

Abstract
Nucleic acid analysis has enhanced our understanding of biological processes and disease progression, elucidated the association of genetic variants and disease, and led to the design and implementation of new treatment strategies. These diverse applications require analysis of a variety of characteristics of nucleic acid molecules: size or length, detection or quantification of specific sequences, mapping of the general sequence structure, full sequence identification, analysis of epigenetic modifications, and observation of interactions between nucleic acids and other biomolecules. Strategies that can detect rare or transient species, characterize population distributions, and analyze small sample volumes enable the collection of richer data from biosamples. Platforms that integrate micro- and nano- fluidic operations with high sensitivity single molecule detection facilitate manipulation and detection of individual nucleic acid molecules. In this review, we will highlight important milestones and recent advances in single molecule nucleic acid analysis in micro- and nano- fluidic platforms. We focus on assessment modalities for single nucleic acid molecules and highlight the role of micro- and nano- structures and fluidic manipulation. We will also briefly discuss future directions and the current limitations and obstacles impeding even faster progress toward these goals.
Graphical Abstract
1. Introduction
Nucleic acid molecules are information rich. They are involved in many critical biological processes including inheritance, cellular activities such as gene expression and cell differentiation, aging, disease progression, and epidemiology. Because nucleic acids are involved in so many aspects of human health, they hold great potential as broad-based biomarkers. For example, the utility of cell-free nucleic acids as biomarkers has been demonstrated for non-invasive diagnosis of fetal aneuploidy1, non-invasive sequencing of the entire prenatal genome2, and is being explored in diseases such as cancer3, 4.
While much progress has been made in the understanding and categorization of nucleic acids based on their structure and function (e.g. DNA, transfer tRNA, messenger mRNA, micro miRNA, etc.), the cellular environment in which they form, act, and from which we sample, is quite complex. Analysis of these diverse species requires tools that are capable of accurate detection and characterization amidst a complex molecular background. Even more complex samples that contain nucleic acid material derived from multiple tissues, such as blood and urine, can provide a snapshot of systemic health for noninvasive health monitoring and diagnostics. In cancer diagnostics, a blood sample may even prove more descriptive than a tissue biopsy5, 6, since branched evolution can introduce intratumor heterogeneity7, 8. Liquid biopsies, therefore, have the potential to enable patient health assessment that is both more complete and less invasive than standard methods, so long as the analysis techniques are capable of accurately probing these highly complex samples. Single molecule detection strategies enable observations of individual molecules, providing unparalleled detection sensitivity and quantification capability, and enabling analysis of subpopulations that are hidden in bulk measurements. Such high sensitivity detection also facilitates analysis of smaller sample sizes, which can be easier to collect and process, potentially be analyzed faster, and minimize the use of precious or rare samples.
Manipulation and detection of single molecules requires a different tool set than bulk sample analysis. Microfluidic devices can play multiple roles in enhancing this particular form of analysis and detection. First, nucleic acid molecules are small, ranging from nm to μm in characteristic dimension. Detection of single molecules requires decreasing the background noise (signal) below the signal emitted by each molecule by limiting the sources of noise. This can be done by decreasing the size of the detection region to a similarly small area on the order of nm to μm in one or more dimensions. Microfluidic devices can be designed to complement high sensitivity single molecule detectors in multiple ways9. First, the sample volume can be confined to match the dimensions of the detection volume, ensuring that the molecule of interest is detected by the single molecule detector for higher mass detection efficiency. Second, micro-and nano- features can be designed to enhance the signal emitted from each molecule. Alternatively, compartmentalization of signal amplification reactions to small micro-reactors such as droplets or wells can be used to increase the local concentration of signal-emitting molecules. Finally, the precise manipulation of individual nucleic acid molecules requires tools and features on the same size scale (nm to μm). Such features can integrated in microfluidic devices.
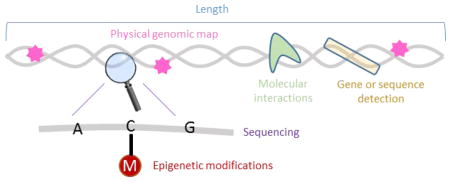
Analysis of single nucleic acid molecules in microfluidic devices is thus poised to both address biological and clinical needs as well as overcome technological barriers that are currently limiting the implementation and use of emerging bioanalytical technologies. This includes, for example, the ability to directly and accurately detect rare molecular species, as well as the ability to perform high throughput analysis to generate large data sets more quickly. Nucleic acid molecules can be characterized according to various attributes, as summarized in Figure 1. From the most global perspective, we can observe the molecule’s size, or length. Length analysis is generally easier, cheaper, and faster than the methods required to analyze other characteristics, and can provide diagnostic and identification information. For example, the size distribution of circulating DNA can serve as a biomarker for cancer detection10, 11. Second, size selection can be an important identification or purification step: for example to isolate miRNA from the total RNA in a cell sample or fetal DNA from a mother’s circulating DNA12, and enzymatic digestion of whole genomes can produce a unique size distribution barcode that can be used to identify the organism13. However, many applications require a deeper analysis of the nucleic acid sequence. Sequence-specific detection to identify particular genes, diseases, or pathogens can be achieved with hybridization-based assays, but requires prior knowledge of the identifying sequence and provides no information on any additional sequences present in the sample. In addition, broad-based detection, in which a single test can be used to diagnose multiple genetic variants, requires highly multiplexed analysis. Optical mapping has been used to generate a physical genomic map of whole nucleic acid molecules or genomes by tabulating locations of specifically marked sequences14–17. Mapping provides important information about structural variants and chromosomal rearrangements that are difficult to detect using other current methods including sequencing. However, mapping cannot be used to identify small structural changes below the optical resolution limit or to obtain sequence information in the untagged regions. Knowledge of the whole sequence in real-time would provide the richest source of information for broad-based analysis. Single molecule sequencing platforms show promise towards this end, but error rates, cost, time, and intense data processing requirements will need to be overcome before this could be used as a routine clinical diagnostic. It is becoming increasingly apparent that many changes in genetic expression are not caused by changes in sequence, but rather epigenetic modifications ranging from DNA methylation to histone modifications18 and miRNA expression. Methods that are sensitive to these changes are important for monitoring their role in conjunction with other nucleic acids analysis techniques. Finally, single molecule studies of nucleic acid interactions with other molecules including nucleic acids, proteins, and small molecules allow researchers to both improve their characterization and understanding of biological interactions and to design effective therapeutic strategies.
Figure 1.

Description of the ways in which nucleic acid molecules are currently characterized in microfluidic single molecule analysis. These include length or size analysis, sequence-specific detection, physical genomic mapping, single molecule sequencing, detection of epigenetic modifications, and characterization of molecular interactions involving nucleic acid molecules.
In this review, we will highlight the important milestones and recent advances in nucleic acid analysis in each of the categories summarized in Fig. 1 specifically through the use of both micro- and nano- fluidics and single molecule detection (SMD) technologies. We will also briefly discuss the current limitations and obstacles to even faster progress and discuss future directions. The development of compartmentalized amplification strategies, such as digital PCR19–21 and enzyme-linked signal amplification22, 23, is a burgeoning field. However, amplification methods do not involve direct observation of single nucleic acid molecules, and thus will not be covered in this review. Readers interested in these topics are referred to other excellent reviews24–27.
2. Assessment Modalities
2.1. Length/Size
The size of a nucleic acid molecule is one of the simplest parameters to measure and is useful for many purposes. First, the size distribution of DNA from a complex sample, such as blood, can be used to assess the origin (maternal or fetal12, cancerous or normal tissue10, 11, 28, 29) or disease status. Second, because gel electrophoresis size separations are relatively cheap, easy and routine benchtop techniques, many assays have been designed to link size analysis with other characteristics, such as restriction enzymatic digestion of genomic DNA for pathogen identification30 and forensic DNA fingerprinting31, and multiplexed ligation-dependent probe amplification (MLPA) for sequence-specific detection and quantification32. Optical mapping (Section 2.3), introduced in 1993, involves optically measuring distances along the length of a single nucleic acid molecule14. Conventional benchtop gel electrophoresis is not capable of handling small sample volumes and lacks the sensitivity for single molecule analysis that is important for increasing the sensitivity, speed, and resolution of population distribution length measurements. Microfluidic technologies enable this single molecule length analysis.
The main strategies for performing single molecule length analysis are outlined in Figure 2. First, the signal accompanying each single molecule detection event can be evaluated to determine its size: either measuring the end-to-end distance (or contour length) of a stretched nucleic acid molecule, or through integration of the total signal intensity generated by each molecule. Alternatively, molecules can be separated by size in solution. These separation-based approaches can be either continuous, where a molecule’s two-dimensional trajectory is dependent on its size, or one-dimensional, where velocity is used to separate molecules by size. Because dsDNA molecules can be ratiometrically labeled with fluorescent intercalating dyes and detected with high signal-to-noise ratio, most single molecule detection methods rely on fluorescence-based optical instruments33. The optical techniques most commonly cited in this review include confocal fluorescence spectroscopy (CFS), fluorescent optical microscopy with high sensitivity intensified iCCD or electron multiplying emCCD cameras, and total internal reflection fluorescence (TIRF). However, we will also discuss alternative electrical single molecule detection methods that are emerging. In this section, we will briefly discuss the background and recent advances for each of the sizing methods, but readers seeking a more comprehensive examination are directed to an excellent recent review of this area34.
Figure 2.

Strategies that have been developed to measure the length of individual nucleic acids can be grouped into four main categories. Direct length measurements can be obtained by (a) stretching the molecules and measuring their end-to-end distance (picture reprinted from ref. 43). (b) Molecules stained ratiometrically with fluorescent dyes can also be sized by their fluorescent burst intensity. Burst sizing with CICS detection is shown to be linear over a size range of 0.6 kbp to 27 kbp (picture reprinted with permission from ref. 4). Alternatively, nucleic acid molecules can be physically separated in solution by their size. One-dimensional separations use microfluidic features to couple a molecule’s velocity or mobility with its size. (c) Free solution hydrodynamic separation coupled to single molecule CICS detection (SML-FSHS) is able to separate a 50 bp dsDNA ladder and accurately identify only 9 molecules sized 1350 bp (picture adapted with permission from ref. 58). Two-dimensional separations relate molecule size and trajectory to enable continuous flow separations. Pulsed electric fields across a micropillar array are used to continuously separate DNA fragments sized 20 kbp to 166 kbp (picture reprinted with permission from ref. 66).
2.1.1. Stretching
In aqueous conditions, DNA molecules are rather flexible and tend to adopt three-dimensional random coil conformations in free solution. Visualization and measurement of full-length molecules requires forcing their extension along one dimension. Microfluidic forces and structures can be used to controllably stretch DNA for analysis in extended conformations. The most commonly-employed methods for stretching nucleic acid samples for sizing and/or optical mapping are shown in Figure 3. One way to stretch DNA is to immobilize one or both ends of the DNA molecule, for example to a surface, and to apply a force to separate the ends. The stretching force can be applied by surface tension at a meniscus, a process called “molecular combing”35 or fluid flow36 (Fig. 3a). An improved deposition efficiency for molecular combing was demonstrated by utilizing recirculating flow within moving droplets and an optimized polymer surface to enable analysis of picograms of DNA within a droplet37. Another approach to analyze dilute DNA solutions uses evaporation on the superhydrophobic surface of a micropillar array to deposit stretched DNA filaments between the pillars38. Techniques have also been developed to stretch molecules without attachment. Shear forces can also be utilized to stretch DNA in fluid flow in microchannel funnels (Fig. 3b-i)39, at stagnation points either hydrodynamically (Fig. 3b-ii)40 or electrokinetically41, or in oscillatory flow42. Alternatively, DNA molecules can be confined in one (nanoslit) or two dimensions (nanochannel) to force their elongation (Fig 3c).
Figure 3.

Methods to stretch DNA for length measurements and optical mapping. In (a), DNA molecules are stretched by capillary flow through a microchannel and adsorb to the bottom glass surface, leaving a highly aligned array of stretched and immobilized molecules (picture reprinted from ref. 16). In (b), shear flows stretch nucleic acid chains in free solutions through either (i) accelerating flow in a microfunnel or (ii) at a stagnation point (pictures reproduced from refs. 39 and 40, respectively). In (c) long nucleic acid molecules are driven electrophoretically into 45nm × 45 nm nanochannels, dimensions smaller than the persistence length of dsDNA. This causes the DNA to elongate along the length of the channel (figure reprinted by permission from Macmillan Publishers Ltd: Nature Biotechnology (ref. 45), copyright 2012). Randomly coiled DNA must overcome a significant entropy barrier to enter the small nanofluidic region (i). Introducing a gradient region with progressively smaller microstructures before the nanochannel entrance (ii) helps to unravel the long molecules into and thread the nanochannels.
DC electric fields can also be used to electrokinetically drive DNA strands into the nanochannels. Then, the field is turned off to create a static array of stretched DNA for imaging. Fig. 2a shows DNA molecules confined in 100 nm by 200 nm nanochannels and imaged with an optical microscope and an intensified CCD camera43. Confocal fluorescence spectroscopy (CFS) can also be used to detect stained DNA molecules as they electrophorese through a nanochannel44. Here, the fluorescent signal obtained from each molecule relates speed and conformation in addition to length44. The main challenges with nanochannel confinement techniques include achieving full stretch (stretching to the full contour length) and preventing the formation of any folds or knots in the elongated molecule within the channel. Folding and looped conformations can be avoided by employing very small (45 nm) nanochannel cross sections45. However, the fabrication of long nanochannels with very small cross sections requires sophisticated nanofabrication techniques46 and result in larger entropic barriers to both entry and transport47 that necessitate a gradient of microstructures to unravel the randomly coiled DNA molecules before entering the small constriction (see Fig. 3c). Larger, more easily fabricated PDMS nanochannels (> 100 nm) have also successfully stretched DNA molecules to high degrees, but this technique requires low ionic strength environments48. Sizing stretched molecules is particularly important for DNA mapping, so the most advanced methods to achieve stretching uniformity and throughput will be discussed further in Section 2.3.
