

Abstract
The inhibitor of apoptosis (IAP) proteins have often been considered inhibitors of cell death due to early studies describing their ability to directly bind and inhibit caspases, the primary factors that implement apoptosis. However, a greater understanding is evolving for the vital roles played by the IAPs as transduction intermediates in a diverse set of signaling cascades that have been associated with functions ranging from the innate immune response to cell migration to cell cycle regulation. In this review, we discuss the functions of the IAPs in signaling, focusing primarily on the cellular IAP (c-IAP) proteins. The c-IAPs are important components in the TNF receptor superfamily signaling cascades, which include the activation of the NF-κB transcription factor family. Since these receptors can modulate cell proliferation and cell death, the roles of the c-IAPs in these pathways provide additional means of controlling cellular fate beyond simply inhibiting caspase activity. Additionally, IAP binding proteins, such as Smac and caspases, which have been described as having cell death-independent roles, may impact c-IAP activity in intracellular signaling. Collectively, the multifaceted functions and complex regulation of the c-IAPs illustrate the importance of the c-IAPs as intracellular signaling intermediates.
Keywords: NF-κB, IAP, apoptosis, TNF, caspases
Introduction
The study of cell death and its regulation led to the discovery and characterization of a family of factors called the inhibitor of apoptosis (IAP) proteins, and over the last few decades, the IAP protein family has been found to play major roles in a multitude of cellular processes. The mammalian IAP family consists of eight members, all of which share the family-defining bacculovirus IAP repeat (BIR) domain (Fig. 1), and while every IAP member possesses at least one BIR domain, many members contain multiple BIRs. Three of the most highly characterized IAPs are X-linked IAP (XIAP), cellular IAP 1 (c-IAP1), and cellular IAP 2 (c-IAP2), each of which possess three BIRs (Fig. 1) that are responsible for protein-protein interactions between the IAPs and other factors, such as caspases [1, 2]. In addition to the BIR motifs, XIAP and the c-IAPs contain a RING domain, which confers E3 ubiquitin ligase activity and plays an important role in the functional activities of these proteins (Fig. 1). The c-IAPs also contain two additional domains: a caspase-associated recruitment domain (CARD) and an ubiquitin-associated (UBA) domain (Fig. 1). While their exact functions remain unresolved, the CARD may allow the c-IAPs to interact with additional CARD-containing proteins and appears to regulate the E3 ubiquitin ligase activity by affecting the conformational status of the protein by interacting with the RING domain [3–5], and the UBA domain has been shown to bind ubiquitin and has recently been found to facilitate recruitment of other factors involved in the ubiquitination process [6–8].
Figure 1. Schematic depiction of the inhibitor of apoptosis (IAP) protein family.
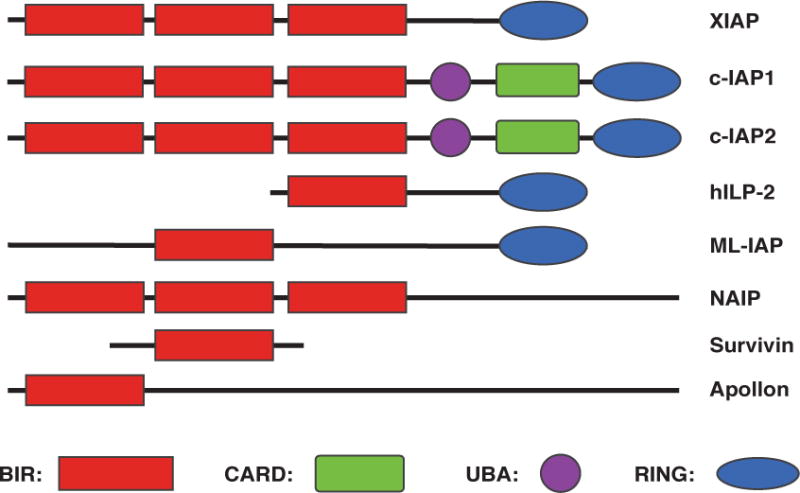
BIR, baculovirus IAP repeat; CARD, caspase-associated recruitment domain; UBA, ubiquitin-associated domain; RING, really interesting new gene.
Early studies with the IAPs were based on the notion that the IAPs regulate cell death through the inhibition of caspases. XIAP is the archetypical IAP protein that directly binds and inhibits caspases, specifically caspases-3, -7, and -9, preventing their downstream effector functions [9, 10]. This binding occurs through the BIR domains of XIAP and has been shown to inhibit cell death [11, 12]. Due to this described role, members of the IAP protein family have been generally considered direct inhibitors of cell death. The caspase inhibition exhibited by XIAP is suppressed following the initiation of apoptotic pathways that progress through the mitochondria. During apoptosis, the mitochondria is permeabilized during a process called mitochondrial outer membrane permeabilization (MOMP), releasing mitochondrial proteins into the cytoplasm, including Smac and the serine protease Omi/HtrA2, that possess IAP binding motifs (IBMs) and bind to XIAP in a manner that liberates the caspases, allowing them to be activated (Fig. 2) [13].
Figure 2. The intrinsic and extrinsic apoptotic pathways.
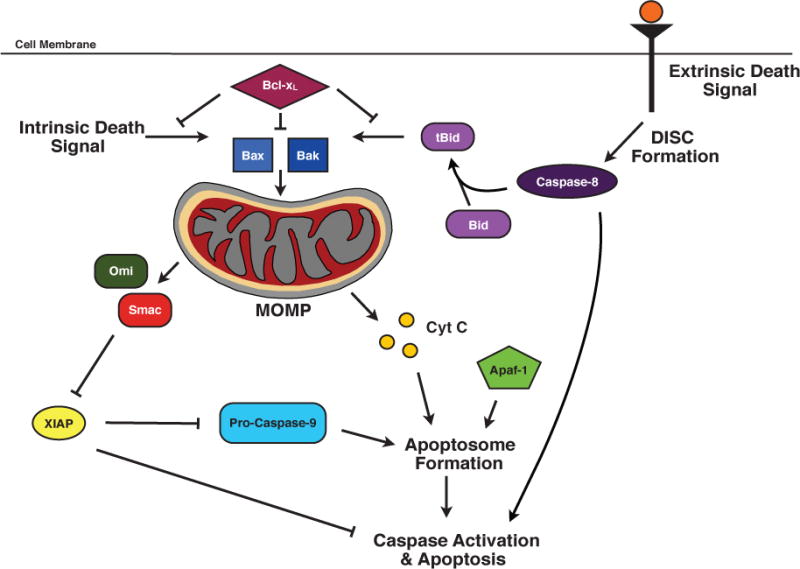
The intrinsic cell death pathway is initiated by an intracellular death signal. This signal results in the oligomerization and translocation of Bax and Bak into the outer membrane of the mitochondria. This triggers mitochondrial outer membrane permeabilization (MOMP) and the release of cytochrome c and IAP binding proteins. Cytochrome c forms the apoptosome with Apaf-1 and pro-caspase-9, which results in cell death. The IAP binding proteins, such as Smac, bind to XIAP and antagonize the caspase-binding function of XIAP. The extrinsic cell death pathway is receptor-mediated and results in the formation of the death inducing signaling complex (DISC). In certain cells, the DISC can directly activate downstream caspases, leading to cell death. However, in most cells, the DISC, through its caspase-8 component, cleaves Bid to form tBid, leading to Bax/Bak oligomerization, MOMP, apoptosome formation, and subsequent cell death.
