

Abstract
Objectives
To investigate the role of the periodontal pathogen Porphyromonas gingivalis in rheumatoid arthritis (RA) etiology, we have analysed the antibody response to P. gingivalis virulence factor arginine gingipainB (RgpB) in relation to anti-citrullinated protein antibodies (ACPA), smoking and HLA-DRB1 shared epitope (SE) alleles, in patients with periodontitis (PD) and RA, and in controls.
Methods
Anti-RgpB IgG was measured by ELISA in 65 PD patients and 59 non-PD controls, and in 1,975 RA cases and 377 non-RA controls from the Swedish population-based case-control study EIRA (Epidemiological Investigation of RA). Autoantibody status, smoking habits and genetic data were retrieved from the EIRA database. Differences in antibody levels were examined using Mann-Whitney U test. Unconditional logistic regression was used to calculate odds ratios (OR) with 95% confidence intervals (CI) for the association of anti-RgpB IgG with different RA subsets.
Results
Anti-RgpB antibody levels were significantly elevated in PD compared to non-PD; in RA compared to non-RA; and in ACPA-positive RA compared to ACPA-negative RA. There was a significant association between anti-RgpB IgG and RA (OR=2.96; 95% CI: 2.00–4.37), which was even stronger than the association between smoking and RA (OR=1.37; 95% CI: 1.07–1.74), and in ACPA-positive RA, there were interactions between anti-RgpB antibodies and both smoking and SE.
Conclusion
Our study suggests that the previously reported link between PD and RA could be accounted for by P. gingivalis infection, and we conclude that P. gingivalis is a credible candidate for triggering and/or driving autoimmunity and autoimmune disease in a subset of RA.
INTRODUCTION
There is accumulating evidence, from a large number of studies, for an association between chronic periodontitis (PD) and rheumatoid arthritis (RA) (1–12). This relationship may be non-causal, based on shared environmental (smoking) (13, 14) and genetic (HLA-DRB1 shared epitope (SE) alleles) (15, 16) risk factors, giving rise to similar pro-inflammatory immune responses, driving bone erosion and tissue destruction in the periodontium and in the synovial joints (12, 17). However, a causal link, where PD triggers and/or drives RA, could also be possible, and was first proposed by Rosenstein et al (18). The fact that the autoimmune response in RA - in the form of autoantibodies targeting citrullinated proteins (ACPA) - often proceeds the clinical symptoms by several years (19) may suggest that RA arise outside the joints, potentially at mucosal sites, such as the lungs or gums.
Chronic periodontitis - the world’s most prevalent inflammatory disease, affecting approximately 30% of the adult population (20) - is initiated by a set of pathogenic bacteria, often Including Porphyromonas gingivalis (P. gingivalis), Tannarella forsythia and Treponema denticola (21). These bacteria are equipped with a wide range of virulence factors, which help to colonize and invade periodontal pockets and disturb the host immune system (22, 23). The most potent virulence factors are the gingipains (24) expressed specifically by P. gingivalis (25). Gingipains are extracellular proteases, which cleave substrates at lysine or arginine residues, “with the precision of a surgeon’s knife” or “with meat chopper-like brutal degradation” (26). Through these actions, P. gingivalis gingipains can break down collagen, interrupt the clotting cascade and degrade and modify immunoglobulins, complement and cytokines (22, 23). Importantly, the arginine gingipains (RgpA and RgpB) act in concert with another major virulence factor unique to P. gingivalis, a peptidylarginine deiminase enzyme denoted P.PAD (27).
Citrullination by P.PAD occurs mainly at C-terminal arginine, which requires the prior actions of arginine gingipains. P.PAD and Rgp co-localise on the bacterial outer membrane, and can also be secreted, hence are perfectly positioned to interact with host proteins (27, 28). P.PAD, which can citrullinate both bacterial and human proteins, after degradation by Rgp (29), could therefore potentially contribute to the generation of de novo epitopes and the formation of autoantigens in RA. The etiological hypothesis suggests that the actions of P.PAD lead to chronic exposure of citrullinated proteins in the inflamed periodontium, which triggers loss of immune tolerance and ACPA-production. Through molecular mimicry and/or epitope spreading, ACPA eventually (cross)-react with citrullinated epitopes in the joint and provoke chronic synovial inflammation in genetically susceptible individuals. P. gingivalis may thus represent a mechanistic link between PD and RA (18, 30, 31).
Some evidence in support of this hypothesis has emerged lately: citrullinated proteins are present in the inflamed periodontium (31–33) and ACPA, although at very low levels, have been detected in sera from patients with PD (33–35). Furthermore, we have previously shown that ACPA targeting human citrullinated α-enolase cross-react with P. gingivalis enolase, forming the basis of a molecular mimicry hypothesis (30), and several studies report on an association between the anti-P. gingivalis antibody response and ACPA-status in RA patients and in individuals at risk of developing RA (36–38). Finally, in a number of mouse models of arthritis, infection with P. gingivalis exacerbates arthritis (39–41).
However, there are also a number of studies that have not been able to demonstrate an association between PD and RA (42–44). These conflicting reports could result from differences in disease classification criteria for PD, missing data on confounding factors, or the selection of controls. Perhaps even more important though, the presence of cultivable P. gingivalis, the detection of bacterial DNA in subgingival plaque, or the presence of antibodies to P. gingivalis in serum, correlate with - but do not confirm - PD (4, 34, 45, 46). Periodontitis may be triggered by periodontal pathogens other than P. gingivalis, (33), and infection by P. gingivalis does not always cause PD (4, 34, 45, 46). Hence, considering the unique ability of P. gingivalis to citrullinate proteins, studies focusing on this specific bacteria rather than PD may give better clues to the potential causal link between oral infection and RA.
In the present study, we thus set out to investigate the role of P. gingivalis in RA etiology, making use of the large and well-characterized Swedish population-based Epidemiological Investigation of RA (EIRA) case-control cohort. Since elevated serum levels of antibodies against P. gingivalis has been described as a good surrogate marker for an invasive, virulent and immunogenic P. gingivalis infection, and since arginine gingipains are among the most potent and specific virulence factors of P. gingivalis (45, 47), we chose to analyse anti-RgpB IgG levels in serum from 1,974 RA cases and 377 controls, in order to determine whether an immune response to the bacteria associates with RA diagnosis, RA-related autoantibodies, genetic- and environmental risk factors.