An alternative to measuring the equilibrium length of stretched molecules in nanochannels longer than the molecule is to analyze the time it takes for a molecule to traverse a short nanochannel or pore49. For example, fragmented genomic DNA can be sized by measuring the translocation time through a nanochannel shorter than the contour length in pulsed electrical fields50. A benefit of this method is that molecules can be detected electronically, obviating the need for molecular labeling and expensive optical equipment that limits portability51. However, improvements in sizing resolution would be necessary, for example by performing multiple measurements on each molecule for statistical averaging51.
2.1.2. Fluorescent Burst Sizing
Single molecule fluorescence intensity can also be used to size nucleic acid fragments. While this can be done for stretched DNA34, it can also be performed on randomly coiled DNA in solution. When using this method sizing resolution is limited by two factors: statistical variation in the ratio of dyes per nucleotide between DNA molecules, and detector sensitivity and uniformity over the excitation volume. The staining ratio inherently limits sizing resolution and makes the method better suited to sizing of larger DNA molecules (kilobasepairs), although sizing of 125 bp fragments has been demonstrated52. The other factor affecting burst size variation is the uniformity of excitation and collection throughout the detection volume. For example, typical confocal spectroscopy systems have very small diffraction-limited observation volumes, smaller than the size of a microfluidic channel, which creates large variations in excitation and collection efficiency across the channel. One way to overcome this is to focus the molecular trajectory through the smaller observation volume using electrokinetic radial migration53 or sheath flow54. Another is to use a cylindrical lens in the optical path to expand the laser beam in one dimension (Cylindrical Illumination Confocal Spectroscopy – CICS) to produce uniform illumination (< 10% variation) across a 5 μm wide channel55. This method demonstrated linear sizing for fragment sizes from 564 bp to 27.5 kbp (Fig. 2b) and was used to size circulating DNA from serum without amplification4.
2.1.3. One-dimensional Separations
Nucleic acid molecules can also be separated by size by differences in mobility prior to detection. The parabolic flow profile of pressure-driven laminar flow in a microchannel has been exploited to separate molecules in solution depending on their favored position in the channel. For channel radii on the order of the molecules to be separated, the hydrodynamic radius of the molecule prevents sampling of the slowest flow near the walls, resulting in an average velocity that is size dependent – where the largest molecules travel faster than smaller molecules, and the solute travels the slowest. When the separation method is combined with CICS detection, individual molecules can be detected and counted with high mass detection efficiency, creating separation chromatograms in terms of single molecule counts56. The sensitivity to detect and identify as few as 9 molecules of a single fragment size (Fig. 2c) and the resolution to distinguish double-stranded and single-stranded DNA fragments of the same length has been demonstrated57–58.
Other separation methods utilize arrays of micro/nanofabricated obstacles to separate DNA by size59, often using electrokinetic as opposed to hydrodynamic forces to guide the DNA into and through the structured array. Microfabrication technologies allow researchers to tightly control the size, spacing, and uniformity of micro and nano structures such as posts, as compared with gels, which have a wide distribution of pore sizes. The electrophoretic mobility of a DNA fragment through a post array depends on the number of collisions and the “hold-up” time spent interacting with the obstacles60, 61. However, one of the limitations of this technology is the size of the array that can be fabricated, limiting the resolution and number of species that can be separated in a single run34. Recently, nanofences have been proposed and demonstrated to increase resolution for electrophoresis of long DNA fragments62. Alternatively, microchannels with a series of nanoslit constrictions have also been shown to form entropic traps to electrophoretically separate kb-sized DNA molecules in DC fields an order of magnitude faster than pulsed-field gel electrophoresis63, 64.
2.1.4. Two-dimensional Separations
Structured arrays can also be used to continuously separate DNA molecules according to size. Variations on this theme include pulsed field electrophoresis through arrays of micropillers65 or nanoparticles (Fig. 2d)66, tilted Brownian ratcheting67, 68, and deterministic lateral displacement69. In general, these methods are limited to large DNA molecules (10’s to 100’s of kb) with limited demonstrated integration with single molecule detection. However, single molecule analysis is useful for mechanistic and optimization studies. For example, analyzing the degree of DNA stretching under various solution conditions was used to improve throughput of deterministic lateral displacement separations with the addition of PEG69. Further improvements in DNA separation and sizing technologies on chip could allow for more efficient measurement and sorting prior to other downstream analyses.
2.2. Sequence-specific analyses
Sequence-specific detection has many applications, ranging from early detection of cancer to identification of pathogens. Recently developed technologies do not necessarily require amplification of nucleic acids for sequence-specific detection70. Bypassing amplification offers several advantages. The amplification process, particularly multiplexed PCR, can produce artifacts71. Second, PCR inhibitors often present in biological samples can hinder amplification72, 73. Single molecule digital PCR has exhibited increased tolerance to interfering substances, conferring advantages over conventional qPCR 74–76. In addition, many clinical applications require rapid processing times. Sensitive detection methods, which require less sample preparation and only a single hybridization step, can generate results more quickly and are thus highly desirable.
One well-documented approach for single molecule nucleic acid detection involves confocal fluorescence spectroscopy77. Here, signal is generated based on the direct hybridization of [unamplified] DNA with fluorescent molecular probes. Examples of probes used with confocal fluorescence spectroscopy are generally based on fluorescence resonance energy transfer (FRET) such as molecular beacons78 and quantum dot nanosensors79. Molecular beacons are hairpin-structured oligonucleotides conjugated with a fluorophore and quencher. Once the molecular beacon hybridizes to the target, the fluorophore and quencher separate, causing a dramatic increase in the quantum yield of the fluorophore. In one study, an on-chip single-molecule assay with molecular beacons was performed53. On-chip electrophoresis and dielectrophoresis were performed to achieve confinement of DNA molecules to a submicron laser focused detection region. Fluorescent signal from as low as 0.7 pM of target was able to be distinguished from the background53. In another study, light sheet-based confocal fluorescence spectroscopy80, coupled with hydrodynamic separation, was applied to maximize the mass detection efficiency and to enhance the sensitivity by separating unbound probes from target-probe hybrids56. Furthermore, droplet confinement through a retractable microfluidic constriction extended droplet duration through the illumination volume, providing the spatial and temporal resolution necessary to detect single biomolecules in a sub-nanoliter droplet81. The tunable nature of the droplet platform that allows attainment of single fluorophore sensitivity was illustrated using a Cy5 molecular beacon complementary to a sequence on 16S rRNA from E. coli82. This platform was extended to a droplet microfluidic chip capable of amplification-free detection of single pathogenic cells (Fig. 4b) 83. Here, a PNA beacon was used that was complementary to a genetic locus within the target bacterial genome. In its natural state, the beacon folds on itself, such that the fluorophore is quenched. PNAs are base sequences attached to an uncharged backbone, which confers stronger binding between complementary PNA/DNA sequences, compared to DNA counterparts. A dilute mixture of pathogenic cells and PNA beacon was encapsulated into picoliter-sized droplets, which were then incubated at elevated temperatures to facilitate cell lysis and beacon-target hybridization. PNA beacons encapsulated in droplets containing cells were hybridized with complementary 16S rRNA targets83. Some drawbacks of molecular beacons include difficulties in removal of excess free fluorophores after synthesis and thermodynamically-driven open and closed states, even at temperatures below the Tm. Therefore at low target concentrations, the number of open hairpins may be comparable to the number of targets, limiting the assay sensitivity.
Figure 4.

Top panel. Single-molecule method for the quantitation of microRNA gene expression 88. (a) miRNAs were hybridized in solution to spectrally distinguishable fluorescent LNA-DNA probes. Complementary DNA probes bearing the fluorescence quencher molecules were hybridized to the remaining unbound fluorescent probes to minimize coincident events that could be created by free probes simulteaneously enter ing the laser interrogation spots. (b) The two fluorophore-labeled miRNAs are flowed by vacuum pressure through a capillary containing a series of femtoliter laser focal volumes. (c) Fluorescence emission was recorded as spikes in signal intensity overtime. Arrows highlight the coincident peaks. (d) The Direct miRNA assay is sensitive to to 500 fM miRNA. A synthetic mir-9 RNA oligonucleotide, serially diluted from 300 pM to 500 fM, was hybridized in the presence of a complex RNA background to its complementary LNA-DNA probes. After a 1-h quenching reaction, the fivefold-diluted hybridization reactions were analyzed on the single-molecule detection platform. Reprinted by permission from Macmillan Publishers Ltd: [Nature Methods] (Neely, Lori A., et al. “A single-molecule method for the quantitation of microRNA gene expression.” Nature Methods 3.1 (2006): 41–46.), copyright (2006) Bottom panel. Droplet microfluidics for amplification-free genetic detection of single cells 83. (a) Schematic of the droplet assay platform for single cell detection. A statistically dilute mixture of pathogenic cells and PNA beacons is encapsulated into picoliter sized droplets, which then are incubated at elevated temperatures to facilitate cell lysis and beacon-target hybridization. PNA beacons encapsulated in a droplet containing a cell start fluorescing after hybridization with complementary 16S rRNA targets released from the cell. The specific cell of interest is detected and quantified by screening the fluorescent droplets using confocal fluorescence spectroscopy (CFS). (b) Fluorescence data collected from droplets generated a low concentration E. coli sample. The green trace shows fluorescence from the indicator dye while the red trace indiciates the fluorescence from the PNA beacon encapsulated within the droplets. The E. coli concentration for this sample was estimated at 1 CFU per 20 droplets. In this case, we expect the majority of droplets to have no E. coli cells with a few droplets having E. coli cells. The inset shows a zoomed in view of a small section of the fluorescence data trace. Reproduced from Ref. 74 with permission from The Royal Society of Chemistry.
The femtoliter detection volume of confocal fluorescence spectroscopy permits coincident single-molecule fluorescence signals that occur when two or more fluorescently-labeled molecules simultaneously pass through the detection volume. This technique is referred to as single-molecule fluorescence burst coincidence detection84–86. By using two differently labeled probes complementary to the same target, coincidence events may be detected. To minimize the probability of two unbound probes simultaneously passing through the detection volume, low concentrations of probes must be used. Coincidence detection has been performed with total internal reflection fluorescence (TIRF) microscopy as well. In one paper, a bioassay was demonstrated for sequence-specific detection of DNA for target concentrations in the pM to fM range. The authors demonstrated quantification of a short DNA sequence via coincidence detection in 30 seconds87. In another approach, a quantitative miRNA assay was demonstrated in which two spectrally distinguishable fluorescent locked nucleic acid (LNA)-DNA oligonucleotide probes were hybridized to the miRNA of interest. Probe/miRNA hybrids were then directly counted on a single-molecule detection instrument (Fig. 4a) 88.
Quantum dots may also be used for single-molecule DNA detection79, 89, 90. Quantum dots can undergo FRET phenomena and be used to investigate interactions between biomolecules91, 92. Good spectral overlap is required for donor and acceptor pairs. QDs boast advantageous photophysical properties, such as size-tunable photoluminescence spectra, broad absorption and narrow emission wavelengths and high quantum yields79, 93. A novel nanosensor-based oligonucleotide ligation assay was used to demonstrate successfully detection of a KRAS point mutation, typical of some ovarian tumors79. The authors of this study purported detection of target concentrations as low as 4.8 fM.
A relatively recent technology, the NanoString nCounter gene expression system, can capture and count specific nucleic acid molecules in a heterogeneous mixture94. Although the NanoString can measure single molecules, the recommended minimum sample input is ~100 ng. The nCounter relies upon development of a probe library with two sequence-specific probes for each gene of interest. The first probe is a capture probe, and contains a 35- to 50-base sequence complementary to a particular target mRNA plus a short common sequence coupled to an affinity tag such as biotin. The second probe is the reporter probe, and contains a second 35- to 50-base sequence complementary to the target mRNA, which is coupled to a color-coded tag that provides the detection signal. The tag consists of a single-stranded DNA molecule, the backbone, annealed to a series of complementary in vitro transcribed RNA segments each labeled with a specific fluorophore. The linear order of these differently colored RNA segments creates a unique code for each gene of interest. The NanoString has been used for a variety of applications, including analysis of formalin-fixed paraffin-embedded tissue obtain in clinical trials 95.
2.3. Physical Genomic Mapping
Although sequence-specific detection strategies can be highly sensitive and specific to the presence of relatively short nucleic acid sequences, detecting large kb to Mb structural changes, such as duplications or inversions is difficult with the methods described in Section 2.2. Optical mapping is a form of physical mapping that can be used to generate a broader picture of the sequence structure of a single nucleic acid molecule or whole genome14. In the simplest terms, a physical map relates the distance, in terms of length or basepairs, between specific sequences in a genome. Because the number and location of these targeted sequences is dependent upon the underlying sequence, a physical map can be used to identify organisms, distinguish bacterial strains, and diagnose diseases caused by structural mutations. To generate an optical map, a single DNA molecule is stretched in one dimension to allow measurement of the physical distance between specific sequence sites. This technique maintains relative locational information along the full length of the molecule to enable assembly of a map that can span the full genome. Conversion of the measured distance to basepairs or kilobasepairs generates a physical map. This system has evolved to construct physical maps of entire genomes de novo, or detect and characterize structural variants, through the analysis of large data sets of individual molecules. These physical mapping scaffolds can serve as a useful companion tool to align next-generation and single molecule sequencing reads or to identify sequence misassemblies96. Optical mapping can also be used in stand-alone applications for pathogen identification97 and strain-typing98 as well as detection of large-scale structural and chromosomal rearrangements that are difficult to detect using other methods99–103. Various techniques can be employed to stretch DNA (see Section 2.1.1), but the methods that are best suited for DNA mapping are ones that are simple to perform and automate, high throughput, and achieve highly repeatability. Uniform stretch efficiency is particularly important for aligning mapped molecules at the highest possible basepair resolution. The current methods that have been utilized for DNA mapping are shown in Fig. 3.