Smac is capable of binding c-IAP1/2 and inducing their autoubiquitination and degradation [14–18]. However, despite their ability to bind caspases, the c-IAPs do not possess the ability to inhibit the apoptotic functions of caspases, unlike XIAP [19]. This suggested that the c-IAPs might regulate cell death through other mechanisms. Recent work has implicated the c-IAPs in regulating other cell death-activating platforms. Stimulation of tumor necrosis factor-receptor 1 (TNF-R1) following the removal of the c-IAPs by chemical, physiological, or genetic means, results in the formation of a complex containing FADD, active caspase-8, and receptor-interacting serine/threonine protein kinase 1 (RIP1) to induce apoptosis [20]. Additionally, the c-IAPs have been associated in a recently described form of cell death, designated necroptosis, which exhibits hallmarks of both apoptosis and the inflammation-inducing necrosis and occurs following the chemical degradation of the c-IAPs in conjunction with the inhibition of caspases [21, 22].
Recently, it has become more apparent that the IAPs play important functions in a host of cellular processes beyond caspase and cell death inhibition, implying that caspase-binding may, perhaps, only represent a minor facet of the IAPs. As such, the IAPs have come to be recognized as important regulators of intracellular signaling cascades, specifically the activation of nuclear factor-κB (NF-κB). The c-IAPs, as well as XIAP, have been implicated in multiple pathways of NF-κB activation [23], a family of transcription factors that play vital roles in a variety of signaling relevant to immunology and cancer as will be described in more detail below. Even though they are poor inhibitors of caspase activity, the c-IAPs may contribute to the determination of cell fate through their regulation of NF-κB.
IAPs as Regulators of Cell Signaling
As mentioned above, the functional scope of the IAPs has expanded beyond caspase-binding and cell death inhibition. The IAPs have been implicated in a diverse spectrum of non-apoptotic signaling, ranging from the regulation of intracellular copper levels [24–26] to the control of cell cycle progression [27, 28] and cell migration [29, 30]. Additionally, IAPs are important signal transduction factors in a variety of receptor-mediated pathways, many of which activate NF-κB [20].
As discussed above, several members of the IAP family, including the c-IAPs and XIAP, possess RING domains, which have been shown to be vital in many of the signaling cascades in which the IAPs participate. In these signaling pathways, the IAPs, through their RING domains, function as E3 ubiquitin ligases, which are the final component in the ubiquitination enzymatic cascade. The consequences of two polyubiquitin chains, the K63-linked chain and the K48-linked chain, have been well studied. These polyubiquitin chains are defined by their linkages at specific lysines within the ubiquitin protein [31], and the IAPs have been shown to mediate both K48 and K63 ubiquitination [32, 33]. The K48 ubiquitin chain is generally considered to be a degradation signal that targets the marked protein for proteasome-mediated destruction. In contrast, K63 polyubiquitination generally does not mediate proteasomal degradation, but can act as a scaffold for recruitment of additional factors to the signaling complex [34]. The c-IAPs are E3 ubiquitin ligases for multiple components in the NF-κB pathways, including RIP1 and NF-κB-inducing kinase (NIK), and the ability to mediate these forms of ubiquitination is integral to the functions of the IAPs in regulating the activation of NF-κB [18, 23].
NF-κB is a family of transcription factors that plays important functions in many cellular responses. The family is comprised of five members: p65 (RelA), RelB, c-Rel, p50 (NF-κB1), and p52 (NF-κB2), and following activation, these factors dimerize to form the active transcription factor [35]. Prior to activation, NF-κB dimers are sequestered in the cytoplasm of the cell, rendered inactive through the binding of IκB proteins. These proteins mask the nuclear localization sequence in NF-κB and prevent DNA binding through their ankyrin repeats [36]. Initiation of the signaling cascade results in IκBα ubiquitination and subsequent degradation by the proteasome following, allowing the NF-κB dimer to translocate into the nucleus.
Two major pathways of NF-κB signaling have been described: the canonical pathway that involves the p65:p50 dimer and the non-canonical pathway that employs the RelB:p52 dimer [35]. Canonical NF-κB can be activated by a wide variety of stimuli, some of which involve the IAPs, and the resulting signaling cascades converge on the IκB kinase (IKK) complex [37]. This complex has three components: IKKα (IKK1), IKKβ (IKK2), and NF-κB essential modulator (NEMO, also known as IKKγ). IKKα and IKKβ are the kinase subunits of the complex, while NEMO is a regulatory factor [38]. Once activated, the IKK complex phosphorylates IκBα, triggering its subsequent ubiquitination and proteasome-mediated degradation. As described above, this allows the canonical NF-κB dimer to translocate to the nucleus and initiate transcription. Non-canonical NF-κB, which is activated by a more limited set of stimuli, relies on a protein called NF-κB-inducing kinase (NIK). Prior to stimulation, NIK is constitutively degraded by a c-IAP-containing complex [39]. Following activation, this complex is inhibited, resulting in an increase in NIK protein levels, which then leads to the activation of IKKα. IKKα phosphorylates the NF-κB precursor protein p100 at Ser-866 and Ser-870 [36], resulting in its proteasome-dependent cleavage and generation of the active p52 subunit. The RelB:p52 dimers can then translocate into the nucleus to modulate gene expression. While the IAPs have been shown to participate in a variety of NF-κB activation pathways, the c-IAPs have been implicated in the regulation of both canonical and non-canonical NF-κB, playing both activating and inhibitory roles [20].
IAPs and the TNF Receptor Superfamily
These contrasting regulatory roles are evident in the signaling cascades of the TNF receptor superfamily, which can be broadly characterized into two major subsets based on their structural domains. Receptors such as Fas and TNF-R1, which can be widely expressed, possess death domains within their cytoplasmic tails [40]. These domains allow the receptor to recruit other DD-containing factors, building a receptor complex that can induce a multiple signaling cascades. These receptors have important roles in inflammation, hematopoiesis, and the immune response [34]. In contrast, certain TNF receptor superfamily members, such as CD30 and CD40, do not possess death domains and instead recruit signaling cofactors via TRAF-binding domains. These domains allow for the recruitment of members of the TNF receptor-associated factor (TRAF) family [20], and while TRAFs are implicated in signaling from both groups of receptors, direct TRAF binding to the receptor results in distinct consequences [41]. Additionally, the expression of these receptors is more limited, and many of these receptors are predominantly found on hematopoietic cells [42], and as such, many of these receptors have been shown to play important roles in immunity.
