

Abstract
Objective
Determine whether repeatedly overloading healthy cartilage disrupts mitochondrial function in a manner similar to that associated with osteoarthritis pathogenesis.
Methods
We exposed normal articular cartilage on bovine osteochondral explants to 1 day or 7 consecutive days of cyclic axial compression (0.25 or 1.0 MPa, 0.5 Hz, 3 hours) and evaluated effects on chondrocyte viability, ATP concentration, reactive oxygen species (ROS) production, indicators of oxidative stress, respiration, and mitochondrial membrane potential.
Results
Neither 0.25 nor 1.0 MPa cyclic compression caused extensive chondrocyte death, macroscopic tissue damage, or overt changes in stress-strain behavior. After one day of loading, differences in respiratory activities between the 0.25 and 1.0 MPa groups were minimal; after 7 loading days, however, respiratory activity and ATP levels were suppressed in the 1.0 MPa group relative to the 0.25 MPa group, an effect prevented with pretreatment with 10 mM N-acetylcysteine. These changes were accompanied by increased proton leakage and decreases in mitochondrial membrane potential as well as by increased ROS formation indicated by dihydroethidium staining and glutathione oxidation.
Conclusion
Repeated overloading leads to chondrocyte oxidant-dependent mitochondrial dysfunction. This mitochondrial dysfunction may contribute to destabilization of cartilage during various stages of OA in distinct ways by disrupting chondrocyte anabolic responses to mechanical stimuli.
Introduction
One of the most widely recognized factors placing otherwise healthy people at risk for osteoarthritis (OA) is joint overuse. Epidemiologic studies have shown that individuals in occupations involving heavy repetitive loading of their joints as well as elite athletes are at increased risk for developing OA [1,2; reviewed 3,4]. Studies suggest that manipulations of key factors including surface mechanics, inflammation, or oxidative injury can alleviate different aspects of cellular injury from overloading [5,6,7]. Among the outcomes, antioxidants appear to combat overactivity after overload of a mechanotransductive pathway whereby loading stimulates rotenone-inhibitable electron transport chain (ETC) activity, subsequent ROS generation as a byproduct of respiration, and, ultimately, increased anabolism by chondrocytes [8,9,10,11,12]. The hypothesis that ROS function as a metabolism-mediating signal in chondrocytes was put forth as early as 1997 by Lee and Urban in studies showing that without oxygen, chondrocyte anabolic function ceases and that this can be restored with addition of exogenous oxidants [13,14]. Following their work in this pathway, we have recently demonstrated that ROS produced as a result of ETC activity mediate cellular injury after excessive loading once intracellular antioxidant defenses are exceeded [10]. We believe this mitochondrial pathway to be of critical importance to early and ongoing pathogenic factors in the natural history of OA.
Growing numbers of studies focused upon metabolic and redox nodes like the mitochondria have identified several proteomic disruptions associated with the OA phenotype [15,16,17]. Alterations in proteins in the tricarboxylic acid cycle, electron transport, and ROS scavenging suggest that large-scale dysregulation of mitochondrial function occurs during OA. It has been shown that OA chondrocytes, as well as healthy chondrocytes with suppressed manganese superoxide dismutase (MnSOD) expression, exhibit depressed mitochondrial membrane potential, increased proton leakage, and lipid peroxidation-associated losses in spare respiratory capacity (SRC), a measure of the difference between basal and maximal respiration [18]. Because this suite of anomalies occurred as a result of impaired mitochondrial redox biology and correlated to OA status, these results strongly support a pathogenic role for increased mitochondrial oxidant production.
We hypothesized that overloading healthy articular cartilage would induce ROS overproduction and mitochondrial dysfunction similar to that observed in OA chondrocytes. Over time this might compromise responses to mechanical stimuli. Such an effect would constitute a critical imbalance in redox and metabolic activities capable of pathogenic disruption of cartilage homeostasis in both the early and late stages of OA. In order to test this hypothesis we exposed matched (ie. from adjacent loaded sites of the same tibial surface) bovine osteochondral explants to cyclic axial compression using normal healthy loads or moderate overloads. Explants were then analyzed for stress-strain mechanical behavior, cell viability, ATP levels, ROS production levels, indications of oxidative stress, respiratory parameters, glycolytic indicators, and mitochondrial membrane potential.
Materials and Methods
Cell culture and reagents
Unless otherwise noted, all reagents used were obtained from Sigma (St. Louis, MO). Bovine stifle joints were obtained from a local abattoir (Bud's Custom Meats, Riverside, IA) and matched 1.2 × 1.2 cm2 osteochondral explants with 10 to 15 mm of subchondral bone attached were prepared from adjacent areas of the central loaded area of the tibial plateau. Each repetition utilized tissue from the same animal at maximally similar anatomical sites to compare the effects of healthy and excessive loading. Following harvest, explants were rinsed in Hank's Balanced Salt Solution (HBSS) (Invitrogen™ Life Technologies, Carlsbad, CA) then cultured in 45% Dulbecco's modified Eagle medium (DMEM), 45% Ham's F-12, and 10% fetal bovine serum (FBS) (Invitrogen). Media contained 100 U/ml penicillin, 100 μg/ml streptomycin, and 2.5 μg/ml amphotericin B with culture conditions of 37°C, 5% CO2 and 5% O2.
For Seahorse Bioscience XF96 experiments, cells were extracted from explants by removing articular cartilage from the subchondral bone and incubating it in 0.25 mg/ml collagenase and pronase (in media) for 8 hours to digest surrounding tissue. Extracted cells were plated in normal growth media on XF96 plates at a density of 30,000 cells/well. This produces a greater than 80% confluent culture of chondrocytes with no cell loss after attachment or doubling prior to assay. For human Seahorse measurements, total knee arthroplasty (TKA) tibial discards were obtained post-op under IACUC approval and cartilage was cut from articular surfaces adjacent to arthritic lesions as well as from corresponding areas of the opposite compartment. Tissue was digested and cells were plated identically to bovine chondrocytes. Upon assay, either five or six days after extraction, media was changed to assay media comprised of RPMI 1640 lacking sodium bicarbonate with added sodium pyruvate (5 mM), penicillin (100 units/ml), and streptomycin (100 μg/ml, all Invitrogen), pH adjusted to 7.4 using HCl. After changing to the assay media lacking sodium bicarbonate one hour prior to assay, cells were transferred to a 37°C CO2-free incubator to equilibrate to Seahorse conditions.
