

Abstract
Clinical presentation of the chronic, heritable condition cystic fibrosis (CF) is complex, with a diverse range of symptoms often affecting multiple organs with varying severity. The primary source of morbidity and mortality is due to progressive destruction of the airways attributable to chronic inflammation arising from microbial colonisation. Antimicrobial therapy combined with practises to remove obstructive mucopurulent deposits form the cornerstone of current therapy. However, new treatment options are emerging which offer, for the first time, the opportunity to effect remission from the underlying cause of CF. Here, we discuss these therapies, their mechanisms of action, and their successes and failures in order to illustrate the shift in the nature of how CF will likely be managed into the future.
Common and Characteristic Pathologies Associated with Cystic Fibrosis (CF)
Cystic fibrosis (CF) is a heritable, chronic condition affecting multiple organs. The loss of or reduction in function of the CF transmembrane regulator (CFTR), a transmembrane ion channel through which chloride and bicarbonate secretion are regulated, interferes with the normal functioning of the epithelia within which it resides.
Functional CFTR is required to adequately hydrate the airway surface liquid (ASL), the continuous transit of which from the respiratory tract into the gastrointestinal tract under ciliary movement aids in the preservation of lung health and removal of inhaled microorganisms and particles. People with CF, however, are unable to clear their dehydrated airway mucous secretions, the prolonged residence of which creates a niche that can be exploited by a number of microorganisms. Many such colonising microorganisms fail to be cleared by the immune system and may persist for many years. Accordingly, this propagates a continuous inflammatory state in the airway which causes bronchiectasis, a progressive destruction of lung tissue architecture. Morbidity of the pulmonary epithelium is most directly responsible for the decreased lifespan experienced by people with CF. Ultimately, mortality results from asphyxiation due to progressive destruction of lung tissue caused by chronic infection and a chronic inflammatory response. Pulmonary insufficiency is the hallmark of CF clinical presentation, and this is reflected by the common use of the FEV1 (forced airway expiratory volume in 1 s) test as an outcome for efficacy of interventions during clinical trials for CF.
In addition to the airway, pathologies are frequently associated with each of the intestinal, biliary and pancreatic mucosa. In the pancreas, CFTR is localised to the apical surface of pancreatic duct cells [1], where it serves to secrete bicarbonate [2], which facilitates raising of the duodenal pH by pancreatic fluid secretions. CFTR deficiency impairs the adequate exocrine functioning of the pancreas [3], reducing fluid throughput, which serves to concentrate secreted digestive enzymes. This, in turn, leads to ductal occlusion and, hence, pancreatic insufficiency [4]. This leads to pancreatic fibrosis with consequent intestinal malabsorption of nutrients, for which therapeutic supplementation of digestive enzymes is required. Approximately 85 % of people with CF have pancreatic insufficiency [4], with 87.3 % of patients in the USA requiring pancreatic enzyme replacement therapy (PERT) in 2014 [5].
In addition to mediating compromised digestion, defective CFTR mediates further pathology through the pancreas: up to 50 % of people diagnosed with CF may suffer from CF-related diabetes (CFRD). CFRD presents atypically with respect to types 1 or 2 diabetes mellitus in non-CF individuals [6], likely due to ancillary nutritional deficiencies, and this can influence the diagnosis, with the prevalence of the condition being approximately 6 % in paediatric patients but rising to ~50 % in adults [7]. Glucose-stimulated electrical conductance has long been known to be involved in insulin secretion from pancreatic β cells [8] and, given that CFTR is expressed on the surface of β cells [9], the absence of CFTR has recently been proposed to result in dysregulation of glucose-mediated electrical activity, hence preventing insulin secretion from pancreatic β cells in some people with CF [10].
Disease of the liver is also a notable contributor to the overall pathology of CF. Epidemiological estimates of its prevalence vary, with incidences of 2–41 % or more having been reported from different studies [11–15]. CFTR in the liver is localised to the luminal surface of intrahepatic cholangiocytes of bile canaliculi [16], where it infuses biliary secretions with bicarbonate in parallel to its function in the pancreas, also serving to induce the efflux of bile acids [17]. Thus, the absence of bicarbonate secretion causes the retention of harmful bile acids within hepatocytes, inducing an inflammatory response, leading to fibrosis analogous to that seen in the pancreas and, ultimately, cirrhosis and portal hypertension [18].
The morbidities associated with pancreatic and hepatic insufficiency can, in turn, cause a number of gastrointestinal sequelae [19], leading to a complex clinical presentation of people with moderate or severe disease requiring multiple concurrent treatment and management strategies. Indeed, several co-morbidities, such as pancreatic insufficiency and CFRD, correlate with lesser lung function [20].
These morbidities and their ensuing therapies can have adverse secondary effects when administered routinely for chronic illness, with secondary kidney dysfunction being an example of this. The kidney can be damaged by successive rounds of non-steroidal anti-inflammatories, aminoglycoside antibacterials, as well as chronic infection itself, or CFRD and insulin treatment [21].
Each of these primary morbidities may individually or interactively engender a number of potential acute or chronic sequelae themselves requiring management for patients [22].
Treatment Strategies
Myriad treatments are prescribed to address the complications engendered by the loss of CFTR function. The majority of these abate the symptoms of this loss but cannot restore the function of the proteins. As our understanding of the structure of native CFTR and its CF-causing mutants has improved, alongside the attendant functional consequences of these, therapeutics that act to express or restore CFTR have begun to emerge. A remarkable diversity of these therapeutics is evident with regard to their mechanisms of action (Fig. 1) and, as discussed here, a number of them are beginning to demonstrate efficacy in trials or in the clinic.
Fig. 1.

Prominent examples of the broad array of therapeutic strategies that are showing promise for the instigation of normal expression and function of cystic fibrosis transmembrane regulator (CFTR), and their cellular targets. Ata ataluren, GP67a-pDNA lipid-enclosed DNA plasmid, Iva ivacaftor, Lum lumacaftor, ZFN zinc finger nuclease
Small-Molecule Therapies for Improving CF Transmembrane Regulator (CFTR) Function
In excess of 2000 variants of the CFTR gene have been described [23], with the consequence of each for the fate of the protein falling into one of six classes (Fig. 2) [24, 25]. The first class results in CFTR simply being absent from the cell by virtue of deletion of all or part of the gene, a premature stop codon being introduced into the gene, a frame-shifted gene or one which produces alternately spliced messenger RNA (mRNA). The second class of mutations comprise those that generate a protein incapable of being trafficked to the membrane. Such misfolded proteins are targeted for proteosomal degradation or remain in a partially mature state in the cytoplasm or endoplasmic reticulum. It is in this class that the common mutant, p.Phe508del CFTR, lies.
Fig. 2.
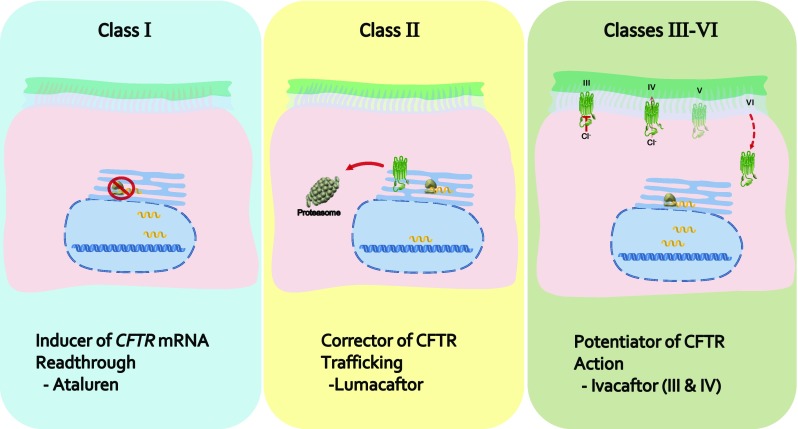
Overview of the classes of defect into which mutant variants of cystic fibrosis transmembrane regulator (CFTR) may be categorised, grouped according to which of the prominent chemotherapeutic agents are designed to compensate for said defect. mRNA messenger RNA
Substitutions of amino acids in either nucleotide-binding domain may yield a protein of sufficient structural normalcy that it is trafficked to the membrane. However, such mutations may result in a third class of defective proteins, so-called gating mutants, whereby the chloride transport capability of the protein is greatly reduced. Alternately, if it possesses one of the fourth class of mutations, then an incorrect amino acid in one of the membrane spanning domains can alter the transmembrane pore sufficiently to prevent conductance of chloride (or bicarbonate).
Normal physiological function requires not only native CFTR but also sufficient levels of the protein. The fifth class of mutations comprise those that result in the degradation of some proportion of the mRNA, thus reducing the quantity of CFTR produced. Similarly, the sixth class of mutations give rise to CFTR that is recycled from the plasma membrane with a frequency that cannot sustain normal levels of chloride throughput.
The first class of mutations that may afflict the CFTR gene include those that result in the inclusion of a premature termination codon (PTC) in the sequence. This will, in most cases, cause the degradation of the PTC-containing mRNA or the truncated protein translated from it. Small-molecule therapeutics have been developed that can induce the read-through of the aberrant mRNA as normal [26]. Assuming that the PTC did not accompany a frameshift of the ensuing mRNA sequence, a protein of potential normalcy may be translated; the utility of such therapeutics extends to improving the likelihood of the correct peptidyl-transfer RNA binding at the A site of the ribosome while the PTC is present, overcoming the potentially deleterious effect of the absence of an amino acid which the PTC would otherwise cause.
One of the furthest-progressed PTC correctors in development is PTC124 (generic name ataluren), being brought to market by PTC Therapeutics (South Plainfield, NJ, USA). It has advantageous potency and tolerability relative to small molecules of comparable function and has been shown to have encouraging efficacy in patients amenable to treatment [27]. In assessing its physiological effect in people with CF harbouring a class I mutation on at least one allele, two open-label clinical trials demonstrated an effect on nasal potential difference—a metric of CFTR function—in the majority of patients over the durations of study (Table 1) [28, 29]. No change in percentage FEV1 was apparent, though this was unsurprising given treatment extended for only 6 weeks.
Table 1.