The c-IAPs are important factors in signaling from both categories of the TNF receptor superfamily, playing distinct roles for each subset of the superfamily. In the context of NF-κB activation by TNF-R1, a DD-containing receptor, the c-IAPs actively participate in the recruitment of proteins for signal transduction. Upon ligand binding to TNF-R1, the adaptor molecule TNF-R1-associated death domain protein (TRADD) is recruited to the DD of TNF-R1 [43]. TRADD binding, in turn, recruits additional proteins, including TRAF2, c-IAP1/2, and RIP1 to form the initial receptor signaling complex (Fig 3) [44, 45]. The c-IAPs become polyubiquitinated in a K63-dependent manner through autoubiquitination and polyubiquitinate RIP1, resulting in K63- and K11-linked ubiquitin chains that form a scaffold for the recruitment of additional factors, including transforming growth factor-β activating kinase 1 (TAK1), TAK1 binding protein 2 (TAB2), and TAB3, which preferentially binds to K63-linked ubiquitin chains (Fig 3) [32–34, 46, 47]. Additionally, the tripartite linear ubiquitin chain assembly complex (LUBAC), comprised of Sharpin, HOIL-1, and HOIP, is recruited to the signaling complex [34, 48, 49]. LUBAC modifies the NEMO subunit of the IKK complex with linear ubiquitin chains, which is believed to impact its function [49]. IKKβ is phosphorylated by the TAK:TAB complex, activating the IKK complex and resulting in the subsequent downstream activation of NF-κB (Fig 3) [50, 51].
Figure 3. The role of c-IAP1/2 in TNF-R1-mediated NF-κB activation.
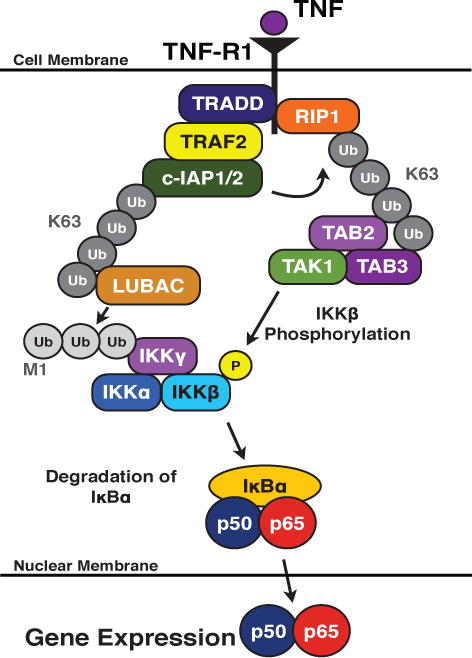
Upon ligand binding to TNR-R1, death domain-containing factors, such as RIP1 and TRADD are recruited to the cytoplasmic tail of the receptor. This, in turn, recruits additional factors, including TRAF2 and the c-IAPs. The c-IAPs undergo autoubiquitination and ubiquitinate RIP1. The ubiquitin chains act as scaffolds to recruit LUBAC and the TAB:TAK complex. The IKK complex is modified with a linear ubiquitin chain by LUBAC and is activated by phosphorylation by the TAB:TAK complex. The IKK complex phosphorylates IκBα, resulting in its subsequent proteasome-dependent degradation and the translocation of the canonical NF-κB dimer into the nucleus.
While the E3 ubiquitin ligase activity of the c-IAPs is involved in the propagation of the TNF-R1 signaling cascade, the c-IAPs also play an inhibitory role in regulating non-canonical NF-κB in the context of CD30-mediated signaling. Prior to receptor activation, the c-IAPs form a complex with TRAF2 and TRAF3. This complex binds NIK, which is constitutively expressed [52, 53]. Both TRAF2 and TRAF3 have been shown to directly bind NIK [54, 55], and the c-IAPs modify NIK with K48-linked ubiquitin chains, resulting in its proteasome-dependent degradation [39, 41, 52, 56], thereby repressing non-canonical NF-κB activation (Fig. 4). Following receptor activation, the TRAF:c-IAP complex is recruited to the cytoplasmic tail of CD30 via its TRAF-binding domains. After this recruitment, the c-IAPs modify TRAF3 with K48-linked ubiquitin chains, leading to the degradation of TRAF3 (Fig. 4) [39, 52, 57]. TRAF2 and c-IAP1/2 are then subsequently degraded following translocation to a detergent insoluble fraction [18, 20, 57–59]. The absence of the c-IAP:TRAF complex allows NIK levels to accumulate, resulting in the activation of non-canonical NF-κB (Fig. 4). This signaling pathway highlights receptor-mediated degradation of the c-IAPs as an important regulatory event for the activation of non-canonical NF-κB.
Figure 4. The role of c-IAP1/2 in CD30-mediated NF-κB activation.
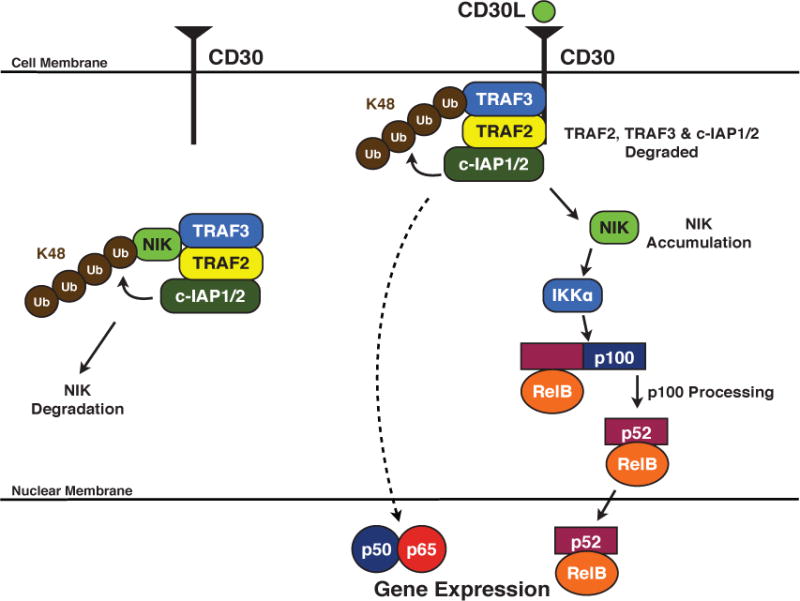
Prior to stimulation, the c-IAPs form a complex with TRAF2 and TRAF3 and ubiquitinate NIK in a K48-dependent manner, resulting in the constitutive degradation of NIK. Following receptor activation, the TRAF:c-IAP complex is recruited to the cytoplasmic tail of CD30, where the c-IAPs ubiquitinate TRAF3, inducing its degradation. TRAF2 and c-IAP1/2 are also degraded, allowing for the accumulation of NIK. NIK activates IKKα, which phosphorylates p100. Subsequently, p100 is cleaved, allowing the active non-canonical NF-κB dimer to translocate into the nucleus. Receptor-mediated canonical NF-κB activation also occurs, albeit through a poorly defined mechanism.
