have a fairly detailed picture of the molecular events that are triggered in cancer cells challenged with chemotherapy [8]. Despite these achievements, from a clinical perspective the analyses of the DDR have historically been confined to a handful of distal effectors acting in the context of specific repair avenues, and they have been overall inconclusive [9]. More recently, novel biomarkers such as RAD51 and the so-called genomic scars, which presumably reflect the underlying state of DNA repair, were proposed, renewing the enthusiasm surrounding the clinical development of DDR-associated endpoints to foresee the efficacy of chemotherapy [10–13].
Phosphorylated (Ser139) H2A Histone Family Member X (γ-H2AX) is an established marker of DNA double-strand breaks (DSBs) [14]. When these lesions occur, the Ataxia-Telangiectasia Mutated (ATM)-Checkpoint Kinase 2 (Chk2) pathway is activated and orchestrates DNA repair [15]. In a treatment-naïve background, elevated γ-H2AX levels might mirror a strategy, namely the activation of the ATM-Chk2 pathway, cancer cells evolved to tolerate endogenous DNA damages arising upon oncogene-induced replication stress [16]. We reasoned that this adaption to deal with replicative stress concomitantly feeds therapeutic resistance.
A second key DDR pathway is the Ataxia Telangiectasia and Rad3-related protein (ATR)-Checkpoint kinase 1 (Chk1)-Wee1-like protein kinase (Wee1) signal [17]. The ATR-Chk1-Wee1 axis, that extensively cooperates with the ATM-Chk2 pathway, is mainly activated by stretched of single-stranded DNAs and governs G2/M transition. By arresting the cell cycle, the ATR-Chk1-Wee1 signal avoids that damaged cells embark into a fatal mitosis.
Given that γ-H2AX and Chk1 operate in the context of major molecular routes deputed to initiate the DDR, their expression might mirror an underlying chemoresistant phenotype. To test this hypothesis, γ-H2AX and phosphorylated Chk1(pChk1) were evaluated by immunohistochemistry (IHC) in pretreatment biopsies related to TNBC patients treated with anthracycline-taxane-based NACT, and their expression analyzed for a potential association with pCR.
RESULTS
Baseline characteristics and treatment outcome of the 66 patients included in the present study are illustrated in Table 1. As showed in Table 2, we observed a significant association between elevated γ-H2AX levels and reduced pCR rate (p = 0.009). In the γ-H2AXlow group we recorded 14 pCRs (43.8%) and 18 (56.2%) residual diseases, whereas in the γ-H2AXhigh group we observed 5 pCR (14.7%) and 29 residual diseases (85.3%). Conversely, pChk1 expression did not appear associated with pCR (p = 0.085), (Table 2). The predictive ability of γ-H2AX levels was observed in the univariate logistic regression model (γ-H2AXhigh vs γ-H2AXlow: Odds Ratio (OR) 4.51, 95% Confidence Interval (CI): 1.39–14.66, p = 0.012) (Table 3), and maintained in the multivariate model (γ-H2AXhigh vs γ-H2AXlow: OR 5.07, 95% CI: 1.28–20.09, p = 0.021) (Table 3). The consistency of the multivariate model was supported by internal validation envisioning a re-sampling without replacement procedure. Median Cohen's Kappa coefficient was 0.492 (moderate agreement), and the replication rate for γ-H2AX was 67%. Sensitivity analysis carried out by removing 13 patients whose tumors changed hormone receptor status during NACT further confirmed the predictive ability of γ-H2AXhigh (univariate and multivariate logistic regression models: γ-H2AXhigh vs γ-H2AXlow: OR 4.71, 95% CI: 1.26–17.66, p = 0.021; and OR 7.07, 95% CI: 1.39–36.02, p = 0.018, respectively) (Table 4). A suggestion for a predictive role of pChk1 stemmed from the 13 tumors that switched hormone receptor expression. In this small subset, we observed 9 residual diseases and 1 pCR in pChk1pos tumors, whereas all the three patients with pChk1neg tumors experienced a pCR (p = 0.014, data available upon request). Finally, analysis of matched pre- and post-treatment tissues showed a significant reduction of both γ-H2AX and Ki-67 expression in residual disease (p < 0.001 and p = 0.012 in Figure 1 and Figure 2, respectively).