PATIENTS AND METHODS
Study populations
We analyzed 1,974 RA cases (diagnosed by rheumatologists according to the 1987 American College of Rheumatology criteria for RA) and 377 controls from the Swedish population-based case-control study EIRA (Epidemiological Investigation of RA) (14). All study subjects donated blood at inclusion (within one month of diagnosis for RA patients). Rheumatoid factor (RF) status was determined at participating clinics. EIRA controls were randomly selected from the population registry, to match EIRA cases on age-, gender- and residential area. Smoking data was collected by questionnaire at baseline. Subjects were categorized as ever-smokers or never-smokers (14). HLA-DRB1 subtyping and genotyping of the protein tyrosine phosphatase gene (PTPN22 rs2476601) was described before (48, 49). Sera from 65 periodontitis patients and 59 gender-matched periodontally healthy controls (all clinically verified) were collected at the Department of Dental Medicine, Karolinska Institutet, Stockholm, Sweden. Clinical criteria for periodontitis included: bone resorption with attachment loss ≥5mm, pocket probing depth ≥4mm, and bleeding on probing. Periodontally healthy controls showed no signs of periodontitis or any other periodontal disease (no gingival inflammation, clinical attachment level ≤3.5 mm, pocket probing depth ≤3.0 mm, and no bleeding on probing). Samples and data were collected with informed consent, in compliance with the Helsinki Declaration. The Regional Ethics Review Board in Stockholm, Sweden, approved the study.
ELISAs
All serum samples were analyzed by ELISA for presence of anti-RgpB IgG (the protocol, modified from Quirke et al, (28), is outlined in detail in the online supplement). The coating antigen, C-terminal hexahistidine-tagged RgpB protein, was purified from the growth medium of genetically modified P. gingivalis strain W83, by affinity chromatography on Ni-Sepharose as previously described (50). Presence/absence of different autoantibodies was determined previously, and information retrieved from the EIRA database. In brief, anti-CCP2 IgG was measured using the Immunoscan CCPlus® assay (Euro-Diagnostica AB, Malmö, Sweden), in accordance with the kit instructions; ACPA targeting citrullinated peptides from α-enolase (CEP-1; amino acid 5–21), vimentin (Cit-vim; amino acid 60–75), fibrinogen (Cit-fib; amino acid 36–52) and collagen type II (Cit-C1; amino acid 359–369) was assayed using in-house peptide ELISAs (51), and; antibodies against carbamylated fibrinogen was analyzed using an in-house protein ELISA (52).
Statistical analyses
Differences in anti-RgpB IgG levels were examined using Mann-Whitney U test for independent groups. The statistical dependence between anti-RgpB IgG levels and PD status was calculated using Spearman’s Rank Correlation Coefficient. Anti-RgpB antibody levels were categorized as positive (i.e. elevated) or negative according to the 95th percentile among the non-PD controls. To determine the association of elevated anti-RgpB IgG levels, smoking, HLA-DRB1 SE and PTPN22 with different RA subsets, odds ratios (OR) with 95% Confidence Intervals (95% CI) were calculated using unconditional logistic regression models, with unexposed cases and controls as reference group. Analyses were adjusted for age, gender and residential area. Further adjustment (smoking habits, alcohol consumption, body mass index) did not alter the results and were not retained in the final analyses. Interaction, defined as departure from additivity of effects (53), was evaluated between smoking (ever vs. never); HLA-DRB1 SE (carriers of any HLA-DRB1*01 (except *0103), *04 or *10 allele vs. non-carriers); PTPN22 polymorphism (carriers of any PTPN22 rs2476601_A alleles vs. non-carriers) and elevated anti-RgpB IgG levels. The attributable proportion due to interaction (AP) with 95% CI was calculated as previously described (54). All analyses were performed using SAS version 9.3.
RESULTS
The anti-RgpB IgG response in PD
Since antibodies to P. gingivalis are known to be elevated in PD patients (34, 46, 55), we first analyzed the anti-RgpB IgG response in a set of serum samples from 65 patients diagnosed with chronic periodontitis and 59 periodontally healthy individuals, in order to set a cut-off value for antibody positivity. Significantly higher anti-RgpB IgG levels (p<0.0001) were detected in the PD subset (median: 260 AU/ml; 75th percentile: 696 AU/ml; 25th percentile: 75 AU/ml) compared to the periodontally healthy subset (median: 89 AU/ml; 75th percentile: 153 AU/ml; 25th percentile: 51 AU/ml) (figure 1A). However, approximately one third of PD patients showed rather low anti-RgpB antibody levels, while a smaller number of periodontally healthy individuals showed elevated antibody levels. These observations were reflected by a weak, though significant, correlation between anti-RgpB IgG levels and periodontitis (r=0.37; p<0.0001), and confirm previous data - that anti-P. gingivalis antibodies should not be used as a serological marker for PD (4, 34, 45, 46). Still, a cut-off value for elevated anti-RgpB antibody levels was set using the 95th percentile among the controls.
Figure 1.
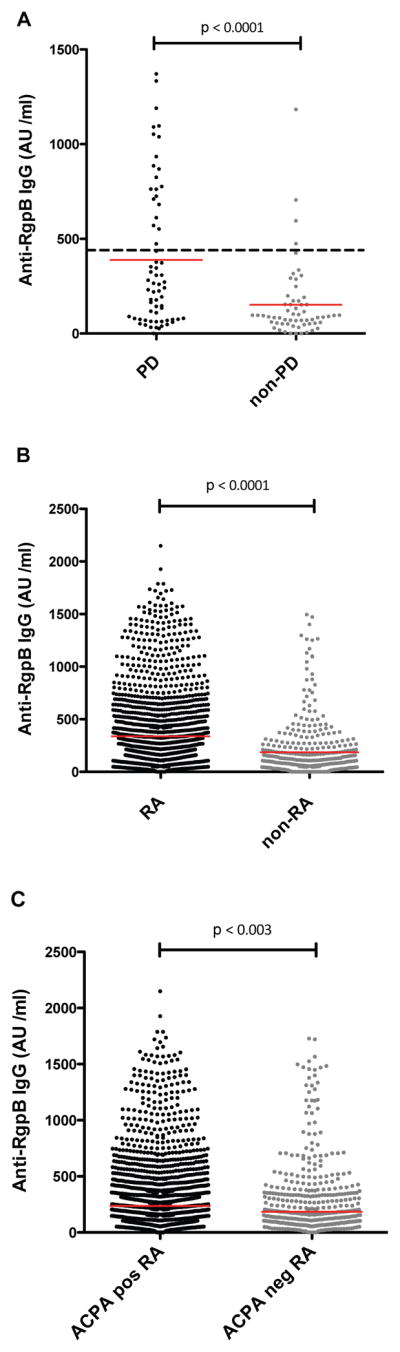
Anti-RgpB IgG levels varied between patient and control populations. Significantly higher anti-RgpB IgG levels were detected in PD patients (n=65) compared to non-PD controls (n=59) (A); in RA (n=1,974) compared to non-RA controls (n=377) (B); and in ACPA-positive RA (n=1,381) compared to ACPA-negative RA (n=593) (C). The dashed line in A indicates the cut-off value (450AU/ml) for elevated anti-RgpB antibody levels, based on the 95th percentile among the non-PD controls. The red solid lines indicate median values. ACPA = anti-citrullinated peptide/protein antibodies; AU = arbitrary units; IgG = immunoglobulin G; PD = periodontitis; RA = rheumatoid arthritis; RgpB = arginine gingipain B.