Tissue Loading Regimen
Explants underwent pre-defined mechanical stimulation using a custom loading apparatus under tissue culture conditions as previously described [10]. The explants were rigidly fixed in a polysulfone/polycarbonate chamber by anchoring the subchondral bone with stainless steel screws and polycaprolactone. Prior to loading, cartilage thickness was measured using an Olympus NDT Sonopen. The loading device imparted cyclic (0.5 Hz) axial compressive stresses of 0.25 MPa or 1.0 MPa to cartilage via an 8 mm diameter stainless steel, flat impermeable platen. Loading was continued for 5400 cycles (three hours +/- 5%). Final engineering strains in the two groups ranged from ∼20% under 0.25 MPa compression, which is well-tolerated by cartilage, to greater than 40% under 1.0 MPa compression, which induces excessive oxidant production and occasional superficial/transitional zone chondrocyte mortality [10,19]. Following loading, explants were returned to normal culture conditions until the following day. Loads were applied either once or daily for 1 week. The antioxidant N-acetylcysteine (NAC) enters cells freely by virtue of its acetylation and is rapidly deacetylated to cysteine thereby supplementing intracellular glutathione (GSH) synthesis initiated by glutamate cysteine ligase. For these treatments, explants were incubated for 2 hours prior to loading in 10 mM NAC to allow time for uptake. NAC was also present during the 5400 cycles of loading but removed afterwards. All NAC experimental runs included healthy and injurious loading controls on each plate. No NAC was present during XF96 analysis.
Measurement of Dihydroethidium Oxidation and Mitochondrial Membrane Potential in situ
Following the loading regimen, full-depth cartilage harvested from the loaded area of each explant was sliced into two sagittal sections. One of these sections was immediately exposed to 1 μM dihydroethidium (DHE, Invitrogen) in PBS, pH 7.4 and placed in an incubator for 30 minutes at 37° C. Calcein AM (1 μM, Invitrogen) was also added to verify chondrocyte viability. Sections were immediately analyzed using an Olympus FV1000 confocal laser scanning microscope (Olympus America, Center Valley, PA) to image tissues to a depth of 80 μm at 20 μm intervals through the sagittal plane. Mean fluorescence intensities of at least three images per specimen roughly representing the three zones of articular cartilage were measured using a custom MATLAB program described previously [19] and the intensities of these images were used for further statistical analysis. This program disregards DHE intensity in the absence of Calcein AM costaining, allowing quantification of live cell DHE intensity through the tissue volume. Because cell microscopy is impossible on the XF96 plates, supplementary measurements of cell viability were also conducted prior to Seahorse measurements as well as immediately after by trypsinizing cells, adding 0.4% trypan blue (Sigma), and counting positive cells per 100 cells. The second of set of slices was exposed to JC-1 (50 μg/ml, Invitrogen) for 60 minutes at 37° C. These samples were rinsed and analyzed via confocal laser scanning to a depth of 100 μm at 20 μm intervals. Chondrocytes were then analyzed for fluorescence intensity in the FL1 channel (λEx 488 nm, λEm 530 nm, 30 nm bandpass) and in the FL2 channel (λEx 488 nm, λEm 585 nm, 42 nm bandpass). The ratio of FL2/(FL1+FL2) was plotted as a measure of mitochondrial membrane potential within cells on a per-cell basis using the custom MATLAB program referenced above. Positive controls for depolarization using carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP) were included to confirm green-only fluorescence.
Measurement of Total Glutathione and Glutathione Disulfide
GSH levels and oxidation status were determined according to the glutathione reductase-recycling method described by Griffith [20]. Briefly, cell samples are homogenized in 5% sulfosalicylic acid and added to a buffer containing dithionitrobenzoic acid (DTNB), glutathione reductase and an excess of NADPH. Sample GSH molecules react with the DTNB to produce a yellow product, whereupon glutathione reductase recycles the GSH. Therefore, the rate of absorbance change at 412 nm is directly proportional to GSH concentration. To measure GSSG, 2-vinylpyridine was added to a sample aliquot for 1 hour prior to analyses rendering reduced GSH unavailable for recycling.
Measurement of Metabolic Flux in Chondrocytes Using the XF96-Extracellular Flux Analyzer
After five or six days culture with regular media changes as described above, a Seahorse Bioscience XF96 Extracellular Flux Analyzer (North Billerica, MA) was used to assay mitochondrial function in cultured chondrocytes from the loaded area of each explant. We chose an XF96 mitochondrial stress test method already characterized by Seahorse Bioscience and others, including some orthopaedic researchers [18]. The XF96 utilizes an individualized-per-well chamber in specialized microplates that allows measurement of oxygen concentrations, and therefore the oxygen consumption rate (OCR), in real time in response to three injections: oligomycin to block complex V, FCCP to uncouple the mitochondria, and a combination of rotenone and antimycin A to block ETC activity. This allows determination of basal respiration, maximal respiration (uncoupled respiration), spare capacity (the difference between the first two quantities), and proton leakage (an expression of that oxygen consumed which does not result in complex V activity). In this manner, the mitochondrial function can be measured across many samples at a time. Inhibitors were mixed in Seahorse medium day of assay from frozen aliquots and concentrations provided were determined through dose response experiments to verify their efficacy and lack of toxicity during the course of the experiment. Basal respiration was calculated from the difference between resting OCR and final, 2 μM rotenone and 5 μM antimycin A -inhibited OCR. Maximum respiration was calculated from the difference between uncoupled respiration in the presence of 0.25 μM FCCP and rotenone/antimycin A-inhibited OCR. Spare respiratory capacity was taken as the difference between the basal and maximum respiration. Data are expressed as the mean rate of oxygen consumption in amol/s/cell based on no less than 4 wells per specimen. Proton leakage was calculated as the difference between 2 μM oligomycin-inhibited OCR and rotenone/antimycin A-inhibited OCR and normalized to basal OCR to conform to similar studies. Extracellular acidification rate (ECAR) data were taken from the identical mitochondrial stress test runs and analyzed only in the basal, uninhibited state. All cells utilized for this study were primary, unpassaged cells and each plate run for this analysis contained all experimental groups.