IAPs and Smac Mimetics
Receptor-mediated degradation of the c-IAPs can be partially mimicked by a class of small molecule compounds based on the IAP binding motif (IBM) of Smac [60–62]. These Smac mimetics (SMs), which are also known as IAP antagonists, similarly bind the IAPs and induce their degradation [56, 63, 64]. Due to the connection between the IAPs and cell death, SMs have been actively studied as potential therapeutics against cancer. While the ability of SMs to liberate caspases from XIAP may contribute to the observed induction of cell death, this is now believed to represent only one aspect of their mechanism. SMs have been shown to preferentially target and degrade the c-IAPs over XIAP [56, 65], and while this does not preclude the inhibition of XIAP, it suggested a more prominent role for the c-IAPs in regulating SM-induced cell death. It is currently thought that SM-induced killing is dependent on TNF [56, 63].
More specifically, following SM treatment, the c-IAPs are degraded, and TNF is produced in an autocrine or paracrine manner, activating TNF-R1. However, in the absence of c-IAP1/2, the ubiquitin scaffolds that help form the receptor signaling complex cannot be properly formed, leading RIP1 to then associate with TRADD, FADD, and caspase-8 to form a death inducing signaling complex, resulting in caspase activation and cell death [32]. Currently, cells can be divided into two major categories: those that are killed by SM treatment alone, and cells that are not killed by SM treatment alone but are sensitized to exogenous TNF [22, 66]. Cells in the former category are able to produce autocrine TNF through a NF-κB-dependent manner [66], while cells in the latter category, conversely, do not produce TNF following SM treatment, and therefore require an additional source of TNF [63]. More recently, another mechanism of cell death has been proposed which involves the formation of the ripoptosome following SM treatment. The formation of the ripoptosome, which is comprised of RIP1, FADD, inactive caspase-8, and RIP3, a related protein to RIP1, is thought to occur in specific cases following genotoxic stress, certain inflammatory stimuli, and c-IAP degradation [21, 22, 67]. Importantly, ripoptosome-mediated cell death is independent of TNF and the mitochondrial death pathways [21, 22], indicating that SM-induced death may not always be reliant on TNF. Collectively, these data illustrate alternative means by which the c-IAPs may inhibit cell death, inhibition that is dependent on their roles in cell signaling rather than through their caspase binding.
In addition to the clinical relevance of studying SMs as potential anti-cancer therapeutics, these compounds may also provide insight into the regulation of the c-IAPs. As noted above, these compounds were based on Smac, a physiological binding partner for the IAPs. In standard models, Smac is sequestered in the mitochondria and is released during mitochondria-mediated cell death. However, recent work has described non-lethal form of mitochondria permeabilization, a process called minority MOMP [68]. During this event, a minority of mitochondria are permeabilized, leading to a limited and sub-lethal activation of downstream caspases. The activation of these caspases results in DNA damage, as well as the promotion of oncogenic transformation [68]. In addition to caspase activation, Smac release was also observed during this process [68]. While the exact role of Smac released during minority MOMP is unclear, Smac may be acting as a signaling cofactor resulting in undefined consequences. In general, this may suggest that novel non-apoptotic functions exist for Smac and other apoptosis-associated proteins, similarly to growing number of non-apoptotic roles for the IAPs that have been described. Furthermore, this may change how we view the relationship between the IAPs and their mitochondrial protein targets. The IAPs may act as sentries to bind and eliminated these proteins, preventing effects caused by their limited release from the mitochondria. Conversely, the mitochondrial proteins may be released in a non-apoptotic manner in order to bind and induce the degradation of the IAPs, thereby regulating their activity. Notably, a similar system has been described for regulating Drosophila IAP activity [69]. These ideas illustrate potential physiological regulatory networks that control IAP activity.
In addition to considering novel means of regulating the IAPs, their overall functional scope may need to be expanded. Recently, we examined the downstream consequences of physiological and synthetic IAP antagonism using transcriptome profiling, identifying potentially novel roles for the c-IAPs [70]. More specifically, we described a role for the c-IAPs in regulating the ribosome and protein synthesis [70]. IAP antagonism by both receptor activation and SM treatment resulted in decreased expression of genes encoding ribosomal proteins, which led to an overall inhibition of protein synthesis. Extrapolating from these results, the c-IAPs may affect sensitivity to cell death by modulating cell cycle progression, since protein synthesis inhibition has been previously associated with both cell cycle arrest [71–73] and increased sensitivity to certain death ligands, such as TRAIL [74, 75]. In addition to highlighting new facets of IAP function, these results may have clinical implications. As described above, SMs are being tested for their therapeutic value and induce a TNF-dependent death in certain cells [56, 63]. These results potentially suggest an additional mechanism of SM killing, wherein SM-induced c-IAP degradation triggers the shutdown of protein synthesis, mimicking the effects of cycloheximide and rendering the cell more susceptible to TNF lethality. The lethality of TNF would then be partially due to the inability to synthesize pro-survival proteins, similar to established mechanisms of TNF killing [67, 76, 77]. Alternatively, c-IAP regulation of protein synthesis may affect sensitivity to cell death by inducing cell cycle arrest and increasing overall sensitivity to death.
These recently defined functions for the c-IAPs, along with the potential regulation of IAP activity by Smac released during a non-apoptotic MOMP event, represent intriguing new avenues of focus and warrant future study. It remains unclear if these effects are isolated to specific stimuli or if the c-IAPs engage in these functions in resting conditions. If the c-IAPs modulate ribosomal gene expression and protein synthesis under normal conditions, Smac could be released through a minority MOMP event to refine this effect. Furthermore, continued analysis of the transcriptome data may reveal more non-traditional roles for the IAPs.
While recent work expands the functional scope of the IAPs, their described role as caspase binding proteins may also be reevaluated. Caspases, though traditionally associated with cell death, have been increasing implicated in non-death related functions. For example, in humans, caspases-1, -4, -5, and -12 are considered inflammatory caspases and are involved in the innate immune response [78]. Caspase-1, more specifically, plays an important role in the processing and maturation of IL-1β during inflammation [79, 80]. Additionally, the apoptotic caspases have also been shown to possess apoptosis-independent functions. For example, caspase-3 plays a role in cell proliferation and differentiation, which may stem from its ability to regulate Akt phosphorylation [78, 81]. Caspase-8, an important factor in apoptosis, has been implicated in the regulation of inflammation in a manner that is independent of apoptosis [82–84], and has been shown to participate in the differentiation of monocytes into macrophages through the regulation of NF-κB via cleavage of RIP1 [85, 86]. Additionally, we found that gene expression regulated by c-IAP degradation was dependent on caspases, illustrating another non-apoptosis-related for these proteins [70]. These data may broaden the functional scope of the IAP:caspase interaction beyond regulation of cell death, with the IAPs acting as general regulators of caspase activity. More specifically, if the IAPs bind and functionally inhibit the caspases, minority MOMP may represent a regulatory event. Minority MOMP could occur, releasing Smac into the cytoplasm, which would then bind and degrade the IAPs, liberating the caspases to perform downstream functions (Fig. 5). As described above, minority MOMP is a recently described event that results in the sub-lethal activation of caspases, and this process also releases the IAP binding protein Smac [68] potentially representing another regulatory Therefore, the IAPs, through their caspase-binding ability, may regulate caspases in a variety of biological functions.
Figure 5. Model of IAP regulation of caspase-dependent signaling.