The anti-RgpB IgG response in RA
We next screened 1,974 RA cases and 377 non-RA controls from the EIRA study for the presence of elevated anti-RgpB antibody levels. The RA subset demonstrated significantly higher anti-RgpB IgG levels compared to the control subset (median: 226 AU/ml; 75th percentile: 429 AU/ml; 25th percentile: 107 AU/ml in RA, and median: 100 AU/ml; 75th percentile: 217 AU/ml; 25th percentile: 42 AU/ml in controls, p<0.0001) (figure 1B). Using the cut-off described above, 23% of RA patients and 9.4% of the non-RA controls showed elevated anti-RgpB antibody levels. Further logistic regression analysis revealed a highly significant odds ratio (OR) of 2.96 (95% CI: 2.00–4.37) for the association between elevated anti-RgpB IgG levels and RA (table 1). This association was even stronger than the well-established association between smoking and RA, which in EIRA demonstrated an odds ratio of 1.37 (95% CI: 1.07–1.74) (table 2).
Table 1.
Association between elevated anti-RgpB IgG levels and RA in in subgroups of patients, divided based on the presence/absence of ACPA
| Subgroups | Anti-RgpB IgG
|
OR* | 95% CI | |
|---|---|---|---|---|
| Negative (%) | Positive (%) | |||
| Controls | 341 (90.45) | 36 (9.55) | 1.0 | ref. |
| All RA | 1518 (76.90) | 456 (23.10) | 2.96 | 2.00–4.37 |
| ACPA-positive RA | 1041 (75.38) | 340 (24.62) | 3.24 | 2.18–4.81 |
| ACPA-negative RA | 477 (80.44) | 116 (19.56) | 2.35 | 1.51–3.65 |
Odds ratios (OR) were adjusted for age, gender and residential area.
Significant ORs are shown in bold. ACPA = anti-citrullinated peptide/protein antibodies; CI = confidence interval; IgG = immunoglobulin G; RA = rheumatoid arthritis; RgpB = arginine gingipains B
Table 2.
Association between smoking and RA in subgroups of patients, divided based on the presence/absence of ACPA
| Groups | Smoking
|
OR* | 95% CI | |
|---|---|---|---|---|
| Never (%) | Ever (%) | |||
| Controls | 137 (36.63) | 237 (63.37) | 1.0 | ref. |
| All RA | 603 (30.61) | 1367 (69.39) | 1.37 | 1.07–1.74 |
| ACPA-positive RA | 331 (26.91) | 899 (73.09) | 1.67 | 1.29–2.17 |
| ACPA-negative RA | 270 (37.60) | 448 (62.40) | 1.00 | 0.75–1.32 |
Odds ratios (OR) were adjusted for age, gender and residential area.
Significant ORs are shown in bold. ACPA= anti-citrullinated peptide/protein antibodies; CI= confidence interval; RA= rheumatoid arthritis
The anti-RgpB IgG response in relation to RA-associated autoantibodies
To investigate whether the antibody response to P. gingivalis RgpB was specifically linked to the ACPA-response, we divided RA patients into ACPA-positive and ACPA-negative subsets. The ACPA-status was determined based on CCP2-reactivity as well as reactivity with specific citrullinated epitopes on α-enolase (CEP-1), vimentin (Cit-vim60-75), fibrinogen (Cit-fib36-52) and collagen type II (Cit-C1). This broader type of ACPA analysis captured more ACPA positive patients (70%), than if only the standard CCP2 ELISA assay had been used (63%).
Subdividing RA based on ACPA-status revealed significantly higher anti-RgpB IgG levels (p<0.003) in the ACPA-positive subset (median: 231 AU/ml; 75th percentile: 514 AU/ml; 25th percentile: 108 AU/ml), compared to the ACPA-negative subset (median: 166 AU/ml; 75th percentile: 348; 25th percentile: 79) (figure 1C). This was also reflected by a significantly stronger association of elevated anti-RgpB IgG levels with ACPA-positive RA than with ACPA-negative RA, with odds ratios of 3.24 (95% CI: 2.18–4.81) and 2.35 (95% CI: 1.51–3.65), respectively (3.24 vs. 2.35, p=0.017) (table 1). Since presence of ACPA has previously been reported in non-RA PD subjects (32–34), we also screened the PD patients and the non-PD controls in our study for presence of ACPA, although no ACPA response could be detected in any of these non-RA individuals (data not shown).
To further investigate the association between the ACPA-response and the anti-RgpB antibody response in RA, we next examined the fine-specificity of the ACPA-response in more detail. Anti-RgpB IgG was analyzed in different subsets of RA, defined by the presence/absence of anti-CEP-1, anti-Cit-vim65-70, anti-Cit-fib36-52 or anti-Cit-C1 antibodies. These analyses were performed in relation to the CCP2 status, which was used as a surrogate marker for the overall ACPA-response. None of the investigated ACPA fine-specificities revealed any significant association with anti-RgpB IgG, beyond the effect of the overall ACPA-response (as measured using the CCP2 test). Similar analyses, based on rheumatoid factor (RF) status, or presence/absence of antibodies to carbamylated fibrinogen, showed no differences between subsets (supplementary table 1).