Table 1. Baseline characteristics and treatment outcome of TNBC patients treated with neoadjuvant chemotherapy (N = 66).
| Characteristics | N (%) |
|---|---|
|
Age at diagnosis Mean ± SD Median (min-max)[IQrange] |
49.6 ± 11.4 48.4 (25.6–76.6) [44.0–57.6] |
|
Stage II III |
23 (34.8) 43 (65.2) |
|
Ki-67 Mean ± SD Median (min-max)[IQrange] |
57.0 ± 25.0 60 (10–90) [40–80] |
|
Grade 1–2 3 |
26 (39.4) 40 (60.6) |
|
Chemotherapy Sequential Concomitant |
56 (84.8) 10 (15.2) |
|
Hormone receptor change No Yes |
53 (80.3) 13 (19.7) |
|
Pathological complete response Yes No |
19 (28.8) 47 (71.2) |
Table 2. Association between biomarkers of DNA damage and repair (γ-H2AX and pChk1) and pathological complete response in TNBC patients treated with neoadjuvant chemotherapy (N = 66).
| Biomarker | Pathological complete response | Chi2 | |
|---|---|---|---|
| No | Yes | ||
| N (%) | N (%) | p-value | |
| γ-H2AXlow | 18 (56.2) | 14 (43.8) | 0.009 |
| γ-H2AXhigh | 29 (85.3) | 5 (14.7) | |
| pChk1neg | 10 (55.6) | 8 (44.4) | 0.085 |
| pChk1pos | 37 (77.1) | 11 (22.9) | |
Table 3. Uni and multivariate logistic regression models of patient- and disease-related features and pathological complete response (N:66).
| Univariate logistic regression model (*) | Multivariate logistic regression model (*) | ||||
|---|---|---|---|---|---|
| OR (95% CI) | p-value | OR (95% CI) | p-value | ||
| Age | >48.4 vs ≤48.4 | 4.13 (1.27–13.37) | 0.018 | 6.15 (1.49– 25.44) | 0.012 |
| Stage | III vs II | 0.81 (0.26–2.54) | 0.723 | 1.23 (0.29–5.14) | 0.780 |
| Grade | III vs I-II | 1.17 (0.40–3.46) | 0.774 | 1.32 (0.34– 5.07) | 0.687 |
| CT | Conc vs Seq | Not applicable | Not applicable | ||
| Ki-67 | ≥60 vs <60 | 0.29 (0.09–0.89) | 0.030 | 0.31 (0.08–1.17) | 0.084 |
| γ-H2AX | high vs low | 4.51 (1.39–14.66) | 0.012 | 5.07 (1.28–20.09) | 0.021 |
| pChk1 | pos vs neg | 2.69 (0.85–8.48) | 0.091 | 2.65 (0.63–11.19) | 0.184 |
Type of chemotherapy (concomitant vs sequential) was not included in uni- and multivariate models given that no pCRs were seen in patients treated with a concomitant schedule. Abbreviations, CT: chemotherapy; Conc: concomitant; Seq: sequential.
Table 4. Uni and multivariate logistic regression models of patient- and disease-related features and pathological complete response after removal of 13 patients whose hormone receptor status changed during neoadjuvant chemotherapy (N:53).
| Univariate logistic regression model (*) | Multivariate logistic regression model (*) | ||||
|---|---|---|---|---|---|
| OR (95% CI) | p-value | OR (95% CI) | p-value | ||
| Age | >48.4 vs ≤48.4 | 3.78 (1.02–14.06) | 0.047 | 4.65 (0.99–21.87) | 0.052 |
| Stage | III vs II | 0.70 (0.19–2.63) | 0.597 | 1.78 (0.30–10.43) | 0.521 |
| Grade | III vs I-II | 1.08 (0.33–3.58) | 0.899 | 1.46 (0.31- 6.84) | 0.627 |
| CT | Conc vs Seq | Not applicable | Not applicable | ||
| Ki-67 | ≥60 vs <60 | 0.35 (0.10–1.19) | 0.092 | 0.25 (0.05–1.19) | 0.082 |
| γ-H2AX | high vs low | 4.71 (1.26–17.66) | 0.021 | 7.07 (1.39–36.02) | 0.018 |
| pChk1 | pos vs neg | 1.40 (0.38–5.10) | 0.610 | 1.10 (0.21–5.67) | 0.909 |
Type of chemotherapy (concomitant vs sequential) was not included in uni- and multivariate models given that no pCRs were seen in patients treated with a concomitant schedule. Abbreviations, CT: chemotherapy; Conc: concomitant; Seq: sequential.