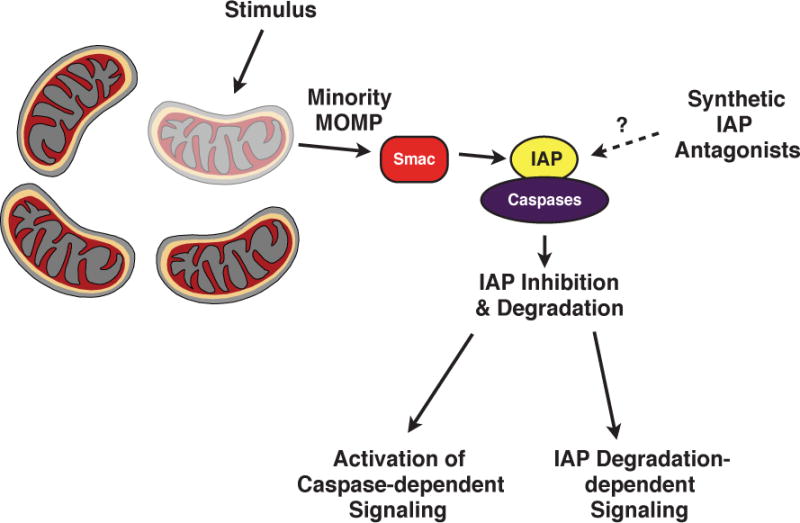
Prior to stimulation, IAPs are bound to caspases, functionally inhibiting them. A stimulus triggers minority MOMP, resulting in the limited release of Smac into the cytoplasm. Smac binds, inhibits, and induces the degradation of the IAPs. The liberated caspases proceed to participate in downstream functions, while additional IAP degradation-dependent signaling pathways are activated in parallel. Synthetic IAP antagonists, or Smac mimetics, may replicate the role of Smac and induce this process in a MOMP-independent manner.
Concluding Remarks
Our understanding of the biological function of the IAPs has expanded beyond their ability to directly bind and inhibit caspase activity, with their functions now encompassing major regulatory roles in a wide range of intracellular signaling pathways. The list of their cellular roles continues to expand as studies implicate the IAPs in a growing number of signaling cascades. Similarly, reexamination of IAP binding proteins, such as Smac and caspases, and their relationships with the IAPs will be warranted as new functions for these proteins emerge. Elucidating the interplay between the IAPs, their binding partners, and their novel functions will expand our understanding of the multifaceted nature of the IAPs as well as the biological impact on their respective signaling cascades.
Acknowledgments
We thank Dr. Stefanie Galbán for her critical reading of the manuscript and our many colleagues and collaborators for their insightful comments and discussions. We apologize to those scientists whose contributions, due to space limitations, we were unable to discuss. Supported in part by NIH 5P30CA46592 and NIH R01CA142809 to CSD and NIH T32AI007413 to AJK.
Abbreviations
- IAP
inhibitor of apoptosis
- XIAP
X-linked inhibitor of apoptosis
- c-IAP
cellular inhibitor of apoptosis
- NF-κB
nuclear factor-κB
- TNF
tumor necrosis factor
Footnotes
Author Contributions
AJK and CSD wrote the paper.
References
- 1.Scott FL, Denault JB, Riedl SJ, Shin H, Renatus M, Salvesen GS. XIAP inhibits caspase-3 and -7 using two binding sites: evolutionarily conserved mechanism of IAPs. EMBO J. 2005;24:645–655. doi: 10.1038/sj.emboj.7600544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wilkinson JC, Wilkinson AS, Galban S, Csomos RA, Duckett CS. Apoptosis-inducing factor is a target for ubiquitination through interaction with XIAP. Mol Cell Biol. 2008;28:237–247. doi: 10.1128/MCB.01065-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dueber EC, Schoeffler AJ, Lingel A, Elliott JM, Fedorova AV, Giannetti AM, Zobel K, Maurer B, Varfolomeev E, Wu P, Wallweber HJ, Hymowitz SG, Deshayes K, Vucic D, Fairbrother WJ. Antagonists induce a conformational change in cIAP1 that promotes autoubiquitination. Science. 2011;334:376–380. doi: 10.1126/science.1207862. [DOI] [PubMed] [Google Scholar]
- 4.Phillips AH, Schoeffler AJ, Matsui T, Weiss TM, Blankenship JW, Zobel K, Giannetti AM, Dueber EC, Fairbrother WJ. Internal motions prime cIAP1 for rapid activation. Nat Struct Mol Biol. 2014;21:1068–1074. doi: 10.1038/nsmb.2916. [DOI] [PubMed] [Google Scholar]
- 5.Lopez J, John SW, Tenev T, Rautureau GJ, Hinds MG, Francalanci F, Wilson R, Broemer M, Santoro MM, Day CL, Meier P. CARD-mediated autoinhibition of cIAP1′s E3 ligase activity suppresses cell proliferation and migration. Mol Cell. 2011;42:569–583. doi: 10.1016/j.molcel.2011.04.008. [DOI] [PubMed] [Google Scholar]
- 6.Budhidarmo R, Day CL. The ubiquitin-associated domain of cellular inhibitor of apoptosis proteins facilitates ubiquitylation. J Biol Chem. 2014;289:25721–25736. doi: 10.1074/jbc.M113.545475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gyrd-Hansen M, Darding M, Miasari M, Santoro MM, Zender L, Xue W, Tenev T, da Fonseca PC, Zvelebil M, Bujnicki JM, Lowe S, Silke J, Meier P. IAPs contain an evolutionarily conserved ubiquitin-binding domain that regulates NF-κB as well as cell survival and oncogenesis. Nat Cell Biol. 2008;10:1309–1317. doi: 10.1038/ncb1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blankenship JW, Varfolomeev E, Goncharov T, Fedorova AV, Kirkpatrick DS, Izrael-Tomasevic A, Phu L, Arnott D, Aghajan M, Zobel K, Bazan JF, Fairbrother WJ, Deshayes K, Vucic D. Ubiquitin binding modulates IAP antagonist-stimulated proteasomal degradation of c-IAP1 and c-IAP2(1) Biochem J. 2009;417:149–160. doi: 10.1042/BJ20081885. [DOI] [PubMed] [Google Scholar]
- 9.Deveraux QL, Roy N, Stennicke HR, Van Arsdale T, Zhou Q, Srinivasula SM, Alnemri ES, Salvesen GS, Reed JC. IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. EMBO J. 1998;17:2215–2223. doi: 10.1093/emboj/17.8.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deveraux QL, Takahashi R, Salvesen GS, Reed JC. X-linked IAP is a direct inhibitor of cell-death proteases. Nature. 1997;388:300–304. doi: 10.1038/40901. [DOI] [PubMed] [Google Scholar]
- 11.Salvesen GS, Duckett CS. IAP proteins: blocking the road to death’s door. Nat Rev Mol Cell Biol. 2002;3:401–410. doi: 10.1038/nrm830. [DOI] [PubMed] [Google Scholar]
- 12.Duckett CS, Li F, Wang Y, Tomaselli KJ, Thompson CB, Armstrong RC. Human IAP-like protein regulates programmed cell death downstream of Bcl-xL and cytochrome c. Mol Cell Biol. 1998;18:608–615. doi: 10.1128/mcb.18.1.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11:621–632. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- 14.Samuel T, Welsh K, Lober T, Togo SH, Zapata JM, Reed JC. Distinct BIR domains of cIAP1 mediate binding to and ubiquitination of tumor necrosis factor receptor-associated factor 2 and second mitochondrial activator of caspases. J Biol Chem. 2006;281:1080–1090. doi: 10.1074/jbc.M509381200. [DOI] [PubMed] [Google Scholar]
- 15.Yang QH, Du C. Smac/DIABLO selectively reduces the levels of c-IAP1 and c-IAP2 but not that of XIAP and livin in HeLa cells. J Biol Chem. 2004;279:16963–16970. doi: 10.1074/jbc.M401253200. [DOI] [PubMed] [Google Scholar]