Selected data reported from clinical trials of ataluren administration in people with cystic fibrosis harbouring a class I mutation in the CFTR gene on at least one allele. Percentage predicted FEV1 is reported at baseline and following treatment for both those in receipt of medication and control subjects who had received placebo treatment, where applicable
| Study type | No. of patients completing (% female) | Median age, years (range) | Treatment regimen | % FEV1 at baseline (range) | Study duration, weeks | Change in % FEV1 (SD) | No. of patients undergoing aminoglycoside treatment [change in % FEV1 (SD)] | References |
|---|---|---|---|---|---|---|---|---|
| Phase III | ||||||||
| Dual-arm RCT | 100 (48) | 22.8 (6–49) | Ataluren 10 mg/kg morning, 10 mg/kg midday and 20 mg/kg evening | 62.1 (38.4–90.3) | 48 | −2.5a (13.3) P = 0.12 |
38 [−0.7 (11.9)] P = 0.0082 |
[30] |
| 103 (50) | 23.2 (8–53) | Matching placebo | 60.2 (36.2–92.6) | −5.5 (12.56) | 40 [−6.4 (12.6)] | |||
| Other | ||||||||
| Single-arm, open-label study | 23 (52) | 25 (18–56) | Ataluren 4 mg/kg morning, 4 mg/kg afternoon and 8 mg/kg evening per day for 14 days Ataluren 10 mg/kg morning, 10 mg/kg afternoon and 20 mg/kg evening per day for 14 days |
65 (41–117) | 6b | –c | 0 | [28] |
| Open-label crossover Study | 14 | 12 (6–18) | Ataluren 4 mg/kg morning, 4 mg/kg afternoon and 8 mg/kg evening per day for 14 days | 90 (40–103) | 6d | –e | 0 | [29] |
| 14 | Ataluren 10 mg/kg morning, 10 mg/kg afternoon and 20 mg/kg evening per day for 14 days | |||||||
CFTR cystic fibrosis transmembrane regulator, FEV 1 forced airway expiratory volume in 1 s, RCT randomised controlled trial, SD standard deviation
aRelative change with respect to baseline
bInclusive of 14-day washout period between treatment cycles
cFigure 5 in original report, no apparent difference day 0 vs. day 42
dCohorts received treatment for 14 days and, following a 14-day washout period, received the other treatment listed
eReported as “no statistically significant change”
Ataluren has recently been subject to a randomised controlled phase III trial in 238 patients who received the drug for 48 weeks [30], with the primary outcome of the study being lessening of the rate of decline of FEV1. Those patients receiving treatment experienced a marginally lesser decline in FEV1 with respect to the control group (2.5 % decline vs. 5.5 % decline; P = 0.12; Table 1). The results of the study do not seem immediately impressive; however, the ability of the drug to preserve patients’ lung function over an extended period of time (years or decades) would be a more informative and useful metric and a halving of the rate of decline over 1 year, as this study demonstrated, is encouraging.
Moreover, post hoc subgroup analysis demonstrated that those patients who had received ataluren, but who were not concurrently taking aminoglycoside antibacterials demonstrated significantly less decline in FEV1 with respect to their counterparts who had received placebo (0.7 vs. 6.4 %; n = 44, P = 0.0082). This subgroup benefit likely stems from the mechanism of aminoglycoside action in which such compounds bind to ribosomal RNA [31]. Here, it may interfere with the ability of ataluren to effect readthrough of PTC-harbouring CFTR mRNA, positing discontinuation of aminoglycoside treatment as an indication for positive response to ataluren. This hypothesis is presently under investigation in the form of a repeat study of the aforementioned phase III trial specifically recruiting patients who are not undergoing treatment with aminoglycoside antibacterials [120].
For the majority of mutations, CFTR protein is produced, albeit in a non-functional form. This has provided the opportunity for development of therapeutics that can restore the normal functioning of the protein. Broadly speaking, such therapeutics fall into one of two categories: potentiators of CFTR open-conformation (enabling throughput of greater quantities of chloride) and correctors of CFTR structure (and, hence, function). Perhaps the most prominent example of a therapeutic targeting the defective CFTR itself is the small molecule, ivacaftor (brand name Kalydeco®), developed by Vertex Pharmaceuticals (Boston, MA, USA).
Ivacaftor was designed to potentiate the channel opening of CFTR bearing a class III single-nucleotide polymorphism, Gly551Asp, and received US FDA approval for the treatment of people with CF bearing this mutation—representing ~5 % of patients—in 2012 [32]. It had demonstrated statistically significant therapeutic efficacy, with respect to control subjects who continued standard treatment, in two randomised controlled phase III trials and a subsequent open-label extension over 48 weeks and up to 144 weeks, respectively (Table 2) [33–35]. Each trial utilised the common primary endpoint of improvement or lesser decline in FEV1. The drug not only halted decline in FEV1 but improved it relative to the baseline established at the trials’ outset, displaying an ~10 % improvement in FEV1 [36].
Table 2.
Selected data reported from clinical trials of ivacaftor administration to people with cystic fibrosis heterozygous or homozygous for alleles giving rise to a gating mutation in CFTR. Percentage predicted FEV1 is reported at baseline and following treatment for both those in receipt of medication and control subjects who had received placebo treatment, where applicable
| Study type | No. of patients enrolled (% female) | Mean age, years (range) [SD] | Treatment regimen | Mean % FEV1 at baseline (range) [SD] | Treatment duration, weeks | Mean absolute change in % FEV1 (95 % CI) [SD] | CFTR genotype | References |
|---|---|---|---|---|---|---|---|---|
| Phase II | ||||||||
| Dual-arm RCT | 8 (62) | 23a (18–40) | 150 mg/12 h | 65 (53–112) | 4 | 8.7b (2.3–31.3) P = 0.56 |
Gly551Asp, ≥1 allele | [75] |
| 4 (25) | 24a (18–42) | Placebo/12 h | 77 (42–122) | 7.3b (5.2–8.2) | ||||
| Dual-arm RCT | 112 (48) | 22.8 (12–52) | 150 mg/12 h | 79.7 (40–129) | 16 | 1.5 (–0.6 to 4.1)c
P = 0.15 |
Phe508del homozygous | DISCOVER; [37] |
| 28 (43) | 25 (12–39) | Placebo/12 h | 74.8 (43–127) | –0.213 | ||||
| Dual-arm RCT–crossoverd | 20 (50) | 16.6 (8–43) | Ivacaftor 150 mg/12 h | 97.2 [9.7] | 4 | 7.1 (1.8–12.2)c
P = 0.0117 |
Gly551Asp, ≥1 allele | [121] |
| Placebo/12 h | –0.213 | |||||||
| Phase III | ||||||||
| Dual-arm RCT | 83 (53) | 26.2 (12–53) | 150 mg once daily | 63.5 (37.3–98.2) | 48e | 10.4 (8.6–12.6)c
P < 0.0001 |
Gly551Asp, ≥1 allele | STRIVE; [33] |
| 78 (51) | 24.7 (12–53) | Placebo once daily | 63.7 (31.6–97.1) | −0.2 | ||||
| Dual-arm RCT | 26 (65) | 8.9 (6–11)f | 150 mg/12 h | 84.7 (52.4–133.8) | 24 | 12.6 (6.6–18.3)c
P < 0.001 |
Gly551Asp, ≥1 allele | ENVISION; [34] |
| 26 (38) | 8.9 (6–11)f | Placebo/12 h | 83.7 (44.0–116.3) | 0.1 | ||||
| Dual-arm RCT | 34 (56) | 29.2 [16.6] | 150 mg/12 h | 70.2 [18.9] | 24 | 2.6 [1.2] P = 0.2 |
Arg117His, ≥1 allele | KONDUCT; [122] |
| 35 (57) | 32.7 [17.4] | Placebo/12 h | 75.7 [19.3] | 0.5 [1.1] | ||||
| Other | ||||||||
| OLEg | 77A (53) | 27.7 [9.8] | 150 mg/12 h | 71.9 [18.5] | 144B | 9.4 [10.8] | Gly551Asp, ≥1 allele | PERSIST; [35] |
| 67B (52) | 26.0 [9.6] | 62.2 [18.7] | 9.5 [11.2] | |||||
| 26C (65) | 9.8 [1.9] | 94.9 [14.5] | 10.3 [12.4] | |||||
| 22D (41) | 9.8 [1.8] | 83.6 [17.4] | 10.5 [11.5] | |||||
| Dual-arm RCT-crossover with OLE | 19i (52.6) | 21.7 (6–47) | 150 mg/12 h | 79.1 (42.9–104.1) | 24 | 13.5 (–6.9 to 36.5) | Non-Gly551Asp gating mutation | KONNECTION; [39] |
| Observational study | 151 (46) | 21.1 [11.4] | 150 mg once daily | 82.6 [25.6] | 26 | 6.7 (4.9–8.5) P < 0.001 |
Gly551Asp, ≥1 allele | GOAL; [38] |
| Case–control study | 21 (52) | 22 (20–31)j | 150 mg once daily | 26.5 [7.2] | 38k | 3.8 (0.2–7.7)j
P = 0.009 |
Gly551Asp, ≥1 allele | [123] |
| 35 (49) | 23 (21–27)j | None | 30.3 [7.5] | 0.6 (–2.1 to 2.8)j | Non-Gly551Asp | |||
CFTR cystic fibrosis transmembrane regulator, CI confidence interval, FEV 1 forced airway expiratory volume in 1 s, OLE open-label extension, RCT randomised controlled trial, SD standard deviation
aMedian
bRelative change with respect to baseline
c95 % confidence interval for difference between arms
dCohort data are collated
ePrimary endpoint considered at 24 weeks
fLimits: 6–11 years
gPrevious cohort: A STRIVE–ivacaftor; B STRIVE–placebo; C ENVISION–ivacaftor; D ENVISION–placebo
hDuration is inclusive of previous enrolment in STRIVE or ENVISION
iPatients who received ivacaftor consecutively during treatment legs
jMedian (interquartile range)
kMedian follow-up
Given its efficacy when administered to patients homozygous for the Gly551Asp mutation, ivacaftor was then trialled in patients homozygous for Phe508del; however, it failed to demonstrate a significant change in FEV1 relative to control subjects [37], reflecting the difference in the nature of the defect affecting CFTR. Nonetheless, ivacaftor treatment appears to be a potent and efficacious therapy for adults with CF who harbour homozygous Gly551Asp mutant CFTR alleles; it improves FEV1 and weight gain as well as reducing incidence of infection by Pseudomonas aeruginosa and prolonging intervals between pulmonary exacerbations [38]. The drugs’ efficacy in patients who have other class III mutations has also been demonstrated clinically [39], leading to its approval as a treatment in patients harbouring such mutations, as well as in patients with p.Arg117His CFTR [32]. It may also prove useful for patients who have other ‘partial function’ mutations (classes III–V), further extending its reach.