This experimental scheme provided excellent reproducibility in bovine and human experiments shown here, with between run coefficients of variance (CV) in the basal OCR measurement, for example, as follows: 0.21 for 0.25 MPa, 0.088 for 1.0 MPa, and 0.03 for 1.0 MPa with NAC. Intra-sample comparisons (in-run CV) were significantly more precise, averaging as follows for basal OCR: 0.029 (+/- 0.017) for 0.25 MPa, 0.047 (+/- 0.009) for 1.0 MPa, and 0.041 (+/- 0.010) for 1.0 MPa with NAC. Human data were much more varied for inter-run comparisons, with inter-run CVs for basal OCR of 0.38 relative to intra-run CVs of 0.043 (+/- 0.006), more in line with the bovine samples. This speaks to the great variety of human samples received as well as the precision of the approach for a given sample. Further information available upon request.
Measurement of ATP Concentration
Sigma bioluminescent ATP assay kits (FLAA, Sigma) were used to determine ATP levels following a similar protocol to Long et al [21]. For normalization, we used tissue wet weight and double stranded DNA content, measured using the Quant-iT™ PicoGreen® dsDNA kit (Invitrogen). One tissue section was immersed in 500 μL of phosphatase inhibitor solution and boiled for 10 minutes to extract the ATP. The other section was digested in 200 μL of papain digest buffer. The DNA concentration of this digested solution was measured and used to determine the nanograms of dsDNA per milligram of tissue. This value was then used to normalize the ATP content measurement to yield ATP/dsDNA (fmol/ng).
Statistical Analyses
All statistical analyses were performed using Graph Pad Prism 5. For microscopy analyses, no fewer than three images from each section were analyzed by custom MATLAB programs as described [19], a mean was calculated from these measurements, and this was repeated for each explant. Seahorse plates containing all experimental groups were run, wells without measurable signal or that did not respond to injections were excluded, and typically more than six wells per explant harvest, never fewer than four wells, were measured and averaged to form a single replicate representative of that explant. Intra-sample (in-run) CVs were calculated as the standard deviation among each specimen's individual wells divided by the mean of those wells. Inter-run CVs were calculated as the standard deviation of each specimen divided by the mean of all specimens. Significance was determined using a repeated measures ANOVA approach given that the healthy and injurious loading groups were always paired.
Results
Repeated Overloading of Bovine Osteochondral Explants
Representative strain profiles in Figure 1 (individual strain data points in black for single day and grey for seven days) demonstrate that normal 0.25 MPa loading and 1.0 MPa overloading induce maximum tissue strains ranging from 15% to 23% and 43% to 56%, respectively. Dynamic strains, occurring over a single cycle, of 0.25 MPa loaded tissue did not exceed 7% of the original thickness while dynamic strains of overloaded 1.0 MPa loaded tissue did not exceed 12%. This mechanical behavior of both healthy 0.25 and 1.0 MPa loaded explants was not significantly altered by the seventh day of loading, Figure 1, though modest swelling of cultured tissue was observed in each explant as expected. Healthy 0.25 MPa-loaded explants reached a steady state with respect to strain prior to completing 5400 cycles of loading; however, some 1.0 MPa-loaded explants continued to strain over the whole 5400 cycles. Whether a steady state was reached or not did not appear to affect mitochondrial function, which was impaired in all overloaded explants.
Figure 1. Representative strain profiles demonstrate no overt changes in stress-strain behavior over one week with healthy or injurious loads.
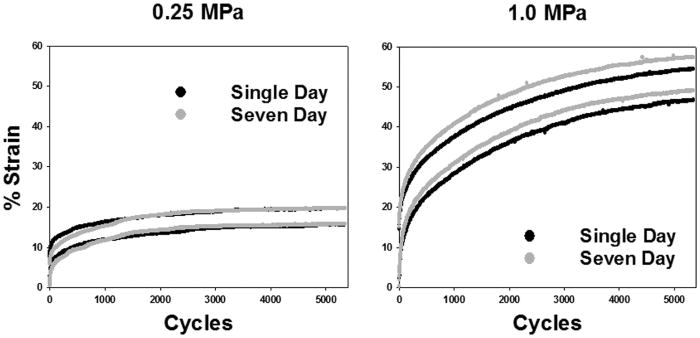
Loading tissue at 0.25 MPa, shown in panel A, or 1.0 MPa, shown in panel B, for 5400 cycles at 0.5 Hz induces maximum tissue strains ranging from 15% to 23% and 43% to 56%, respectively, in both single day and seven day loading specimens. Strain occurring over a single cycle during 0.25 MPa loading did not exceed 7% of the original thickness while these strains in 1.0 MPa loaded tissue did not exceed 12%. This mechanical behavior of both 0.25 and 1.0 MPa loaded explants was not significantly altered by the seventh day of loading, shown in grey, relative to single day loading, shown in black. This suggests that the loading procedure utilized did not overtly alter the mechanical properties of the bovine cartilage.