Figure 1. Box plot showing the distribution of γ-H2AX values in pre and post-neoadjuvant chemotherapy samples.
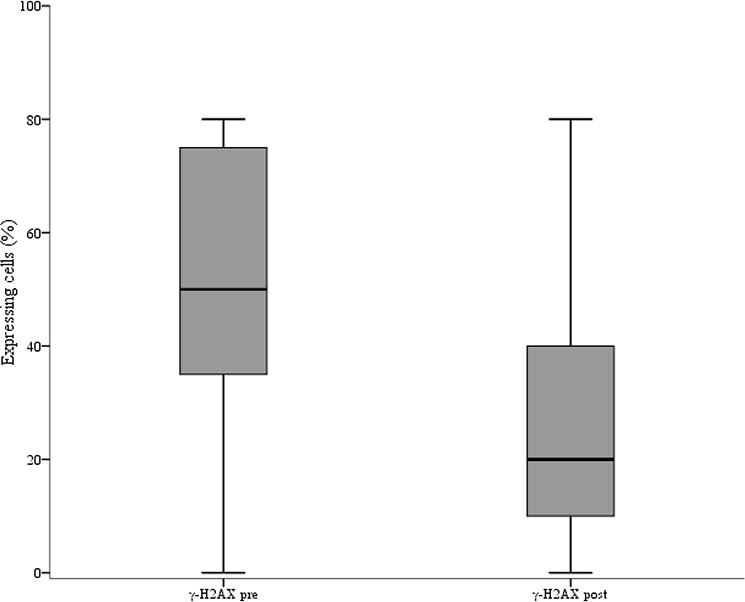
The figure shows the median values (horizontal bars within boxes), 25th and 75th percentile (lower and upper horizontal lines of the boxes), and minimum and maximum values (lower and upper horizontal bars outside the boxes).
Figure 2. Box plot of the distribution of Ki-67 values in pre and post-neoadjuvant chemotherapy samples.
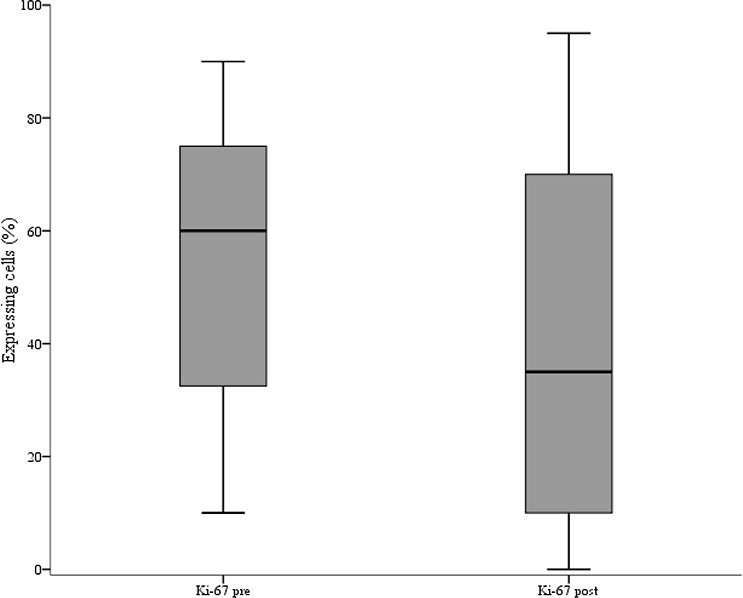
The figure shows the median values (horizontal bars within boxes), 25th and 75th percentile (lower and upper horizontal lines of the boxes), and minimum and maximum values (lower and upper horizontal bars outside the boxes).
DISCUSSION
In the present study, we investigated the predictive ability of γ-H2AX and pChk1 expression in TNBC patients treated with NACT. To our knowledge, this is the first study pointing on these DNA damage and repair biomarkers as candidate predictive factors in TNBC. Overall, we observed a significant association between elevated levels of γ-H2AX and reduced pCR rate, whereas a similar association did not emerge for pChk1. We also observed a significant reduction in γ-H2AX levels when comparing primary and residual cancers. Considering the retrospective nature, in our opinion this study has some important strengths.