There are also various methods for producing an optical DNA map or barcode (Fig. 5). Denaturation mapping, shown in Figure 5a, utilizes differences in local melting temperatures along the length of a long dsDNA molecule to probe generalities about the underlying structure. AT-rich regions tend to have lower double helix stability than GC-rich regions, causing AT-rich regions to begin melting at lower temperatures. A dsDNA molecule stained with intercalating dye that is treated with chemical and heat denaturants will partially melt in accordance with the underlying sequence. The intercalating dye diffuses away from the melted regions, leaving a fluorescent barcode along the length of the DNA molecule104. This technique can be performed with relatively little sample preparation, but cannot identify small differences (basepairs) between similar sequences. In addition, DNA melting is a complicated process that strongly depends on temperature and chemical conditions, which can be difficult to precisely control or account for, and can consequently affect quantitative agreement between experiment and theory104.
Figure 5.

Three main techniques have been utilized to generate optical mapping barcodes. (a) Denaturation mapping exploits differences in melting temperature along a single DNA molecule to create a fluorescent barcode when heated to elevated temperatures (figure reproduced from ref. 104). (b) Ordered restriction maps are generated by exposing stretched DNA molecules to sequence-specific restriction enzymes. Double stranded breaks are seen as small holes in the stretched molecule (figure reproduced with permission from ref. 210 Copyright © 2013, American Society for Microbiology). (c) Fluorescent tags can be generated in multiple ways. Fluorescently labeled probes can be used to hybridize to specific sequences. Alternatively, nicking enzymes can be used to generate single stranded breaks in a sequence-specific manner. Then, DNA polymerase can incorporate fluorescent nucleotides at the nicking site (picture reprinted by permission from Macmillan Publishers Ltd: Nature Biotechnology (ref. 45), copyright 2012).
Alternatively, restriction endonucleases can be employed to cut dsDNA fragments at a specific recognition site to generate a restriction map (Fig. 5b). DNA profiling techniques that employ enzymatic digestion prior to electrophoretic separation sizing have been used for decades for bacterial strain typing, forensic identification, and paternity testing105–107. In contrast, an ordered optical restriction map is obtained by stretching locally isolated DNA molecules prior to digestion so that the relative location of each restriction site is retained along the length of each molecule. This can be performed in a nanochannel108, but requires careful control of buffer conditions to prevent digestion from occurring before the enzyme-bound DNA molecule is stretched within the nanochannel. Additionally, long observations of single molecules can result in photo-induced cutting109, so care must be taken to ensure that cuts are enzymatic and not a result of photodamage. Alternatively, stretched DNA molecules adsorbed to a surface can also be used to generate restriction maps. Microchannel-based adaptations are now commercially available and have been used to generate dense and highly aligned genetic maps16. Single molecule restriction mapping has been used for pathogen identification and monitoring applications, including strain typing of bacterial outbreaks110 and monitoring the genomic stability and evolution of laboratory bacterial strains including Staphyloccus aureus111 and Escherichia coli112. It has also been used to generate reference maps for large eukaryotic genomes such as mouse113, maize114, and rice115. The creation of reference restriction maps has enabled the detection of numerous structural variants within the human genome100–103 and cancer116, as well as facilitated de novo genome sequence assembly and validation with companion data from pyrosequencing117, sequencing-by-synthesis118, 119, and SMRT sequencing (see Section 2.4)120, 121 platforms.
In another approach, a microchannel funnel was used to generate elongational flow that stretches DNA molecules while traversing consecutive confocal detection volumes (Fig. 3b-i)39. Fluorescently labeled PNA probes were designed to hybridize to 7–8nt long sequences within a dsDNA molecule stained with intercalating dye39. This method, termed GSS, Genome Sequence Scanning (previously DLA, Direct Linear Analysis) generates a fluorescent barcode along a single DNA molecule that is read as it flows through the multicolor confocal detection region39. Improved consistency in both intercalation dye labeling and DNA stretching efficiency was achieved by performing DNA intercalation staining on the device122. The device was further optimized to increase throughput and stretching consistency by redesigning the funnel geometry to maintain constant shear through the detection region123. The platform has been used for bacterial genotyping124 and strain typing125. The same platform was also used to detect non-nucleic acid antigens along a DNA carrier molecule for uses in pathogen identification for food safety testing and outbreak investigations126. Target antigens were detected in the presence of 100-fold excess background bacterial mixture127 on a device capable of testing air samples.
Elongational flows can also be used to stretch DNA at a stagnation point (Fig 3b-ii)128, 129. This has been used for restriction mapping by binding the restriction enzyme prior to mapping and introducing the cofactor Mg++ during stretching, allowing the location of cutting and kinetics to be studied simultaneously130. This method has also been combined with denaturation mapping131 for identification and analysis of DNA molecules of over 1Mb is length. Denaturation mapping was used to identify its chromosomal origin as well as any insertions, deletions, inversions, ranging in size from a few kb to Mb. Although this method is not easily parallelizable, by limiting the throughput (only 1 molecule can be investigated at a time), the authors demonstrated the ability to capture the molecule after mapping for further PCR and sequencing analysis. The authors have also demonstrated important advances in automation132 including image filtering and analysis as well as automated individual DNA molecule selection, manipulation, and stretching of long DNA molecules for mapping. This is important because all mapping methods suffer in throughput in the presence of smaller DNA fragments. Either improved sample preparation methods that reduce the number of small fragments introduced to the mapping device, or devices which incorporate some form of size selection or size filtering prior to analysis will be important for alleviating these issues.
BioNano Genomics’ Irys Technology17 also utilizes fluorescent tags to generate an optical map (Fig. 5c), but uses different approaches to generate the tags, stretch the molecules, and image the mapped DNA. A double-stranded molecule is nicked in sequence-specific locations with nicking enzyme Nb.BbvCI, producing single stranded breaks to serve as labeling sites. In nick labeling, fluorescent nucleotides are incorporated into the nick sites by a DNA polymerase molecule17, 133. In flap labeling, the single stranded flap generated by the extension reaction is hybridized with a fluorescently tagged DNA probe134. By using a gradient of nanostructures before the 45 × 45 nm channels, DNA is threaded into the channels to avoid any folds in the DNA that might affect analysis. This approach has been used in haplotyping and scaffolding for de novo genome assembly45. The authors have also demonstrated that the use of super-resolution imaging techniques can improve resolution between fluorescent nicking sites to 100 bp135. They demonstrated the use of mapping as scaffolding to complete de novo sequence assembly of new genomes136, to finish particularly difficult and repetitive regions of the human genome137, to detect structural variations (insertions, deletions, and inversions)138, 139 in an individual human genome140, and for strain typing of bacteriophages λ and T7 from a background phage library by covalently labeling at methyltransferase recognition sites141. Techniques for mapping epigenetic markers will be further discussed in Section 2.5.
2.4. Single Molecule Sequencing
The full de novo generated sequence of a nucleic acid molecule provides more information than physical genomic mapping or sequence-specific detection because it does not require prior knowledge of the sequence. Second-generation sequencing methods are helping to lower the price and increase the speed of sequencing analysis. However, most utilize PCR to clone copies for analysis, which can introduce amplification bias in the sequenced reads. Additionally, sequencing information is attained from the simultaneous read of a colony of synchronized molecules, which increases the signal generated from each population, but limits the read length due to desynchronization142. Short read lengths require in depth data analysis to assemble the genome and deep sequencing (many reads of the same sequence). Repetitive sections of the genome longer than the read lengths are difficult to piece together, creating gaps in the assembled genome and making certain large-scale rearrangements difficult to detect. Sequencing of single molecules can have longer read lengths and can avoid the use of PCR altogether.
An optical method has been developed by Pacific Biosciences using zero-mode wave guides to increase signal detection sensitivity from single fluorophores even at very high concentrations of background fluorophores143. This method, termed Single Molecule Real Time (SMRT) sequencing144, uses a high density of zero-mode waveguides patterned into a surface thereby allowing for efficient sequence data collection, as shown in Figure 6a. A single polymerase enzyme attached to a DNA molecule template occupies the bottom of a waveguide. Fluorescently-tagged nucleotides can diffuse into the waveguide and generate a fluorescent signal upon incorporation. Nucleotides are labeled with color fluorophores to allow discrimination between the bases. Single molecule sequencing has specific advantages for particular applications including bacterial strain evolution analysis145, 146, de novo gene assembly without a reference genome147, 148, finishing particularly difficult gaps in genome assembly149, accurate identification of structural150 and mRNA splicing151 variants, and analysis of methylation patterns152–154. Although error rates for individual reads are relatively high compared to other methods, the error sources are random and therefore require only sufficient read redundancy (recommended depth > 8 reads) to overcome stochastic errors. Other sequencing methods with systematic errors, on the other hand, require the use of additional complementary techniques to resolve the errors155. To correct incorrect base calls from a single sequence read, the double-stranded template can be converted into a circular template to generate multiple reads of both the sense and anti-sense sequences156, 157. However, this error-checking method is most useful for shorter sample fragments. Very long circularized molecules may only be ready once since the maximum continuous read length is limited by other factors in the system. Therefore, the user must choose whether high accuracy single reads or very long read lengths are most important when choosing the fragment insert size.
Figure 6.

Two current technologies for sequencing single nucleic acid molecules are single molecule real-time (SMRT) sequencing commercialized by Pacific Biosciences and nanopore sequencing commercialized by Oxford Nanopore Technologies. In SMRT sequencing (a), the sequence is read by polymerizing a complementary sequence using fluorescently-tagged nucleotides. When a polymerase enzyme incorporates a new nucleotide, a fluorescent signal is released and detected using zero-mode waveguide technology. The color of the fluorescent signal relays the nucleotide identity. In nanopore sequencing (b), a voltage applied across a nanopore in an insulating membrane generates a current flow of ions through the nanopore. When a single stranded nucleic acid molecule is threaded through a nanopore, the nanopore is partially blocked and the measured current flow drops. An additional protein is used to ratchet the nucleic acid molecule through the pore a single base at a time so that the current trace can distinguish individual molecules sequentially along the length of a single nucleic acid molecule. Figure (a) was reproduced from ref 144. Reprinted with permission from AAAS. Figure (b) was redrawn from images on Oxford Nanopore’s website 55.
SMRT sequencing does require fluorescently labeled dNTPs and sequencing-by-synthesis, which relies on the activity of a single polymerase enzyme for each molecule read. An alternative label-free approach detects changes in current flow through a nanopore when a nucleic acid molecule partially blocks the nanopore158. The principle is illustrated in Figure 6b. A voltage across a nanopore drives charged ions to traverse through the pore to carry the electrical current. However, if a large molecule blocks some of the pore, there is increased resistance to ion flow and therefore a smaller current is measured. As a nucleic acid molecule traverses the pore, its primary sequence can be ascertained by measuring the characteristic current signal that is ostensibly determined by the nucleotide sequence. Emerging commercial technologies include the products from Oxford Nanopore, who uses a biological nanopore in addition to a helicase protein to unwind dsDNA and ratchet a single strand through the nanopore159,160.
The realization of single nucleotide discrimination in nanopores has been difficult for numerous reasons. First, single-stranded nucleic acid molecules have very small radii (<1 nm)161 and are charged themselves, causing them to move very quickly through the nanopores. Controlling the speed of translation through the pore is one important aspect to improving base identification162. Second, the pores are fairly deep compared to the length of each nucleotide along a DNA molecule. Therefore, reading the resistance/current change from a single nucleotide, not a series of nucleotides, is also difficult. Modifications to the pore structure, such as multiple “reading heads”163 or an engineered mutation at a reading head164 have already been demonstrated and implemented in Oxford Nanopore’s technology. Sequencing via exonuclease activity165 could help to address both issues. Sequencing speed is currently 30 bases per second for each nanopore, but faster modes will be available by choosing different temperatures, buffers, or enzymes166.
Currently there are three products based on nanopore sequencing in development, but only the small, portable minION is presently available, with the larger prometheION access program starting soon. Since the minION was released in the early access program, the long read length, extremely low cost (only $1000), and portability have attracted researchers pursuing diverse projects. These include analysis of viral diversity and evolution167, bacterial strain typing and identification of antibiotic/antimicrobial resistance168–171, identification of alternatively spliced mRNA isoforms172, de novo bacterial genome assembly without a companion scaffolding technology173, rapid viral identification from clinical samples 174 and noninvasive prenatal testing175. RNA sequencing is currently performed after first generating complementary cDNA through reverse transcription, but new workflows that will allow direct sequencing of mRNA as well as miRNA are in development166. Since the minION’s release in late 2013, updates in chemistry and data processing algorithms have already improved error rates176, though performance still lags behind the SMRT sequencing platform. The long read length offered by both platforms will benefit from the development of new data algorithms to help overcome the error rates177, 178. At present, high error rates, time consuming sample library preparation 179, and resource intensive data acquisition and analysis have limited the use of single molecule sequencing to mostly research applications, rather than personalized medicine.