- 16.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 17.Wu G, Chai J, Suber TL, Wu JW, Du C, Wang X, Shi Y. Structural basis of IAP recognition by Smac/DIABLO. Nature. 2000;408:1008–1012. doi: 10.1038/35050012. [DOI] [PubMed] [Google Scholar]
- 18.Csomos RA, Wright CW, Galban S, Oetjen KA, Duckett CS. Two distinct signalling cascades target the NF-κB regulatory factor c-IAP1 for degradation. Biochem J. 2009;420:83–91. doi: 10.1042/BJ20082140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eckelman BP, Salvesen GS. The human anti-apoptotic proteins cIAP1 and cIAP2 bind but do not inhibit caspases. J Biol Chem. 2006;281:3254–3260. doi: 10.1074/jbc.M510863200. [DOI] [PubMed] [Google Scholar]
- 20.Silke J, Brink R. Regulation of TNFRSF and innate immune signalling complexes by TRAFs and cIAPs. Cell Death Differ. 2010;17:35–45. doi: 10.1038/cdd.2009.114. [DOI] [PubMed] [Google Scholar]
- 21.Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M, Cain K, MacFarlane M, Hacker G, Leverkus M. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell. 2011;43:449–463. doi: 10.1016/j.molcel.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, Zachariou A, Lopez J, MacFarlane M, Cain K, Meier P. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell. 2011;43:432–448. doi: 10.1016/j.molcel.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 23.Gyrd-Hansen M, Meier P. IAPs: from caspase inhibitors to modulators of NF-κB, inflammation and cancer. Nat Rev Cancer. 2010;10:561–574. doi: 10.1038/nrc2889. [DOI] [PubMed] [Google Scholar]
- 24.Burstein E, Ganesh L, Dick RD, van De Sluis B, Wilkinson JC, Klomp LW, Wijmenga C, Brewer GJ, Nabel GJ, Duckett CS. A novel role for XIAP in copper homeostasis through regulation of MURR1. EMBO J. 2004;23:244–254. doi: 10.1038/sj.emboj.7600031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mufti AR, Burstein E, Csomos RA, Graf PC, Wilkinson JC, Dick RD, Challa M, Son JK, Bratton SB, Su GL, Brewer GJ, Jakob U, Duckett CS. XIAP Is a copper binding protein deregulated in Wilson’s disease and other copper toxicosis disorders. Mol Cell. 2006;21:775–785. doi: 10.1016/j.molcel.2006.01.033. [DOI] [PubMed] [Google Scholar]
- 26.Brady GF, Galban S, Liu X, Basrur V, Gitlin JD, Elenitoba-Johnson KS, Wilson TE, Duckett CS. Regulation of the copper chaperone CCS by XIAP-mediated ubiquitination. Mol Cell Biol. 2010;30:1923–1936. doi: 10.1128/MCB.00900-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Samuel T, Okada K, Hyer M, Welsh K, Zapata JM, Reed JC. cIAP1 localizes to the nuclear compartment and modulates the cell cycle. Cancer Res. 2005;65:210–218. [PubMed] [Google Scholar]
- 28.Cartier J, Berthelet J, Marivin A, Gemble S, Edmond V, Plenchette S, Lagrange B, Hammann A, Dupoux A, Delva L, Eymin B, Solary E, Dubrez L. Cellular inhibitor of apoptosis protein-1 (cIAP1) can regulate E2F1 transcription factor-mediated control of cyclin transcription. J Biol Chem. 2011;286:26406–26417. doi: 10.1074/jbc.M110.191239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oberoi TK, Dogan T, Hocking JC, Scholz RP, Mooz J, Anderson CL, Karreman C, Meyer zu Heringdorf D, Schmidt G, Ruonala M, Namikawa K, Harms GS, Carpy A, Macek B, Koster RW, Rajalingam K. IAPs regulate the plasticity of cell migration by directly targeting Rac1 for degradation. EMBO J. 2012;31:14–28. doi: 10.1038/emboj.2011.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kenneth NS, Duckett CS. IAP proteins: regulators of cell migration and development. Curr Opin Cell Biol. 2012;24:871–875. doi: 10.1016/j.ceb.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 31.Heride C, Urbe S, Clague MJ. Ubiquitin code assembly and disassembly. Curr Biol. 2014;24:R215–R220. doi: 10.1016/j.cub.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 32.Varfolomeev E, Goncharov T, Fedorova AV, Dynek JN, Zobel K, Deshayes K, Fairbrother WJ, Vucic D. c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor α (TNFα)-induced NF-κB activation. J Biol Chem. 2008;283:24295–24299. doi: 10.1074/jbc.C800128200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, Gillard JW, Jaquith JB, Morris SJ, Barker PA. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell. 2008;30:689–700. doi: 10.1016/j.molcel.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 34.Walczak H. TNF and ubiquitin at the crossroads of gene activation, cell death, inflammation, and cancer. Immunol Rev. 2011;244:9–28. doi: 10.1111/j.1600-065X.2011.01066.x. [DOI] [PubMed] [Google Scholar]
- 35.Hayden MS, Ghosh S. Shared principles in NF-κB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 36.Sun SC. The noncanonical NF-κB pathway. Immunological Reviews. 2012;246:125–140. doi: 10.1111/j.1600-065X.2011.01088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perkins ND. Integrating cell-signalling pathways with NF-κB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- 38.Scheidereit C. IκB kinase complexes: gateways to NF-κB activation and transcription. Oncogene. 2006;25:6685–6705. doi: 10.1038/sj.onc.1209934. [DOI] [PubMed] [Google Scholar]
- 39.Vallabhapurapu S, Matsuzawa A, Zhang W, Tseng PH, Keats JJ, Wang H, Vignali DA, Bergsagel PL, Karin M. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-κB signaling. Nat Immunol. 2008;9:1364–1370. doi: 10.1038/ni.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wertz IE, Dixit VM. Regulation of death receptor signaling by the ubiquitin system. Cell Death Differ. 2010;17:14–24. doi: 10.1038/cdd.2009.168. [DOI] [PubMed] [Google Scholar]
- 41.Hacker H, Tseng PH, Karin M. Expanding TRAF function: TRAF3 as a tri-faced immune regulator. Nat Rev Immunol. 2011;11:457–468. doi: 10.1038/nri2998. [DOI] [PubMed] [Google Scholar]