Strain-Responsive Reactive Oxygen Species Production in Live Chondrocytes
DHE oxidation in live chondrocytes following a week of cyclic loading was significantly higher in explants loaded with 1.0 MPa than in explants loaded with 0.25 MPa, Figure 2A (red) quantified in 2B. Z-stacked images, shown in composite, were analyzed for live-cell DHE intensity following exclusion of Calcein Green AM negative areas from analysis. No observable differences in the number of viable chondrocytes were detected by Calcein Green AM staining, indicating that chondrocytes survived both stress conditions equally well, Figure 2A (green). There were also no significant indications of cell death under any culture conditions using trypan blue exclusion testing in vitro as well as after XF96 analyses, Supplemental Figure 1. Oxidized glutathione levels were significantly increased after a week of overloading, Figure 2C, especially after normalization to matched controls, Figure 2D. One week 1.0 MPa overloading caused an increase in GSSG levels relative to the normally loaded control in each matched sample set, suggesting a consistent increase in oxidative stress. These increases in GSSG during 1.0 MPa loading were reversed with NAC addition prior to and during loading, confirming that NAC treatment could supplement GSH levels in our culture system.
Figure 2. One week of heavy loading increases endpoints indicative of oxidative stress in chondrocytes.
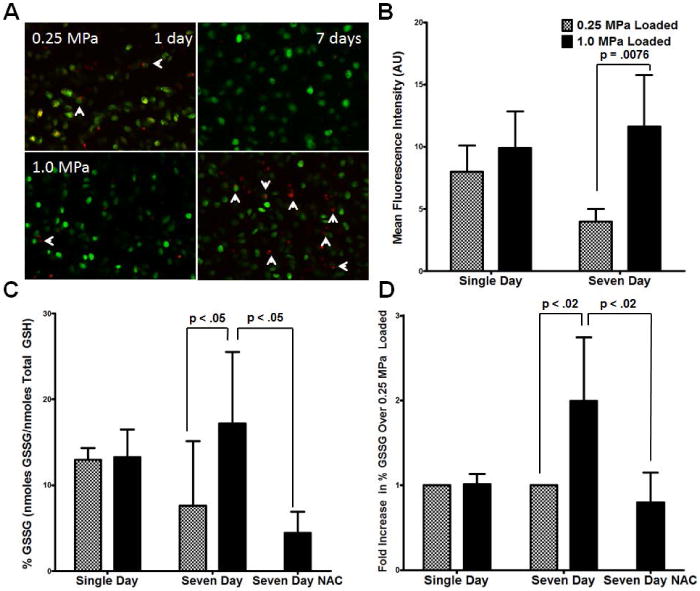
DHE oxidation in live chondrocytes following a week of cyclic loading was significantly higher in explants loaded with 1.0 MPa than in explants loaded with 0.25 MPa, (A, red, quantified in B). Of particular interest are chondrocytes staining more lightly for Calcein with significant DHE staining (A, arrows). No observable changes in the viable chondrocyte density were detected as indicated by Calcein Green AM staining (A, green). In association with increased live cell oxidation of DHE, GSSG levels were significantly increased after a week of overloading (C normalized to matched controls in D), an effect reversed by NAC addition. Data represent the mean of at least three images per sample, standard deviation shown is for n = 5 for all groups except NAC where n = 4, p-values as indicated. These data indicate NAC-responsive cellular oxidative stress following repeated injurious loading.
Mitochondrial Function of Overloaded Articular Chondrocytes
We chose to verify that our Seahorse Bioscience XF96 techniques were operating similarly to previous studies' by measuring mitochondrial function in primary chondrocytes harvested under IRB approval from TKA procedures. Chondrocytes harvested from articular cartilage in direct proximity to arthritic lesions were compared to chondrocytes from the opposite compartment when lesions were not present in that compartment. Similar to previous studies [18], we found increases in chondrocyte basal and maximum oxygen consumption as well as a strong trend towards a decrease in spare respiratory capacity, Figure 3A, and a modest increase in proton leakage when expressed as a % of the basal respiratory rate, shown in Figure 3B. It is worth noting that this difference in proton leak is significant without normalization to the basal rate. The magnitudes of these measurements were very similar to previous results.
Figure 3. Repeated injurious loading compromises mitochondrial function to a similar but not identical degree to changes observed in osteoarthritis.
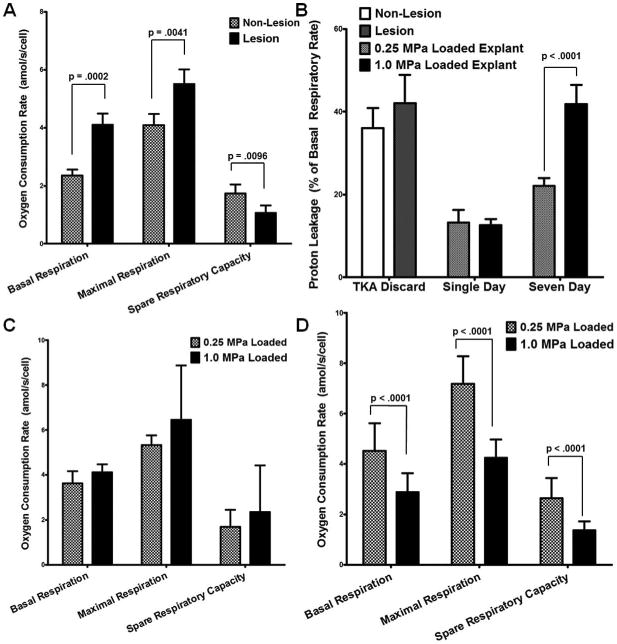
As previously shown, human osteoarthritic chondrocytes display increased basal and maximum mitochondrial oxygen consumption, relative to intact tissue, as well as a strong trend towards a decrease in spare respiratory capacity (A) and an increase in proton leakage (B), indicating mitochondrial dysfunction. After single day cyclic loading, bovine chondrocytes demonstrate no significant differences between 0.25 and 1.0 MPa loading (C). After 7 loading sessions, significant losses in basal respiration, maximum respiration, and spare respiratory capacity were observed in the overloaded explants (D). We also observed significant increases in proton leakage associated with overloading, similar to those observed previously in arthritic chondrocytes (18) though less significant (B). We expressed proton leakage as a % of basal function, a likely source of this distinction. Data represent the mean of at least 4 wells per explant, standard error of the mean of n = 5 for bovine data and n = 18 for human data, p-values as indicated. These data suggest that damage similar to that observed in human arthritic chondrocytes is occurring within the mitochondria of overloaded cartilage.