The anti-RgpB IgG response in relation to cigarette smoking
Since PD patients showed elevated anti-RgpB antibody levels (figure 1A), and since smoking is a well-established risk factor for PD (13), we first examined the relationship between smoking status and anti-RgpB IgG exposure in EIRA. Potentially, the positive association between anti-RgpB IgG and ACPA-positive RA (figure 1C and table 1) could result from smoking as a confounding factor. However, we could not identify any association between smoking and elevated anti-RgpB IgG levels in EIRA RA cases. Among EIRA controls, we could even observe an inverse association (supplementary table 2). Instead, smoking and elevated anti-RgpB IgG levels were independently associated with ACPA-positive RA, with odds ratios of 1.36 (95% CI: 1.01–1.83) and 2.40 (95% CI: 1.29–4.46), respectively. Moreover, a significant additive interaction was observed between the two factors, with an odds ratio of 5.35 (95% CI: 3.07–9.33), and a significant attributable proportion (AP) due to the interaction of 0.48 (95% CI: 0.12–0.85). In ACPA-negative RA on the other hand, neither smoking nor elevated anti-RgpB IgG levels showed any independent associations, and there was no interaction (table 3).
Table 3.
Additive Interaction between smoking and elevated anti-RgpB IgG levels in subgroups of RA, divided based on the presence/absence of ACPA
| Factors | Cases (%) | Controls (%) | OR* (95% CI) | |
|---|---|---|---|---|
|
| ||||
| Smoking | Anti-RgpB IgG | |||
| ACPA-positive RA | ||||
| − | − | 289 (70.83) | 119 (29.17) | 1.0 ref. |
| + | − | 751 (77.42) | 219 (22.58) | 1.36 (1.01–1.83) |
| − | + | 99 (84.62) | 18 (15.38) | 2.40 (1.29–4.46) |
| + | + | 241 (93.05) | 18 (6.95) | 5.35 (3.07–9.33) |
| AP | 0.48 (0.12–0.85) | |||
| ACPA-negative RA | ||||
| + | − | 299 (57.72) | 219 (42.28) | 1.0 ref. |
| − | − | 176 (59.66) | 119 (40.34) | 1.09 (0.78–1.52) |
| − | + | 39 (68.42) | 18 (31.58) | 1.67 (0.86–3.24) |
| + | + | 76 (80.85) | 18 (19.15) | 3.01 (1.69–5.35) |
| AP | 0.42 (−0.07–0.91) | |||
Odds ratios (OR) were adjusted for age, gender and residential area.
Significant ORs and AP values are shown in bold. ACPA = anti-citrullinated peptide/protein antibodies; AP = attributable proportion due to additive interaction; IC = confidence interval; IgG = immunoglobulin G; RA = rheumatoid arthritis; RgpB = arginine gingipain B
The anti-RgpB IgG response in relation to RA risk genes
We also investigated the association between elevated anti-RgpB IgG levels and HLA-DRB1 SE alleles and PTPN22 polymorphism. None of these genetic risk factors associated with elevated anti-RgpB IgG levels (supplementary table 2). Instead, HLA-DRB1 SE and anti-RgpB antibodies showed independent associations with ACPA-positive RA (ORs = 5.66 (95% CI: 4.22–7.59) and 4.11 (95% CI: 2.3–7.35), respectively), and to a minor extent also with ACPA-negative RA (ORs = 1.56 (95% CI: 1.14–2.13) and 2.77 (95% CI: 1.52–5.03), respectively). In ACPA-positive RA, there was more than an additive effect among double exposed cases (OR = 16.62; 95% CI: 9.26–29.83), and a significant interaction (AP = 0.47; 95% CI: 0.15–0.79), not present in ACPA-negative RA (table 4). No interaction could be observed between anti-RgpB IgG exposure and PTPN22 polymorphism, but the number of double exposed subjects was small among controls, which hampers the interpretation (table 5).
Table 4.
Additive Interaction between HLA DRB1 SE allele and elevated anti-RgpB IgG levels in subgroups of RA, divided based on the presence/absence of ACPA
| Factors
|
Cases (%) | Controls (%) | OR* (95%CI) | |
|---|---|---|---|---|
| Any SE | Anti-RgpB IgG | |||
| ACPA-positive RA | ||||
| − | − | 185 (51.53) | 174 (48.47) | 1.0 ref. |
| + | − | 841 (85.12) | 147 (14.88) | 5.66 (4.22–7.59) |
| − | + | 74 (79.57) | 19 (20.43) | 4.11 (2.30–7.35) |
| + | + | 262 (94.93) | 14 (5.07) | 16.62 (9.26–29.83) |
| AP | 0.47 (0.15–0.79) | |||
| ACPA-negative RA | ||||
| − | − | 209 (54.57) | 174 (45.43) | 1.0 ref. |
| + | − | 263 (64.15) | 147 (35.85) | 1.56 (1.14–2.13) |
| − | + | 60 (75.95) | 19 (24.05) | 2.77 (1.52–5.03) |
| + | + | 56 (80.00) | 14 (20.00) | 3.10 (1.62–5.92) |
| AP | −0.07 (−0.89–0.75) | |||
Odds ratios were adjusted for age, gender and residential area.
Significant ORs and AP values are shown in bold. ACPA = anti-citrullinated peptide/protein antibodies; AP = attributable proportion due to additive interaction; IC = confidence interval; IgG = immunoglobulin G; RA = rheumatoid arthritis; RgpB = arginine gingipain B; SE: shared epitope
Table 5.
Additive Interaction between PTPN22 polymorphism and elevated anti-RgpB IgG levels in subgroups of RA, divided based on the presence/absence of ACPA
| Factors
|
Cases (%) | Controls (%) | OR* (95%CI) | |
|---|---|---|---|---|
| PTPN22 | Anti-RgpB IgG | |||
| ACPA-positive RA | ||||
| − | − | 704 (73.95) | 248 (26.05) | 1.0 ref. |
| + | − | 311 (79.54) | 80 (20.46) | 1.38 (1.01–1.90) |
| − | + | 231 (88.51) | 30 (11.49) | 2.98 (1.92–4.64) |
| + | + | 103 (95.37) | 5 (4.63) | 6.08 (2.42–15.23) |
| AP | 0.47 (−0.10–0.99) | |||
| ACPA-negative RA | ||||
| − | − | 334 (57.39) | 248 (42.61) | 1.0 ref. |
| + | − | 133 (62.44) | 80 (37.56) | 1.27 (0.89–1.83) |
| − | + | 89 (74.79) | 30 (25.21) | 2.22 (1.36–3.62) |
| + | + | 27 (84.38) | 5 (15.63) | 4.00 (1.46–10.91) |
| AP | 0.38 (−0.29–1.05) | |||
Odds ratios (OR) were adjusted for age, gender and residential area.