Moreover, lesser doses are being trialled in paediatric patients to assess tolerability, as CF pathology is often apparent in infancy [40, 41]; ivacaftor treatment is presently approved to commence from 2 years of age [32]. While the requirement for early intervention is apparent, this is accompanied by several caveats. As mentioned, tolerability is an issue, given the load placed particularly on the hepatic cytochrome, cytochrome P450 3A (CYP3A), by ivacaftor [40]. In addition, the yearly cost per patient of ivacaftor treatment is considerable—£182,000 per patient per year in the UK—leading to stringent assessment of its clinical benefit [42, 43], which may limit the breadth of patients to whom it will be administered.
Concern over the costings of new investigational drugs for ostensibly orphan conditions may be tempered somewhat by the nature of the re-distribution of revenue. The arrangement in place between Vertex Pharmaceuticals and the Cystic Fibrosis Foundation—the US-based not-for-profit CF research organisation, whose model of venture philanthropy has seen it receive ~US$3.3 billion in drug royalties in 2014 [44]—may help to ensure continued high levels of research into the condition.
Vertex Pharmaceuticals has also progressed to clinical trials with a therapeutic, generic name lumacaftor, designed to assist in the trafficking of Phe508del CFTR to the epithelial membrane [45]. Despite in vitro mechanistic evidence, phase II trials failed to show a benefit of taking lumacaftor for patients homozygous for the Phe508del mutation (Table 3) [46], which represents the genotype of approximately 50 % of CF patients in both the EU and USA [22]. Accordingly, it was decided to combine lumacaftor treatment with ivacaftor treatment on the basis that ivacaftor may prove effective once Phe508del CFTR had been trafficked. Phase II trials of several dosage permutations of this combined therapy showed a modest improvement in FEV1 at high doses of each therapy: 600 mg of lumacaftor once daily and 250 mg of ivacaftor once daily [47]. Patients in this treatment arm demonstrated a 5.6 % improvement in % FEV1, and, as such, this regimen was progressed to two phase III trials which are currently undergoing open-label extensions having compared 600 mg lumacaftor once daily vs placebo and 400 mg lumacaftor twice daily vs placebo, respectively [48]. The aforementioned phase III trials each demonstrated significant improvement in percentage FEV1 and other metrics (Table 3). Accordingly, licensing for a combination therapy of lumacaftor and ivacaftor (trade name Orkambi™) has been approved by the FDA [49].
Table 3.
Selected data reported from clinical trials of lumacaftor administration to people with cystic fibrosis homozygous or heterozygous for the c.1521_1523delCTT CFTR allele which gives rise to mistrafficked p.Phe508del CFTR. The percentage predicted FEV1 is reported at baseline and following treatment for both those in receipt of medication and control subjects who had received placebo treatment, where applicable
| Study type | No. of patients enrolled (% female) | Mean age, years (range) [SD] | Treatment regimen | % FEV1 at baseline (range) | Treatment duration, weeks | Mean absolute change in % FEV1 (95 % CI) | Phe508del status | References |
|---|---|---|---|---|---|---|---|---|
| Phase II RCT, multiple armsa | 20 (40) | 28.5 [9.8] | Lumacaftor 200 mg once daily + ivacaftor 150 mg/12 h | 75.1 (42.4–117.1) | 1 | 3.1 (0.1–6.1) P = 0.176 |
Homozygous | [47] |
| 21 (62) | 28.6 [9.1] | Lumacaftor 200 mg once daily + ivacaftor 250 mg/12 h | 57.0 (39.1–93.3) | 1 | 0.5 (−2.8 to 3.8) P = 0.908 |
Homozygous | ||
| 21 (48) | 30.1 [10.3] | Placebo once daily + placebo/12 h | 69.1 (32.8–100.8) | 1 | 0.3 (−2.6 to 3.1) | Homozygous | ||
| 23 (48) | 28.1 [9.0] | Lumacaftor 200 mg once daily + ivacaftor 250 mg/12 h | 72.4 (43.3–99.1) | 4 | 2.0 (−0.8 to 4.8) P = 0.072 |
Homozygous | ||
| 21 (43) | 29.2 [8.5] | Lumacaftor 400 mg once daily + ivacaftor 250 mg/12 h | 67.4 (40.4–91.1) | 4 | 2.0 (−0.9 to 4.8) P = 0.074 |
Homozygous | ||
| 21 (52) | 26.7 [6.5] | Lumacaftor 600 mg once daily + ivacaftor 250 mg/12 h | 65.6 (37.0–98.4) | 4 | 6.2 (3.3–9.0) P < 0.001 |
Homozygous | ||
| 11 (45) | 25.5 [6.7] | Lumacaftor 400 mg/12 h + ivacaftor 250 mg/12 h | 63.7 (41.8–93.4) | 4 | 6.1 (2.0–10.2) P = 0.003 |
Homozygous | ||
| 17 | 30.8 [12.4] | Placebo once daily + placebo/12 h | 72.0 (36.4–107.1) | 4 | −1.6 (−4.2 to 1.1) | Homozygous | ||
| 4 | Placebo/12 h + placebo/12 h | Homozygous | ||||||
| 6 | Placebo once daily + placebo/12 h | Heterozygous | ||||||
| 21 (38) | 27.5 [7.2] | Lumacaftor 600 mg once daily + ivacaftor 250 mg/12 h | 68.5 (38.3–101.7) | 4 | 2.3 (−0.8 to 5.4) P = 0.067 |
Heterozygous | ||
| Phase III triple-arm RCTb | 368 (49.3) | 25.3 (12–54) | Lumacaftor 600 mg once daily + ivacaftor 250 mg/12 h | 60.8 (31.1–92.3) | 24 | 5.6 (3.8–7.3)c
P < 0.001 |
Homozygous | Traffic and Transport; [48] |
| 369 (49.5) | 24.5 (12–57) | Lumacaftor 400 mg/12 h + ivacaftor 250 mg/12 h | 60.5 (31.3–96.5) | 4.8 (3.0–6.6)c
P < 0.001 |
||||
| 371 (48.8) | 25.4 (12–64) | Placebo/12 h | 60.4 (33.9–99.8) | NA |
CFTR cystic fibrosis transmembrane regulator, CI confidence interval, FEV 1 forced airway expiratory volume in 1 s, NA not applicable, RCT randomised controlled trial, SD standard deviation
aAll patients had received the indicated dose of lumacaftor or placebo for 14 days (group 1–3) or 28 days (all other groups) immediately prior to commencing dual treatment
bReported here as pooled data arising from TRAFFIC and TRANSPORT studies
cDifference in percentage change in % FEV1 from baseline with respect to placebo
There are other novel therapeutics following in the developmental pipeline of Vertex Pharmaceuticals, focused on the major mutations in CF. The furthest progressed of these is another corrector of Phe508del CFTR, designated VX-661. This has shown an ability to improve CFTR operation in vitro and seems to be well-tolerated in patients with some efficacy when administered in conjunction with ivacaftor [50].
The same defects are currently being targeted by other companies or institutions. For example, Novartis have developed a series of correctors for Phe508del CFTR that possess similar functional activity to those of the Vertex therapeutics [51]. Many other candidate chemical entities have so far been declared to have efficacy in restoring CFTR function in vitro and, undoubtedly, further useful therapies will emerge during clinical trials [36].
The current high cost of the ivacaftor regimen had raised doubts as to whether it would be supported by public health schemes or insurance providers. However, given the larger cohort of patients eligible to receive Orkambi™, the latter regimen has been offered at a cost lower than that for the ivacaftor monotherapy [52], which may facilitate its prescription to patients. Longer term, however, a proliferation of personalised medicines for CF seems unsustainable from an economic standpoint and there are numerous calls for action to be taken to ensure that costs are controlled, while preserving research investment [43, 53].
Gene Therapy for the Restoration of Normal CFTR Function
While antibacterial use is frequent in clinical treatment of CF, arguably the most direct route to treating CF, as well as CFTR-related diseases, would be to integrate the native gene into airway stem cells in vivo. Such integrating vectors exist [54] but have been demonstrated to be oncogenic in some settings, justifiably giving rise to concerns over the use of integrating gene therapy [55]. The next best option would be to introduce copies of the native CFTR gene into the cells of the affected epithelia. Encouraging work has been carried out in vitro in this regard. Introduction of the native CFTR gene into CFTR-deficient airway epithelial cell line cells—via a transformed human parainfluenza virus—has been shown to result in the expression of the protein in those cells [56]. Furthermore, that study highlighted that normal ASL height was restored above the cell layer following induction of expression in as few as 25 % of the cells, lessening the issues of bioavailability and transfection efficiency of the vector.
The utility of virally mediated gene delivery has potential issues of toxicity or interference with cell functioning derived from the virus’ actions. These problems were encountered during efforts to use respiratory adenoviruses as delivery vectors; while they possessed a natural ability to traverse the airway epithelium, immune responses curtailed the expression of adenoviral transgenes [57].
As such, design of a viral delivery vector should account for these factors and further modifications to the virus, such as rendering it more immunotolerable, may be necessary to lessen its negative impact [58]. Even overcoming these issues, the turnover of epithelial cells requires the repeated administration of the vector throughout the life of the patient, ultimately resulting in failure of the virus due to its recognition and destruction by the immune system. This would, in turn, place a burden on the development of these viral vectors as they would require continual re-engineering and, hence, renewed regulatory approval.
Tolerance by the immune system for a given virus would render it amenable to repeated administration, but a virus possessing this property alongside an ability to infect the airway epithelium, without adverse consequences, has not been described. This has, however, been overcome by the generation of chimeric viruses possessing each of these traits: the well-tolerated lentivirus, simian immunodeficiency virus, was transformed with coat proteins of the respiratory infectious agent, Sendai virus, producing a virus that possessed the favourable traits of both [59].