Chondrocytes were harvested from the loaded area of explants after a single day of loading and subjected to the same Seahorse protocol as TKA patients' chondrocytes. These chondrocytes demonstrate no significant differences between 0.25 and 1.0 MPa loading, Figure 3C. However, following week long loading regimens, significant losses in basal respiration, maximum respiration, and spare respiratory capacity were observed in the overloaded explants relative to the normally loaded controls, Figure 3D. Further, there were significant increases in proton leakage associated with overloading, similar to those observed previously [18] in arthritic chondrocytes, Figure 3B.
Given the changes observed in mitochondrial function, sections of the loaded area of each explant were assayed for mitochondrial membrane potential using JC-1 confocal microscopy. As shown in Figure 4A and (images in 4B; positive control with FCCP in Supplemental Figure 2), single day overloading has no impact upon mitochondrial membrane potential compared to normally loaded controls. However, overloading for a week appears to depress mitochondrial membrane potential to levels consistent with the losses in mitochondrial function described above. In order to confirm that losses in metabolic function occurred as a result of increased cellular oxidative stress, we repeated our respiratory measurements after NAC treatment during loading. This prevented loss of basal respiration, shown in Figure 5A, as well as prevented increases in proton leakage, Figure 5B, as a result of cyclic overloading.
Figure 4. Mitochondrial membrane potential is depressed after one week of injurious loading.
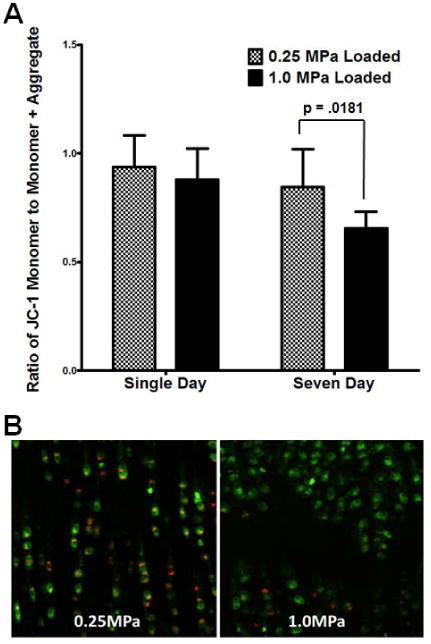
Sagittal sections of the loaded area of each explant were assayed immediately after harvest for mitochondrial membrane potential using JC-1 confocal microscopy. Similar to respiratory effects, single day overloading has no impact upon mitochondrial membrane potential compared to normally loaded controls (quantified in A, representative pictures in B). By contrast, overloading for one week appears to depress mitochondrial membrane potential to levels consistent with the losses in mitochondrial function described above (18). Data represent the mean of at least three images per sample, standard error of the mean of n = 4 is shown, p-values as indicated. These data suggest that the losses in respiration observed in extracellular flux measurements are present immediately at harvest and also indicate a disruption of overall mitochondrial function consistent with osteoarthritis.
Figure 5. N-acetylcysteine protects against metabolic dysfunction associated with overloading.
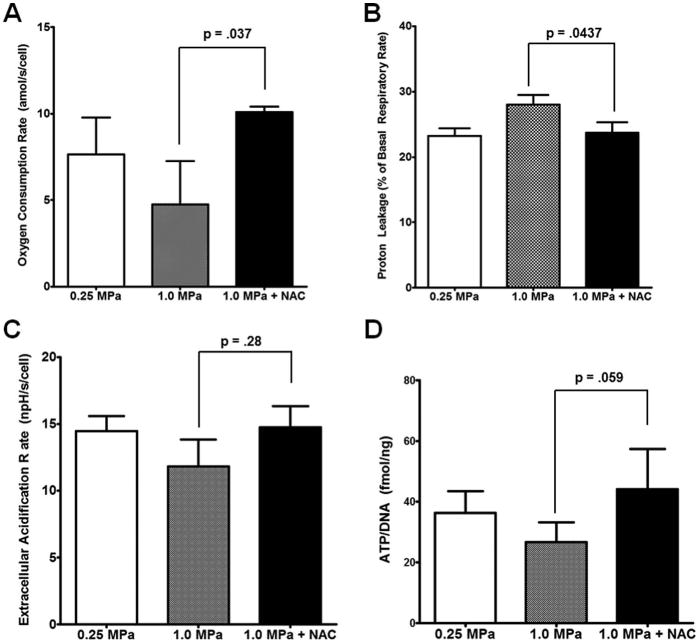
Pretreatment with 10 mM NAC 2 hours prior to loading with NAC maintained during loading was able to protect against loss of basal mitochondrial respiration (A). This was associated with prevention of increases in proton leakage caused by overloading (B). ECAR data obtained from this study suggest a trend towards decreased in glycolytic function (C); however, ATP content, a stronger indicator of anabolic activity, was decreased with overloading (D). In both ECAR and ATP measurements, NAC prevented deficits incurred by overloading. Seahorse XF96 data represent the mean of at least 4 wells per sample, standard error of the mean of n = 4 is shown, p-values as indicated. ATP measurement shown with standard deviations of n = 4, p-value as indicated. These data suggest that the losses in respiration observed are occurring as a result of oxidative damage to the chondrocytes.