Significant ORs and AP values are shown in bold. ACPA = anti-citrullinated peptide/protein antibodies; AP = attributable proportion due to additive interaction; IC = confidence interval; IgG = immunoglobulin G; RA = rheumatoid arthritis; RgpB = arginine gingipain B; PTPN22: protein tyrosine phosphatase non-receptor type 22
DISCUSSION
Here we present data in support of a role for the oral pathogen P. gingivalis in the etiology of RA. To our knowledge, this is the largest epidemiological investigation of an immune response to P. gingivalis in patients with RA, and to our knowledge this is the strongest association described to date. The association between RA and elevated anti-RgpB antibody levels was stronger than the association between RA and the well-established risk factor smoking. Notably, the association with anti-RgpB IgG was independent of smoking, and in EIRA controls, we could even observe an inverse association, in line with previous reports demonstrating lower anti-P. gingivalis antibody levels in smokers compared to non-smokers (34, 55).
Smoking is a well-known risk factor for PD (13), but importantly, we did not investigate PD in the present study. We investigated the antibody response to a P. gingivalis-specific antigen. While antibodies to P. gingivalis clearly associate with PD, as shown by us (figure 1A) and by others (34, 46, 55), these antibodies should not be used as surrogate markers for diagnosing PD. P. gingivalis does not have to be present for PD to develop (34) and many individuals carry P. gingivalis without any symptoms of PD (46).
Our data, demonstrating a heightened antibody response to a P. gingivalis-specific antigen in RA compared to controls, is in agreement with a number of previous reports (36–38). However more recently, Mikuls and colleagues were unable to detect any differences in anti-P. gingivalis antibody concentrations between RA and controls (2), and in a separate study, the frequency of anti-P. gingivalis antibodies was actually found to be non-significantly lower in patients with RA compared to controls (6). These conflicting reports may result from using different P. gingivalis-derived antigens when analyzing the anti-P. gingivalis antibody response. We choose RgpB because it is one of the most potent virulence factors described for P. gingivalis (24). Accordingly, we anticipate that a strong anti-RgpB IgG response will reflect infection, current or historical, with a pathogenic, virulent, invasive and immunogenic strain of the bacteria (23, 45), likely to promote increased protein citrullination in vivo, in line with the etiological hypothesis (18, 30, 31). Moreover, by using purified hexahistidine-tagged P. gingivalis-specific RgpB protein as coating antigen in our ELISA assay, rather than whole bacterial lysates or the outer membrane, as used by others (4, 36–38), we also avoid citrullinated epitopes, and thereby the potential false positive result due to cross-reactive ACPA. The discrepant results between the different studies may also be explained by differences between RA cohorts. Factors such as disease duration, treatment, smoking history, ethnicity, age and gender of the study population may all affect the outcome. Moreover, bacterial strain diversity may influence the data. Different clonal types of P. gingivalis with differences in expression of virulence factors exist (56, 57), perhaps with variations in P. PAD activity. Additionally, geographical differences in the profiles of periodontal microbiota have been reported (58).
Interestingly, we could observe interactions between elevated anti-RgpB IgG levels and both smoking and HLA-DRB1 SE, only in ACPA-positive RA. These data supports the hypothesis of P. gingivalis as an etiological agent in the development of ACPA-positive RA, where smoking and HLA-DRB1 SE alleles already constitute well-established risk factors. However, anti-RgpB IgG exposure itself associated with both ACPA-positive and ACPA-negative disease (although to a significantly lesser extent with ACPA-negative disease). This observation could be due to the presence of other ACPA fine-specificities (not investigated here) in the subset that we have defined as ACPA-negative RA. We have previously shown that the CCP2 assay does not capture all ACPA fine-specificities (51), and even though we expanded our current analyses to also include antibodies targeting CEP-1, Cit-vim65-70, Cit-fib36-52 and Cit-C1, we have by no means covered all possible ACPA fine-specificities. Alternatively, P. gingivalis may be triggering/driving RA through other mechanisms than solely via P. PAD and ACPA production. For example, P. gingivalis may trigger/drive RA by inducing IL-1 and IL-6 production, giving rise to a pathogenic Th17 response, as suggested from animal studies (39, 41).
Additionally, we cannot rule out the possibility that the enhanced antibody response to P. gingivalis arginine gingipain B is a consequence, rather than a cause, of RA. Although, we would argue against this, since the patients in EIRA comprise only newly diagnosed RA cases, enrolled within one month of diagnosis, and with symptoms for less than one year. Moreover, in studies of individuals at increased risk of developing RA, but with no apparent symptoms, significant associations between the anti-P. gingivalis antibody response and the presence of RA-related autoantibodies have been reported (36, 59), supporting a causative link between P. gingivalis infection and the development of RA. However, in conflict with this hypothesis, is the recent study of arthralgia patients by de Smit and colleagues, which concluded that elevated anti-P. gingivalis antibody levels could not predict RA development within 12 months (60). Notably, though, that study also failed to confirm known associations between CRP, age, gender and smoking with future development of RA, hence de Smit’s data needs to be reproduced.
In summary, we can demonstrate an epidemiological association between elevated anti-P. gingivalis arginine gingipain B antibody levels and RA diagnosis, that is even stronger than the well-known association between smoking and RA. Our study also reveals statistically significant interactions, which can be interpreted as biological interactions (53), between elevated anti-RgpB IgG levels and smoking, as well as HLA-DRB1 SE, in ACPA-positive RA. Hence, based on the data presented herein, we conclude that the oral pathogen Porphyromonas gingivalis remains a credible candidate for triggering and/or driving autoimmunity and autoimmune disease in a subset of RA.
Supplementary Material
Acknowledgments
The authors wish to thank: Study participants, research nurses, and the EIRA study group for their contributions; Professor Lars Klareskog, for support and scientific input and for establishing the EIRA study; scientists previously involved in the generation of data for the EIRA database, specifically: Associate Professor Leonid Padyukov, Dr Patrick Stolt, Dr Camilla Bengtsson and Professor Rikard Holmdahl. This work was supported by grants from the Swedish Research Council, the Swedish Rheumatic Foundation and the EU-funded FP7 projects Gums & Joints (FP7-Health-2010-261460) and TRIGGER (FP7-Health-2013-306029) and the ITN project Rapid (290246). The work of BP and JP was additionally supported by grants from NIH (DE022597) and NCN (2011/01/B/NZ6/00268).