Given the complexities inherent in developing viruses as vectors for gene therapy, alternate vectors for the delivery of CFTR DNA are under investigation. For example, pH-responsive peptides may be used. These peptides are bound to nucleic acids and remain so during endocytosis, subsequently enabling the escape of the nucleic acid from the endosome through a pH-dependent conformational change which disrupts the endosomal membrane [60]. Such a delivery vector avoids much of the immunogenicity inherent in a virus.
It should be noted that, while CF is a disease affecting multiple organs, correction of defects in the airway will provide a significant improvement in the quality of patients’ lives as well as an extension of their lifespan and has been pursued most extensively for this reason, in conjunction with the supposed ease of delivery of therapies via inhalation. However, the mucous layer presents an obstacle to delivery of therapeutics in this setting. For (relatively) small-molecule therapeutics, access to the epithelium may be improved by coating them in a non-mucous-adhesive compound such as polyethylene glycol (PEG), the efficacy of which can be further improved by coadministration with a mucolytic agent such as N-acetyl cysteine [61].
Combining a non-viral vector with each of these traits would represent a promising gene therapy for the treatment of CF lung disease. To that end, the UK CF Gene Therapy Consortium have collectively progressed research into such a vector. A plasmid harbouring CFTR was depleted of pro-inflammatory CpG motifs and encapsulated within a proprietary cationic lipid, GL67 (developed by Genzyme, Cambridge, MA, USA), in conjunction with the neutral lipid dioleoylphosphatidylethanolamine and a PEG-containing fusion lipid (Fig. 1). This has been shown in a murine study to have a favourable tolerability and induce persistent expression of CFTR, and each of these characteristics were maintained throughout the repeated-dose study [62].
The outcome of this study gave rise to a phase IIb clinical trial of the formulation. It was demonstrated that administration of a once-monthly dose of the formulation detailed above over 12 months arrested the decline in FEV1 percentage for treated patients with respect to those who had received nebulised saline as placebo (−0.4 vs. −4.0 %, respectively; Table 4) [63]. While modest in size, it is notable that some treated patients had better responses than others, which indicates that a study powered to identify subgroup effects may lead to further improvement in the utility of the therapy. Moreover, adverse events were comparable to the placebo-treated cohort and response did not depend on the type of CFTR mutation borne by patients (with respect to Phe508del status).
Table 4.
Selected data from prominent clinical trials of other interventions, curative or palliative, against cystic fibrosis lung disease
| Study type | No. of patients completing (% female) | Mean age, years (SD) | Treatment regimen | % FEV1 at baseline (range) | Study duration, weeks | Mean response (95 % CI) | References |
|---|---|---|---|---|---|---|---|
| Phase II dual-arm RCT | 62 (50) | 23.6 (10.8) | pGM169/GL67A 5 mL/28 daysa | 69.9 (49.6–89.9) | 52 | −0.4b (−2.8 to 2.1) P = 0.046 |
[63] |
| 54 (46) | 26 (13) | Placebo: 0.9 % saline/28 days | 69 (49.6–89.9) | −4.0b (−6.6 to −1.4) | |||
| Phase III dual-arm RCT | 334 (44.6) | 21.3 (10.7) | Inhaled mannitol 400 mg twice daily | 63.6 (25–105) | 26 | 3.6c | [78] |
| 232 (49.8) | 21.6 (10.5) | Inhaled mannitol 50 mg twice daily | 61.9 (30–100) | NA |
CI confidence interval, FEV 1 forced airway expiratory volume in 1 s, NA not applicable, RCT randomised controlled trial, SD standard deviation
a±5 days
bRelative change with respect to baseline
cDifference between groups in absolute % FEV1
Successful transfection of airway cells with the CFTR gene subsequently requires it to be expressed. The plasmid being utilised by the UK CF Gene Therapy Consortium harbours a humanised promoter to ensure expression, but a number of other groups have sought to correct the sequence that already exists. The recent advent of genome editing tools has made this feasible. One such tool, which has been applied to the introduction of CFTR into the genome, is zinc finger nuclease (ZFN) technology. This comprises a fusion protein, one domain of which specifically recognises short (9–12 bp) sequences of DNA, while the other possesses non-specific endonuclease activity capable of introducing double-strand breaks into the DNA at the targeted site, allowing the sequence of interest to insert [64]. Such ZFN complexes form heterodimers to carry out this activity, enabling the recognition of ~18 bp sequences, hence providing a high degree of specificity [65]. This approach has been demonstrated in vitro to correct the defective CFTR sequence in epithelial cells having the c.1521_1523delCTT genotype, which gives rise to the Phe508del mutant CFTR [66].
Indirect Therapies for CF Management
Putative Mediators of Improved Lung Physiology
Concurrent efforts are also ongoing to correct the defective physiology evident in CF by compensating for the lack of functional CFTR, a strategy that has the advantage of being applicable to all patients with CF. Most directly, increasing the activity of alternative chloride channels could, in principle, restore the native phenotype of the mucosal epithelium. For example, the calcium-activated chloride channel, TMEM16A, is expressed on airway goblet cells (which secrete mucins) and contributes to mucin release [67]. Mouse ASL height is also regulated by TMEM16A [68]. A direct-acting, long duration pharmacological modulator of TMEM16A is currently being sought [69].
Alternatively, modulation of sodium absorption, which is hyperactive in CF, may be considered [22]. The epithelial sodium channel (ENaC) has been explored in this context, with some therapeutics having reached clinical trials before ultimately being rejected on the basis of lack of efficacy due to insufficient potency or duration of effect or adverse effects such as pulmonary oedema [70, 71]. Efforts in this regard are continuing to pursue longer-acting modulators of ENaC, with some demonstrating increased ASL height and mucociliary clearance in animal models [72]. More recently, however, additional regulators of ENaC have been described, offering targets for candidate drugs with more finely tuned effects [73]. Similarly, the chaperones that guide the folding and trafficking of CFTR itself have been proposed for therapeutic action, which would be of particular benefit to class II mutations that do not reach the plasma membrane and class V mutations where CFTR is recycled from the membrane too quickly [74].
Given the attractiveness of ostensibly curing a chronic, debilitating, life-shortening condition, combined with the apparent requirement to take the aforementioned therapeutics for the duration of life [75], there is considerable research endeavour taking place into their further improvement. Presumably, the combined efforts of various entities in this regard will eventually yield an effective, persistent correctional treatment, but in the intervening years palliative therapy remains the best available option.
One such therapy concerns emulating the effect of ASL hydration, which is lacking in CF, by inducing osmosis of water from the airway cells into the luminal space through the inhalation of nebulised hyperosmolar solutions or dry powders. Two such osmolytes are in use: hypertonic saline, which overcomes ENaC-mediated hyper-absorption of sodium, and mannitol, which also establishes an osmotic driving force; both of which also induce coughing, thereby helping to dislodge deposited mucous. In concentrations as high as 6 %, hypertonic saline has been demonstrated to improve lung function after 16 weeks of therapy [76]; extension of therapy to 48 weeks failed to confirm improved lung function but did extend the duration between pulmonary exacerbations [77], highlighting the utility of clearing the lungs of deposited material. By comparison, two recent phase III trials of inhaled mannitol demonstrated improved lung function for up to 26 weeks (the duration of the studies) and prolonged the interval between exacerbations (Table 4) [78].
There is potential for incidental effects of mannitol administration, with in vitro evidence suggesting that the presence of mannitol can induce sensitivity to the antibacterial tobramycin in P. aeruginosa biofilm persister cells [79], presumably due to carbon source recognition by the bacteria leading to their ‘awakening’. Similarly, mannitol may enhance adhesion by CF-associated Burkholderia multivorans strains [80], illustrating a potential need for concurrent antibacterial treatment with mannitol therapy.
Treatment with recombinant human deoxyribonuclease (rhDNase), also known as dornase alfa or pulmozyme, represents another measure commonly employed to aid breathing by clearance of pulmonary obstructions. rhDNase treatment degrades the DNA derived from lysed neutrophil infiltrates, enabling easier clearance by airway clearance techniques such as saline therapy or thoracic agitation to induce coughing [81]. This treatment can improve lung function, though its efficacy in reducing exacerbations is less clear [82]. It may also be useful for disruption of microbial biofilms in the airways [83].
Given that lung damage driven by inflammation is the major contributor to mortality in people with CF, inhaled corticosteroids are often prescribed to lessen the extent of inflammation experienced by patients. Oral prednisone had been tested under clinical trials, lasting up to 48 months, and has shown improvements in patients’ lung function [84, 85]. However, an increase in adverse events compared with the placebo group was evident and age-matched growth impairment was noted among males for up to several years following discontinuation of treatment [86].
A recent systematic review of trials of inhaled corticosteroids (n = 13) failed to find a significant improvement in patients’ health while participating in those studies [87]. The authors noted that withdrawal from corticosteroids often took place without issue and highlighted possible negative side effects from taking them, in a parallel to the experience with oral prednisone. As a counterargument to the lack of trial evidence, such trials were conducted for only a limited period of time in patients who have already experienced prolonged inflammation. They cannot reflect any benefit that early, prolonged intervention with corticosteroids may have.
Improvement of patient health can not only enable better outcomes to treatment and immunity to exacerbations but can improve the quality of patients’ lives. To that end, pancreatic or hepatobiliary insufficiency are often compensated for by supplementation with digestive enzymes and vitamins specific to the needs of the patient, alongside maintenance of a high-calorie diet that emphasises high intake of fat and protein [88].
PERT is widely used; porcine-derived pancrelipase has demonstrated tolerability and mediates improvements to absorption of fats and nitrogenous compounds, hence contributing to restoration of normal growth and maintenance of adequate weight [89]. Porcine-derived native enzymes do, however, present challenges regarding dosage and efficacy during gastrointestinal transit [90]. Accordingly, non-animal-derived PERT agents are being pursued with liprotamase, for example, having demonstrated clinical efficacy and presently undergoing further investigation [91, 92]. Similarly, microbially derived lipases may be effective as PERT agents and are under clinical investigation [93]. Bile acids are also administered for those patients experiencing CF liver disease, though their efficacy is questionable [94] and probably dependent on the residual functioning of the liver among other factors such as age at intervention [95].