The neoadjuvant setting is ideal when the scope is the identification and development of predictive biomarkers. This is related to the short time span required to obtain efficacy data, the association between pCR and long-term survival outcomes, and the availability of pre- and post-treatment tumor tissues suitable for molecular analyses, at least for non-responders [4].
The logic behind this study was to investigate two cooperating DNA repair avenues, representing master regulators of the DDR machinery. The increased therapeutic resistance observed in tumors characterized by high levels of γ-H2AX raised the hypothesis that the ATM-Chk2 pathway is crucial for initiating DNA repair in TNBC cells.
Even though our data did not support a predictive role for pChk1 in TNBC, in our opinion the G2/M checkpoint should not be underestimated for different reasons. First, TP53 mutations are extremely common in TNBC [18]. p53-defective tumors are known to be extremely dependent on G2/M checkpoint activation to arrest the cell cycle and initiate DNA repair upon exposure to chemotherapy [17]. This form of “addiction” might therefore be exploited to look at potential G2/M checkpoint-associated biomarkers. Second, the use of carboplatin in the neoadjuvant setting has been recently found to achieve a greater rate of pCR in TNBC patients [19, 20], and platinum compounds represented the preferred partners for the development of Chk1 and Wee1 antagonists [17]. Thus, the impact of G2/M checkpoint-related molecular determinants on therapeutic outcomes in TNBC patients treated with carboplatin deserve to be investigated. Nonetheless, the involvement of the ATR-Chk1 and ATM-Chk2 pathways in the intra-S and G2/M checkpoints, and the connection between Chk1 and the spindle checkpoint, raised the hypothesis that their activation may confer chemoresistant features independently on the chemotherapy regimen used [17]. Overall, the lack of association between Chk1 and pCR, and the observation that ∼15% of patients whose tumors displayed elevated γ-H2AX levels experienced a pCR, encouraged us to initiate a more comprehensive analysis. To this end, our strategy for the development of a DDR signature envisions: i) The combined assessment of key components of the ATR-Chk1 and ATM-Chk2 pathways, e.g. pATM, pChk2, pATR, pWee1, pRPA32, together with genetic alterations that activate the DDR cascade, such as TP53 mutations and MYC amplification [17], ii) Deeper characterization of the heterogeneity of TNBC, with a specific focus on the basal-like subtype, together with the assessment of androgen receptor expression (luminal androgen receptor subtype) given its potential as therapeutic target [21–23], and iii) The evaluation of multiple clinical outcomes, even including disease-free and overall survival. This second step will be instrumental for our prospective validation efforts. Moreover, the suggestion for an association between pChk1 and pCR in the subgroup of tumors that underwent a conversion in hormone receptor status was hypothesis-generating, and prompted us to undertake DDR analysis in luminal-type BC.
A further point that deserves mention relates to the analysis of residual disease. We would have expected an increase in γ-H2AX levels, as a consequence of the accumulation of DSBs following chemotherapy. Conversely, an opposite phenomenon was recorded. We can speculate that NACT operated an enrichment for slowly-cycling, chemotherapy-resistant cancer stem cells (CSCs) [24–27]. Considering that a series of studies, though retrospective yet, connected CSC-related endpoints with poorer survival outcomes [28], we envision that changes in γ-H2AX levels between pre- and post-NACT tissues might affect survival outcomes. An ad hoc study was designed to test this hypothesis.
In conclusion, γ-H2AX expression showed ability to foresee pCR in TNBC patients treated with anthracycline-taxane-based NACT. The results herein presented support the concept that DDR-related endpoints deserve further studies in TNBC.