- 42.Croft M, Duan W, Choi H, Eun SY, Madireddi S, Mehta A. TNF superfamily in inflammatory disease: translating basic insights. Trends Immunol. 2012;33:144–152. doi: 10.1016/j.it.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hsu H, Xiong J, Goeddel DV. The TNF receptor 1-associated protein TRADD signals cell death and NF-κB activation. Cell. 1995;81:495–504. doi: 10.1016/0092-8674(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 44.Micheau O, Tschopp J. Induction of TNF receptor-I mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 45.Vince JE, Pantaki D, Feltham R, Mace PD, Cordier SM, Schmukle AC, Davidson AJ, Callus BA, Wong WW, Gentle IE, Carter H, Lee EF, Walczak H, Day CL, Vaux DL, Silke J. TRAF2 must bind to cellular inhibitors of apoptosis for tumor necrosis factor (tnf) to efficiently activate nf-{κ}b and to prevent tnf-induced apoptosis. J Biol Chem. 2009;284:35906–35915. doi: 10.1074/jbc.M109.072256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dynek JN, Goncharov T, Dueber EC, Fedorova AV, Izrael-Tomasevic A, Phu L, Helgason E, Fairbrother WJ, Deshayes K, Kirkpatrick DS, Vucic D. c-IAP1 and UbcH5 promote K11-linked polyubiquitination of RIP1 in TNF signaling. EMBO J. 2010;29:4198–4209. doi: 10.1038/emboj.2010.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang C, Deng L, Hong M, Akkaraju GR, Inoue JI, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412:346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- 48.Haas TL, Emmerich CH, Gerlach B, Schmukle AC, Cordier SM, Rieser E, Feltham R, Vince J, Warnken U, Wenger T, Koschny R, Komander D, Silke J, Walczak H. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell. 2009;36:831–844. doi: 10.1016/j.molcel.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 49.Gerlach B, Cordier SM, Schmukle AC, Emmerich CH, Rieser E, Haas TL, Webb AI, Rickard JA, Anderton H, Wong WW, Nachbur U, Gangoda L, Warnken U, Purcell AW, Silke J, Walczak H. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature. 2011;471:591–596. doi: 10.1038/nature09816. [DOI] [PubMed] [Google Scholar]
- 50.Yang J, Lin Y, Guo Z, Cheng J, Huang J, Deng L, Liao W, Chen Z, Liu ZG, Su B. The essential role of MEKK3 in TNF-induced NF-κB activation. Nat Immunol. 2001;2:620–624. doi: 10.1038/89769. [DOI] [PubMed] [Google Scholar]
- 51.Takaesu G, Surabhi RM, Park KJ, Ninomiya-Tsuji J, Matsumoto K, Gaynor RB. TAK1 is critical for IκB kinase-mediated activation of the NF-κB pathway. J Mol Biol. 2003;326:105–115. doi: 10.1016/s0022-2836(02)01404-3. [DOI] [PubMed] [Google Scholar]
- 52.Zarnegar BJ, Wang Y, Mahoney DJ, Dempsey PW, Cheung HH, He J, Shiba T, Yang X, Yeh WC, Mak TW, Korneluk RG, Cheng G. Noncanonical NF-κB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat Immunol. 2008;9:1371–1378. doi: 10.1038/ni.1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vallabhapurapu S, Karin M. Regulation and function of NF-κB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 54.Malinin NL, Boldin MP, Kovalenko AV, Wallach D. MAP3K-related kinase involved in NF-κB induction by TNF, CD95, and IL-1. Nature. 1997;385:540–544. doi: 10.1038/385540a0. [DOI] [PubMed] [Google Scholar]
- 55.Liao G, Zhang M, Harhaj EW, Sun SC. Regulation of the NF-κB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J Biol Chem. 2004;279:26243–26250. doi: 10.1074/jbc.M403286200. [DOI] [PubMed] [Google Scholar]
- 56.Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P, Zobel K, Dynek JN, Elliott LO, Wallweber HJ, Flygare JA, Fairbrother WJ, Deshayes K, Dixit VM, Vucic D. IAP antagonists induce autoubiquitination of c-IAPs, NF-κB activation, and TNFα-dependent apoptosis. Cell. 2007;131:669–681. doi: 10.1016/j.cell.2007.10.030. [DOI] [PubMed] [Google Scholar]
- 57.Duckett CS, Thompson CB. CD30-dependent degradation of TRAF2: implications for negative regulation of TRAF signaling and the control of cell survival. Genes Dev. 1997;11:2810–2821. doi: 10.1101/gad.11.21.2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Varfolomeev E, Goncharov T, Maecker H, Zobel K, Komuves LG, Deshayes K, Vucic D. Cellular inhibitors of apoptosis are global regulators of NF-κB and MAPK activation by members of the TNF family of receptors. Sci Signal. 2012;5 doi: 10.1126/scisignal.2001878. [DOI] [PubMed] [Google Scholar]
- 59.Wright CW, Rumble JM, Duckett CS. CD30 activates both the canonical and alternative NF-κB pathways in anaplastic large cell lymphoma cells. J Biol Chem. 2007;282:10252–10262. doi: 10.1074/jbc.M608817200. [DOI] [PubMed] [Google Scholar]
- 60.Schimmer AD, Welsh K, Pinilla C, Wang Z, Krajewska M, Bonneau MJ, Pedersen IM, Kitada S, Scott FL, Bailly-Maitre B, Glinsky G, Scudiero D, Sausville E, Salvesen GS, Nefzi A, Ostresh JM, Houghten RA, Reed JC. Small-molecule antagonists of apoptosis suppressor XIAP exhibit broad antitumor activity. Cancer Cell. 2004;5:25–35. doi: 10.1016/s1535-6108(03)00332-5. [DOI] [PubMed] [Google Scholar]
- 61.Li L, Thomas RM, Suzuki H, de Brabander JK, Wang X, Harran PG. A small molecule Smac mimetic potentiates TRAIL- and TNFα-mediated cell death. Science. 2004;305:1471–1474. doi: 10.1126/science.1098231. [DOI] [PubMed] [Google Scholar]
- 62.Sun H, Nikolovska-Coleska Z, Yang CY, Xu L, Liu M, Tomita Y, Pan H, Yoshioka Y, Krajewski K, Roller PP, Wang S. Structure-based design of potent, conformationally constrained Smac mimetics. J Am Chem Soc. 2004;126:16686–16687. doi: 10.1021/ja047438+. [DOI] [PubMed] [Google Scholar]
- 63.Vince JE, Wong WW, Khan N, Feltham R, Chau D, Ahmed AU, Benetatos CA, Chunduru SK, Condon SM, McKinlay M, Brink R, Leverkus M, Tergaonkar V, Schneider P, Callus BA, Koentgen F, Vaux DL, Silke J. IAP antagonists target cIAP1 to induce TNFα-dependent apoptosis. Cell. 2007;131:682–693. doi: 10.1016/j.cell.2007.10.037. [DOI] [PubMed] [Google Scholar]