Antimicrobial Agents
Strategically related to amelioration of inflammation is the removal of its cause, namely the colonisation of patients’ lungs by microorganisms. Numerous species of microorganisms are frequently isolated from CF lung sputum samples, most commonly P. aeruginosa, Staphylococcus aureus, Haemophilus influenzae, as well as the fungal pathogen Aspergillus fumigatus and members of the Burkholderia cepacia complex of bacteria (Bcc), but a host of others are also frequently detected by routine culture including Stenotrophomonas maltophilia, non-tuberculous mycobacteria, etc. [96, 97].
P. aeruginosa, for example, has a prevalence ranging from 13 to 75 % of CF patients, depending on age and geographic location [96, 98]. Its prevalence increases steadily with age (from ~25 % at the onset of adolescence to ~75 % at adulthood in the US population), reflecting the tendency for many patients to become chronically colonised with a given strain with worsening airway conditions, often defined as greater than 50 % of their sputum samples being culture positive over a 12-month period [99]. A recent study in the USA indicated that as many as 48 % of people with CF become colonised with P. aeruginosa by the age of 6 years (n = 3608) [100]. Concurrently, the rate of pulmonary exacerbations significantly increases in patients following acquisition [101].
The overall prevalence of P. aeruginosa is falling, as evident in US patient registry data, which can be attributed to the success of early and frequent intervention with antibacterials such as inhaled tobramycin or aztreonam [102, 103]. A number of trials have demonstrated the utility of inhaled antibacterials for improving lung function and lessening the rate of pulmonary exacerbations in patients with long-term colonisation by P. aeruginosa [104]. Moreover, if treatment with antibacterials of proven efficacy is commenced soon after acquisition of the bacterium, it is possible to effectively eradicate P. aeruginosa [103].
While many patients for whom P. aeruginosa is successfully eradicated following early intervention were shown to sustain the absence of the bacterium for at least 5 years, many failed to demonstrate an improvement in their rate of pulmonary exacerbation [105]. This may suggest that other microorganisms then dominate, emphasising that effective antimicrobial regimens against a broad range of CF-associated pathogens is needed. This presents a challenge where microorganisms of low prevalence are concerned, as trials of the efficacy of candidate therapies may be under-powered.
Considering the example of colonisation by species of Burkholderia bacteria, their multi-drug-resistant status has precluded the instigation of a standard course of treatment. This is brought into sharp relief by the failure of a recent review, carried out by the Cochrane collaboration, to find even a single eligible trial of effective interventions against Bcc bacteria [106]. Clinical trials are also lacking interventions against the increasingly prevalent methicillin-resistant S. aureus (MRSA) [107], S. maltophilia [108] or non-tuberculous mycobacteria [109].
Such endeavours are not trivial, however, as evidenced by findings from systematic reviews of trials of antibacterial interventions against chronic P. aeruginosa colonisation. A recent review of trials of oral antibacterials for the treatment of P. aeruginosa failed to find a notable improvement to either lung function or quality of life [110]. The authors highlighted both the subjectivity of quality-of-life assessments, as well as the differential response of different patients to a given treatment (into which contribute myriad factors). This situation is echoed in a review of studies of inhaled tobramycin for treatment of chronic P. aeruginosa colonisation [102], in which the ability to improve clinical condition in the majority of patients was clear, but the failure of some patients to respond confounded large-scale analyses of antibacterial efficacy.
In a more general sense, the preferential route of antibacterial administration has proven equally difficult to confirm, with none of oral, intravenous or nebulised administration emerging as clearly more efficacious in treating pulmonary exacerbations [111]. However, for long-term therapy, inhalation may have an advantage, particularly in the case of inhaled tobramycin [112, 113]. It is also recognised that the propensity of successful colonisers to form biofilms may necessitate the administration of types and concentrations of antibacterials with anti-biofilm efficacy, though solid confirmation of this has not yet been provided [114].
Hence, treatment decisions are informed by prior experience and case reports. In the case of Burkholderia bacteria, there are isolated case studies of successful eradication of Burkholderia from CF patients and from settings other than CF, such as successful intervention with aztreonam in non-CF bronchiectasis [115]. However, while early eradication is often attempted, using combined intravenous and nebulised antibacterials, the success rate is low, with many infections remaining in situ [116].
Transplant
Management of the CF pulmonary physiology and microbiome is a matter of delaying the decline in lung function, but inevitably patients are rendered unable to achieve sufficient oxygen intake through breathing and, as they approach this point, bilateral lung transplantation is considered. In a retrospective study of 101 transplantations in CF patients in Italy, the 1-, 5- and 10-year survival rates for patients post-transplantation were 79, 58 and 42 %, respectively, while FEV1 increased from a pre-operative mean of 22 to 85 % 1 year after the operation [117]. Interestingly, these figures match those of a UK cohort of transplant recipients, with mean FEV1 for those patients improving from 21 to 78 % at 1 year, while survival rates were 82 % at 1 year, 62 % at 5 years and 51 % at 10 years (n = 176) [118].
Hence, there is a substantial survival and life-quality advantage to be gained through transplantation. Although there was sizable post-operative mortality (14.8 % of patients in the Italian clinic), the potential benefits may be decisive in listing patients for transplant. Supplementary to this, many clinics view colonisation by certain bacteria, such as Burkholderia, as a contra-indication for transplantation. In a UK CF clinic, ~10 % of CF patients who underwent lung transplantation had colonising Burkholderia. Of these, ~40 % died within a year following the operation, with the most frequent cause of death being sepsis [119]. This greatly exceeded the mortality rate for patients who did not harbour these bacteria, and these outcomes led to that clinic ceasing to list CF patients who harbour Burkholderia for transplantation.
Concluding Remarks
CF causes chronic morbidity in multiple organs, leading to a substantial reduction in the lifespan of those affected. Loss of lung function, mediated by tissue destruction pursuant to microbial colonisation and the attendant inflammatory response, is the most pronounced driver of mortality for people with CF. As such, multiple avenues of therapeutic interventions are being pursued. Ultimately, restoration of the airways to a normal phenotype holds the most promise for meaningful extensions of survival for people with CF, whether by allowing the epithelium to produce normal CFTR or by restoring the normal function of existing CFTR. In the meantime, diminishing the microbial burden in the airways is an important focus of research, but requires potent antibacterials and better delivery of them. These interventions are greatly aided by a therapeutic strategy that addresses as many of the symptoms of CF as possible; the resulting improvements to organ function and, hence, quality of life are instrumental in continuing to extend the lives of people with CF.
Compliance with Ethical Standards
MPM was supported by a Science Foundation Ireland (SFI) Research Frontiers Programme grant (Grant Number RFP2816) and has no conflicts of interest to declare. EC is the recipient of an SFI Stokes Lectureship Programme award and has no conflicts of interest to declare. No funding was received in the preparation of this review.
Contributor Information
Mark P. Murphy, Email: murphym8@tcd.ie
Emma Caraher, Phone: (+353) 01 4042296, Email: emma.caraher@ittdublin.ie.
References
- 1.Marino CR, Matovcik LM, Gorelick FS, Cohn JA. Localization of the cystic fibrosis transmembrane conductance regulator in pancreas. J Clin Invest. 1991;88:712–716. doi: 10.1172/JCI115358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ishiguro H, Steward MC, Naruse S, Ko SB, Goto H, Case RM, et al. CFTR functions as a bicarbonate channel in pancreatic duct cells. J Gen Physiol. 2009;133:315–326. doi: 10.1085/jgp.200810122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahmed N, Corey M, Forstner G, Zielenski J, Tsui LC, Ellis L, et al. Molecular consequences of cystic fibrosis transmembrane regulator (CFTR) gene mutations in the exocrine pancreas. Gut. 2003;52:1159–1164. doi: 10.1136/gut.52.8.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilschanski M, Durie PR. Patterns of GI disease in adulthood associated with mutations in the CFTR gene. Gut. 2007;56:1153–1163. doi: 10.1136/gut.2004.062786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cystic Fibrosis Foundation. Patient registry: annual data report. Bethesda: Cystic Fibrosis Foundation: 2014. https://www.cff.org/2014_CFF_Annual_Data_Report_to_the_Center_Directors.pdf/. Accessed 29 Nov 2015.