Anabolic Function of Overloaded Articular Chondrocytes
Because mitochondrial ETC activity precedes chondrocyte glycolytic and ATP synthetic activity, we have also included the ECAR data from these Seahorse XF96 runs. Overloading has a much more modest effect upon ECAR than upon respiration, but still showed a trend towards decreased metabolic flux in a NAC-preventable fashion relative to healthy loaded controls, Figure 5C. To support the hypothesis that these changes in ECAR are representative of overall anabolic function, we also analyzed ATP concentrations. ATP is the primary mediator of chondrocyte anabolic activity, and overloading has reduced that output by 30%, Figure 5D, an effect that is prevented with the addition of NAC.
Discussion
In order to recreate physiologically relevant cell phenomena in response to mechanical loads in situ, we must account for several factors. We have shown that explants in our model system undergoing healthy or injurious loading demonstrated no aberrant mechanical behavior indicative of structural failure over the course of any experiment. Overall axial strains after 3 hours of cyclic compression with 1.0 MPa (40-50%) were clearly elevated above estimates of in vivo strains in normal joints (10-15%) [1,2,4,22]. However, it is possible that contact stress elevations associated with unstable or incongruent joints, or normal contact stresses on degenerated cartilage lead to strain values in the range we imposed by 1.0 MPa compression in our system [22]. Further, our loading device puts a flat, non-porous platen in contact with the surface of a larger diameter osteochondral explant. This configuration exaggerates shear stresses in cartilage at the edges of the platen relative to cartilage under the same load in intact, congruent joints [22]. This may exaggerate chondrocyte responses to a given load compared to what would be expected normally in vivo. Alternatively, excessive shear stress comparable to our system may be found in joints with meniscus and ACL deficiency, or at step-offs in cartilage surfaces after intra-articular fracture, all of which are known to increase the risk for OA. Thus, though our strain values for overloaded tissue may be considered excessive for average joint-wide strains in vivo, it is not our intention to model the entirety of a joint. Given that OA begins as a focal disease, we have focused on in vitro mechanical modeling of in vivo loci at risk for lesion formation, such as those near joint incongruities or other physiological mechanical overloads [24, 25].
Another concern we did not rule out is that some of the distinctions between human physiologic loading of cartilage and the harm done to bovine tissue here by overloading arise from differences in nutrient diffusion. However, glucose and other small molecules diffuse readily in resting cartilage and diffusion rates are not improved with cyclic loading [23]. Moreover, culture medium is extremely nutrient-rich, making it unlikely that bioavailability is limiting.
As expected, oxidative stress appears to be indicated by increases in DHE and GSH oxidation after one week of injurious loading, despite the absence of these indications in single day loading specimens. After seven consecutive injurious loading days, chondrocytes oxidize more DHE than healthy loaded controls in a relatively unstimulated state. This increase mirrors stable increases in oxidant production published in human OA patients post-TKA [15,17,18, and others]. This similarity suggests a role for mechanically-initiated oxidative stress in OA; however, making blanket statements about oxidative stress parameters in this system is difficult. Previous studies have shown distinct roles for different free radicals and ROS in specific processes associated with OA ranging from cellular inflammation to mechanical overload [26,27,28].
Indeed, pilot studies with other antioxidants were not promising (data not shown, pilot studies only). Given the benefits of NAC shown here, the GSH pathway and its intersection with glucose metabolism appear to be a powerful node for chondrocyte redox metabolism. Because NAC is rapidly converted to GSH upon uptake, protection by NAC suggests peroxidation and thiol chemistry may be key mediators of load-induced damage. As oxidative stress increases, NADPH, made via pentose phosphate activity, is required to recycle intracellular glutathione and maintain cellular reduction potential [20]. It is possible that by supplementing GSH with NAC, thereby preserving NADPH, chondrocytes can make ATP from a larger portion of their glucose, avoiding the pentose shunt.
The persistence of the respiratory phenotype resulting from this oxidative damage described both in situ and in monolayer and through multiple species is noteworthy. Extracellular flux data obtained via Seahorse XF96 adheres closely to previously published results in chondrocytes and are similar to those obtained using other techniques [18, 29]. With regard to phenotype stability, following overloading in bovine or human TKA, chondrocytes were cultured for as many as 6 days prior to respiratory measurements. During that time where cells were unloaded in monolayer, deficits in respiration indicated at harvest by low JC-1 staining for mitochondrial membrane potential persisted over the course of as many as 6 days in culture before the extracellular flux measurements. This suggests that changes present during the Seahorse run did not arise out of common tissue culture artifacts when dealing with primary chondrocytes. The damaged bovine chondrocytes also show an increase in proton leakage coherent with lipid peroxidation as suggested by previous studies [18]. Similarly, both show limitations in chondrocytes' capacity to increase ETC activity in response to metabolic stimuli suggesting a compromise of cellular metabolism that could alter tissue anabolic activities.
There are key distinctions between loss of spare capacity in overloaded intact tissue and in human TKA tissue. A significant increase in basal respiration relative to a modest increase in maximum respiration in OA cultures characterizes human OA tissue. This is in contrast to the significant decrease in all respiratory measures in overloaded but previously healthy bovine chondrocytes. In this case the one week mitochondrial phenotype observed appears to be a direct result of injurious loading whereas the phenotype observed in patients has likely been developed with numerous other pathogenic factors including matrix degradation, shifting loading conditions, inflammation, cellular adaptations or changes in gene expression, etc. Further, contrasts in mechanobiology between intact bovine articular cartilage and TKA specimens should be expected given descriptions of radical local changes in the loading environment of chondrocytes proximal to lesions [30]. It seems likely that chondrocyte mitochondrial dysfunction originally at critical regions of high strain, such as at joint incongruence, might present quite differently at these sites once prevailing mechanical conditions have caused lesions like the post-symptomatic TKA tissue utilized for this study.