2.5. Epigenetic Modifications
Genetic mutations may result in changes to the sequence of a DNA molecule. DNA can undergo additional modifications that are not detected as changes in the sequence, but can still affect gene expression. These types of non-sequence alterations are called epigenetic modifications. Epigenetic changes encompass covalent changes to DNA, chromatin proteins and remodeling of nucleosome positioning on DNA. Environmental agents can catalyze epigenetic changes. These modifications influence differentiation during embryonic development and may affect transcription. Epigenetic changes such as DNA methylation have also been implicated in diseases such as cancer and mental retardation18, 180–182. Several comprehensive review articles on micro- and nano-scale devices for studying epigenetic modifications of chromosomes have recently been published183, 184.
Different sequencing approaches have been employed to assess epigenetic modifications. One approach is bisulfite conversion followed by sequencing (BSC-seq)185. This method is used to study DNA cytosine methylation and operates by treatment of purified DNA with a bisulphite salt186. Bisulphite treatment converts unmethylated cytosines to uracil, thereby translating an epigenetic alteration to a change in DNA sequence. Another approach, single-molecule, real-time (SMRT) sequencing, previously discussed in Section 2.4, exploits differences in polymerase kinetics to directly detect base modifications without bisulfite conversion152. In this approach, DNA polymerases catalyze the incorporation of fluorescently labeled nucleotides into complementary nucleic acid strands. The arrival times and durations of the fluorescence pulses enable detection of epigenetic modifications of nucleotides, including N6-methyladenine, 5-methylcytosine and 5-hydroxymethylcytosine152. Finally, chromatin immunoprecipitation followed by sequencing (ChIP-seq)187 incorporates the use of antibodies to capture chromatin fragments bearing specific modifications. Following release, the DNA from the captured chromatin fragments can be used to identify genetic sequences associated with the selected modification183. This technique requires alignment to a known reference sequence to map the location of the histone modification within the genome.
Modified bases can be fluorescently tagged and located along the length of a single DNA molecule using optical mapping techniques such as those discussed in Section 2.3. Stretching and linearization of DNA is key for continuous mapping between spatial location and genomic location188. In one study, capillary assembly was demonstrated, in which a liquid droplet was dragged over a microstructured piece of silicone rubber (Fig. 7a,b). The topography induced molecular elongation in ordered arrays containing more than 250000 immobilized DNA molecules. The methylation state of the DNA was then detected and mapped by binding fluorescently labeled methyl-CpG binding domain peptides to the elongated dsDNA molecules and imaging their distribution189.
Figure 7.

Epigenetic modifications. Top panel. Ordered arrays of native chromatin molecules for high-resolution imaging and analysis 189. (a) Schematic representation of the experimental methodology to generate stretched and oriented chromatin arrays on a solid support. (1,2) Assembly and stretching process by capillarity. (1) Microstructured PDMS stamp is placed on a translation stage controlled in speed. A droplet of extracted chromatin in solution is deposited on the stamp. (2) Liquid meniscus of the solution containing the extracted chromatin is dragged over the microstructured PDMS stamp at controlled speed. The evaporation phenomenon is represented in red. The molecules are physically trapped and stretched as the meniscus is displaced across the substrate. (3) Transfer printing of the obtained chromatin array on an APTES-coated coverslip by contacting the PDMS stamp with the APTES-coated surface for 2 min and then peeling it away. (b) Fluorescence images of an array of stretched and oriented M091 chromatin molecules transferred onto an APTES-coated coverslip (excitation at 475 nm). The molecules are YOYO-1 stained. (B) A 1.5× zoom of image (A). Reprinted (adapted) with permission from (Cerf, Aline, Harvey C. Tian, and Harold G. Craighead. “Ordered arrays of native chromatin molecules for high-resolution imaging and analysis.” ACS nano 6.9 (2012): 7928–7934.). Copyright (2012) American Chemical Society. Bottom panel. SCAN workflow. (A) Native chromatin bearing epigenetic marks is mixed with fluorophore (e.g., AlexaFluor488) labeled antibody specific to a given mark. After binding, the chromatin is labeled with an intercalator (e.g., TOTO-3). Finally, the chromatin is driven by voltage through a nanoscale channel fabricated in fused silica and fluorescent measurements of individual molecules are taken in a 150-aL inspection volume. A more detailed schematic of laser setup can be found in our previous publication (10). (B) Probability of erroneously interrogating more than a single molecule increases with analyte concentration according to a Poisson distribution less than 0.5% at the concentrations used here (≤1 nM). Pc(m), probability of m molecules residing in the 150-aL interrogation volume at any one time, given concentration c of fluorescent molecules in analyte, expressed as molecule count x in 150 aL, where NA is Avogadro’s number. The curve shows the probability that more than one molecule is in the inspection volume. Reprinted (adapted) with permission from (Cipriany, Benjamin R., et al. “Single molecule epigenetic analysis in a nanofluidic channel.” Analytical chemistry 82.6 (2010): 2480–2487.). Copyright (2010) American Chemical Society.
Another optical mapping method for epigenetic analysis uses nanoconfinement in a channel with width and depth smaller than the DNA persistence length to stretch and interrogate individual molecules190, 191. Single Chromatin Molecule Analysis in Nanochannels (SCAN) enables high-throughput fluorescent measurements of single DNA and chromatin molecules188, 191 (Fig. 7c). SCAN was used to detect methylated DNA by a fluorescently tagged methyl binding protein-1 in the presence of unmethylated DNA 192 and to demonstrate the interdependence of histone modifications and DNA methylation status 193. For example, in primary cultured cells, 5-methylcytosine (5mC) was shown to be necessary for proper placement of a certain histone marker, while antagonizing the placement of others. However, the effects of 5mC were reversed in immortalized cells where methylation had the opposite effects. This suggests a mechanism for aberrant placement of gene silencing marks on tumor suppressors in disease progression. This platform can be applied to studies to investigate the mechanism of epigenetic marks and their role in disease.
Nanopore-based sensing, discussed in Section 2.4, offers an alternative non-optical method for profiling covalent DNA modifications on a single molecule. Nanopores use the principle of ionic current spectroscopy 194 to electrically interrogate structural motifs of individual DNA molecules. Recently, electrical discrimination between unmethylated and methylated DNA was demonstrated in solid-state nanopores. This technique does not require bisulfite conversion, but does require labeling CpG dinucleotides with a 75 amino acid region of the methyl DNA binding protein MBD1. The presence of the MBD1 results in a 3-fold increase in the measured blockage current. This technique has demonstrated the capability of providing single CpG dinucleotide sensitivity195.
2.6. Conformation and Intermolecular Interactions
Single molecule studies of interactions between molecules allows for more accurate kinetic and thermodynamic characterization as well as analysis of population distributions. Microfluidic manipulation provides precise control over experimental conditions: e.g. forming stable concentration gradients, rapidly mixing reagents, and rapidly switching between conditions for sequential operations or high throughput testing. In this section, we provide examples of how micro and nanofluidic devices, such as those shown in Figure 8, have been utilized for studying interactions between individual nucleic acid molecules.
Figure 8.

Microfluidic tools and operations are used to enhance observation and analysis of interactions involving single nucleic acid molecules. Stable concentration gradients, generated by diffusion across flow streams in a microchannel, enable analysis of protein/DNA interactions on a DNA curtain over a continuous range of protein concentrations in a single experiment (picture reprinted from ref. 198 copyright 2013, with permission from Elsevier). Rapid mixing at a channel constriction coupled with convective flow can be used to observe fast kinetics through spatial separation (figure reprinted by permission from Macmillan Publishers Ltd: Nature Protocols ref. 203, copyright 2013). Rapid pump mixing can also be used for high throughput combinatorial screening separation (figure reprinted by permission from Macmillan Publishers Ltd: Nature Methods ref.204, copyright 2011). Microchannel constrictions can also be used to enhance detection efficiency by matching the microfluidic chamber dimensions to the optical detection volume. This has been demonstrated in both z-dimension with compression of channel height (figure reprinted by permission from Macmillan Publishers Ltd: Nature Methods ref. 205, copyright 2014), and x-dimension through microfabricated cross-section constriction (figure reproduced from ref. 206).
Microfluidic operations and features can be used to hold long nucleic acid molecules in place in particular conformations for further analysis. Azad et al. demonstrated the ability to capture a DNA molecule at a Y-shaped nanochannel intersection196 and generate stable loops in single DNA molecules by balancing confinement, self-avoidance, and flow forces. They also demonstrated that this method could be used to bring two molecules into close contact to increase the effective concentrations for analysis of DNA-DNA or DNA-protein or DNA-DNA-protein interactions in uniform or concentration gradient conditions. Elongational flows at a stagnation point that are used to stretch DNA molecules, as mentioned in previous sections, can also be used to monitor DNA-protein interactions and restriction enzyme cleavage kinetics by adding the cofactor in the opposing flow at the junction (see Fig. 3b)130.
A long line of stretched DNA molecules can be used for high throughput analysis of intermolecular interactions. Such DNA curtains can be generated by attaching one end of the DNA molecules to a surface and applying flow to uniformly stretch the molecules in parallel36. This technique is particularly useful to study protein interactions and movements along DNA molecules197. A DNA curtain situated in a Y-shaped laminar diffusion-mixing channel can be used to investigate the effect of concentration on DNA-protein interactions. For example, Frykholm et al. looked at Rad51 binding on lambda DNA curtains as a function of protein concentration (Fig. 8a)198. This work used surface functionalization through supported lipid bilayer (SLB) to avoid non-specific adsorption, and individually adjustable syringe-pump driven flow rates to optimally stretch the DNA and provide a concentration gradient over the field of view. Robinson and Finkelstein showed that microfluidic devices with DNA curtains can be made without expensive specialized photolithographic equipment, demonstrating that these devices could be available even for labs without these resources199. A thorough review of protein studies on DNA curtains is available elsewhere197.
Interactions that occur on a size scale smaller than the diffraction limit (1–10 nm) can be investigated with single molecule FRET (smFRET)-based techniques200, 201. Typical CFS requires the use of relatively low concentrations to ensure that only one molecule is present in the observation volume. However, the thermodynamics of interactions between many biological molecules would require higher concentrations to drive the equilibrium toward interaction. Observation of these interactions at lower concentrations would thus occur not at equilibrium. For interactions with fast kinetics, this requires the ability to rapidly mix and dilute before dissociation. A microfluidic device with two inlets was designed to generate rapid microfluidic dilution up to 1:10000 for the purpose of observations before dissociation 202. Observation of fast association kinetics can also be achieved using microfluidic devices. A microfluidic constriction or elongated pinchpoint in a PDMS device (Fig. 8b) was designed to decrease the time required to fully mix to species to sub millisecond203. A long observation channel with length markings then allows observation of kinetic dissociation and unfolding processes spanning a time range of 1 ms to seconds. The design could be further optimized to observe even earlier timepoints for even faster kinetic studies.
While flow in microfluidic devices can allow for the generation of continuous and controllable chemical gradients or states, it is often desirable to investigate processes in the absence of flow, as these hydrodynamic forces can affect the measured interaction. Microfluidic devices can still be used to quickly start and stop flow to increase testing throughput over a wide range of conditions in a single device. Kim et al. incorporated valves and a peristaltic pump, as shown in Figure 8c, to accomplish automated titration and mixing for smFRET measurements over a wide range of chemical conditions. Seven individually addressable inlet valves that allowed precision to tens of picoliters were used for combinatorial testing of various inputs over a wide of concentrations204. This allowed simultaneous analysis of the effects of both ionic strength and hybridization on the end-to-end distance of an ssDNA probe, demonstrating the ability to characterize processes in up to 6 chemical dimensions concurrently. The use of this scheme allows for fast characterization and optimization of experimental and reaction conditions, such as the enzymatic activity of RNA polymerase. In this device, many molecules could be analyzed rapidly to create a population distribution under each experimental condition. However, the ability to track an individual molecule or complex over longer time periods would allow observation of intermediate structures and kinetics of an entire process. Typically, these long-term observations would require immobilization of one molecule to prevent diffusion away from the detection region as well as an oxygen scavenging method to reduce photobleaching and damage from extended high-intensity exposure. However, this immobilization can be difficult to design and perform for some molecules, and could affect the process itself. Tyagi et al demonstrated a method to allow for long-time smFRET observations without immobilization (SWIFT – single molecule without immobilization for TIRF)205. A control layer in a two-layer microfluidic device was used to partition one 1 μm tall channel into two smaller channels with a height < 100 nm (see Fig. 8d). The smaller nanochannels confine the molecules to allow for longer observation, but the ability to rapidly change size to larger microchannels allows for quick transfer of buffers and reaction conditions without the requirement for high pressures (which can distort channels made from PDMS and also result in debonding) to overcome the high fluidic resistance. It is also easier to passivate the surfaces of larger channels. Through the use of PDMS and nitrogen gas, the authors were able to prevent photobleaching without the addition of oxygen scavengers to the reaction. This enabled continuous observation of a single molecule throughout an entire process: such as diffusion trajectory, protein folding, and Holiday Junction folding process. Further confinement to the detection region through feedback control, a revised channel structure, or a larger detection region would allow for even longer measurements. An alternative method to coordinate channel dimensions with the detection method is to incorporate a microfluidic constriction matched to the size of expanded observation volume of Cylindrical Illumination Confocal Spectroscopy (Fig. 8e). This was used previously to increase fluorescence detection uniformity for DNA sizing 4, and was recently applied to analyze the DNA content of polymer nanoparticles206. Even more recently, CICS fluorescent burst analysis coupled to hydrodynamic separation was used to distinguish DNA conformational differences including topology and hybridization as well as fluctuations in hydrodynamic shape including elongation and compaction within a microcapillary58. Optical tweezers207 can be used to precisely control the forces on and conformation of a DNA molecule independently of flow conditions. In this technique, a laser beam focused to a diffraction limited spot forms an optical trap that can capture and stably hold a microparticle. A DNA dumbbell is formed by attaching both ends of a DNA molecule to separate microparticles that can be independently controlled with separate optical traps. This technique can be used to precisely manipulate the conformation and extension length without flow. Forget et al. designed a method to perform DNA barbell assembly in a microfluidic device, and subsequently transfer the assembly to a flow-free detection chamber to analyze protein-DNA interactions as a function of DNA stretch208. DNA barbells can also be used to force-melt dsDNA into ssDNA. Flow can then be used to remove the unattached single strand, leaving one long single stranded molecule for analysis with protein binding to ssDNA209.