MATERIALS AND METHODS
This retrospective study has been conducted in accordance with the ethical standards and according to the Declaration of Helsinki and according to national and international guidelines and has been approved by the Ethic Committee of “Regina Elena” National Cancer Institute of Rome, the coordinating centre. Written informed consents were obtained before chemotherapy. Sixty-six patients treated with NACT were included in this retrospective analysis. Patients were considered eligible if the treatment was completed, data on clinical-pathological features were available, and tumors did not show HER2 overexpression/amplification according to ASCO-CAP guidelines. Concerning the expression of the estrogen receptor (ER) and progesterone receptor (PgR), 53 patients had TNBC in both diagnostic biopsies and in residual cancers when present, whereas 13 tumors switched their hormone-receptor status from weak positivity (ER or PgR ≤ 10%) in diagnostic biopsies to negativity in surgical samples (N: 10) or vice versa (N: 3). These patients were included based on the clinical plausibility of a basal-like molecular portrait, considering that up to 20% of basal-like cancers are not “pure” TNBC and express the ER [1]. Analyses were initially run in the entire cohort, and then repeated upon removal of these 13 samples. All patients had received anthracycline-taxane-based chemotherapy, either according to a concomitant or sequential approach. Of the 10 patients treated with concomitant chemotherapy, 9 received epirubicin 80 mg/m2 plus docetaxel 80 mg/m2 administered intravenously (IV) on day 1 every 3 weeks for four cycles, and 1 patient epirubicin 75 mg/m2 plus docetaxel 75 mg/m2 plus cyclophosphamide 500 mg/m2 IV on day 1 every 3 weeks for six cycles. In the 56 patients treated with sequential chemotherapy, epirubicin was given at 90 mg/m2 on day 1 every two weeks or 100–120 mg/m2 on day 1 every three weeks in 29 and 27 patients, respectively. In these patients, epirubicin was administered in association with cyclophosphamide 600 mg/m2 IV for four cycles, followed by docetaxel 100 mg/m2 IV on day 1 every 3 weeks for four cycles. pCR was defined as no residual invasive tumor in both breast and axilla, irrespective of the presence of ductal carcinoma in situ (ypT0/is ypN0), and was assessed by local pathologists. The immunohistochemical assessment of γ-H2AX and pChk1 was performed in formalin-fixed paraffin-embedded tissues using the anti-phospho-H2AX (Ser139) (clone JBW301) mouse monoclonal antibody (MAb) (Upstate) at the dilution of 1:500, and the anti-phospho-Chk1 (Ser345) (clone 133D3) rabbit MAb (Cell Signaling) at the dilution of 1:150. γ-H2AX expression was considered as the percentage of nuclear-expressing tumor cells and analyzed as a categorical variable, using the median score of all tumors to define high and low expressing samples (γ-H2AXlow and γ-H2AXhigh). pChk1 was graded based on nuclear staining intensity (0: negative, 1+: weak, 2+: moderate, 3+: strong), and it was considered as negative (0: pChk1neg) or positive (1–3: pChk1pos). Two investigators (ADB and CE) blinded to the outcome independently evaluated immunoreactivity. A third investigator (MM) reviewed discordant cases.
Statistical analysis
Clinical, pathological and molecular features were descriptively characterized for all the patients included in the present analysis. Continuous variables were reported as medians and ranges, and categorical variables were expressed by frequencies and percentage values. The Pearson's Chi-squared test of independence (2-tailed) and the Fisher Exact test were used to assess the relationship between categorical variables. Univariate logistic regression models were used to identify variables impacting treatment outcome, and multivariate logistic regression models were built by including variables significant at the univariate assessment or based on their clinical or biological plausibility in influencing pCR. Internal validation was conducted through a re-sampling procedure without replacement in order to estimate the risk of an overfitted model [29]. By randomly removing ∼20% of the original sample, one hundred less-powered datasets were created and, for each simulation, the multivariate logistic regression model was carried out. For each simulation we calculated the Cohen's Kappa coefficient. The replication rate was also calculated. The Wilcoxon test was used to evaluate pre- and post-NACT changes in γ-H2AX and Ki-67. We considered statistically significant p values less than 0.05. Statistical analyses were carried out using SPSS software (SPSS version 21, SPSS Inc., Chicago, IL, USA).
Acknowledgments
We thank Tania Merlino and Ana Maria Edlisca for editorial assistance.
Footnotes
FINANCIAL SUPPORT
IV is supported by the Italian Association for Cancer Research (AIRC, MFAG), Ministero Italiano della Salute, and the Programma per i Giovani Ricercatori “Rita Levi Montalcini” 2011. This study was supported by the AIRC Investigator Grant (RDM), and the Consorzio Interuniversitario Nazionale per la Bio-Oncologia (CINBO) (CN).