Footnotes
COMPETING INTERESTS
None of the authors have any conflicts of interest in relation to this publication.
The authors declare that they have no conflicting financial interest and have not received funding from commercial sources in the context of this study.
References
- 1.Dissick A, Redman RS, Jones M, Rangan BV, Reimold A, Griffiths GR, et al. Association of periodontitis with rheumatoid arthritis: a pilot study. J Periodontol. 2010;81:223–30. doi: 10.1902/jop.2009.090309. [DOI] [PubMed] [Google Scholar]
- 2.Mikuls T, Payne JB, Yu F, Thiele GM, Reynolds RJ, Cannon GW, et al. Periodontitis and Porphyromonas gingivalis in Patients With Rheumatoid Arthritis. Arthtitis & Rheumatology. 2014;66:1090–1100. doi: 10.1002/art.38348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Pablo P, Dietrich T, McAlindon T. Association of periodontal disease and tooth loss with rheumatoid arthritis in the US population. J Rheumatol. 2008;35:70–76. [PubMed] [Google Scholar]
- 4.de Smit Menke, Westra Johanna, Vissink Arjan, Doornbos-van der Meer Berber, Brouwer Elisabeth, Winkelhoff AJv. Periodontitis in established rheumatoid arthritis patients: a cross-sectional clinical, microbiological and serological study. Arthritis Research & Therapy. 2012;14(R222):1–10. doi: 10.1186/ar4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mercado F, Marshall R, Klestov A, Bartold P. Relationship between rheumatoid arthritis and periodontitis. J Periodontol. 2001;72:779–787. doi: 10.1902/jop.2001.72.6.779. [DOI] [PubMed] [Google Scholar]
- 6.Scher J, Ubeda C, Equinda M, Khanin R, Buischi Y, Viale A, et al. Periodontal disease and the oral microbiota in new-onset rheumatoid arthritis. Arthritis Rheum. 2012;64:3083–3094. doi: 10.1002/art.34539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pischon N, Pischon T, Kroger J, Gülmez E, Kleber B, Bernimoulin J, et al. Association among rheumatoid arthritis, oral hygiene, and periodontitis. J Periodontol. 2008;79:979–986. doi: 10.1902/jop.2008.070501. [DOI] [PubMed] [Google Scholar]
- 8.Mercado F, Marshall R, Klestov A, Bartold P. Is there a relationship between rheumatoid arthritis and periodontal disease? J Clin Periodontol. 2000;27:267–272. doi: 10.1034/j.1600-051x.2000.027004267.x. [DOI] [PubMed] [Google Scholar]
- 9.Chen H, Nicole H, Chen Y, Chen T, Chou P, Lee Y, et al. Association between a history of periodontitis and the risk of rheumatoid arthritis: a nationwide, population-based, case–control study. Ann Rheum Dis. 2013;72:1206–1211. doi: 10.1136/annrheumdis-2012-201593. [DOI] [PubMed] [Google Scholar]
- 10.Koziel J, Mydel P, Potempa J. The Link Between Periodontal Disease and Rheumatoid Arthritis. Curr Rheumatol Rep. 2014;16(408):1–7. doi: 10.1007/s11926-014-0408-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Pablo P, Chapple I, Buckley C, Dietrich T. Periodontitis in systemic rheumatic diseases. Nat Rev Rheumatol. 2009;5:218–224. doi: 10.1038/nrrheum.2009.28. [DOI] [PubMed] [Google Scholar]
- 12.Mercado F, Marshall R, Bartold P. Inter-relationships between rheumatoid arthritis and periodontal disease. J Clin Periodontol. 2003;30:761–772. doi: 10.1034/j.1600-051x.2003.00371.x. [DOI] [PubMed] [Google Scholar]
- 13.Tomar S, Asma S. Smoking-attributable periodontitis in the United States: Findings from NHANES III. J Periodontol. 2000;71:743–751. doi: 10.1902/jop.2000.71.5.743. [DOI] [PubMed] [Google Scholar]
- 14.Stolt P, Bengtsson C, Nordmark B, Lindblad S, Lundberg I, Klareskog L, et al. Quantification of the influence of cigarette smoking on rheumatoid arthritis: results from a population based case-control study, using incident cases. Ann Rheum Dis. 2003;62:835–841. doi: 10.1136/ard.62.9.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bonfil J, Dillier F, Mercier P, Reviron D, Foti B, Sambuc R, et al. A “case control” study on the role of HLA DR4 in severe periodontitis and rapidly progressive periodontitis: identification of types and subtypes using molecular biology (PCR.SSO) J Clin Periodontol. 1999;26:77–84. doi: 10.1034/j.1600-051x.1999.260203.x. [DOI] [PubMed] [Google Scholar]
- 16.Gregersen P, Silver J, Winchester R. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987;30:1205–1213. doi: 10.1002/art.1780301102. [DOI] [PubMed] [Google Scholar]
- 17.Snyderman R, McCarty G. Analogous mechanism of tissue destruction in rheumatoid arthritis and periodontal disease. In: Genco R, Mergenhagen S, editors. Host-parasite interactions in periodontal diseases. American Society for Microbiology; 1982. pp. 354–362. [Google Scholar]
- 18.Rosenstein E, Greenwald R, Kushner L, Weissmann G. Hypothesis: the humoral immune response to oral bacteria provides a stimulus for the development of rheumatoid arthritis. Inflammation. 2004;28:311–318. doi: 10.1007/s10753-004-6641-z. [DOI] [PubMed] [Google Scholar]
- 19.Rantapää-Dahlqvist S, de Jong B, Berglin E, Hallmans G, Wadell G, Stenlund H, et al. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. 2003;48:2741–2749. doi: 10.1002/art.11223. [DOI] [PubMed] [Google Scholar]
- 20.Brown L, Loe H. Prevalence, extent, severity and progression of periodontal disease. Periodontol 2000. 1993;2:57–71. doi: 10.1111/j.1600-0757.1993.tb00220.x. [DOI] [PubMed] [Google Scholar]
- 21.Socransky S, Haffajee A, Cugini M, Smith C, Kent R. Microbial complexes in subgingival plaque. J Clin Periodontol. 1998;25:134–144. doi: 10.1111/j.1600-051x.1998.tb02419.x. [DOI] [PubMed] [Google Scholar]