Of particular interest for therapeutic extension of these findings, induction of mitochondrial turnover using rapamycin recently showed ameliorating effects upon OA development [31], possibly as a result of removing malfunctional mitochondria. By adding NAC in excess during loading, we may be activating or protecting from deactivation members of the mTOR pathway or other mitochondrial regulatory pathways. Many members of these signaling cascades have sensitivities to oxidation. Future experimentation will be focused upon identifying specific targets that NAC protected in this manner. In conclusion, the study presented here provides significant mechanobiological insight into the effects of mechanical overloading upon chondrocyte mitochondria. Our data suggest a persistent disruption of chondrocyte mitochondrial physiology may be playing a role during OA pathogenesis as a result of mechanical overload.
Supplementary Material
Supplementary Figure 1. No differences in cell viability were observed after different loading regimens or as a result of culture conditions. Calcein AM showed no significant decrease in viable cell density with 1.0 MPa loading for one week. Because XF96 plates are not amenable to microscopy, we measured trypan blue exclusion prior to XF96 measurements and immediately afterwards. No differences were observed as a result of loading. All measurements normalized to fresh tissue and shown with standard deviations of n = 4, p-value as indicated. These data support the assertion that changes in respiration observed occur independent of chondrocyte viability.
Supplementary Figure 2. Uncoupling chondrocyte mitochondria mutes JC-1 aggregation in situ. Top down views of full thickness cartilage demonstrate normal JC-1 staining in both the red (aggregate, polarized) and green (monomer, nonpolarized) channel, indicating the presence of polarized mitochondria in the left, control panel. In the presence of 10 μM FCCP, right panel, the same specimen shows that the mitochondrial electrochemical gradient has been destroyed by FCCP, leading to only green channel staining. This shows that the excitation/emission settings described do not lead to any fluorescent artifact as may be common in articular cartilage in some settings.
Acknowledgments
This work was supported by US DHHS NIH/NIAMS (P50 ARO55533; P30 CA086862) and a Department of Defense CDMRP Award (W81XWH-11-1-0583).
We would like to thank the Free Radical and Radiation Biology Core at the University of Iowa for their assistance with the GSH/GSSG and Seahorse XF96 assays. In particular, Brett Wagner and Garry Buettner were crucial in providing assistance in development of the Seahorse XF96 assays. We would also like to thank Saran Tantavisut who coordinated surgical discard collection.
Footnotes
Mitchell Coleman, James Martin, Marc Brouillette and Prem Ramakrishnan have no conflicts of interest or other disclosures.
References
- 1.Anderson DD, Marsh JL, Brown TD. The pathomechanical etiology of post-traumatic osteoarthritis following intraarticular fractures. Iowa Orthop J. 2011;31:1–20. [PMC free article] [PubMed] [Google Scholar]
- 2.Segal NA, Anderson DD, Iyer KS, Baker J, Torner JC, Lynch JA, et al. Baseline articular contact stress levels predict incident symptomatic knee osteoarthritis development in the MOST cohort. J Orthop Res. 2009;27:1562–8. doi: 10.1002/jor.20936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buckwalter JA, Martin JA. Sports and osteoarthritis. Curr Opin Rheumatol. 2004;16:634–9. doi: 10.1097/01.bor.0000132647.55056.a9. Review. [DOI] [PubMed] [Google Scholar]
- 4.Egloff C, Hugle T, Valderrabano V. Biomechanics and pathomechanisms of osteoarthritis. Swiss Med Wkly. 2012;142:w13583. doi: 10.4414/smw.2012.13583. Review. [DOI] [PubMed] [Google Scholar]
- 5.Baars DC, Rundell SA, Haut RC. Treatment with the non-ionic surfactant poloxamer P188 reduces DNA fragmentation in cells from bovine chondral explants exposed to injurious unconfined compression. Biomech Model Mechanobiol. 2006;5:133–9. doi: 10.1007/s10237-006-0024-3. [DOI] [PubMed] [Google Scholar]
- 6.Kraus VB, Birmingham J, Stabler TV, Feng S, Taylor DC, Moorman CT, 3rd, et al. Effects of intraarticular IL1-Ra for acute anterior cruciate ligament knee injury: a randomized controlled pilot trial ( NCT00332254) Osteoarthritis Cartilage. 2012;20:271–8. doi: 10.1016/j.joca.2011.12.009. [DOI] [PubMed] [Google Scholar]
- 7.Martin JA, McCabe D, Walter M, Buckwalter JA, McKinley TO. N-acetylcysteine inhibits post-load chondrocyte death in osteochondral explants. J Bone Joint Surg Am. 2009;91:1890–7. doi: 10.2106/JBJS.H.00545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goodwin W, McCabe D, Sauter E, Reese E, Walter M, Buckwalter JA, et al. Rotenone prevents load-induced chondrocyte death. J Orthop Res. 2010;28:1057–63. doi: 10.1002/jor.21091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martin JA, Martini A, Molinari A, Morgan W, Ramalingam W, Buckwalter JA, et al. Mitochondrial electron transport and glycolysis are coupled in articular cartilage. Osteoarthritis Cartilage. 2012;20:323–9. doi: 10.1016/j.joca.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wolff KJ, Ramakrishnan PS, Brouillette MJ, Journot BJ, McKinley TO, Buckwalter JA, et al. Mechanical stress and ATP synthesis are coupled by mitochondrial oxidants in articular cartilage. J Orthop Res. 2013;31:191–6. doi: 10.1002/jor.22223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boveris A, Cadenas E, Stoppani AO. Role of ubiquinone in the mitochondrial generation of hydrogen peroxide. Biochem J. 1976;156:435–44. doi: 10.1042/bj1560435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev. 1979;59:527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- 13.Lee RB, Urban JP. Evidence for a negative Pasteur effect in articular cartilage. Biochem J. 1997;321:95–102. doi: 10.1042/bj3210095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee RB, Urban JP. Functional replacement of oxygen by other oxidants in articular cartilage. Arthritis Rheum. 2002;46:3190–200. doi: 10.1002/art.10686. [DOI] [PubMed] [Google Scholar]