3. Summary and Future Directions
We have covered recent improvements in microfabrication combined with single molecule detection strategies to enable detection and characterization of individual nucleic acid molecules and their interactions. The integration of single molecule analysis with microfluidics has improved upon conventional practices for assessment of nucleic acid characteristics. Length analysis can be performed with higher sensitivity, smaller sample volumes, and in less time through the coupled use of micro- and nano- structures with single molecule detection. Sequence-specific detection can be performed without amplification for enhanced quantification capabilities and streamlined assay design. Sequencing of single molecules enables longer read-lengths for real-time de novo sequence assembly as compared with colony-based approaches. Combinatorial and high-throughput testing is faster and cheaper through the use of microfluidic devices. Examination of individual nucleic acid molecules with microfluidic operation has also enabled the emergence of new analysis techniques. Optical mapping without single molecule detection would require sequence alignment between molecules. Similarly, observations of fast kinetics or multi-step processes would require synchronization of multiple interactions. The complementarity of single molecule detection and micro- and nano- fluidics therefore fosters the generation of new and improved single nucleic acid molecule analysis.
Improvements in support technologies would enhance the utility of single molecule microfluidic methods. Many of the methods described above require complicated and time-consuming sample preparation techniques. For example, nucleic acids must be isolated through extraction, a process which can compromise DNA integrity. This is troublesome for applications which require high DNA integrity, such as optical mapping and long-read single molecule sequencing. Furthermore, the loss of DNA integrity because of the extraction process limits the utility of DNA integrity as a biomarker. A second consequence of harsh sample treatment is DNA damage, which can result in analysis errors. Analyzing multiple molecules can average out these errors. However, in applications where maintaining the integrity of every molecule is important, such as single cell analysis, this issue will need to be addressed. Standardizing sample collection and processing techniques is important for the development of novel diagnostic biomarkers. Furthermore, faster and gentler processing and extraction techniques that help to maintain sample integrity will be crucial to realize the full potential of DNA mapping and single molecule sequencing technologies. Universal sample preparation protocols would allow for multi-parametric analysis (e.g. mapping and sequencing, epigenetic analysis and binding kinetic analysis).
Sample introduction and device interfacing offer additional areas for improvement. The most common method for introducing samples into microfluidic devices involve creating access holes that intersect with a reservoir or channel. This results in large dead volumes and trapped air bubbles that result in sample waste and losses, and reduced functionality of the microfluidic device. Furthermore, although small volumes can be handled on-chip, handling those same sized volumes off-device (either before injection, or to collect and process with another technique) is difficult if not impossible due to the size constraints of typical methods (e.g. pipettes) as well as rapid evaporation from smaller volumes. Novel approaches to device interfacing that limit dead volume and sample waste as well as new sample handling techniques that enable efficient collection and transfer of small volumes would help to bridge this gap and increase the utility of microfluidic technologies. To further enhance sample handling, versatile devices which are capable of multi-parametric analyses would potentially eliminate external sample transfer steps.
Furthermore, multi-parametric analysis on a single device would enhance user-friendliness. Nucleic acids are a rich information source; however, each of the characteristic measurements described above acquires just a snapshot of the information contained in that molecule. Combining complementary techniques onto a single device is highly desirable. For example, long nucleic acid molecules are ideal for optical mapping and sequencing. Short, information-poor molecules, waste resources and space on the device. By implementing an on-chip DNA separation mechanism, the unnecessary analysis of information-poor molecules could be alleviated, further increasing throughput and data quality. The ability to perform multiple analyses sequentially or in parallel requires a device that can perform multiple analyses, including intermediary sample preparation steps. Modular designs have a wider audience and enable the collection of richer information from each sample. Applications that require high sensitivity and throughput, such as single cell analysis, may also benefit greatly from such modular designs.
Moreover, enhancing user-friendliness would expedite the adoption of microfluidic devices in research laboratories and clinical settings. Manual operations that require trained technicians for operation can limit the speed and extent to which these techniques are implemented in clinical settings. The utility of particular single molecule analysis techniques including physical genomic mapping and sequencing has already been demonstrated for applications including epidemiology of outbreak analysis. However, to engender widespread use and broaden their clinical uses to such fields as personalized medicine and real-time diagnostics would require increasing the speed of response from sample collection to answer, including streamlined sample preparation strategies, and in some cases decreasing the cost-per-test. Overcoming these barriers would give clinicians and patients access to the knowledge necessary to enable truly personalized treatments.
We have highlighted recent microfluidic technologies which are capable of characterizing single nucleic acids. With further improvements to sample processing and real-world interfacing to microfluidic devices, the utility of such devices will be enhanced. Furthermore, the ability to perform multi-parametric analysis on individual samples would increase adoption of microfluidic devices in basic research and clinical settings. In the future, microfluidic devices may become essential in the discovery of novel biomarkers and precision medicine.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
Authors also would like to thank funding source from National Institutes of Health (R01CA155305, R21CA173390 and R21CA186809).
Biographies
Sarah Friedrich is a Ph.D. candidate at the Johns Hopkins University in the Department of Biomedical Engineering. She is working in the BioMEMS lab of Prof. Tza-Huei Wang in the field of single molecule DNA analysis in microchannels. Previously, she received her Bachelors of Mechanical Engineering degree from University of Delaware in 2011.
Helena Zec is a postdoctoral researcher at the Johns Hopkins University in the Department of Mechanical Engineering. She develops miniaturized devices for high throughput genotyping. She received her PhD from Johns Hopkins in Biomedical Engineering in 2015. She received her Masters in Biomedical Engineering from ETH Zürich in 2008.
Tza-Huei Wang is a Professor in the departments of Mechanical Engineering and Biomedical Engineering at Johns Hopkins University. He received his PhD in 2002 from the University of California, Los Angeles. His current research focuses on the use and integration of microfluidic platforms, biomolecular sensors, and single molecule spectroscopy to analyze nucleic acids for use in disease diagnostics.
References
- 1.Fan HC, Quake SR. Anal Chem. 2007;79:7576–7579. doi: 10.1021/ac0709394. [DOI] [PubMed] [Google Scholar]
- 2.Fan HC, Gu W, Wang J, Blumenfeld YJ, El-Sayed YY, Quake SR. Nature. 2012;487:320–324. doi: 10.1038/nature11251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schwarzenbach H, Hoon DS, Pantel K. Nature Reviews Cancer. 2011;11:426–437. doi: 10.1038/nrc3066. [DOI] [PubMed] [Google Scholar]
- 4.Liu KJ, Brock MV, Shih IM, Wang TH. J Am Chem Soc. 2010;132:5793–5798. doi: 10.1021/ja100342q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Diaz LA, Jr, Bardelli A. J Clin Oncol. 2014;32:579–586. doi: 10.1200/JCO.2012.45.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pisanic TR, Athamanolap P, Poh W, Chen C, Hulbert A, Brock MV, Herman JG, Wang TH. Nucleic acids research. 2015:gkv795. doi: 10.1093/nar/gkv795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P. New England Journal of Medicine. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marusyk A, Polyak K. Biochimica et biophysica acta. 2010;1805:105–117. doi: 10.1016/j.bbcan.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vasdekis AE, Laporte GPJ. Int J Mol Sci. 2011;12:5135–5156. doi: 10.3390/ijms12085135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mouliere F, Rosenfeld N. Proceedings of the National Academy of Sciences. 2015;112:3178–3179. doi: 10.1073/pnas.1501321112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elshimali Y, Khaddour H, Sarkissyan M, Wu Y, Vadgama J. International Journal of Molecular Sciences. 2013;14:18925–18958. doi: 10.3390/ijms140918925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu SCY, Chan KCA, Zheng YWL, Jiang P, Liao GJW, Sun H, Akolekar R, Leung TY, Go ATJI, van Vugt JMG, Minekawa R, Oudejans CBM, Nicolaides KH, Chiu RWK, Lo YMD. Proceedings of the National Academy of Sciences. 2014;111:8583–8588. doi: 10.1073/pnas.1406103111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hyytia-Trees EK, Cooper K, Ribot EM, Gerner-Smidt P. Future microbiology. 2007;2:175–185. doi: 10.2217/17460913.2.2.175. [DOI] [PubMed] [Google Scholar]
- 14.Schwartz DC, Li X, Hernandez LI, Ramnarain SP, Huff EJ, Wang YK. Science. 1993;262:110–114. doi: 10.1126/science.8211116. [DOI] [PubMed] [Google Scholar]
- 15.Lim A, Dimalanta ET, Potamousis KD, Yen G, Apodoca J, Tao CH, Lin JY, Qi R, Skiadas J, Ramanathan A, Perna NT, Plunkett G, Burland V, Mau B, Hackett J, Blattner FR, Anantharaman TS, Mishra B, Schwartz DC. Genome Res. 2001;11:1584–1593. doi: 10.1101/gr.172101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dimalanta ET, Lim A, Runnheim R, Lamers C, Churas C, Forrest DK, de Pablo JJ, Graham MD, Coppersmith SN, Goldstein S, Schwartz DC. Anal Chem. 2004;76:5293–5301. doi: 10.1021/ac0496401. [DOI] [PubMed] [Google Scholar]
- 17.Xiao M, Phong A, Ha C, Chan TF, Cai DM, Leung L, Wan E, Kistler AL, DeRisi JL, Selvin PR, Kwok PY. Nucleic Acids Res. 2007:35. doi: 10.1093/nar/gkl1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Egger G, Liang G, Aparicio A, Jones PA. Nature. 2004;429:457–463. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- 19.Ottesen EA, Hong JW, Quake SR, Leadbetter JR. science. 2006;314:1464–1467. doi: 10.1126/science.1131370. [DOI] [PubMed] [Google Scholar]
- 20.Du W, Li L, Nichols KP, Ismagilov RF. Lab on a Chip. 2009;9:2286–2292. doi: 10.1039/b908978k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, Bright IJ, Lucero MY, Hiddessen AL, Legler TC. Analytical chemistry. 2011;83:8604–8610. doi: 10.1021/ac202028g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Song LA, Shan DD, Zhao MW, Pink BA, Minnehan KA, York L, Gardel M, Sullivan S, Phillips AF, Hayman RB, Walt DR, Duffy DC. Anal Chem. 2013;85:1932–1939. doi: 10.1021/ac303426b. [DOI] [PubMed] [Google Scholar]