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- 1.Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010;363:1938–1948. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- 2.von Minckwitz G, Martin M. Neoadjuvant treatments for triple-negative breast cancer (TNBC) Ann Oncol. 2012;23:vi35–9. doi: 10.1093/annonc/mds193. [DOI] [PubMed] [Google Scholar]
- 3.von Minckwitz G, Fontanella C. Comprehensive Review on the Surrogate Endpoints of Efficacy Proposed or Hypothesized in the Scientific Community Today. J Natl Cancer Inst Monogr. 2015;2015:29–31. doi: 10.1093/jncimonographs/lgv007. [DOI] [PubMed] [Google Scholar]
- 4.Bardia A, Baselga J. Neoadjuvant therapy as a platform for drug development and approval in breast cancer. Clin Cancer Res. 2013;19:6360–6370. doi: 10.1158/1078-0432.CCR-13-0916. [DOI] [PubMed] [Google Scholar]
- 5.Maugeri-Saccà M, Vigneri P, De Maria R. Cancer stem cells and chemosensitivity. Clin Cancer Res. 2011;17:4942–4947. doi: 10.1158/1078-0432.CCR-10-2538. [DOI] [PubMed] [Google Scholar]
- 6.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 7.Maugeri-Saccà M, Bartucci M, De Maria R. DNA damage repair pathways in cancer stem cells. Mol Cancer Ther. 2012;11:1627–1636. doi: 10.1158/1535-7163.MCT-11-1040. [DOI] [PubMed] [Google Scholar]
- 8.Khanna A. DNA Damage in Cancer Therapeutics: A Boon or a Curse? Cancer Res. 2015;75:2133–2138. doi: 10.1158/0008-5472.CAN-14-3247. [DOI] [PubMed] [Google Scholar]
- 9.Friboulet L, Olaussen KA, Pignon JP, Shepherd FA, Tsao MS, Graziano S, Kratzke R, Douillard JY, Seymour L, Pirker R, Filipits M, André F, Solary E, et al. ERCC1 isoform expression and DNA repair in non-small-cell lung cancer. N Engl J Med. 2013;368:1101–1110. doi: 10.1056/NEJMoa1214271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Graeser M, McCarthy A, Lord CJ, Savage K, Hills M, Salter J, Orr N, Parton M, Smith IE, Reis-Filho JS, Dowsett M, Ashworth A, Turner NC. A marker of homologous recombination predicts pathologic complete response to neoadjuvant chemotherapy in primary breast cancer. Clin Cancer Res. 2010;16:6159–6168. doi: 10.1158/1078-0432.CCR-10-1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Birkbak NJ, Wang ZC, Kim JY, Eklund AC, Li Q, Tian R, Bowman-Colin C, Li Y, Greene-Colozzi A, Iglehart JD, Tung N, Ryan PD, Garber JE, et al. Telomeric allelic imbalance indicates defective DNA repair and sensitivity to DNA-damaging agents. Cancer Discov. 2012;2:366–375. doi: 10.1158/2159-8290.CD-11-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abkevich V, Timms KM, Hennessy BT, Potter J, Carey MS, Meyer LA, Smith-McCune K, Broaddus R, Lu KH, Chen J, Tran TV, Williams D, Iliev D, et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br J Cancer. 2012;107:1776–1782. doi: 10.1038/bjc.2012.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Popova T, Manié E, Rieunier G, Caux-Moncoutier V, Tirapo C, Dubois T, Delattre O, Sigal-Zafrani B, Bollet M, Longy M, Houdayer C, Sastre-Garau X, Vincent-Salomon A, et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012;72:5454–5462. doi: 10.1158/0008-5472.CAN-12-1470. [DOI] [PubMed] [Google Scholar]
- 14.Sedelnikova OA, Bonner WM. GammaH2AX in cancer cells: a potential biomarker for cancer diagnostics, prediction and recurrence. Cell Cycle. 2006;5:2909–2913. doi: 10.4161/cc.5.24.3569. [DOI] [PubMed] [Google Scholar]
- 15.Reinhardt HC, Yaffe MB. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr Opin Cell Biol. 2009;21:245–255. doi: 10.1016/j.ceb.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bartek J, Bartkova J, Lukas J. DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene. 2007 Dec 10;26:7773–7779. doi: 10.1038/sj.onc.1210881. [DOI] [PubMed] [Google Scholar]