- 15.Blanco FJ, Lopez-Armada MJ, Maneiro E. Mitochondrial dysfunction in osteoarthritis. Mitochondrion. 2004;4:715–28. doi: 10.1016/j.mito.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 16.Maneiro E, Martin MA, de Andres MC, Lopez-Armada MJ, Fernandez-Sueiro JL, del Hoyo P, et al. Mitochondrial respiratory activity is altered in osteoarthritic human articular chondrocytes. Arthritis Rheum. 2003;48:700–8. doi: 10.1002/art.10837. [DOI] [PubMed] [Google Scholar]
- 17.Ruiz-Romero C, Calamia V, Mateos J, Carreira V, Martinez-Gomariz M, Fernandez M, et al. Mitochondrial dysregulation of osteoarthritic human articular chondrocytes analyzed by proteomics: a decrease in mitochondrial superoxide dismutase points to a redox imbalance. Mol Cell Proteomics. 2009;8:172–89. doi: 10.1074/mcp.M800292-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gavriilidis C, Miwa S, von Zglinicki T, Taylor RW, Young DA. Mitochondrial dysfunction in osteoarthritis is associated with down-regulation of superoxide dismutase 2. Arthritis Rheum. 2013;65:378–87. doi: 10.1002/art.37782. [DOI] [PubMed] [Google Scholar]
- 19.Brouillette MJ, Ramakrishnan PS, Wagner VM, Sauter EE, Journot BJ, McKinley TO, et al. Strain-dependent oxidant release in articular cartilage originates from mitochondria. Biomech Model Mechanobiol. 2013;13:565–72. doi: 10.1007/s10237-013-0518-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Griffith OW. Determination of glutathione and glutathione disulfide using glutathione reductase and 2-vinylpyridine. Anal Biochem. 1980;106:207–212. doi: 10.1016/0003-2697(80)90139-6. [DOI] [PubMed] [Google Scholar]
- 21.Long JA, Guthrie HD. Validation of a rapid, large-scale assay to quantify ATP concentration in spermatozoa. Theriogenology. 2006;65:1620–1630. doi: 10.1016/j.theriogenology.2005.06.020. [DOI] [PubMed] [Google Scholar]
- 22.Brown TD. Techniques for mechanical stimulation of cells in vitro: a review. J Biomech. 2000;33:3–14. doi: 10.1016/s0021-9290(99)00177-3. [DOI] [PubMed] [Google Scholar]
- 23.O'Hara BP, Urban JP, Maroudas A. Influence of cyclic loading on the nutrition of articular cartilage. Ann Rheum Dis. 1990;49:536–9. doi: 10.1136/ard.49.7.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McKinley TO, Tochigi Y, Rudert MJ, Brown TD. Instability-associated changes in contact stress and contact stress rates near a step-off incongruity. J Bone Joint Surg Am. 2008;90:375–83. doi: 10.2106/JBJS.G.00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tochigi Y, Rudert MJ, McKinley TO, Pedersen DR, Brown TD. Correlation of dynamic cartilage contact stress aberrations with severity of instability in ankle incongruity. J Orthop Res. 2008;26:1186–93. doi: 10.1002/jor.20589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Whiteman M, Armstrong JS, Cheung NS, Siau JL, Rose P, Schantz JT, et al. Peroxynitrite mediates calcium-dependent mitochondrial dysfunction and cell death via activation of calpains. FASEB J. 2004;18:1395–7. doi: 10.1096/fj.03-1096fje. [DOI] [PubMed] [Google Scholar]
- 27.Regan E, Flannelly J, Bowler R, Tran K, Nicks M, Carbone BD, et al. Extracellular superoxide dismutase and oxidant damage in osteoarthritis. Arthritis Rheum. 2005;52:3479–91. doi: 10.1002/art.21387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Regan EA, Bowler RP, Crapo JD. Joint fluid antioxidants are decreased in osteoarthritic joints compared to joints with macroscopically intact cartilage and subacute injury. Osteoarthritis Cartilage. 2008;16:515–21. doi: 10.1016/j.joca.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 29.Wagner BA, Venkataraman S, Buettner GR. The rate of oxygen utilization by cells. Free Radic Biol Med. 2011;51:700–12. doi: 10.1016/j.freeradbiomed.2011.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moo EK, Han SK, Federico S, Sibole SC, Jinha A, Abu Osman NA, et al. Extracellular matrix integrity affects the mechanical behaviour of in-situ chondrocytes under compression. J Biomech. 2014;47:1004–13. doi: 10.1016/j.jbiomech.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 31.Carames B, Hasegawa A, Taniguchi N, Miyaki S, Blanco FJ, Lotz M. Autophagy activation by rapamycin reduces severity of experimental osteoarthritis. Ann Rheum Dis. 2012;71:575–81. doi: 10.1136/annrheumdis-2011-200557. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. No differences in cell viability were observed after different loading regimens or as a result of culture conditions. Calcein AM showed no significant decrease in viable cell density with 1.0 MPa loading for one week. Because XF96 plates are not amenable to microscopy, we measured trypan blue exclusion prior to XF96 measurements and immediately afterwards. No differences were observed as a result of loading. All measurements normalized to fresh tissue and shown with standard deviations of n = 4, p-value as indicated. These data support the assertion that changes in respiration observed occur independent of chondrocyte viability.
Supplementary Figure 2. Uncoupling chondrocyte mitochondria mutes JC-1 aggregation in situ. Top down views of full thickness cartilage demonstrate normal JC-1 staining in both the red (aggregate, polarized) and green (monomer, nonpolarized) channel, indicating the presence of polarized mitochondria in the left, control panel. In the presence of 10 μM FCCP, right panel, the same specimen shows that the mitochondrial electrochemical gradient has been destroyed by FCCP, leading to only green channel staining. This shows that the excitation/emission settings described do not lead to any fluorescent artifact as may be common in articular cartilage in some settings.