- 22.Holt S, Kesavalu L, Walker S, Genco C. Virulence factors of Porphyromonas gingivalis. Periodontology. 2000;20:168–238. doi: 10.1111/j.1600-0757.1999.tb00162.x. [DOI] [PubMed] [Google Scholar]
- 23.Mysak J, Podzimek S, Sommerova P, Lyuya-Mi Y, Bartova U, Janatova T, et al. Porphyromonas gingivalis: Major Periodontopathic Pathogen Overview. Journal of Immunology Research. 2014:1–8. doi: 10.1155/2014/476068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kadowaki T, Nakayama K, Yoshimura F, Okamoto K, Abe N, Yamamoto K, et al. Arg-gingipain Acts as a Major Processing Enzyme for Various Cell Surface Proteins in Porphyromonas gingivalis. J Biol Chem. 1998;273:29072–29076. doi: 10.1074/jbc.273.44.29072. [DOI] [PubMed] [Google Scholar]
- 25.Chen Z, Potempa J, Polanowski A, WikstromII M, Travis J. Purification and Characterization of a 50-kDa Cysteine Proteinase (Gingipain) from Porphyromonas gingivalis. The journal of Biological Chemistry. 1992;267:18895–18901. [PubMed] [Google Scholar]
- 26.Guo Y, Nguyen K, Potempa J. Dichotomy of gingipains action as virulence factors: from cleaving substrates with the precision of a surgeon's knife to a meat chopper-like brutal degradation of proteins. Periodontology. 2010;54:15–44. doi: 10.1111/j.1600-0757.2010.00377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McGraw W, Potempa J, Farley D, Travis J. Purification, characterization, and sequence analysis of a potential virulence factor from Porphyromonas gingivalis, peptidylarginine deiminase. Infect Immun. 1999;67:3248–3256. doi: 10.1128/iai.67.7.3248-3256.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quirke A-M, Lugli E, Wegner N, Hamilton B, Charles P, Chowdhury M, et al. Heightened immune response to autocitrullinated Porphyromonas gingivalis peptidylarginine deiminase: a potential mechanism for breaching immunologic tolerance in rheumatoid arthritis. Ann Rheum Dis. 2014;73:263–269. doi: 10.1136/annrheumdis-2012-202726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wegner N, Wait R, Sroka A, Eick S, Nguyen K, Lundberg K, et al. Peptidylarginine deiminase from Porphyromonas gingivalis citrullinates human fibrinogen and alpha-enolase: implications for autoimmunity in rheumatoid arthritis. Arthritis Rheum. 2010;62:2662–2672. doi: 10.1002/art.27552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lundberg K, Kinloch A, Fisher B, Wegner N, Wait R, Charles P, et al. Antibodies to citrullinated alpha-enolase peptide 1 are specific for rheumatoid arthritis and cross-react with bacterial enolase. Arthritis Rheum. 2008;58:3009–3019. doi: 10.1002/art.23936. [DOI] [PubMed] [Google Scholar]
- 31.Lundberg K, Wegner N, Yucel-Lindberg T, Venables P. Periodontitis in RAthe citrullinated enolase connection. Nat Rev Rheumatol. 2010;6:727–730. doi: 10.1038/nrrheum.2010.139. [DOI] [PubMed] [Google Scholar]
- 32.Nesse W, Westra J, van der Wal J, Abbas F, Nicholas A, Vissink A, et al. The periodontium of periodontitis patients contains citrullinated proteins which may play a role in ACPA (anti-citrullinated protein antibody) formation. J Clin Periodontol. 2012;39:599–607. doi: 10.1111/j.1600-051X.2012.01885.x. [DOI] [PubMed] [Google Scholar]
- 33.Harvey G, Fitzsimmons T, Dhamarpatni A, Marchant C, Haynes D, Bartold P. Expression of of peptidylarginine deiminase-2 and -4, citrullinated proteins and anti-citrullinated protein antibodies in human gingiva. J Periodontal Res. 2013;48:252–261. doi: 10.1111/jre.12002. [DOI] [PubMed] [Google Scholar]
- 34.Lappin D, Apatzidou D, Quirke A-M, Oliver-Bell J, Butcher J, Kinane F, et al. Influence of periodontal disease, Porphyromonas gingivalis and cigarette smoking on systemic anti-citrullinated peptide antibody titres. J Clin Periodontol. 2013;40:907–915. doi: 10.1111/jcpe.12138. [DOI] [PubMed] [Google Scholar]
- 35.Hendler A, Mulli T, Hughes F, Perrett D, Bombardieri M, Houri-Haddad Y, et al. Involvement of autoimmunity in the pathogenesis of aggressive periodontitis. Journal of Dental Research. 2010;89:1389–1394. doi: 10.1177/0022034510381903. [DOI] [PubMed] [Google Scholar]
- 36.Hitchon C, Chandad F, Ferucci E, Willemze A, Ioan-Facsinay A, van der Woude D, et al. Antibodies to Porphyromonas gingivalis are associated with anticitrullinated protein antibodies in patients with rheumatoid arthritis and their relatives. J Rheumatol. 2010;37:1105–1112. doi: 10.3899/jrheum.091323. [DOI] [PubMed] [Google Scholar]
- 37.Mikuls T, Payne J, Reinhardt R, Thiele G, Maziarz E, Cannella A, et al. Antibody responses to Porphyromonas gingivalis (P. gingivalis) in subjects with rheumatoid arthritis and periodontitis. Int Immunopharmacol. 2009;9:38–42. doi: 10.1016/j.intimp.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ogrendik M, Kokino S, Ozdemir F, Bird F, Hamlet S. Serum antibodies to oral anaerobic bacteria in patients with rheumatoid arthritis. MedGenMed. 2005:7–2. [PMC free article] [PubMed] [Google Scholar]
- 39.Marchesan J, Gerow E, Schaff R, Taut A, Shin S, Sugai J, et al. Porphyromonas gingivalis oral infection exacerbates the development and severity of collagen-induced arthritis. Arthritis ResTher. 2013;15:R186. doi: 10.1186/ar4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maresz K, Hellvard A, Sroka A, Adamowicz K, Bielecka E, Koziel J, et al. Porphyromonas gingivalis facilitates the development and progression of destructive arthritis through its unique bacterial peptidylarginine deiminase (PAD) PLoS Pathog. 2013;9(e1003627):1–10. doi: 10.1371/journal.ppat.1003627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.de Aquino S, Abdollahi-Roodsaz S, Koenders M, Loo F, Pruijn G, Marijnissen R, et al. Periodontal pathogens directly promote autoimmune experimental arthritis by inducing a TLR2- and IL-1-driven Th17 response. J Immunology. 2014;192(9):4103–4111. doi: 10.4049/jimmunol.1301970. [DOI] [PubMed] [Google Scholar]