- 17.Maugeri-Saccà M, Bartucci M, De Maria R. Checkpoint kinase 1 inhibitors for potentiating systemic anticancer therapy. Cancer Treat Rev. 2013;39:525–533. doi: 10.1016/j.ctrv.2012.10.007. [DOI] [PubMed] [Google Scholar]
- 18.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.von Minckwitz G, Schneeweiss A, Loibl S, Salat C, Denkert C, Rezai M, Blohmer JU, Jackisch C, Paepke S, Gerber B, Zahm DM, Kümmel S, Eidtmann H, et al. Neoadjuvant carboplatin in patients with triple-negative and HER2-positive early breast cancer (GeparSixto; GBG 66): a randomised phase 2 trial. Lancet Oncol. 2014;15:747–756. doi: 10.1016/S1470-2045(14)70160-3. [DOI] [PubMed] [Google Scholar]
- 20.Sikov WM, Berry DA, Perou CM, Singh B, Cirrincione CT, Tolaney SM, Kuzma CS, Pluard TJ, Somlo G, Port ER, Golshan M, Bellon JR, Collyar D, et al. Impact of the addition of carboplatin and/or bevacizumab to neoadjuvant once-per-week paclitaxel followed by dose-dense doxorubicin and cyclophosphamide on pathologic complete response rates in stage II to III triple-negative breast cancer: CALGB 40603 (Alliance) J Clin Oncol. 2015;33:13–21. doi: 10.1200/JCO.2014.57.0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121:2750–2767. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gucalp A, Tolaney S, Isakoff SJ, Ingle JN, Liu MC, Carey LA, Blackwell K, Rugo H, Nabell L, Forero A, Stearns V, Doane AS, Danso M, et al. Phase II trial of bicalutamide in patients with androgen receptor-positive, estrogen receptor-negative metastatic Breast Cancer. Clin Cancer Res. 2013;19:5505–12. doi: 10.1158/1078-0432.CCR-12-3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Traina TA, Miller K, Yardley DA, O'Shaughnessy J, Cortes J, Awada A, Kelly CM, Trudeau ME, Schmid P, Gianni L, García-Estevez L, Nanda R, Ademuyiwa FO, et al. Results from a phase 2 study of enzalutamide (ENZA), an androgen receptor (AR) inhibitor, in advanced AR+ triple-negative breast cancer (TNBC) J Clin Oncol. 2015 (suppl; abstr 1003) [Google Scholar]
- 24.Freitas DP, Teixeira CA, Santos-Silva F, Vasconcelos MH, Almeida GM. Therapy-induced enrichment of putative lung cancer stem-like cells. Int J Cancer. 2014;134:1270–1278. doi: 10.1002/ijc.28478. [DOI] [PubMed] [Google Scholar]
- 25.Abubaker K, Latifi A, Luwor R, Nazaretian S, Zhu H, Quinn MA, Thompson EW, Findlay JK, Ahmed N. Short-term single treatment of chemotherapy results in the enrichment of ovarian cancer stem cell-like cells leading to an increased tumor burden. Mol Cancer. 2013;12:24. doi: 10.1186/1476-4598-12-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, Bruns CJ, Heeschen C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1:313–323. doi: 10.1016/j.stem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 27.Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, Hilsenbeck SG, Pavlick A, Zhang X, Chamness GC, Wong H, Rosen J, Chang JC. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst. 2008;100:672–679. doi: 10.1093/jnci/djn123. [DOI] [PubMed] [Google Scholar]
- 28.Maugeri-Saccà M, Vici P, Di Lauro L, Barba M, Amoreo CA, Gallo E, Mottolese M, De Maria R. Cancer stem cells: are they responsible for treatment failure? Future Oncol. 2014;10:2033–2044. doi: 10.2217/fon.14.126. [DOI] [PubMed] [Google Scholar]
- 29.Bria E, Milella M, Sperduti I, Alessandrini G, Visca P, Corzani F, Giannarelli D, Cerasoli V, Cuppone F, Cecere FL, Marchetti A, Sacco R, Mucilli F, et al. A novel clinical prognostic score incorporating the number of resected lymph-nodes to predict recurrence and survival in non-small-cell lung cancer. Lung Cancer. 2009;66:365–371. doi: 10.1016/j.lungcan.2009.02.024. [DOI] [PubMed] [Google Scholar]