- 64.Darding M, Feltham R, Tenev T, Bianchi K, Benetatos C, Silke J, Meier P. Molecular determinants of Smac mimetic induced degradation of cIAP1 and cIAP2. Cell Death Differ. 2011;18:1376–1386. doi: 10.1038/cdd.2011.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lu J, Bai L, Sun H, Nikolovska-Coleska Z, McEachern D, Qiu S, Miller RS, Yi H, Shangary S, Sun Y, Meagher JL, Stuckey JA, Wang S. SM-164: a novel, bivalent Smac mimetic that induces apoptosis and tumor regression by concurrent removal of the blockade of cIAP-1/2 and XIAP. Cancer Res. 2008;68:9384–9393. doi: 10.1158/0008-5472.CAN-08-2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Petersen SL, Wang L, Yalcin-Chin A, Li L, Peyton M, Minna J, Harran P, Wang X. Autocrine TNFα signaling renders human cancer cells susceptible to Smac-mimetic-induced apoptosis. Cancer Cell. 2007;12:445–456. doi: 10.1016/j.ccr.2007.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Darding M, Meier P. IAPs: guardians of RIPK1. Cell Death Differ. 2012;19:58–66. doi: 10.1038/cdd.2011.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ichim G, Lopez J, Ahmed SU, Muthalagu N, Giampazolias E, Delgado ME, Haller M, Riley JS, Mason SM, Athineos D, Parsons MJ, van de Kooij B, Bouchier-Hayes L, Chalmers AJ, Rooswinkel RW, Oberst A, Blyth K, Rehm M, Murphy DJ, Tait SW. Limited Mitochondrial Permeabilization Causes DNA Damage and Genomic Instability in the Absence of Cell Death. Mol Cell. 2015 doi: 10.1016/j.molcel.2015.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ditzel M, Wilson R, Tenev T, Zachariou A, Paul A, Deas E, Meier P. Degradation of DIAP1 by the N-end rule pathway is essential for regulating apoptosis. Nat Cell Biol. 2003;5:467–473. doi: 10.1038/ncb984. [DOI] [PubMed] [Google Scholar]
- 70.Kocab AJ, Veloso A, Paulsen MT, Ljungman M, Duckett CS. Effects of physiological and synthetic IAP antagonism on c-IAP-dependent signaling. Oncogene. 2015 doi: 10.1038/onc.2015.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Saracino F, Bassler J, Muzzini D, Hurt E, Agostoni Carbone ML. The yeast kinase Swe1 is required for proper entry into cell cycle after arrest due to ribosome biogenesis and protein synthesis defects. Cell Cycle. 2004;3:648–654. [PubMed] [Google Scholar]
- 72.Verbin RS, Farber E. Effect of cycloheximide on the cell cycle of the crypts of the small intestine of the rat. J Cell Biol. 1967;35:649–658. doi: 10.1083/jcb.35.3.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Medrano EE, Pardee AB. Prevalent deficiency in tumor cells of cycloheximide-induced cell cycle arrest. Proc Natl Acad Sci U S A. 1980;77:4123–4126. doi: 10.1073/pnas.77.7.4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Darzynkiewicz Z, Williamson B, Carswell EA, Old LJ. Cell cycle-specific effects of Tumor Necrosis Factor. Cancer Res. 1984;44:83–90. [PubMed] [Google Scholar]
- 75.Ehrhardt H, Wachter F, Grunert M, Jeremias I. Cell cycle-arrested tumor cells exhibit increased sensitivity towards TRAIL-induced apoptosis. Cell Death Dis. 2013;4:e661. doi: 10.1038/cddis.2013.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 77.Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-κB signaling pathways. Nat Immunol. 2011;12:695–708. doi: 10.1038/ni.2065. [DOI] [PubMed] [Google Scholar]
- 78.Shalini S, Dorstyn L, Dawar S, Kumar S. Old, new and emerging functions of caspases. Cell Death Differ. 2014 doi: 10.1038/cdd.2014.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, Miller DK, Molineaux SM, Weidner JR, Aunins J, Elliston KO, Ayala JM, Casano FJ, Chin J, Ding GJF, Egger LA, Gaffney EP, Limjuco G, Palyha OC, Raju SM, Rolando AM, Salley JP, Yamin TT, Lee TD, Shively JE, MacCross M, Mumford RA, Schmidt JA, Tocci MJ. A novel heterodimeric cysteine protease is required for interleukin-1β processing in monocytes. Nature. 1992;356:768–774. doi: 10.1038/356768a0. [DOI] [PubMed] [Google Scholar]
- 80.Ghayur T, Banerjee S, Hugunin M, Butler D, Herzog L, Carter A, Quintal L, Sekut L, Talanian R, Paskind M, Wong W, Kamen R, Tracey D, Allen H. Caspase-1 processes IFN-γ-inducing factor and regulates LPS-induced IFN-γ production. Nature. 1997;286:619–623. doi: 10.1038/386619a0. [DOI] [PubMed] [Google Scholar]
- 81.Fernando P, Kelly JF, Balazsi K, Slack RS, Megeney LA. Caspase 3 activty is required for skeletal muscle differentiation. Proc Natl Acad Sci U S A. 2002;99:11025–11030. doi: 10.1073/pnas.162172899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Maelfait J, Vercammen E, Janssens S, Schotte P, Haegman M, Magez S, Beyaert R. Stimulation of Toll-like receptor 3 and 4 induces interleukin-1β maturation by caspase-8. J Exp Med. 2008;205:1967–1973. doi: 10.1084/jem.20071632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vince JE, Wong WW, Gentle I, Lawlor KE, Allam R, O’Reilly L, Mason K, Gross O, Ma S, Guarda G, Anderton H, Castillo R, Hacker G, Silke J, Tschopp J. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity. 2012;36:215–227. doi: 10.1016/j.immuni.2012.01.012. [DOI] [PubMed] [Google Scholar]
- 84.Allam R, Lawlor KE, Yu EC, Mildenhall AL, Moujalled DM, Lewis RS, Ke F, Mason KD, White MJ, Stacey KJ, Strasser A, O’Reilly LA, Alexander W, Kile BT, Vaux DL, Vince JE. Mitochondrial apoptosis is dispensable for NLRP3 inflammasome activation but non-apoptotic caspase-8 is required for inflammasome priming. EMBO Rep. 2014;15:982–990. doi: 10.15252/embr.201438463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rebe C, Cathelin S, Launay S, Filomenko R, Prevotat L, L’Ollivier C, Gyan E, Micheau O, Grant S, Dubart-Kupperschmitt A, Fontenay M, Solary E. Caspase-8 prevents sustained activation of NF-κB in monocytes undergoing macrophagic differentiation. Blood. 2007;109:1442–1450. doi: 10.1182/blood-2006-03-011585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kang TB, Ben-Moshe T, Varfolomeev EE, Pewzner-Jung Y, Yogev N, Jurewicz A, Waisman A, Brenner O, Haffner R, Gustafsson E, Ramakrishnan P, Lapidot T, Wallach D. Caspase-8 serves both apoptotic and nonapoptotic roles. J Immunol. 2004;173:2976–2984. doi: 10.4049/jimmunol.173.5.2976. [DOI] [PubMed] [Google Scholar]