- 6.Konrad K, Scheuing N, Badenhoop K, Borkenstein MH, Gohlke B, Schofl C, et al. Cystic fibrosis-related diabetes compared with type 1 and type 2 diabetes in adults. Diabetes Metab Res Rev. 2013;29:568–575. doi: 10.1002/dmrr.2429. [DOI] [PubMed] [Google Scholar]
- 7.Moran A, Dunitz J, Nathan B, Saeed A, Holme B, Thomas W. Cystic fibrosis-related diabetes: current trends in prevalence, incidence, and mortality. Diabetes Care. 2009;32:1626–1631. doi: 10.2337/dc09-0586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ammala C, Larsson O, Berggren PO, Bokvist K, Juntti-Berggren L, Kindmark H, et al. Inositol trisphosphate-dependent periodic activation of a Ca(2+)-activated K+ conductance in glucose-stimulated pancreatic beta-cells. Nature. 1991;353:849–852. doi: 10.1038/353849a0. [DOI] [PubMed] [Google Scholar]
- 9.Boom A, Lybaert P, Pollet JF, Jacobs P, Jijakli H, Golstein PE, et al. Expression and localization of cystic fibrosis transmembrane conductance regulator in the rat endocrine pancreas. Endocrine. 2007;32:197–205. doi: 10.1007/s12020-007-9026-x. [DOI] [PubMed] [Google Scholar]
- 10.Guo JH, Chen H, Ruan YC, Zhang XL, Zhang XH, Fok KL, et al. Glucose-induced electrical activities and insulin secretion in pancreatic islet beta-cells are modulated by CFTR. Nat Commun. 2014;5:4420. doi: 10.1038/ncomms5420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scott-Jupp R, Lama M, Tanner MS. Prevalence of liver disease in cystic fibrosis. Arch Dis Child. 1991;66:698–701. doi: 10.1136/adc.66.6.698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lamireau T, Monnereau S, Martin S, Marcotte JE, Winnock M, Alvarez F. Epidemiology of liver disease in cystic fibrosis: a longitudinal study. J Hepatol. 2004;41:920–925. doi: 10.1016/j.jhep.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 13.Bhardwaj S, Canlas K, Kahi C, Temkit M, Molleston J, Ober M, et al. Hepatobiliary abnormalities and disease in cystic fibrosis: epidemiology and outcomes through adulthood. J Clin Gastroenterol. 2009;43:858–864. doi: 10.1097/MCG.0b013e31819e8bbd. [DOI] [PubMed] [Google Scholar]
- 14.Costa PC, Barreto CC, Pereira L, Lobo ML, Costa MA, Lopes AI. Cystic fibrosis-related liver disease: a single-center experience. Pediatr Rep. 2011;3:e21. doi: 10.4081/pr.2011.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kobelska-Dubiel N, Klincewicz B, Cichy W. Liver disease in cystic fibrosis. Prz Gastroenterol. 2014;9:136–141. doi: 10.5114/pg.2014.43574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cohn JA, Strong TV, Picciotto MR, Nairn AC, Collins FS, Fitz JG. Localization of the cystic fibrosis transmembrane conductance regulator in human bile duct epithelial cells. Gastroenterology. 1993;105:1857–1864. doi: 10.1016/0016-5085(93)91085-v. [DOI] [PubMed] [Google Scholar]
- 17.Boyer JL. Bile formation and secretion. Compr Physiol. 2013;3:1035–1078. doi: 10.1002/cphy.c120027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Colombo C, Russo MC, Zazzeron L, Romano G. Liver disease in cystic fibrosis. J Pediatr Gastroenterol Nutr. 2006;43(Suppl 1):S49–S55. doi: 10.1097/01.mpg.0000226390.02355.52. [DOI] [PubMed] [Google Scholar]
- 19.Li L, Somerset S. Digestive system dysfunction in cystic fibrosis: challenges for nutrition therapy. Dig Liver Dis. 2014;46:865–874. doi: 10.1016/j.dld.2014.06.011. [DOI] [PubMed] [Google Scholar]
- 20.Kerem E, Viviani L, Zolin A, MacNeill S, Hatziagorou E, Ellemunter H, et al. Factors associated with FEV1 decline in cystic fibrosis: analysis of the ECFS patient registry. Eur Respir J. 2014;43:125–133. doi: 10.1183/09031936.00166412. [DOI] [PubMed] [Google Scholar]
- 21.Nazareth D, Walshaw M. A review of renal disease in cystic fibrosis. J Cyst Fibros. 2013;12:309–317. doi: 10.1016/j.jcf.2013.03.005. [DOI] [PubMed] [Google Scholar]
- 22.Bell SC, De Boeck K, Amaral MD. New pharmacological approaches for cystic fibrosis: promises, progress, pitfalls. Pharmacol Ther. 2015;145:19–34. doi: 10.1016/j.pharmthera.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 23.CFTR2. The Clinical and Functional TRanslation of CFTR (CFTR2). http://www.cftr2.org. Accessed 29 Nov 2015.
- 24.Fanen P, Wohlhuter-Haddad A, Hinzpeter A. Genetics of cystic fibrosis: CFTR mutation classifications toward genotype-based CF therapies. Int J Biochem Cell Biol. 2014;52:94–102. doi: 10.1016/j.biocel.2014.02.023. [DOI] [PubMed] [Google Scholar]
- 25.Wang Y, Wrennall JA, Cai Z, Li H, Sheppard DN. Understanding how cystic fibrosis mutations disrupt CFTR function: from single molecules to animal models. Int J Biochem Cell Biol. 2014;52:47–57. doi: 10.1016/j.biocel.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 26.Sermet-Gaudelus I, Renouil M, Fajac A, Bidou L, Parbaille B, Pierrot S, et al. In vitro prediction of stop-codon suppression by intravenous gentamicin in patients with cystic fibrosis: a pilot study. BMC Med. 2007;5:5. doi: 10.1186/1741-7015-5-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447:87–91. doi: 10.1038/nature05756. [DOI] [PubMed] [Google Scholar]
- 28.Kerem E, Hirawat S, Armoni S, Yaakov Y, Shoseyov D, Cohen M, et al. Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: a prospective phase II trial. Lancet. 2008;372:719–727. doi: 10.1016/S0140-6736(08)61168-X. [DOI] [PubMed] [Google Scholar]
- 29.Sermet-Gaudelus I, Boeck KD, Casimir GJ, Vermeulen F, Leal T, Mogenet A, et al. Ataluren (PTC124) induces cystic fibrosis transmembrane conductance regulator protein expression and activity in children with nonsense mutation cystic fibrosis. Am J Respir Crit Care Med. 2010;182:1262–1272. doi: 10.1164/rccm.201001-0137OC. [DOI] [PubMed] [Google Scholar]
- 30.Kerem E, Konstan MW, De Boeck K, Accurso FJ, Sermet-Gaudelus I, Wilschanski M, et al. Ataluren for the treatment of nonsense-mutation cystic fibrosis: a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Respir Med. 2014;2:539–547. doi: 10.1016/S2213-2600(14)70100-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Keeling K, Wang D, Conard S, Bedwell D. Suppression of premature termination codons as a therapeutic approach. Crit Rev Biochem Mol. 2012;47:444–463. doi: 10.3109/10409238.2012.694846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kalydeco® (ivacaftor) [prescribing information]. Boston: Vertex; 2015. http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/207925s000lbl.pdf. Accessed 29 Nov 2015.
- 33.Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Drevinek P, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365:1663–1672. doi: 10.1056/NEJMoa1105185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davies JC, Wainwright CE, Canny GJ, Chilvers MA, Howenstine MS, Munck A, et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am J Respir Crit Care Med. 2013;187:1219–1225. doi: 10.1164/rccm.201301-0153OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McKone EF, Borowitz D, Drevinek P, Griese M, Konstan MW, Wainwright C, et al. Long-term safety and efficacy of ivacaftor in patients with cystic fibrosis who have the Gly551Asp-CFTR mutation: a phase 3, open-label extension study (PERSIST) Lancet Respir Med. 2014;2:902–910. doi: 10.1016/S2213-2600(14)70218-8. [DOI] [PubMed] [Google Scholar]
- 36.Pettit RS, Fellner C. CFTR modulators for the treatment of cystic fibrosis. P T. 2014;39:500–511. [PMC free article] [PubMed] [Google Scholar]
- 37.Flume PA, Liou TG, Borowitz DS, Li H, Yen K, Ordonez CL, et al. Ivacaftor in subjects with cystic fibrosis who are homozygous for the F508del-CFTR mutation. Chest. 2012;142:718–724. doi: 10.1378/chest.11-2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rowe SM, Heltshe SL, Gonska T, Donaldson SH, Borowitz D, Gelfond D, et al. Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis. Am J Respir Crit Care Med. 2014;190:175–184. doi: 10.1164/rccm.201404-0703OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De Boeck K, Munck A, Walker S, Faro A, Hiatt P, Gilmartin G, et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J Cyst Fibros. 2014;13:674–680. doi: 10.1016/j.jcf.2014.09.005. [DOI] [PubMed] [Google Scholar]
- 40.Sly PD, Brennan S, Gangell C, de Klerk N, Murray C, Mott L, et al. Lung disease at diagnosis in infants with cystic fibrosis detected by newborn screening. Am J Respir Crit Care Med. 2009;180:146–152. doi: 10.1164/rccm.200901-0069OC. [DOI] [PubMed] [Google Scholar]
- 41.Vertex Pharmaceuticals Incorporated. Study of ivacaftor in cystic fibrosis subjects 2 through 5 years of age with a CFTR gating mutation [ClinicalTrials.gov identifier NCT01705145]. US National Institutes of Health, ClinicalTrials.gov. 2012. https://clinicaltrials.gov. . Accessed 29 Nov 2015.
- 42.Whiting P, Al M, Burgers L, Westwood M, Ryder S, Hoogendoorn M, et al. Ivacaftor for the treatment of patients with cystic fibrosis and the G551D mutation: a systematic review and cost-effectiveness analysis. Health Technol Assess. 2014;18:1–106. doi: 10.3310/hta18180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Balfour-Lynn IM. Personalised medicine in cystic fibrosis is unaffordable. Paediatr Respir Rev. 2014;15:2–5. doi: 10.1016/j.prrv.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 44.Senior M. Foundation receives $3.3-billion windfall for Kalydeco. Nat. Biotech. 2014;33:8–9. doi: 10.1038/nbt0115-8. [DOI] [PubMed] [Google Scholar]
- 45.Van Goor F, Hadida S, Grootenhuis PD, Burton B, Stack JH, Straley KS, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci USA. 2011;108:18843–18848. doi: 10.1073/pnas.1105787108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clancy JP, Rowe SM, Accurso FJ, Aitken ML, Amin RS, Ashlock MA, et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax. 2012;67:12–18. doi: 10.1136/thoraxjnl-2011-200393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boyle MP, Bell SC, Konstan MW, McColley SA, Rowe SM, Rietschel E, et al. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. Lancet Respir Med. 2014;2:527–538. doi: 10.1016/S2213-2600(14)70132-8. [DOI] [PubMed] [Google Scholar]
- 48.Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M, et al. Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med. 2015;373:220–231. doi: 10.1056/NEJMoa1409547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Orkambi™ (lumacaftor/ivacaftor) [prescribing information]. Boston: Vertex; 2015. http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/206038Orig1s000lbl.pdf. Accessed 29 Nov 2015.