- 23.Guan WH, Chen LB, Rane TD, Wang TH. Scientific Reports. 2015:5. doi: 10.1038/srep13795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang C, Da X. Chem Rev. 2010;110:4910–4947. doi: 10.1021/cr900081z. [DOI] [PubMed] [Google Scholar]
- 25.Streets AM, Huang Y. Curr Opin Biotechnol. 2014;25:69–77. doi: 10.1016/j.copbio.2013.08.013. [DOI] [PubMed] [Google Scholar]
- 26.Zec H, Shin DJ, Wang TH. Expert Rev Mol Diagn. 2014;14:787–801. doi: 10.1586/14737159.2014.945437. [DOI] [PubMed] [Google Scholar]
- 27.Taly V, Pekin D, El Abed A, Laurent-Puig P. Trends Mol Med. 2012;18:405–416. doi: 10.1016/j.molmed.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 28.Wang BG, Huang HY, Chen YC, Bristow RE, Kassauei K, Cheng CC, Roden R, Sokoll LJ, Chan DW, Shih IM. Cancer research. 2003;63:3966–3968. [PubMed] [Google Scholar]
- 29.Jahr S, Hentze H, Englisch S, Hardt D, Fackelmayer FO, Hesch RD, Knippers R. Cancer research. 2001;61:1659–1665. [PubMed] [Google Scholar]
- 30.Danna K, Nathans D. Proceedings of the National Academy of Sciences. 1971;68:2913–2917. doi: 10.1073/pnas.68.12.2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jeffreys AJ, Wilson V, Thein SL. Nature. 1985;314:67–73. doi: 10.1038/314067a0. [DOI] [PubMed] [Google Scholar]
- 32.Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Nucleic acids research. 2002;30:e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dittrich PS, Manz A. Anal Bioanal Chem. 2005;382:1771–1782. doi: 10.1007/s00216-005-3335-9. [DOI] [PubMed] [Google Scholar]
- 34.Dorfman KD, King SB, Olson DW, Thomas JD, Tree DR. Chem Rev. 2013;113:2584–2667. doi: 10.1021/cr3002142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bensimon A, Simon A, Chiffaudel A, Croquette V, Heslot F, Bensimon D. Science. 1994;265:2096–2098. doi: 10.1126/science.7522347. [DOI] [PubMed] [Google Scholar]
- 36.Fazio T, Visnapuu ML, Wind S, Greene EC. Langmuir. 2008;24:10524–10531. doi: 10.1021/la801762h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deen J, Sempels W, De Dier R, Vermant J, Dedecker P, Hofkens J, Neely RK. Acs Nano. 2015;9:809–816. doi: 10.1021/nn5063497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.De Angelis F, Gentile F, Mecarini F, Das G, Moretti M, Candeloro P, Coluccio ML, Cojoc G, Accardo A, Liberale C, Zaccaria RP, Perozziello G, Tirinato L, Toma A, Cuda G, Cingolani R, Di Fabrizio E. Nat Photonics. 2011;5:683–688. [Google Scholar]
- 39.Chan EY, Goncalves NM, Haeusler RA, Hatch AJ, Larson JW, Maletta AM, Yantz GR, Carstea ED, Fuchs M, Wong GG, Gullans SR, Gilmanshin R. Genome Res. 2004;14:1137–1146. doi: 10.1101/gr.1635204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu W, Muller SJ. Lab on a chip. 2012;12:647–651. doi: 10.1039/c2lc20880f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang J, Doyle PS. Appl Phys Lett. 2007:90. [Google Scholar]
- 42.Jo K, Chen YL, de Pablo JJ, Schwartz DC. Lab Chip. 2009;9:2348–2355. doi: 10.1039/b902292a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tegenfeldt JO, Prinz C, Cao H, Chou S, Reisner WW, Riehn R, Wang YM, Cox EC, Sturm JC, Silberzan P, Austin RH. Proc Natl Acad Sci U S A. 2004;101:10979–10983. doi: 10.1073/pnas.0403849101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reccius CH, Stavis SM, Mannion JT, Walker LP, Craighead HG. Biophys J. 2008;95:273–286. doi: 10.1529/biophysj.107.121020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lam ET, Hastie A, Lin C, Ehrlich D, Das SK, Austin MD, Deshpande P, Cao H, Nagarajan N, Xiao M, Kwok PY. Nat Biotechnol. 2012;30:771–776. doi: 10.1038/nbt.2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marie R, Kristensen A. Journal of biophotonics. 2012;5:673–686. doi: 10.1002/jbio.201200050. [DOI] [PubMed] [Google Scholar]
- 47.Menard LD, Ramsey JM. Anal Chem. 2013;85:1146–1153. doi: 10.1021/ac303074f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim Y, Kim KS, Kounovsky KL, Chang R, Jung GY, dePablo JJ, Jo K, Schwartz DC. Lab Chip. 2011;11:1721–1729. doi: 10.1039/c0lc00680g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Storm AJ, Storm C, Chen JH, Zandbergen H, Joanny JF, Dekker C. Nano Lett. 2005;5:1193–1197. doi: 10.1021/nl048030d. [DOI] [PubMed] [Google Scholar]
- 50.Gupta C, Liao WC, Gallego-Perez D, Castro CE, Lee LJ. Biomicrofluidics. 2014;8 doi: 10.1063/1.4871595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sen YH, Jain T, Aguilar CA, Karnik R. Lab Chip. 2012;12:1094–1101. doi: 10.1039/c2lc20771k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ferris MM, Yan X, Habbersett RC, Shou Y, Lemanski CL, Jett JH, Yoshida TM, Marrone BL. Journal of clinical microbiology. 2004;42:1965–1976. doi: 10.1128/JCM.42.5.1965-1976.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang TH, Peng YH, Zhang CY, Wong PK, Ho CM. J Am Chem Soc. 2005;127:5354–5359. doi: 10.1021/ja042642i. [DOI] [PubMed] [Google Scholar]
- 54.Castro A, Fairfield FR, Shera EB. Analytical Chemistry. 1993;65:849–852. [Google Scholar]
- 55.Liu KJ, Wang TH. Biophys J. 2008;95:2964–2975. doi: 10.1529/biophysj.108.132472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu KJ, Rane TD, Zhang Y, Wang TH. Journal of the American Chemical Society. 2011;133:6898–6901. doi: 10.1021/ja200279y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Friedrich S, Liu K, Wang T-H. presented in part at the Proceedings of The 17th International Conference on Miniaturized Systems for Chemistry and Life Sciences; Freiburg, Germany. 27–31 October, 2013. [Google Scholar]
- 58.Friedrich SM, Liu KJ, Wang T-H. J Am Chem Soc. 2015 doi: 10.1021/jacs.5b10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xuan J, Lee ML. Anal Methods-Uk. 2014;6:27–37. [Google Scholar]
- 60.Olson DW, Ou J, Tian MW, Dorfman KD. Electrophoresis. 2011;32:573–580. doi: 10.1002/elps.201000466. [DOI] [PubMed] [Google Scholar]
- 61.Olson DW, Dutta S, Laachi N, Tian MW, Dorfman KD. Electrophoresis. 2011;32:581–587. doi: 10.1002/elps.201000467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Park SG, Olson DW, Dorfman KD. Lab Chip. 2012;12:1463–1470. doi: 10.1039/c2lc00016d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Han J, Turner SW, Craighead HG. Phys Rev Lett. 1999;83:1688–1691. [Google Scholar]
- 64.Han J, Craighead HG. Science. 2000;288:1026–1029. doi: 10.1126/science.288.5468.1026. [DOI] [PubMed] [Google Scholar]
- 65.Huang LR, Tegenfeldt JO, Kraeft JJ, Sturm JC, Austin RH, Cox EC. Nat Biotechnol. 2002;20:1048–1051. doi: 10.1038/nbt733. [DOI] [PubMed] [Google Scholar]
- 66.Nazemifard N, Bhattacharjee S, Masliyah JH, Harrison DJ. Electrophoresis. 2013;34:2453–2463. doi: 10.1002/elps.201300152. [DOI] [PubMed] [Google Scholar]
- 67.Huang LR, Silberzan P, Tegenfeldt JO, Cox EC, Sturm JC, Austin RH, Craighead H. Phys Rev Lett. 2002;89 doi: 10.1103/PhysRevLett.89.178301. [DOI] [PubMed] [Google Scholar]
- 68.Huang LR, Cox EC, Austin RH, Sturm JC. Anal Chem. 2003;75:6963–6967. doi: 10.1021/ac0348524. [DOI] [PubMed] [Google Scholar]
- 69.Chen Y, Abrams ES, Boles TC, Pedersen JN, Flyvbjerg H, Austin RH, Sturm JC. Phys Rev Lett. 2015;114 doi: 10.1103/PhysRevLett.114.198303. [DOI] [PubMed] [Google Scholar]
- 70.Ranasinghe RT, Brown T. Chemical Communications. 2011;47:3717–3735. doi: 10.1039/c0cc04215c. [DOI] [PubMed] [Google Scholar]
- 71.Edwards MC, Gibbs RA. Genome Research. 1994;3:S65–S75. doi: 10.1101/gr.3.4.s65. [DOI] [PubMed] [Google Scholar]
- 72.Al-Soud WA, Rådström P. Journal of Clinical Microbiology. 2000;38:4463–4470. doi: 10.1128/jcm.38.12.4463-4470.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Al-Soud WA, Rådström P. Applied and environmental microbiology. 1998;64:3748–3753. doi: 10.1128/aem.64.10.3748-3753.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sedlak RH, Kuypers J, Jerome KR. Diagnostic microbiology and infectious disease. 2014;80:285–286. doi: 10.1016/j.diagmicrobio.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 75.Dingle TC, Sedlak RH, Cook L, Jerome KR. Clinical chemistry. 2013;59:1670–1672. doi: 10.1373/clinchem.2013.211045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.White RA, Quake SR, Curr K. Journal of virological methods. 2012;179:45–50. doi: 10.1016/j.jviromet.2011.09.017. [DOI] [PubMed] [Google Scholar]
- 77.Wang T-H, Puleo CM, Yeh H-C. Integrated Biochips for DNA Analysis. Springer; 2007. pp. 139–150. [Google Scholar]
- 78.Tyagi S, Kramer FR. Nature biotechnology. 1996;14:303–308. doi: 10.1038/nbt0396-303. [DOI] [PubMed] [Google Scholar]
- 79.Zhang CY, Yeh HC, Kuroki MT, Wang TH. Nature materials. 2005;4:826–831. doi: 10.1038/nmat1508. [DOI] [PubMed] [Google Scholar]
- 80.Liu KJ, Wang TH. Biophysical journal. 2008;95:2964–2975. doi: 10.1529/biophysj.108.132472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rane T, Puleo C, Liu K, Zhang Y, Lee A, Wang T. Lab on a Chip. 2010;10:161–164. doi: 10.1039/b917503b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Puleo CM, Wang TH. Lab on a Chip. 2009;9:1065–1072. doi: 10.1039/b819605b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rane TD, Zec HC, Puleo C, Lee AP, Wang TH. Lab Chip. 2012;12:3341–3347. doi: 10.1039/c2lc40537g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Castro A, Williams JG. Analytical chemistry. 1997;69:3915–3920. doi: 10.1021/ac970389h. [DOI] [PubMed] [Google Scholar]
- 85.Li H, Ying L, Green JJ, Balasubramanian S, Klenerman D. Analytical chemistry. 2003;75:1664–1670. doi: 10.1021/ac026367z. [DOI] [PubMed] [Google Scholar]
- 86.Yeh HC, Ho YP, Wang TH. Nanomedicine: Nanotechnology, Biology and Medicine. 2005;1:115–121. doi: 10.1016/j.nano.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 87.Hollars CW, Puls J, Bakajin O, Olsan B, Talley CE, Lane SM, Huser T. Analytical and bioanalytical chemistry. 2006;385:1384–1388. doi: 10.1007/s00216-006-0561-8. [DOI] [PubMed] [Google Scholar]
- 88.Neely LA, Patel S, Garver J, Gallo M, Hackett M, McLaughlin S, Nadel M, Harris J, Gullans S, Rooke J. Nat Methods. 2006;3:41–46. doi: 10.1038/nmeth825. [DOI] [PubMed] [Google Scholar]
- 89.Ho YP, Kung MC, Yang S, Wang TH. Nano Letters. 2005;5:1693–1697. doi: 10.1021/nl050888v. [DOI] [PubMed] [Google Scholar]
- 90.Puleo C, Yeh H, Liu K, Wang T. Lab on a Chip. 2008;8:822–825. doi: 10.1039/b717941c. [DOI] [PubMed] [Google Scholar]
- 91.Pisanic T, II, Zhang Y, Wang T. Analyst. 2014;139:2968–2981. doi: 10.1039/c4an00294f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang Y, Wang TH. Theranostics. 2012;2:631. doi: 10.7150/thno.4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Medintz IL, Uyeda HT, Goldman ER, Mattoussi H. Nature materials. 2005;4:435–446. doi: 10.1038/nmat1390. [DOI] [PubMed] [Google Scholar]
- 94.Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL, Fell HP, Ferree S, George RD, Grogan T. Nature biotechnology. 2008;26:317–325. doi: 10.1038/nbt1385. [DOI] [PubMed] [Google Scholar]
- 95.Veldman-Jones MH, Brant R, Rooney C, Geh C, Emery H, Harbron CG, Wappett M, Sharpe A, Dymond M, Barrett JC. Cancer research. 2015;75:2587–2593. doi: 10.1158/0008-5472.CAN-15-0262. [DOI] [PubMed] [Google Scholar]
- 96.Muggli MD, Puglisi SJ, Ronen R, Boucher C. Bioinformatics. 2015;31:80–88. doi: 10.1093/bioinformatics/btv262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chapleau RR. Journal of Clinical and Diagnostic Research. 2015 doi: 10.7860/JCDR/2015/13983.6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bosch T, Verkade E, van Luit M, Pot B, Vauterin P, Burggrave R, Savelkoul P, Kluytmans J, Schouls L. PLoS ONE. 2013;8 doi: 10.1371/journal.pone.0066493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Feuk L, Carson AR, Scherer SW. Nat Rev Genet. 2006;7:85–97. doi: 10.1038/nrg1767. [DOI] [PubMed] [Google Scholar]