- 42.Arkema E, Karlson E, Costenbader K. A prospective study of periodontal disease and risk of rheumatoid arthritis. J Rheumatol. 2010;37:1800–1804. doi: 10.3899/jrheum.091398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Demmer R, Molitor J, Jacobs D, Michalowicz B. Periodontal disease, tooth loss and incident rheumatoid arthritis: results from the First National Health and Nutrition Examination Survey and its epidemiological follow-up study. J Clin Periodontol. 2011;38:998–1006. doi: 10.1111/j.1600-051X.2011.01776.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pedersen M, Jacobsen S, Klarlund M, Pedersen B, Wiik A, Wohlfahrt J, et al. Environmental risk factors differ between rheumatoid arthritis with and without auto-antibodies against cyclic citrullinated peptides. Arthritis Res Ther. 2006;8(R133):1–15. doi: 10.1186/ar2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haffajee A, Socransky S. Microbial etiological agents of destructive periodontal diseases. Periodontology. 2000;5:78–111. doi: 10.1111/j.1600-0757.1994.tb00020.x. [DOI] [PubMed] [Google Scholar]
- 46.Pussinen P, Könönen E, Paju S, Hyvärinen K, Gursoy U, Huumonen S, et al. Periodontal pathogen carriage, rather than periodontitis, determines the serum antibody levels. J Clin Periodontol. 2011;38:405–411. doi: 10.1111/j.1600-051X.2011.01703.x. [DOI] [PubMed] [Google Scholar]
- 47.Potempa J, Banbula A, Travis J. Role of bacterial proteinases in matrix destruction and modulation of host responses. Periodontol 2000. 2000;24:153–159. doi: 10.1034/j.1600-0757.2000.2240108.x. [DOI] [PubMed] [Google Scholar]
- 48.Padyukov L, Silva C, Stolt P, Alfredsson L, Klareskog L. A gene-environment interaction between smoking and shared epitope genes in HLA-DR provides a high risk of seropositive rheumatoid arthritis. Arthritis Rheum. 2004;50:3085–3092. doi: 10.1002/art.20553. [DOI] [PubMed] [Google Scholar]
- 49.Kallberg H, Padyukov L, Plenge R. Gene-gene and gene-environment interactions involving HLA-DRB1, PTPN22, and smoking in two subsets of rheumatoid arthritis. Am J Hum Genet. 2007;80:867–875. doi: 10.1086/516736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Potempa J, Nguyen K. Purification and characterization of gingipains. Curr Protoc Protein Sci. 2007:1–27. doi: 10.1002/0471140864.ps2120s49. [DOI] [PubMed] [Google Scholar]
- 51.Lundberg K, Bengtsson C, Kharlamova N, Reed E, Jiang X, Kallberg H, et al. Genetic and environmental determinants for disease risk in subsets of RA defined by the ACPA fine specificity profile. Ann Rheum Dis. 2013;72:652–658. doi: 10.1136/annrheumdis-2012-201484. [DOI] [PubMed] [Google Scholar]
- 52.Jiang X, Trouw L, van Wesemael T, Shi J, Bengtsson C, Källberg H, et al. Anti-CarP antibodies in two large cohorts of patients with rheumatoid arthritis and their relationship to genetic risk factors, cigarette smoking and other autoantibodies. Ann Rheum Dis. 2014;73:1761–1768. doi: 10.1136/annrheumdis-2013-205109. [DOI] [PubMed] [Google Scholar]
- 53.Rothman K, Greenland S, Walker A. Concepts of interaction. Am J Epidemiol. 1980;112:467–470. doi: 10.1093/oxfordjournals.aje.a113015. [DOI] [PubMed] [Google Scholar]
- 54.Hosmer D, Lemeshow S. Confidence interval estimation of interaction. Epidemiology. 1992;3:452–455. doi: 10.1097/00001648-199209000-00012. [DOI] [PubMed] [Google Scholar]
- 55.Vlachojannis C, Dye B, Herrera-Abreu M, Pikdöken L, Lerche-Sehmm J, Pretzl B, et al. Determinants of serum IgG responses to periodontal bacteria in a nationally representative sample of US adults. Journal of Clinical Periodontology. 2010;37:685–689. doi: 10.1111/j.1600-051X.2010.01592.x. [DOI] [PubMed] [Google Scholar]
- 56.Tachibana-Ono M, Yoshida A, Kataoka S, Ansai T, Shintani Y, Takahashi Y, et al. Identification of the genes associated with a virulent strain of Porphyromonas gingivalis using the subtractive hybridization technique. Oral Microbiology Immunology. 2008;23:84–87. doi: 10.1111/j.1399-302X.2007.00396.x. [DOI] [PubMed] [Google Scholar]
- 57.Sundqvist G, Figdor D, Hänström L, Sörlin S, Sandström G. Phagocytosis and virulence of different strains of Porphyromonas gingivalis. Scand J Dent Res. 1991;99:117–129. doi: 10.1111/j.1600-0722.1991.tb01874.x. [DOI] [PubMed] [Google Scholar]
- 58.Haffajeem A, Bogren A, Hasturk H, Feres M, Lopez N, Socransky S. Subgingival microbiota of chronic periodontitis subjects from different geographic locations. J Clin Periodontol. 2004;31:996–1002. doi: 10.1111/j.1600-051X.2004.00597.x. [DOI] [PubMed] [Google Scholar]
- 59.Mikuls T, Thiele G, Deane K, Payne J, O'Dell J, Yu F, et al. Porphyromonas gingivalis and disease-related autoantibodies in individuals at increased risk for rheumatoid arthritis. Arthritis Rheum. 2012;64:3522–3530. doi: 10.1002/art.34595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.de Smit M, van de Stadt L, Janssen K, Doornbos-van der Meer B, Vissink A, van Winkelhoff A, et al. Antibodies against Porphyromonas gingivalis in seropositive arthralgia patients do not predict development of rheumatoid arthritis. Ann Rheum Dis. 2014:1–2. doi: 10.1136/annrheumdis-2013-204594. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.