- 50.Donaldson S, Pilewski J, Griese M, Dong Q, Lee PS. VX-661, an investigational CFTR corrector, in combination with ivacaftor, a CFTR potentiator, in patients with CF and homozygous for the F508Del-CFTR mutation: interim analysis. J Cyst Fibros. 2013;12:S14. doi: 10.1016/S1569-1993(13)60042-9. [DOI] [Google Scholar]
- 51.Norman P. Novel picolinamide-based cystic fibrosis transmembrane regulator modulators: evaluation of WO2013038373, WO2013038376, WO2013038381, WO2013038386 and WO2013038390. Expert Opin Ther Pat. 2014;24:829–837. doi: 10.1517/13543776.2014.876412. [DOI] [PubMed] [Google Scholar]
- 52.Silverman E. Orkambi’s slick unveiling puts insurers in a bind. Manag Care. 2015;24:16–17. [PubMed] [Google Scholar]
- 53.Godman B, Malmström RE, Diogene E, Gray A, Jayathissa S, Timoney A, et al. Are new models needed to optimize the utilization of new medicines to sustain healthcare systems? Expert Rev Clin Pharmacol. 2015;8:77–94. doi: 10.1586/17512433.2015.990380. [DOI] [PubMed] [Google Scholar]
- 54.Sinn PL, Burnight ER, Hickey MA, Blissard GW, McCray PB., Jr Persistent gene expression in mouse nasal epithelia following feline immunodeficiency virus-based vector gene transfer. J Virol. 2005;79:12818–12827. doi: 10.1128/JVI.79.20.12818-12827.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118:3132–3142. doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang L, Button B, Gabriel SE, Burkett S, Yan Y, Skiadopoulos MH, et al. CFTR delivery to 25 % of surface epithelial cells restores normal rates of mucus transport to human cystic fibrosis airway epithelium. PLoS Biol. 2009;7:e1000155. doi: 10.1371/journal.pbio.1000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gregory SM, Nazir SA, Metcalf JP. Implications of the innate immune response to adenovirus and adenoviral vectors. Future Virol. 2011;6:357–374. doi: 10.2217/fvl.11.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Granio O, Ashbourne Excoffon KJ, Henning P, Melin P, Norez C, Gonzalez G, et al. Adenovirus 5-fiber 35 chimeric vector mediates efficient apical correction of the cystic fibrosis transmembrane conductance regulator defect in cystic fibrosis primary airway epithelia. Hum Gene Ther. 2010;21:251–269. doi: 10.1089/hum.2009.056. [DOI] [PubMed] [Google Scholar]
- 59.Griesenbach U, Inoue M, Meng C, Farley R, Chan M, Newman NK, et al. Assessment of F/HN-pseudotyped lentivirus as a clinically relevant vector for lung gene therapy. Am J Respir Crit Care Med. 2012;186:846–856. doi: 10.1164/rccm.201206-1056OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liang W, Kwok PC, Chow MY, Tang P, Mason AJ, Chan HK, et al. Formulation of pH responsive peptides as inhalable dry powders for pulmonary delivery of nucleic acids. Eur J Pharm Biopharm. 2014;86:64–73. doi: 10.1016/j.ejpb.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Suk JS, Lai SK, Boylan NJ, Dawson MR, Boyle MP, Hanes J. Rapid transport of muco-inert nanoparticles in cystic fibrosis sputum treated with N-acetyl cysteine. Nanomedicine (London). 2011;6:365–375. doi: 10.2217/nnm.10.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Alton EW, Boyd AC, Cheng SH, Davies JC, Davies LA, Dayan A, et al. Toxicology study assessing efficacy and safety of repeated administration of lipid/DNA complexes to mouse lung. Gene Ther. 2014;21:89–95. doi: 10.1038/gt.2013.61. [DOI] [PubMed] [Google Scholar]
- 63.Alton EW, Armstrong DK, Ashby D, Bayfield KJ, Bilton D, Bloomfield EV, et al. UK Cystic Fibrosis Gene Therapy Consortium. Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet. Respir Med. 2015;3(9):684–691. doi: 10.1016/S2213-2600(15)00245-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Moehle EA, Rock JM, Lee YL, Jouvenot Y, DeKelver RC, Gregory PD, et al. Targeted gene addition into a specified location in the human genome using designed zinc finger nucleases. Proc Natl Acad Sci USA. 2007;104:3055–3060. doi: 10.1073/pnas.0611478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Doyon Y, Vo TD, Mendel MC, Greenberg SG, Wang J, Xia DF, et al. Enhancing zinc-finger-nuclease activity with improved obligate heterodimeric architectures. Nat Methods. 2011;8:74–79. doi: 10.1038/nmeth.1539. [DOI] [PubMed] [Google Scholar]
- 66.Lee CM, Flynn R, Hollywood JA, Scallan MF, Harrison PT. Correction of the deltaF508 mutation in the cystic fibrosis transmembrane conductance regulator gene by zinc-finger nuclease homology-directed repair. Biores Open Access. 2012;1:99–108. doi: 10.1089/biores.2012.0218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Scudieri P, Caci E, Bruno S, Ferrera L, Schiavon M, Sondo E, et al. Association of TMEM16A chloride channel overexpression with airway goblet cell metaplasia. J Physiol. 2012;590:6141–6155. doi: 10.1113/jphysiol.2012.240838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tarran R, Loewen ME, Paradiso AM, Olsen JC, Gray MA, Argent BE, et al. Regulation of murine airway surface liquid volume by CFTR and Ca2+-activated Cl- conductances. J Gen Physiol. 2002;120:407–418. doi: 10.1085/jgp.20028599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sondo E, Caci E, Galietta LJV. The TMEM16A chloride channel as an alternative therapeutic target in cystic fibrosis. Int J Biochem Cell Biol. 2014;52:73–76. doi: 10.1016/j.biocel.2014.03.022. [DOI] [PubMed] [Google Scholar]
- 70.Althaus M, Clauss WG, Fronius M. Amiloride-sensitive sodium channels and pulmonary edema. Pulm Med. 2011;2011:830320. doi: 10.1155/2011/830320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ratjen F, Durham T, Navratil T, Schaberg A, Accurso FJ, Wainwright C, et al. Long term effects of denufosol tetrasodium in patients with cystic fibrosis. J Cyst Fibros. 2012;11:539–549. doi: 10.1016/j.jcf.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 72.Astrand AB, Hemmerling M, Root J, Wingren C, Pesic J, Johansson E, et al. Linking increased airway hydration, ciliary beating and mucociliary clearance through ENaC inhibition. Am J Physiol Lung Cell Mol Physiol. 2015;308(1):L22–L32. doi: 10.1152/ajplung.00163.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Almaça J, Faria D, Sousa M, Uliyakina I, Conrad C, Sirianant L, et al. High-content siRNA screen reveals global ENaC regulators and potential cystic fibrosis therapy targets. Cell. 2013;154:1390–1400. doi: 10.1016/j.cell.2013.08.045. [DOI] [PubMed] [Google Scholar]
- 74.Chanoux RA, Rubenstein RC. Molecular chaperones as targets to circumvent the CFTR defect in cystic fibrosis. Front Pharmacol. 2012;3:137. doi: 10.3389/fphar.2012.00137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Accurso FJ, Rowe SM, Clancy JP, Boyle MP, Dunitz JM, Durie PR, et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N Engl J Med. 2010;363:1991–2003. doi: 10.1056/NEJMoa0909825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dwyer T, Elkins M, Dentice R, Forbes S, McArthur M, Cooper P, et al. Saline at Lower Tonicity in Cystic Fibrosis (SALTI-CF) trial: a randomised, controlled trial comparing 0.9 % v 3 % v 6 % nebulised saline [abstract no. WS9.5] J Cyst Fibros. 2013;12:S19. doi: 10.1016/S1569-1993(13)60056-9. [DOI] [Google Scholar]
- 77.Wark P, McDonald VM. Nebulised hypertonic saline for cystic fibrosis. Cochrane Database Syst Rev. 2009;(2):CD001506. [DOI] [PubMed]
- 78.Bilton D, Bellon G, Charlton B, Cooper P, De Boeck K, Flume PA, et al. Pooled analysis of two large randomised phase III inhaled mannitol studies in cystic fibrosis. J Cyst Fibros. 2012;12:367–376. doi: 10.1016/j.jcf.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 79.Barraud N, Buson A, Jarolimek W, Rice SA. Mannitol enhances antibiotic sensitivity of persister bacteria in Pseudomonas aeruginosa biofilms. PLoS One. 2013;8:e84220. doi: 10.1371/journal.pone.0084220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Denman CC, Brown AR. Mannitol promotes adherence of an outbreak strain of Burkholderia multivorans via an exopolysaccharide-independent mechanism that is associated with upregulation of newly identified fimbrial and afimbrial adhesins. Microbiology. 2013;159:771–781. doi: 10.1099/mic.0.064832-0. [DOI] [PubMed] [Google Scholar]
- 81.Shak S. Aerosolized recombinant human DNase I for the treatment of cystic fibrosis. Chest. 1995;107:65S–70S. doi: 10.1378/chest.107.2_Supplement.65S. [DOI] [PubMed] [Google Scholar]
- 82.Jones AP, Wallis C. Dornase alfa for cystic fibrosis. Cochrane Database Syst Rev. 2010;(3):CD001127. [DOI] [PubMed]
- 83.Konstan MW, Ratjen F. Effect of dornase alfa on inflammation and lung function: potential role in the early treatment of cystic fibrosis. J Cyst Fibros. 2012;11:78–83. doi: 10.1016/j.jcf.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Auerbach H, Kirkpatrick J, Williams M, Colten H. Alternate-day prednisone reduces morbidity and improves pulmonary function in cystic fibrosis. Lancet. 1985;326:686–688. doi: 10.1016/S0140-6736(85)92929-0. [DOI] [PubMed] [Google Scholar]
- 85.Eigen H, Rosenstein BJ, FitzSimmons S, Schidlow DV. A multicenter study of alternate-day prednisone therapy in patients with cystic fibrosis. Cystic Fibrosis Foundation Prednisone Trial Group. J Pediatr. 1995;126:515–23. [DOI] [PubMed]
- 86.Lai HC, FitzSimmons SC, Allen DB, Kosorok MR, Rosenstein BJ, Campbell PW, et al. Risk of persistent growth impairment after alternate-day prednisone treatment in children with cystic fibrosis. N Engl J Med. 2000;342:851–859. doi: 10.1056/NEJM200003233421204. [DOI] [PubMed] [Google Scholar]
- 87.Balfour-Lynn IM, Welch K. Inhaled corticosteroids for cystic fibrosis. Cochrane Database Syst Rev. 2014;10:CD001915. [DOI] [PubMed]
- 88.Schindler T, Michel S, Wilson AWM. Nutrition management of cystic fibrosis in the 21st century. Nutr Clin Pract. 2015;30:488–500. doi: 10.1177/0884533615591604. [DOI] [PubMed] [Google Scholar]
- 89.Kuhn RJ, Gelrud A, Munck A, Caras S. CREON (pancrelipase delayed-release capsules) for the treatment of exocrine pancreatic insufficiency. Adv Ther. 2010;27:895–916. doi: 10.1007/s12325-010-0085-7. [DOI] [PubMed] [Google Scholar]
- 90.Kuhn RJ, Eyting S, Henniges F, Potthoff A. In vitro comparison of physical parameters, enzyme activity, acid resistance, and pH dissolution characteristics of enteric-coated pancreatic enzyme preparations: implications for clinical variability and pharmacy substitution. J Pediatr Pharmacol Ther. 2007;12:115–128. doi: 10.5863/1551-6776-12.2.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Borowitz D, Stevens C, Brettman LR, Campion M, Chatfield B, Cipolli M. International phase III trial of liprotamase efficacy and safety in pancreatic-insufficient cystic fibrosis patients. J Cyst Fibros. 2011;10:443–452. doi: 10.1016/j.jcf.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 92.Anthera Pharmaceuticals. SOLUTION: study of oral liprotamase unit-matched therapy of non-porcine origin in patients with cystic fibrosis [ClinicalTrials.gov identifier NCT02279498]. US National Institutes of Health, ClinicalTrials.gov; 2015. https://clinicaltrials.gov. Accessed 29 Nov 2015.