- 100.Kidd JM, Cooper GM, Donahue WF, Hayden HS, Sampas N, Graves T, Hansen N, Teague B, Alkan C, Antonacci F, Haugen E, Zerr T, Yamada NA, Tsang P, Newman TL, Tuzun E, Cheng Z, Ebling HM, Tusneem N, David R, Gillett W, Phelps KA, Weaver M, Saranga D, Brand A, Tao W, Gustafson E, McKernan K, Chen L, Malig M, Smith JD, Korn JM, McCarroll SA, Altshuler DA, Peiffer DA, Dorschner M, Stamatoyannopoulos J, Schwartz D, Nickerson DA, Mullikin JC, Wilson RK, Bruhn L, Olson MV, Kaul R, Smith DR, Eichler EE. Nature. 2008;453:56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Teague B, Waterman MS, Goldstein S, Potamousis K, Zhou SG, Reslewic S, Sarkar D, Valouev A, Churas C, Kidd JM, Kohn S, Runnheim R, Lamers C, Forrest D, Newton MA, Eichler EE, Kent-First M, Surti U, Livny M, Schwartz DC. Proc Natl Acad Sci U S A. 2010;107:10848–10853. doi: 10.1073/pnas.0914638107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cao HZ, Hastie AR, Cao DD, Lam ET, Sun YH, Huang HD, Liu X, Lin LY, Andrews W, Chan S, Huang SJ, Tong X, Requa M, Anantharaman T, Krogh A, Yang HM, Cao H, Xu X. Gigascience. 2014;3 doi: 10.1186/2047-217X-3-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lam ET, Hastie A, Lin C, Ehrlich D, Das SK, Austin MD, Deshpande P, Cao H, Nagarajan N, Xiao M, Kwok PY. Nat Biotechnol. 2012;30:771–776. doi: 10.1038/nbt.2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Reisner W, Larsen NB, Silahtaroglu A, Kristensen A, Tommerup N, Tegenfeldt JO, Flyvbjerg H. Proc Natl Acad Sci U S A. 2010;107:13294–13299. doi: 10.1073/pnas.1007081107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Botstein D, White RL, Skolnick M, Davis RW. Am J Hum Genet. 1980;32:314–331. [PMC free article] [PubMed] [Google Scholar]
- 106.Narayanan S. Ann Clin Lab Sci. 1991;21:291–296. [PubMed] [Google Scholar]
- 107.Li WJ, Raoult D, Fournier PE. Fems Microbiol Rev. 2009;33:892–916. doi: 10.1111/j.1574-6976.2009.00182.x. [DOI] [PubMed] [Google Scholar]
- 108.Riehn R, Lu M, Wang YM, Lim SF, Cox EC, Austin RH. Proc Natl Acad Sci U S A. 2005;102:10012–10016. doi: 10.1073/pnas.0503809102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tycon MA, Dial CF, Faison K, Melvin W, Fecko CJ. Anal Biochem. 2012;426:13–21. doi: 10.1016/j.ab.2012.03.021. [DOI] [PubMed] [Google Scholar]
- 110.Fey PD, Iwen PC, Zentz EB, Briska AM, Henkhaus JK, Bryant KA, Larson MA, Noel RK, Hinrichs SH. J Clin Microbiol. 2012;50:3063–3065. doi: 10.1128/JCM.01320-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sabirova JS, Xavier BB, Ieven M, Goossens H, Malhotra-Kumar S. BMC Res Notes. 2014;7:704. doi: 10.1186/1756-0500-7-704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Raeside C, Gaffe J, Deatherage DE, Tenaillon O, Briska AM, Ptashkin RN, Cruveiller S, Medigue C, Lenski RE, Barrick JE, Schneider D. Mbio. 2014;5 doi: 10.1128/mBio.01377-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Church DM, Goodstadt L, Hillier LW, Zody MC, Goldstein S, She XW, Bult CJ, Agarwala R, Cherry JL, DiCuccio M, Hlavina W, Kapustin Y, Meric P, Maglott D, Birtle Z, Marques AC, Graves T, Zhou SG, Teague B, Potamousis K, Churas C, Place M, Herschleb J, Runnheim R, Forrest D, Amos-Landgraf J, Schwartz DC, Cheng Z, Lindblad-Toh K, Eichler EE, Ponting CP MGS Consortium. PLoS Biol. 2009;7 [Google Scholar]
- 114.Zhou SG, Wei FS, Nguyen J, Bechner M, Potamousis K, Goldstein S, Pape L, Mehan MR, Churas C, Pasternak S, Forrest DK, Wise R, Ware D, Wing RA, Waterman MS, Livny M, Schwartz DC. PLoS Genet. 2009;5 doi: 10.1371/journal.pgen.1000711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhou S, Bechner MC, Place M, Churas CP, Pape L, Leong SA, Runnheim R, Forrest DK, Goldstein S, Livny M, Schwartz DC. BMC Genomics. 2007;8 doi: 10.1186/1471-2164-8-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gupta A, Place M, Goldstein S, Sarkar D, Zhou SG, Potamousis K, Kim J, Flanagan C, Li Y, Newton MA, Callander NS, Hematti P, Bresnick EH, Ma J, Asimakopoulos F, Schwartz DC. Proc Natl Acad Sci U S A. 2015;112:7689–7694. doi: 10.1073/pnas.1418577112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Onmus-Leone F, Hang J, Clifford RJ, Yang Y, Riley MC, Kuschner RA, Waterman PE, Lesho EP. PLoS ONE. 2013;8 doi: 10.1371/journal.pone.0061762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dong Y, Xie M, Jiang Y, Xiao NQ, Du XY, Zhang WG, Tosser-Klopp G, Wang JH, Yang S, Liang J, Chen WB, Chen J, Zeng P, Hou Y, Bian C, Pan SK, Li YX, Liu X, Wang WL, Servin B, Sayre B, Zhu B, Sweeney D, Moore R, Nie WH, Shen YY, Zhao RP, Zhang GJ, Li JQ, Faraut T, Womack J, Zhang YP, Kijas J, Cockett N, Xu X, Zhao SH, Wang J, Wang W. Nat Biotechnol. 2013;31:135–141. doi: 10.1038/nbt.2478. [DOI] [PubMed] [Google Scholar]
- 119.Ganapathy G, Howard JT, Ward JM, Li JW, Li B, Li YR, Xiong YQ, Zhang Y, Zhou SG, Schwartz DC, Schatz M, Aboukhalil R, Fedrigo O, Bukovnik L, Wang T, Wray G, Rasolonjatovo I, Winer R, Knight JR, Koren S, Warren WC, Zhang GJ, Phillippy AM, Jarvis ED. Gigascience. 2014;3 doi: 10.1186/2047-217X-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lim YL, Ee R, How KY, Lee SK, Yong D, Tee KK, Yin WF, Chan KG. Peerj. 2015;3 doi: 10.7717/peerj.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pendleton M, Sebra R, Pang AWC, Ummat A, Franzen O, Rausch T, Stutz AM, Stedman W, Anantharaman T, Hastie A, Dai H, Fritz MHY, Cao H, Cohainl A, Deikusl G, Durrett RE, Blanchard SC, Altman R, Chin CS, Guo Y, Paxinos EE, Korbe JO, Darne RB, McCombiemii WR, Kwok PY, Mason CE, Schadt EE, Bashirl A. Nat Methods. 2015;12:780–U140. doi: 10.1038/nmeth.3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Griffis JW, Safranovitch MM, Vyas SP, Gerrin S, Protozanova E, Malkin G, Meltzer RH. Lab Chip. 2014;14:3881–3893. doi: 10.1039/c4lc00781f. [DOI] [PubMed] [Google Scholar]
- 123.Griffis JW, Protozanova E, Cameron DB, Meltzer RH. Lab Chip. 2013;13:240–251. doi: 10.1039/c2lc40943g. [DOI] [PubMed] [Google Scholar]
- 124.Protozanova E, Zhang M, White EJ, Mollova ET, Broeck DT, Fridrikh SV, Cameron DB, Gilmanshin R. Analytical biochemistry. 2010;402:83–90. doi: 10.1016/j.ab.2010.03.024. [DOI] [PubMed] [Google Scholar]
- 125.White EJ, Fridrikh SV, Chennagiri N, Cameron DB, Gauvin GP, Gilmanshin R. Clin Chem. 2009;55:2121–2129. doi: 10.1373/clinchem.2009.128967. [DOI] [PubMed] [Google Scholar]
- 126.Burton RE, White EJ, Foss TR, Phillips KM, Meltzer RH, Kojanian N, Kwok LW, Lim A, Pellerin NL, Mamaeva NV, Gilmanshin R. Lab Chip. 2010;10:843–851. doi: 10.1039/b922106a. [DOI] [PubMed] [Google Scholar]
- 127.Meltzer RH, Krogmeier JR, Kwok LW, Allen R, Crane B, Griffis JW, Knaian L, Kojanian N, Malkin G, Nahas MK, Papkov V, Shaikh S, Vyavahare K, Zhong Q, Zhou Y, Larson JW, Gilmanshin R. Lab on a Chip. 2011;11:863. doi: 10.1039/c0lc00477d. [DOI] [PubMed] [Google Scholar]
- 128.Dylla-Spears R, Townsend JE, Jen-Jacobson L, Sohn LL, Muller SJ. Lab Chip. 2010;10:1543–1549. doi: 10.1039/b926847b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Perkins TT, Smith DE, Chu S. Science. 1997;276:2016–2021. doi: 10.1126/science.276.5321.2016. [DOI] [PubMed] [Google Scholar]
- 130.Xu WL, Muller SJ. Lab Chip. 2011;11:435–442. doi: 10.1039/c0lc00176g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Marie R, Pedersen JN, Bauer DL, Rasmussen KH, Yusuf M, Volpi E, Flyvbjerg H, Kristensen A, Mir KU. Proc Natl Acad Sci U S A. 2013;110:4893–4898. doi: 10.1073/pnas.1214570110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sorensen KT, Lopacinska JM, Tommerup N, Silahtaroglu A, Kristensen A, Marie R. Rev Sci Instrum. 2015;86:063702. doi: 10.1063/1.4922068. [DOI] [PubMed] [Google Scholar]
- 133.Jo K, Dhingra DM, Odijk T, de Pablo JJ, Graham MD, Runnheim R, Forrest D, Schwartz DC. Proc Natl Acad Sci U S A. 2007;104:2673–2678. doi: 10.1073/pnas.0611151104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Das SK, Austin MD, Akana MC, Deshpande P, Cao H, Xiao M. Nucleic Acids Res. 2010;38 doi: 10.1093/nar/gkq673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Baday M, Cravens A, Hastie A, Kim H, Kudeki DE, Kwok PY, Xiao M, Selvin PR. Nano Lett. 2013;13:1365–1365. doi: 10.1021/nl302069q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hastie AR, Dong L, Smith A, Finklestein J, Lam ET, Huo N, Cao H, Kwok PY, Deal KR, Dvorak J, Luo MC, Gu Y, Xiao M. PloS one. 2013;8:e55864. doi: 10.1371/journal.pone.0055864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.O’Bleness M, Searles VB, Dickens CM, Astling D, Albracht D, Mak AC, Lai YY, Lin C, Chu C, Graves T, Kwok PY, Wilson RK, Sikela JM. BMC genomics. 2014;15:387. doi: 10.1186/1471-2164-15-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Cao H, Hastie AR, Cao D, Lam ET, Sun Y, Huang H, Liu X, Lin L, Andrews W, Chan S, Huang S, Tong X, Requa M, Anantharaman T, Krogh A, Yang H, Cao H, Xu X. GigaScience. 2014;3:34. doi: 10.1186/2047-217X-3-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Mak AC, Lai YY, Lam ET, Kwok TP, Leung AK, Poon A, Mostovoy Y, Hastie AR, Stedman W, Anantharaman T, Andrews W, Zhou X, Pang AW, Dai H, Chu C, Lin C, Wu JJ, Li CM, Li JW, Yim AK, Chan S, Sibert J, Dzakula Z, Cao H, Yiu SM, Chan TF, Yip KY, Xiao M, Kwok PY. Genetics. 2016;202:351–362. doi: 10.1534/genetics.115.183483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.English AC, Salerno WJ, Hampton OA, Gonzaga-Jauregui C, Ambreth S, Ritter DI, Beck CR, Davis CF, Dahdouli M, Ma S, Carroll A, Veeraraghavan N, Bruestle J, Drees B, Hastie A, Lam ET, White S, Mishra P, Wang M, Han Y, Zhang F, Stankiewicz P, Wheeler DA, Reid JG, Muzny DM, Rogers J, Sabo A, Worley KC, Lupski JR, Boerwinkle E, Gibbs RA. BMC genomics. 2015;16:286. doi: 10.1186/s12864-015-1479-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Grunwald A, Dahan M, Giesbertz A, Nilsson A, Nyberg LK, Weinhold E, Ambjornsson T, Westerlund F, Ebenstein Y. Nucleic Acids Res. 2015 doi: 10.1093/nar/gkv563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Fuller CW, Middendorf LR, Benner SA, Church GM, Harris T, Huang X, Jovanovich SB, Nelson JR, Schloss JA, Schwartz DC, Vezenov DV. Nat Biotechnol. 2009;27:1013–1023. doi: 10.1038/nbt.1585. [DOI] [PubMed] [Google Scholar]
- 143.Levene MJ, Korlach J, Turner SW, Foquet M, Craighead HG, Webb WW. Science. 2003;299:682–686. doi: 10.1126/science.1079700. [DOI] [PubMed] [Google Scholar]
- 144.Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B, Bibillo A, Bjornson K, Chaudhuri B, Christians F, Cicero R, Clark S, Dalal R, Dewinter A, Dixon J, Foquet M, Gaertner A, Hardenbol P, Heiner C, Hester K, Holden D, Kearns G, Kong XX, Kuse R, Lacroix Y, Lin S, Lundquist P, Ma CC, Marks P, Maxham M, Murphy D, Park I, Pham T, Phillips M, Roy J, Sebra R, Shen G, Sorenson J, Tomaney A, Travers K, Trulson M, Vieceli J, Wegener J, Wu D, Yang A, Zaccarin D, Zhao P, Zhong F, Korlach J, Turner S. Science. 2009;323:133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 145.Chin CS, Sorenson J, Harris JB, Robins WP, Charles RC, Jean-Charles RR, Bullard J, Webster DR, Kasarskis A, Peluso P, Paxinos EE, Yamaichi Y, Calderwood SB, Mekalanos JJ, Schadt EE, Waldor MK. New Engl J Med. 2011;364:33–42. doi: 10.1056/NEJMoa1012928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Rasko DA, Webster DR, Sahl JW, Bashir A, Boisen N, Scheutz F, Paxinos EE, Sebra R, Chin CS, Iliopoulos D, Klammer A, Peluso P, Lee L, Kislyuk AO, Bullard J, Kasarskis A, Wang S, Eid J, Rank D, Redman JC, Steyert SR, Frimodt-Moller J, Struve C, Petersen AM, Krogfelt KA, Nataro JP, Schadt EE, Waldor MK. New Engl J Med. 2011;365:709–717. doi: 10.1056/NEJMoa1106920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Koren S, Schatz MC, Walenz BP, Martin J, Howard JT, Ganapathy G, Wang Z, Rasko DA, McCombie WR, Jarvis ED, Phillippy AM. Nat Biotechnol. 2012;30:692. doi: 10.1038/nbt.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Koren S, Harhay GP, Smith TP, Bono JL, Harhay DM, McVey SD, Radune D, Bergman NH, Phillippy AM. Genome Biol. 2013;14:R101. doi: 10.1186/gb-2013-14-9-r101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Huddleston J, Ranade S, Malig M, Antonacci F, Chaisson M, Hon L, Sudmant PH, Graves TA, Alkan C, Dennis MY, Wilson RK, Turner SW, Korlach J, Eichler EE. Genome Res. 2014;24:688–696. doi: 10.1101/gr.168450.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Chaisson MJP, Huddleston J, Dennis MY, Sudmant PH, Malig M, Hormozdiari F, Antonacci F, Surti U, Sandstrom R, Boitano M, Landolin JM, Stamatoyannopoulos JA, Hunkapiller MW, Korlach J, Eichler EE. Nature. 2015;517:608–U163. doi: 10.1038/nature13907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Treutlein B, Gokce O, Quake SR, Sudhof TC. Proc Natl Acad Sci U S A. 2014;111:E1291–E1299. doi: 10.1073/pnas.1403244111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Flusberg BA, Webster DR, Lee JH, Travers KJ, Olivares EC, Clark TA, Korlach J, Turner SW. Nat Methods. 2010;7:461–U472. doi: 10.1038/nmeth.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Song CX, Clark TA, Lu XY, Kislyuk A, Dai Q, Turner SW, He C, Korlach J. Nat Methods. 2012;9:75–U188. doi: 10.1038/nmeth.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Grad YH, Lipsitch M, Feldgarden M, Arachchi HM, Cerqueira GC, FitzGerald M, Godfrey P, Haas BJ, Murphy CI, Russ C, Sykes S, Walker BJ, Wortman JR, Young S, Zeng QD, Abouelleil A, Bochicchio J, Chauvin S, DeSmet T, Gujja S, McCowan C, Montmayeur A, Steelman S, Frimodt-Moller J, Petersen AM, Struve C, Krogfelt KA, Bingen E, Weill FX, Lander ES, Nusbaum C, Birren BW, Hung DT, Hanage WP. Proc Natl Acad Sci U S A. 2012;109:3065–3070. doi: 10.1073/pnas.1121491109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Roberts RJ, Carneiro MO, Schatz MC. Genome Biol. 2013;14:405. doi: 10.1186/gb-2013-14-7-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Travers KJ, Chin CS, Rank DR, Eid JS, Turner SW. Nucleic Acids Res. 2010;38 doi: 10.1093/nar/gkq543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Westbrook CJ, Karl JA, Wiseman RW, Mate S, Koroleva G, Garcia K, Sanchez-Lockhart M, O’Connor DH, Palacios G. Hum Immunol. 2015 doi: 10.1016/j.humimm.2015.03.022. [DOI] [PubMed] [Google Scholar]
- 158.Feng Y, Zhang Y, Ying C, Wang D, Du C. Genomics Proteomics Bioinformatics. 2015;13:4–16. doi: 10.1016/j.gpb.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Stoddart D, Heron AJ, Mikhailova E, Maglia G, Bayley H. Proc Natl Acad Sci U S A. 2009;106:7702–7707. doi: 10.1073/pnas.0901054106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Karlsson E, Lärkeryd A, Sjödin A, Forsman M, Stenberg P. Scientific Reports. 2015;5:11996. doi: 10.1038/srep11996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Akahori R, Haga T, Hatano T, Yanagi I, Ohura T, Hamamura H, Iwasaki T, Yokoi T, Anazawa T. Nanotechnology. 2014;25:275501. doi: 10.1088/0957-4484/25/27/275501. [DOI] [PubMed] [Google Scholar]
- 162.Branton D, Deamer DW, Marziali A, Bayley H, Benner SA, Butler T, Di Ventra M, Garaj S, Hibbs A, Huang X, Jovanovich SB, Krstic PS, Lindsay S, Ling XS, Mastrangelo CH, Meller A, Oliver JS, Pershin YV, Ramsey JM, Riehn R, Soni GV, Tabard-Cossa V, Wanunu M, Wiggin M, Schloss JA. Nat Biotechnol. 2008;26:1146–1153. doi: 10.1038/nbt.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Stoddart D, Maglia G, Mikhailova E, Heron AJ, Bayley H. Angew Chem Int Edit. 2010;49:556–559. doi: 10.1002/anie.200905483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Stoddart D, Heron AJ, Klingelhoefer J, Mikhailova E, Maglia G, Bayley H. Nano Lett. 2010;10:3633–3637. doi: 10.1021/nl101955a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Clarke J, Wu HC, Jayasinghe L, Patel A, Reid S, Bayley H. Nat Nanotechnol. 2009;4:265–270. doi: 10.1038/nnano.2009.12. [DOI] [PubMed] [Google Scholar]
- 166.Ehrlich SD, Bierne H, d’Alencon E, Vilette D, Petranovic M, Noirot P, Michel B. Gene. 1993;135:161–166. doi: 10.1016/0378-1119(93)90061-7. [DOI] [PubMed] [Google Scholar]
- 167.Wang J, Moore NE, Deng YM, Eccles DA, Hall RJ. Front Microbiol. 2015;6:766. doi: 10.3389/fmicb.2015.00766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Cao MD, Ganesamoorthy D, Elliott A, Zhang H, Cooper M, Coin L. Real-time strain typing and analysis of antibiotic resistance potential using Nanopore MinION sequencing. 2015 [Google Scholar]
- 169.Judge K, Harris SR, Reuter S, Parkhill J, Peacock SJ. Journal of Antimicrobial Chemotherapy. 2015;70:2775–2778. doi: 10.1093/jac/dkv206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Ashton PM, Nair S, Dallman T, Rubino S, Rabsch W, Mwaigwisya S, Wain J, O’Grady J. Nat Biotechnol. 2015;33:296–300. doi: 10.1038/nbt.3103. [DOI] [PubMed] [Google Scholar]
- 171.Quick J, Ashton P, Calus S, Chatt C, Gossain S, Hawker J, Nair S, Neal K, Nye K, Peters T, De Pinna E, Robinson E, Struthers K, Webber M, Catto A, Dallman TJ, Hawkey P, Loman NJ. Genome Biol. 2015;16:114. doi: 10.1186/s13059-015-0677-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Bolisetty MT, Rajadinakaran G, Graveley BR. Genome Biol. 2015;16:204. doi: 10.1186/s13059-015-0777-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Loman NJ, Quick J, Simpson JT. Nat Methods. 2015;12:733–U751. doi: 10.1038/nmeth.3444. [DOI] [PubMed] [Google Scholar]
- 174.Greninger AL, Naccache SN, Federman S, Yu G, Mbala P, Bres V, Stryke D, Bouquet J, Somasekar S, Linnen JM, Dodd R, Mulembakani P, Schneider BS, Muyembe-Tamfum JJ, Stramer SL, Chiu CY. Genome Med. 2015;7:99. doi: 10.1186/s13073-015-0220-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Cheng SH, Jiang P, Sun K, Cheng YK, Chan KC, Leung TY, Chiu RW, Lo YM. Clin Chem. 2015;61:1305–1306. doi: 10.1373/clinchem.2015.245076. [DOI] [PubMed] [Google Scholar]
- 176.Jain M, Fiddes IT, Miga KH, Olsen HE, Paten B, Akeson M. Nat Methods. 2015;12:351–356. doi: 10.1038/nmeth.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Warren RL, Yang C, Vandervalk BP, Behsaz B, Lagman A, Jones SJ, Birol I. Gigascience. 2015;4:35. doi: 10.1186/s13742-015-0076-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Koren S, Phillippy AM. Curr Opin Microbiol. 2015;23:110–120. doi: 10.1016/j.mib.2014.11.014. [DOI] [PubMed] [Google Scholar]
- 179.Laver T, Harrison J, O’Neill PA, Moore K, Farbos A, Paszkiewicz K, Studholme DJ. Biomolecular Detection and Quantification. 2015;3:1–8. doi: 10.1016/j.bdq.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Feinberg AP. Nature. 2007;447:433–440. doi: 10.1038/nature05919. [DOI] [PubMed] [Google Scholar]
- 181.Herman JG, Baylin SB. New England Journal of Medicine. 2003;349:2042–2054. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 182.Jones PA, Baylin SB. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Aguilar CA, Craighead HG. Nature nanotechnology. 2013;8:709–718. doi: 10.1038/nnano.2013.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Hyun BR, McElwee JL, Soloway PD. Methods. 2015;72:41–50. doi: 10.1016/j.ymeth.2014.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Li Y, Tollefsbol TO. Epigenetics Protocols. Springer; 2011. pp. 11–21. [Google Scholar]
- 186.Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB. Proceedings of the National Academy of Sciences. 1996;93:9821–9826. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Goren A, Ozsolak F, Shoresh N, Ku M, Adli M, Hart C, Gymrek M, Zuk O, Regev A, Milos PM. Nature methods. 2010;7:47–49. doi: 10.1038/nmeth.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Streng DE, Lim SF, Pan JH, Karpusenka A, Riehn R. Lab Chip. 2009;9:2772–2774. doi: 10.1039/b909217j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Cerf A, Tian HC, Craighead HG. ACS nano. 2012;6:7928–7934. doi: 10.1021/nn3023624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Lim SF, Karpusenko A, Sakon JJ, Hook JA, Lamar TA, Riehn R. Biomicrofluidics. 2011;5:034106. doi: 10.1063/1.3613671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Cipriany BR, Zhao R, Murphy PJ, Levy SL, Tan CP, Craighead HG, Soloway PD. Anal Chem. 2010;82:2480–2487. doi: 10.1021/ac9028642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Cipriany BR, Murphy PJ, Hagarman JA, Cerf A, Latulippe D, Levy SL, Benítez JJ, Tan CP, Topolancik J, Soloway PD. Proceedings of the National Academy of Sciences. 2012;109:8477–8482. doi: 10.1073/pnas.1117549109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Murphy PJ, Cipriany BR, Wallin CB, Ju CY, Szeto K, Hagarman JA, Benitez JJ, Craighead HG, Soloway PD. Proceedings of the National Academy of Sciences. 2013;110:7772–7777. doi: 10.1073/pnas.1218495110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Shim J, Humphreys GI, Venkatesan BM, Munz JM, Zou X, Sathe C, Schulten K, Kosari F, Nardulli AM, Vasmatzis G. Scientific reports. 2013;3 doi: 10.1038/srep01389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Shim J, Kim Y, Humphreys GI, Nardulli AM, Kosari F, Vasmatzis G, Taylor WR, Ahlquist DA, Myong S, Bashir R. Acs Nano. 2015;9:290–300. doi: 10.1021/nn5045596. [DOI] [PubMed] [Google Scholar]
- 196.Azad Z, Roushan M, Riehn R. Nano Lett. 2015;15:5641–5646. doi: 10.1021/acs.nanolett.5b02476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Silverstein TD, Gibb B, Greene EC. DNA Repair. 2014;20:94–109. doi: 10.1016/j.dnarep.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Frykholm K, Freitag C, Persson F, Tegenfeldt JO, Graneli A. Anal Biochem. 2013;443:261–268. doi: 10.1016/j.ab.2013.08.023. [DOI] [PubMed] [Google Scholar]
- 199.Robison AD, Finkelstein IJ. Anal Chem. 2014;86:4157–4163. doi: 10.1021/ac500267v. [DOI] [PubMed] [Google Scholar]
- 200.Ha T, Enderle T, Ogletree DF, Chemla DS, Selvin PR, Weiss S. Proc Natl Acad Sci U S A. 1996;93:6264–6268. doi: 10.1073/pnas.93.13.6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Deniz AA, Dahan M, Grunwell JR, Ha TJ, Faulhaber AE, Chemla DS, Weiss S, Schultz PG. Proc Natl Acad Sci U S A. 1999;96:3670–3675. doi: 10.1073/pnas.96.7.3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Horrocks MH, Rajah L, Jonsson P, Kjaergaard M, Vendruscolo M, Knowles TPJ, Klenerman D. Anal Chem. 2013;85:6855–6859. doi: 10.1021/ac4010875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Wunderlich B, Nettels D, Benke S, Clark J, Weidner S, Hofmann H, Pfeil SH, Schuler B. Nat Protoc. 2013;8:1459–1474. doi: 10.1038/nprot.2013.082. [DOI] [PubMed] [Google Scholar]
- 204.Kim S, Streets AM, Lin RR, Quake SR, Weiss S, Majumdar DS. Nat Methods. 2011;8:242–U283. doi: 10.1038/nmeth.1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Tyagi S, Vandelinder V, Banterle N, Fuertes G, Milles S, Agez M, Lemke E. Mol Biol Cell. 2014;25 doi: 10.1038/nmeth.2809. [DOI] [PubMed] [Google Scholar]
- 206.Beh CW, Pan D, Lee J, Jiang X, Liu KJ, Mao HQ, Wang TH. Nano Lett. 2014;14:4729–4735. doi: 10.1021/nl5018404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Ashkin A, Dziedzic JM, Bjorkholm JE, Chu S. Opt Lett. 1986;11:288–290. doi: 10.1364/ol.11.000288. [DOI] [PubMed] [Google Scholar]
- 208.Forget AL, Dombrowski CC, Amitani I, Kowalczykowski SC. Nat Protoc. 2013;8:525–538. doi: 10.1038/nprot.2013.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Candelli A, Holthausen JT, Depken M, Brouwer I, Franker MAM, Marchetti M, Heller I, Bernard S, Garcin EB, Modesti M, Wyman C, Wuite GJL, Peterman EJG. Proc Natl Acad Sci U S A. 2014;111:15090–15095. doi: 10.1073/pnas.1307824111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Miller JM. J Clin Microbiol. 2013;51:1066–1070. doi: 10.1128/JCM.00093-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.