- 93.Nordmark Arzneimittel GmbH & Co. KG. Efficacy and tolerability of NM-BL in patients with exocrine pancreatic insufficiency due to cystic fibrosis [ClinicalTrials.gov identifier NCT01710644]. US National Institutes of Health. 2014. https://clinicaltrials.gov. Accessed 29 Nov 2015.
- 94.Cheng K, Ashby D, Smyth RL. Ursodeoxycholic acid for cystic fibrosis-related liver disease. Cochrane Database Syst Rev. 2012;10:CD000222. [DOI] [PubMed]
- 95.Siano M, De Gregorio F, Boggia B, Sepe A, Ferri P, Buonpensiero P, et al. Ursodeoxycholic acid treatment in patients with cystic fibrosis at risk for liver disease. Dig Liver Dis. 2010;42:428–431. doi: 10.1016/j.dld.2009.07.022. [DOI] [PubMed] [Google Scholar]
- 96.LiPuma JJ. The changing microbial epidemiology in cystic fibrosis. Clin Microbiol Rev. 2010;23:299–323. doi: 10.1128/CMR.00068-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.de Vrankrijker AM, Wolfs TF, van der Ent CK. Challenging and emerging pathogens in cystic fibrosis. Paediatr Respir Rev. 2010;11:246–254. doi: 10.1016/j.prrv.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 98.Zolin A, McKone EF, van Rens J, et al. 2010 ECFS patient registry annual data report. https://www.ecfs.eu/files/webfm/webfiles/File/ecfs_registry/ECFSPR_Report10_v12014_final_020617.pdf. Accessed 29 Nov 2015.
- 99.Lee TWR, Brownlee KG, Conway SP, Denton M, Littlewood JM. Evaluation of a new definition for chronic Pseudomonas aeruginosa infection in cystic fibrosis patients. J Cyst Fibros. 2003;2:29–34. doi: 10.1016/S1569-1993(02)00141-8. [DOI] [PubMed] [Google Scholar]
- 100.Psoter KJ, Rosenfeld M, De Roos AJ, Mayer JD, Wakefield J. Differential geographical risk of initial Pseudomonas aeruginosa acquisition in young US children with cystic fibrosis. Am J Epidemiol. 2014;179:1503–1513. doi: 10.1093/aje/kwu077. [DOI] [PubMed] [Google Scholar]
- 101.Zemanick ET, Emerson J, Thompson V, McNamara S, Morgan W, Gibson RL, et al. Clinical outcomes after initial Pseudomonas acquisition in cystic fibrosis. Pediatr Pulmonol. 2015;50:42–48. doi: 10.1002/ppul.23036. [DOI] [PubMed] [Google Scholar]
- 102.Máiz L, Giron RM, Olveira C, Quintana E, Lamas A, Pastor D, et al. Inhaled antibiotics for the treatment of chronic bronchopulmonary Pseudomonas aeruginosa infection in cystic fibrosis: systematic review of randomised controlled trials. Expert Opin Pharmacother. 2013;14:1135–1149. doi: 10.1517/14656566.2013.790366. [DOI] [PubMed] [Google Scholar]
- 103.Langton Hewer SC, Smyth AR. Antibiotic strategies for eradicating Pseudomonas aeruginosa in people with cystic fibrosis. Cochrane Database Syst Rev. 2014;11:CD004197. [DOI] [PubMed]
- 104.Quon BS, Goss CH, Ramsey BW. Inhaled antibiotics for lower airway infections. Ann Am Thorac Soc. 2014;11:425–434. doi: 10.1513/AnnalsATS.201311-395FR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mayer-Hamblett N, Kloster M, Rosenfeld M, Gibson RL, Retsch-Bogart GZ, Emerson J, et al. Impact of sustained eradication of new Pseudomonas aeruginosa infection on long-term outcomes in cystic fibrosis. Clin Infect Dis. 2015;61:707–715. doi: 10.1093/cid/civ377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Regan KH, Bhatt J. Eradication therapy for Burkholderia cepacia complex in people with cystic fibrosis. Cochrane Database Syst Rev. 2014;10:CD009876. [DOI] [PubMed]
- 107.Lo DK, Hurley MN, Muhlebach MS, Smyth AR. Interventions for the eradication of methicillin-resistant Staphylococcus aureus (MRSA) in people with cystic fibrosis. Cochrane Database Syst Rev. 2013;2:CD009650. [DOI] [PubMed]
- 108.Amin R, Waters V. Antibiotic treatment for Stenotrophomonas maltophilia in people with cystic fibrosis. Cochrane Database Syst Rev. 2012;5:CD009249. [DOI] [PubMed]
- 109.Waters V, Ratjen F. Antibiotic treatment for nontuberculous mycobacteria lung infection in people with cystic fibrosis. Cochrane Database Syst Rev. 2012;12:CD010004. [DOI] [PubMed]
- 110.Remmington T, Jahnke N, Harkensee C. Oral anti-pseudomonal antibiotics for cystic fibrosis. Cochrane Database Syst Rev. 2013;10:CD005405. [DOI] [PubMed]
- 111.Ryan G, Jahnke N, Remmington T. Inhaled antibiotics for pulmonary exacerbations in cystic fibrosis. Cochrane Database Syst Rev. 2012;12:CD008319. [DOI] [PubMed]
- 112.Ryan G, Singh M, Dwan K. Inhaled antibiotics for long-term therapy in cystic fibrosis. Cochrane Database Syst Rev. 2011;(3):CD001021. [DOI] [PubMed]
- 113.Das RR, Kabra SK, Singh M. Treatment of Pseudomonas and Staphylococcus bronchopulmonary infection in patients with cystic fibrosis. ScientificWorldJournal. 2013;2013:645653. doi: 10.1155/2013/645653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Waters V, Ratjen F. Standard versus biofilm antimicrobial susceptibility testing to guide antibiotic therapy in cystic fibrosis. Cochrane Database Syst Rev. 2012;11:CD009528. [DOI] [PubMed]
- 115.Iglesias A, Artiles I, Cabanillas JJ, Alvarez-Sala R, Prados C. Eradication of Burkholderia cepacia using inhaled aztreonam lysine in two patients with bronchiectasis. Case Rep Pulmonol. 2014;192146. [DOI] [PMC free article] [PubMed]
- 116.Horsley A, Webb K, Bright-Thomas R, Govan J, Jones A. Can early Burkholderia cepacia complex infection in cystic fibrosis be eradicated with antibiotic therapy? Front Cell Infect Microbiol. 2011;1:18. doi: 10.3389/fcimb.2011.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Diso D, Anile M, Patella M, Pecoraro Y, Rendina EA, Carillo C, et al. Lung transplantation for cystic fibrosis: outcome of 101 single-center consecutive patients. Transplant Proc. 2013;45:346–348. doi: 10.1016/j.transproceed.2012.08.010. [DOI] [PubMed] [Google Scholar]
- 118.Meachery G, De Soyza A, Nicholson A, Parry G, Hasan A, Tocewicz K, et al. Outcomes of lung transplantation for cystic fibrosis in a large UK cohort. Thorax. 2008;63:725–731. doi: 10.1136/thx.2007.092056. [DOI] [PubMed] [Google Scholar]
- 119.De Soyza A, Meachery G, Hester KL, Nicholson A, Parry G, Tocewicz K, et al. Lung transplantation for patients with cystic fibrosis and Burkholderia cepacia complex infection: a single-center experience. J Heart Lung Transplant. 2010;29:1395–1404. doi: 10.1016/j.healun.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 120.PTC Therapeutics. Study of ataluren in nonsense mutation cystic fibrosis (ACT CF) [ClinicalTrials.gov identifier NCT02139306]. US National Institutes of Health, ClinicalTrials.gov. https://clinicaltrials.gov. Accessed 15 Dec 2015.
- 121.Davies J, Sheridan H, Bell N, Cunningham S, Davis SD, Elborn JS, et al. Assessment of clinical response to ivacaftor with lung clearance index in cystic fibrosis patients with a G551D-CFTR mutation and preserved spirometry: a randomised controlled trial. Lancet Respir Med. 2013;1:630–638. doi: 10.1016/S2213-2600(13)70182-6. [DOI] [PubMed] [Google Scholar]
- 122.Moss RB, Flume PA, Elborn JS, Cooke J, Rowe SM, McColley SA, et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis who have an Arg117His-CFTR mutation: a double-blind, randomised controlled trial. Lancet Respir Med.2015;3:524-33. [DOI] [PMC free article] [PubMed]
- 123.Barry PJ, Plant BJ, Nair A, Bicknell S, Simmonds NJ, Bell NJ, et al. Effects of ivacaftor in patients with cystic fibrosis who carry the G551D mutation and have severe lung disease. Chest. 2014;146:152-8. [DOI] [PubMed]